Introduction
The enhancement of re-endothelialization is a
critical therapeutic option to repair injured blood vessels.
Regeneration of the injured endothelium is linked to the
proliferation and migration of neighboring endothelial cells (ECs)
(1). Mature ECs are a group of
cells with low proliferative potential and a limited capacity to
substitute the damaged endothelium. Accumulating evidence has
indicated that endothelial progenitor cells (EPCs), which can home
to sites of tissue injury and differentiate into mature ECs and
participate in re-endothelialization after vascular injury, may be
an endogenous repair mechanism to maintain the integrity of the
endothelial monolayer by replacing denuded areas of the artery
(2–4). The discovery of new methods to
improve the EPC re-endothelialization process is currently the
subject of intensive investigation.
Growth arrest-specific gene 6 (Gas6), is a member of
the vitamin K-dependent family of proteins, which includes the
procoagulant factors II, VII, IX, and X, and the anticoagulant
factors, protein C and S, as well as protein Z (5). Even though Gas6 was discovered as a
homolog of protein S more than a decade ago, it plays no role in
the generation of fibrin (6).
Instead, Gas6 exerts several other functions that belong to the
repertoire of growth or survival factors. Firstly, the original
observation that Gas6 is upregulated in growth-arrested cells
suggests a role in the protection from certain cellular stresses,
such as apoptosis (7).
Subsequently, a number of studies have demonstrated the ability of
Gas6 to promote either cell survival (8) and/or proliferation (9). Additional growth factor-like
properties of Gas6 have been reported, including the stimulation of
cell migration and cell-cell adhesion (10).
The first hint that Gas6 may be important in the
vasculature came from the purification of Gas6 from the conditioned
medium of vascular smooth muscle cells (VSMCs) that potentiated the
growth response of VSMCs treated with angiotensin II (11). It was subsequently discovered that
the expression of Gas6 was increased in the injured rat carotid,
with a time course paralleling that of neointima formation
(12). Further experimental
evidence indicated that ECs in culture express and release Gas6,
and that it promotes cell survival, possibly through an autocrine
pathway (13–16). These findings highlight the
importance of Gas6 in vascular function. Gas6 was found to bind to
the extracellular regions of three distinct receptor tyrosine
kinases, namely Axl, Mertk and Tyro3. A recent study demonstrated
that Axl may be a potential angiogenic target (17). Considering that Gas6 has various
potential bioactivities, it is of great interest to identify those
that have a significant impact on EPCs.
Materials and methods
Isolation and culture of late EPCs
Ethical approval was granted by the Institutional
Review Board of Tongji Medical College, Hubei, China. Informed
consent was obtained from healthy donors prior to the collection of
umbilical cord blood. The mononuclear cells were isolated from
umbilical cord blood by Ficoll density gradient centrifugation with
Histopaque 1077 (Sigma, St. Louis, MO, USA). The isolated cells
were resuspended using an EGM-2 BulletKit system (catalog number
CC-3202; Lonza) consisting of endothelial basal medium, 5% fetal
bovine serum, human epidermal growth factor (hEGF), vascular
endothelial growth factor (VEGF), human fibroblast growth factor
(hFGF), hFGF-B, insulin-like growth factor (IGF)-1 and ascorbic
acid. Mononuclear cells were seeded on fibronectin-coated (Sigma)
dishes and maintained in a 5% CO2 incubator at 37°C.
Three days after planting, the non-adherent cells were removed and,
thereafter, the medium was changed every 2 days. Cobblestone-like
cell colonies were observed after 2 weeks.
Characterization of EPCs
Direct fluorescent staining was used to detect the
dual binding of 1, 1-dioctadecyl-3, 3, 3,
3-tetramethylindocarbocyanine-labeled acetylated low-density
lipoprotein (Dil-ac-LDL; molecular probe) and fluorescein
isothiocyanate (FITC)-conjugated Ulex europaeus agglutinin
lectin (UEA-1; Sigma). The cells were incubated with 2.4 μg/ml
Dil-ac-LDL for 4 h in a cell incubator. The cells were then washed
and fixed with 4% paraformaldehyde for 10 min and incubated with 10
μg/ml FITC-labeled UEA-1 for 1 h. Subsequently, the cells were
washed and incubated with Hoechst 33258. Double-positive cells were
observed under a laser confocal microscope (FV500; Olympus, Tokyo,
Japan). To assess the expression of surface antigen on the cells,
we performed fluorescence-activated cell sorter (FACS) analysis.
Five million EPCs per sample were stained for 30 min at 4°C with
fluorescein isothiocyanate-conjugated monoclonal mouse anti-human
CD34 (BD Pharmingen, San Diego, CA, USA) antibody,
phycoerythin-conjugated monoclonal mouse anti-human VEGFR2 (BD
Pharmingen) antibody and mouse anti-human CD133 antibody conjugated
to allophyocyanin (Miltenyi Biotec, Auburn, CA, USA). Data were
processed using FlowJo software (version 7.6).
Proliferation assay
The effects of Gas6 on EPC proliferation were
determined by MTT assay. A total of 5×103 cells/well
were seeded on 96-well culture plates and then deprived of serum
for 12 h to achieve cell cycle synchronization. The dose range (25,
50, 100 and 200 ng/ml) of Gas6 (R&D Systems, Minneapolis, MN,
USA) was the same as that used in previous studies (18,19). The control groups received a
dilution of water equivalent to Gas6 at the highest concentration.
The cells were cultured and treated for 24 and 48 h, respectively.
MTT solution (5 mg/ml) was added to each well. Following 4 h of
incubation at 37°C, the supernatant was discarded and the EPC
preparation was shaken with dimethyl sulfoxide (DMSO) for 10 min
before the optical density measurement (490 nm) was taken. The
results were calculated from 4 experiments with 5 replicates of
each. To investigate the effects of PI3K and ERK inhibitors on the
viability of EPCs, the cells were pre-treated in the absence or
presence of LY294002 (10, 20 and 30 μmol/l) (Cell Signaling
Technology, Inc., Danvers or Beverly, MA, USA) or PD98059 (5, 10
and 20 μmol/l) (Cell Signaling Technology, Inc.) for 30 min. After
being cultured for 48 h, the EPCs were supplemented with MTT (5
mg/ml) and incubated for another 4 h. The EPC preparation was then
shaken with DMSO for 10 min, before the OD measurement at 490 nm
was taken.
Migration assay
The migration ability of the EPCs was evaluated
using a Transwell migration assay (Costar, Cambridge, MA, USA) with
6.5-mm-diameter polycarbonate filters (8 μm pore size). Gas6 with
various concentrations plus endothelial basal medium-2 and 0.2% FBS
were placed in the lower wells. EPCs (4×104 cells/well)
were seeded onto the upper chamber supplemented with serum-free
endothelial growth medium. After 12 h of incubation in the cell
incubator, the upper chamber was removed and wiped clean with a
cotton swab; the lower side of the filter was washed with PBS and
fixed with 4% paraformaldehyde for 10 min. For quantification, the
cell nucleus was stained with crystal violet. Cell migration into
the lower chamber and attachment to the lower side of the filter
were manually counted in 16 fixed microscopic fields
(magnification, ×400) by independent investigators blinded to the
treatment regimen, randomly. Each test was performed in triplicate,
and assays were repeated 3 times with individual EPCs. When
investigating the effects of PI3K and ERK inhibitors on the
migration ability of the cells, the EPCs were cultured in the
absence or presence of 200 ng/ml Gas6 and the indicated
concentrations of 20 μmol/l LY294002 or 5 μmol/l PD98059 for 24 h.
The following steps were the same as those described above.
In vitro tube-formation assay
In vitro tube-formation assay was performed
using Matrigel (BD Biosciences, San Jose, CA, USA) according to the
manufacturer’s instructions. Passage-3 EPCs were incubated with an
additional 50, 100 and 200 ng/ml Gas6 and 10 ng/ml VEGF or 200
ng/ml Gas6 plus 10 ng/ml VEGF for 24 h. The Matrigel solution was
thawed at 4°C for 30 min to allow gelation, then 1×104
EPCs with the previous treatment were placed on top of the
Matrigel. After 12 h of incubation, the mean tube length was
calculated in 3 randomly selected fields from each well (x100)
using Image-Pro Plus software and was calculated against the value
of the control groups. The experiment was repeated 5 times.
Western blot analysis
The EPCs pre-treated with 200 ng/ml Gas6 were lysed
with RIPA buffer and electrophoresed on 10% SDS-PAGE gels at 100 V
for 2 h, and electroblotted onto a PVDF membrane at 275 mA for 12
h. The membrane was incubated with 5% fat-free milk PBS for 2 h at
room temperature. The membrane was then incubated with anti-AKT,
anti-ERK (1:500; Cell Signaling Technology, Inc.), anti-phospho-AKT
and anti-phospho-ERK (1:500; Cell Signaling Technology, Inc.)
rabbit monoclonal antibodies followed by the addition of a goat
anti-rabbit peroxidase-conjugated secondary antibody (Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA). The immunoreactive bands
were then visualized with a chemiluminescence reagent (Thermo
Fisher Scientific Inc., Rockford, IL, USA) and exposed to X-ray
film. The density of each band was quantified using ImageJ
software. All the assays were performed in triplicate with
individual EPCs.
Statistical analysis
Results were obtained from at least 3 independent
experiments and data are presented as the means ± SD. The Student’s
t-test was performed for statistical comparisons between 2 groups
and ANOVA was used for comparisons between >2 groups. A P-value
<0.05 was considered to indicate a statistically significant
difference.
Results
Characterization of EPCs
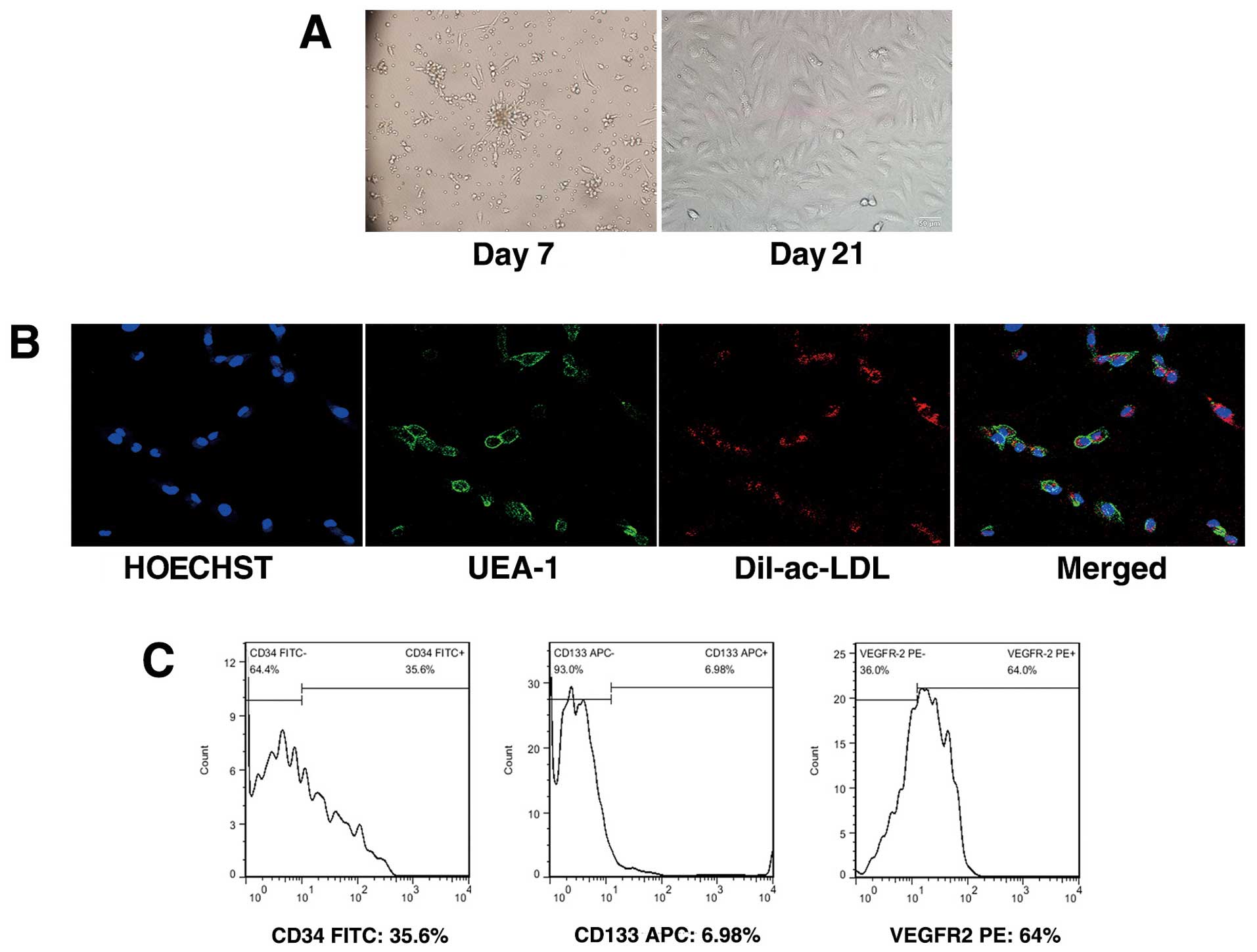
Late EPCs appeared after 1–2 weeks as small colonies
in cultures of mononuclear cells (MNCs) and developed a
cobblestone-like cell morphology over time (Fig. 1A). To confirm the EPC phenotype,
attached mononuclear cells were incubated with Dil-ac-LDL and
FITC-UEA-1. Cells demonstrating double-positive fluorescence were
identified as differentiated EPCs. Almost all adherent cells were
shown to endocytose Dil-ac-LDL and bind FITC-UEA-1 (Fig. 1B). Flow cytometric analysis for
positive staining with CD34, VEGFR2 and CD133 further confirmed the
EPC characteristics (Fig.
1C).
Gas6 stimulates the proliferation of
human EPCs
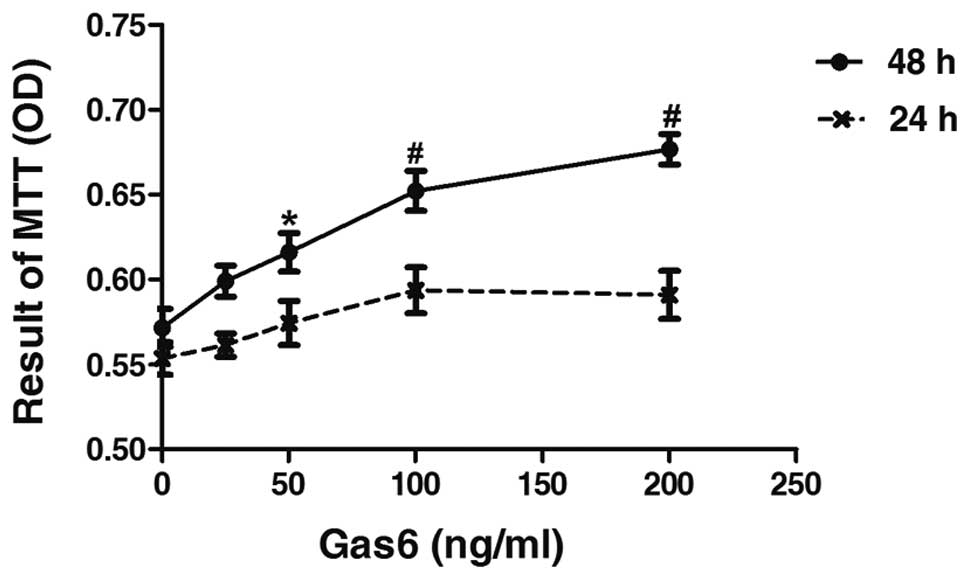
To determine the effects of Gas6 on the growth of
human EPCs, we performed a time- and dose-response experiment. Gas6
at concentrations of 50, 100 and 200 ng/ml induced an increase in
the proliferation of EPCs of 7.26% (P<0.05 vs. control), 14.10%
(P<0.001 vs. control) and 18.40% (P<0.001 vs. control),
respectively, after 48 h of culture (Fig. 2). However, treatment with
increased concentrations of Gas6 for 24 h did not affect the
proliferative ability of the EPCs compared with the control. The
effects on cell proliferation induced by Gas6 occurred in a dose-
and time-dependent manner.
Gas6 stimulates EPC migration
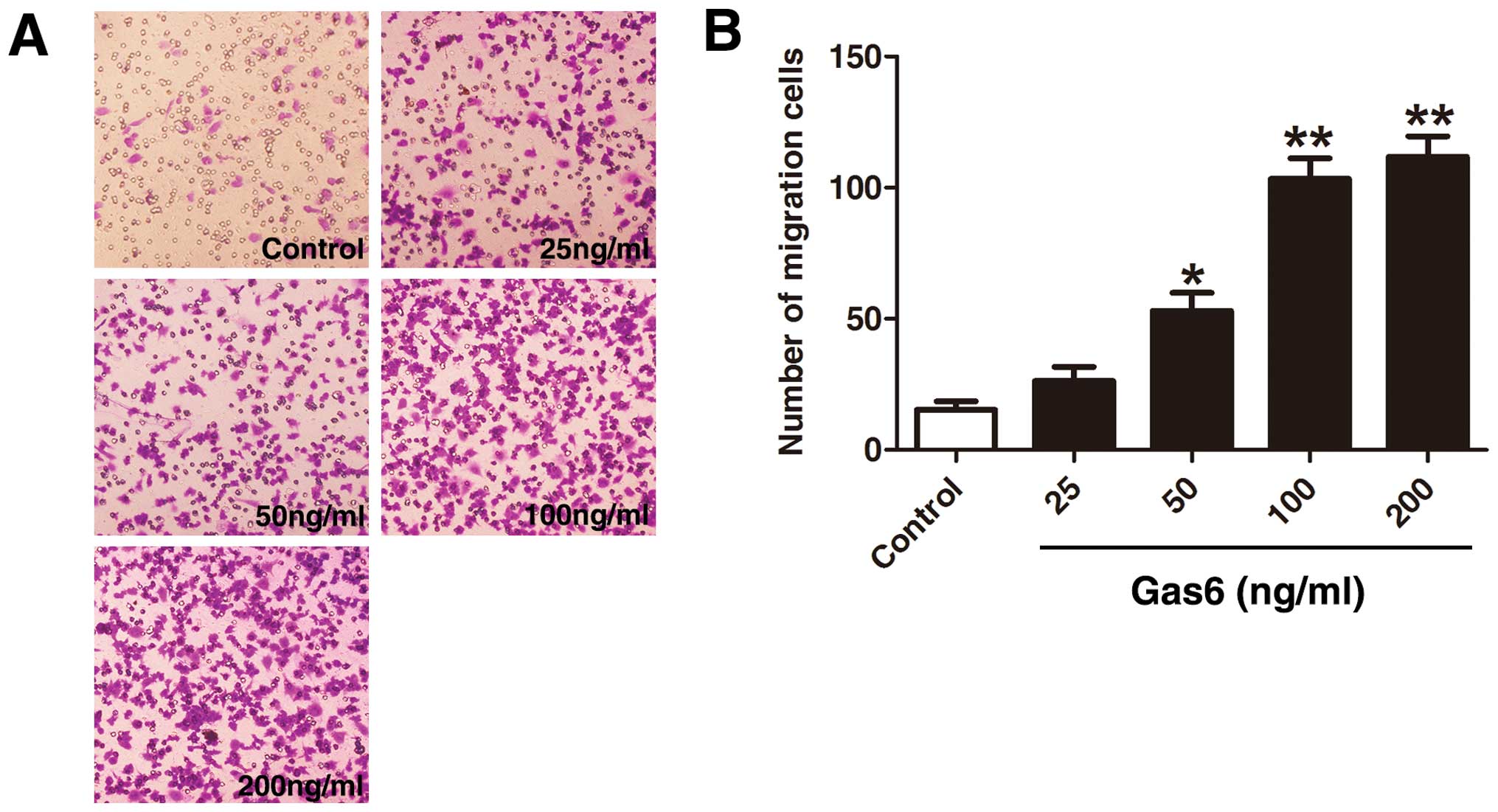
The effects of Gas6 on late EPC migration were
determined by a Transwell assay (Fig.
3). A large number of cells treated with Gas6 had migrated to
the lower side of the membrane in the Transwell chamber. Treatment
with Gas6 increased EPC migration in a dose-dependent manner
(control, 15.33±5.50; 25 ng/ml, 36.00±6.55; 50 ng/ml, 53.00±12.00;
100 ng/ml, 103.33±13.57; and 200 ng/ml, 111.66±13.57 cells,
respectively), with a significant effect at a dose of 50 ng/ml
(P<0.05 vs. control); the most significant effect was observed
with the highest Gas6 dose used (200 ng/ml; P<0.01 vs. control).
However, there was no statistically significant difference between
the 100 and 200 ng/ml groups (P=0.384).
Gas6 does not promote EPC differentiation
on Matrigel
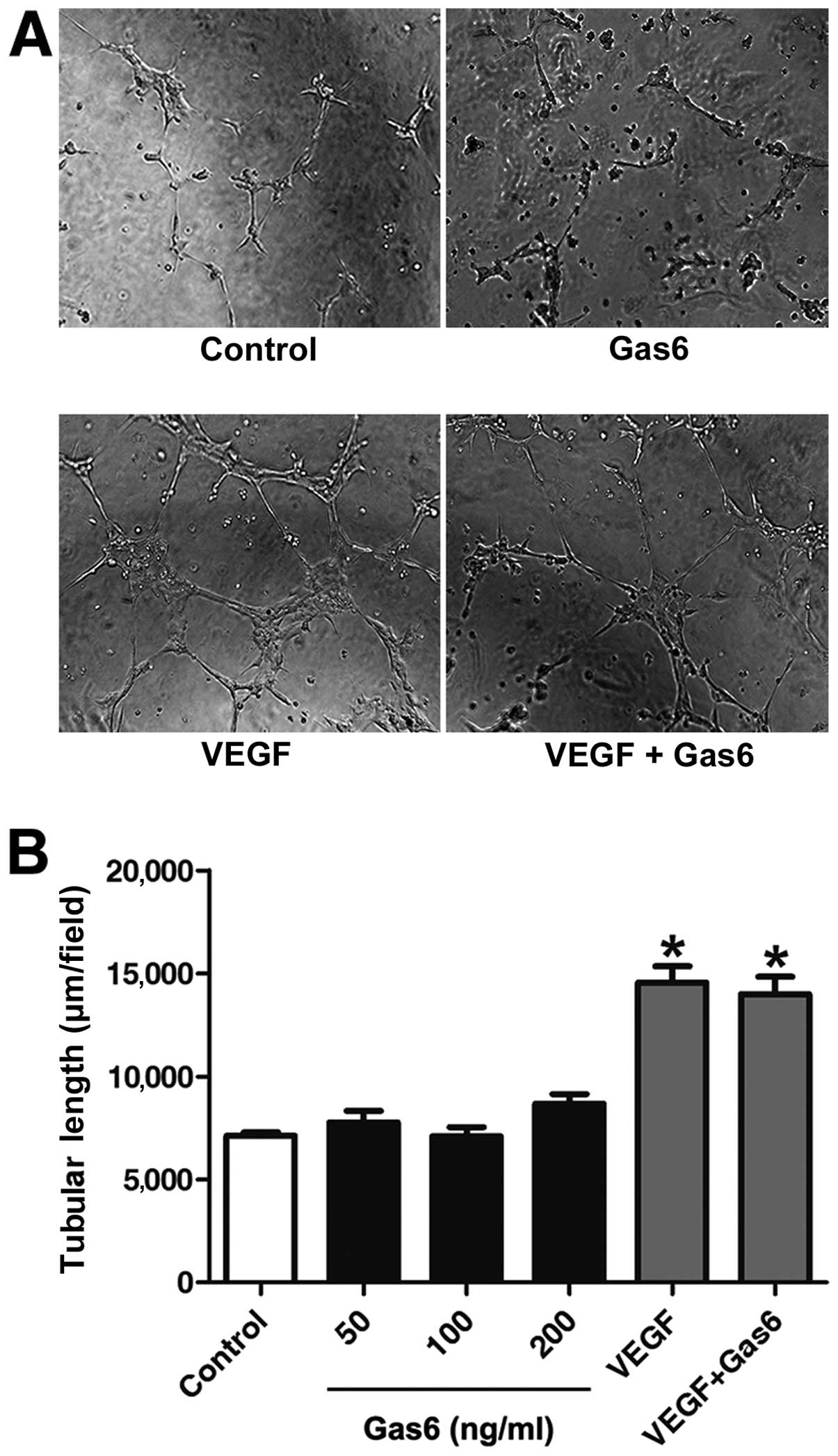
Gas6 promoted the proliferation and migration of
EPCs; we thus investigated whether this protein affects the
capillary-like structure formation of EPCs. Following stimulation
for 12 h, Gas6 at 50, 100 and 200 ng/ml did not ameliorate the
capacity of the cells to form capillary-like structures compared
with the control. VEGF-A is recognized as a key regulator of tube
formation in the process of angiogenesis (35). Further investigation indicated
that Gas6 did not alter VEGF-A-dependent tube formation (Fig. 4).
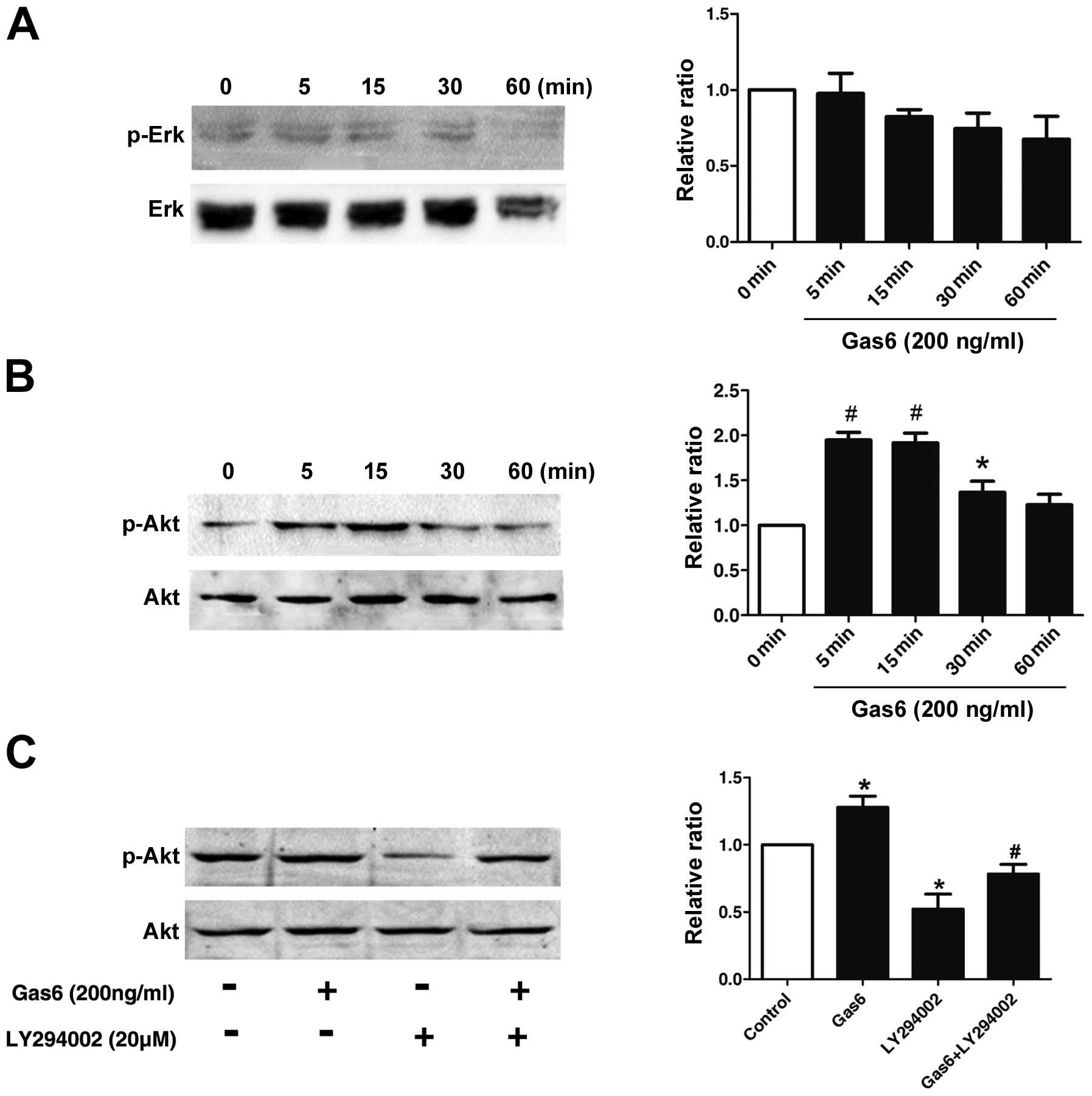
Effects of Gas6 on the phosphorylation of
AKT
To elucidate the molecular mechanisms underlying the
effects of Gas6 on EPCs, the phosphorylation status of the MAP
kinases and AKT, which are implicated in EPC proliferation and
function, was examined by western blot analysis (20,21). As shown in Fig. 5B, AKT phosphorylation in the EPCs
was markedly induced at 5 min and persisted for up to the 30-min
time point following treatment with Gas6. However, as demonstrated
in Fig. 5A, Gas6 did not cause
any significant change in the phopho-ERK/ERK level over 1-h period,
indicating that Gas6 had no effect on ERK activation. To confirm
that Gas6 promotes EPC viability and motility through the AKT
pathways, we treated the EPCs with LY294002 (PI3K inhibitor) alone
or in combination with Gas6. LY294002 inhibited the phosphorylation
of AKT; when used in combination with Gas6, Gas6 partly reversed
the inhibition of phospho-AKT induced by LY294002 (Fig. 5C). These results indicate that
Gas6 promotes EPC proliferation and migration, most likely through
the AKT signaling pathway.
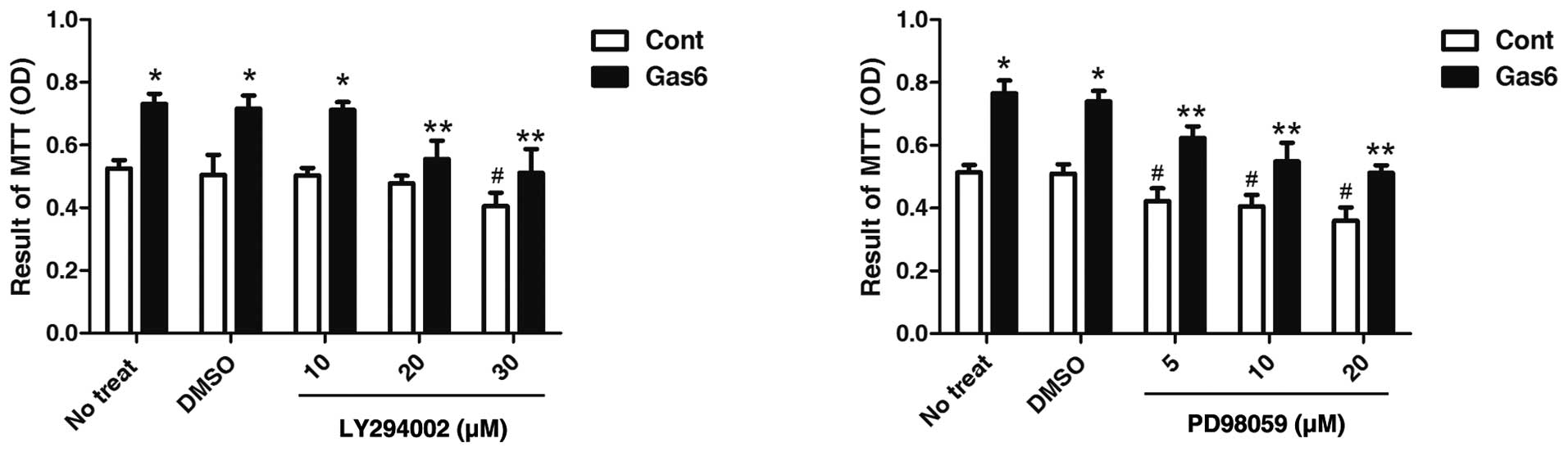
Effects of PI3K or ERK inhibitor on
Gas6-induced EPC proliferation
To further confirm the roles of AKT in the
Gas6-induced effects on EPCs, we determined the effects of AKT
inhibition on EPC proliferation. As shown in Fig. 6, the PI3K inhibitor, LY294002, at
dose 20 μmol/l did not alter the viability of the control EPCs, but
markedly attenuated the effects of Gas6 on EPC proliferation.
However, PD98059, an ERK inhibitor, at a concentration of 5 μmol/l,
exhibited a similar effect between the absence and presence of
Gas6-induced EPC proliferation vs. their own control. The decrease
in EPC proliferation in both the Gas6 group and the control group
treated with PD98059 was due to ERK inhibition. These results
suggest that AKT, but not ERK, is involved in the Gas6-induced EPC
proliferation.
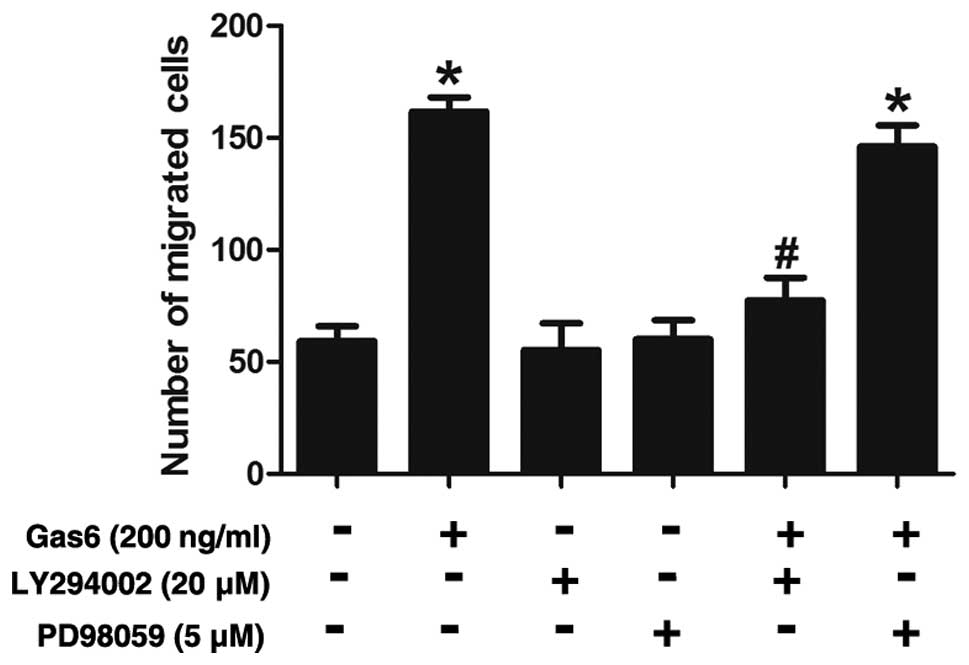
Effects of PI3K or ERK inhibitor on
Gas6-induced EPC migration
The possible roles of AKT in the Gas6-induced
augmentation of the EPC migration were also assessed. The results
revealed that the Gas6-induced increase in EPC migration was
substantially attenuated by LY294002 (a PI3K inhibitor) (Fig. 7). By contrast, PD98059 (an ERK
inhibitor) did not seem to have any significant effect on the
migratory activity of these cells, suggesting that ERK is not
involved in the stimulatory effects of Gas6 on EPC migration.
Discussion
To the best of our knowledge, the present study is
the first to describe a novel effect by which Gas6 is engaged in
the biological function of EPCs. The major findings of this study
were the following: a) Gas6 significantly stimulated EPC
proliferation and migration in vitro; b) Gas6 upregulated
phospho-AKT but not phospho-ERK expression; c) Gas6 did not promote
EPC differentiation on Matrigel; and d) the positive effects on
proliferation and migration were abrogated in the presence of the
PI3K-specific inhibitor, LY294002.
In recent years, it has become apparent that
circulating EPCs are crucial to maintaining cardiovascular
homeostasis and vascular integrity compared with mature ECs. It has
been found that EPCs contribute up to 25% of ECs in newly formed
vessels (22,23). Cell therapy using EPCs in ischemic
heart disease has been evaluated and proven to be safe and
effective in a number of pre-clinical studies (24,25). Accordingly, it is important to
investigate the endogenous or exogenous factors that affect these
cells. In the present study, we investigated whether the treatment
of EPCs with Gas6 can improve the the proliferation, migration and
tube formation of EPCs.
Gas6 is widely expressed and has been found in the
lungs, heart, kidneys, intestine, ECs, bone marrow, VSMCs and
monocytes and, at a very low level, in the liver. With the use of
an ELISA-based method, Gas6 was detected in human plasma. It
regulates homotypic and heterotypic adhesion (10), promotes proliferation (26,27), survival (15,28,29) and motility (30,31) and amplifies the activity of
extracellular stimuli (11,32). In addition to the general effects
of Gas6 mentioned above, we focused on its role in the
cardiovascular system. Our previous study demonstrated that, in
patients with acute coronary syndrome, Gas6 plasma levels at
admission reflect the presence of common cardiovascular risk
factors and can independently predict cardiovascular events
(33). These data indicate that
Gas6 may play an additional role in the vascular system. Therefore,
on an experimental ground, we investigated the possible roles of
Gas6 in the biological function of EPCs.
Firstly, we demonstrated the biological function of
EPCs treated with Gas6. We concluded that Gas6 promotes EPC
proliferation and migration. These results are in accordance with
those of a previous study by Holland et al, who found that
Gas6 silencing reduces EC haptotaxis towards vitronectin (34). Conversely, Gallicchio et al
found that Gas6 stimulates Axl and inhibits the ligand-dependent
activation of VEGF receptor 2 (VEGFR2) and the consequent
activation of an angiogenic program in vascular ECs (35). In a recent study, Ruan et
al found that Gas6 did not significantly alter the
VEGF-A-dependent activation of VEGFR-2 (17). It is very interesting that the two
groups produced different results. We did not investigate
VEGF-A-triggered signaling with Gas6; however, we found that Gas6
did not interfere with VEGF-A-induced tube formation in EPCs.
It has been previously demonstrated that Axl
knockdown in ECs impaires tube formation (36). Further research has demonstrated
that Axl regulates tube formation by the modulation of signaling
through the angiopoietin/Tie2 and Dickkopf pathways. In addition,
Axl is essential for the VEGF-A-dependent activation of PI3K/AKT
(17). Yet, we, as well as others
have observed that Gas6 does not promote tube formation (35). Axl is an angiogenic receptor
tyrosine kinase that can be engaged by multiple stimuli, including
Gas6, VEGF, lactate or hypoxia (37). Thus, we consider that the
angiogenic role of Axl may be independent of Gas6 administration.
More complex mechanisms may be involved in the bioactivities of
Gas6 in EPCs. Angiogenesis is a complex process, requiring the
coordinated action of a variety of growth factors in ECs. A recent
study demonstrated that, although Gas6 and Ang1 alone did not
promote tube formation in ECs, the combination of Gas6 and Ang1 did
(37). We thus consider the
possibility that Gas6 alone may not promote tube formation, but it
may do so when combined with other factors.
We extended our investigation with the aim of
determining the mechanisms associated with the effects of Gas6 on
EPCs. Previous studies have indicated that Gas6 plays an important
role in some cell types through its regulation of the AKT and ERK
signaling pathways following the initial effects on the cellular
survival and proliferation (9,38,39). Additionally, the AKT and ERK
signaling pathways are well-documented signaling pathways involved
in EPC biology (40,41). To explore whether these same
kinases play a role in the Gas6-induced proliferation and migration
of EPCs, their phosphorylated status following Gas6 treatment was
assessed. Our data demonstrated that Gas6 induced the transient
activation of AKT but not the ERK kinases.
To further demonstrate the role of AKT and ERK in
the Gas6-induced proliferation and migration of EPCs, we performed
additional experiments in which AKT and ERK activation was blocked
by the pharmacological agents, LY294002 and PD98059, respectively.
Our data demonstrated that at concentrations that did not affect
the growth of the control EPCs, LY294002 markedly attenuated the
cell proliferation and migration induced by Gas6. The decrease in
EPC proliferation in both the GAS6 and control groups treated with
PD98059 was due to ERK inhibition and not the blockade of
GAS6-mediated ERK activation. Taken together, these findings,
indicate that Gas6 promotes the EPC proliferation and migration
through the PI3K/AKT pathway. It should be noted that the
activation of PI3K/AKT is not sufficient to drive angiogenesis,
while PI3K/AKT is required in regulating cell angiogenesis
(41). The finding that Gas6 did
not engage certain downstream signaling effectors, such as ERK, may
account for the inability of Gas6 to promote angiogenic
responses.
The Gas6-induced growth and differentiation of
hematopoietic cells, particularly the erythroid progenitor cells,
has been well documented (42,43). The effects of Gas6 on EPCs have
not been demonstrated, although EPCs and hematopoietic cells are
derived from the same ancestor, i.e., the hemangioblast (44). It appears that Gas6 regulates EPC
growth and differentiation through mechanisms that are distinct
from those observed in erythroid progenitor cells. In conclusion,
the present study demonstrates that Gas6 promotes EPC proliferation
and migration through the PI3K/AKT signaling pathway. This poses an
interesting question on the manipulation of EPCs with Gas6 to
enhance the therapeutic effects of cell therapy in regenerating the
endothelium. However, the results presented in this study are
preliminary; therefore, further investigation is required in order
for our data to be used as a basis for the development of a
therapeutic strategy for re-endothelialization. Moreover, further
stuides are required in order to explore the effects of Gas6 on
animals.
Acknowledgements
This study was supported by a grant from the
National Natural Science Foundation of China, no. 81370468.
References
|
1
|
Walter DH, Rittig K, Bahlmann FH, et al:
Statin therapy accelerates reendothelialization: a novel effect
involving mobilization and incorporation of bone marrow-derived
endothelial progenitor cells. Circulation. 105:3017–3024. 2002.
View Article : Google Scholar
|
|
2
|
He T, Smith LA, Harrington S, Nath KA,
Caplice NM and Katusic ZS: Transplantation of circulating
endothelial progenitor cells restores endothelial function of
denuded rabbit carotid arteries. Stroke. 35:2378–2384. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Werner N, Junk S, Laufs U, et al:
Intravenous transfusion of endothelial progenitor cells reduces
neointima formation after vascular injury. Circ Res. 93:e17–e24.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li DW, Liu ZQ, Wei J, Liu Y and Hu LS:
Contribution of endothelial progenitor cells to neovascularization
(Review). Int J Mol Med. 30:1000–1006. 2012.PubMed/NCBI
|
|
5
|
Hafizi S and Dahlback B: Gas6 and protein
S. Vitamin K-dependent ligands for the Axl receptor tyrosine kinase
subfamily. FEBS J. 273:5231–5244. 2006.PubMed/NCBI
|
|
6
|
Stitt TN, Conn G, Gore M, et al: The
anticoagulation factor protein S and its relative, Gas6, are
ligands for the Tyro 3/Axl family of receptor tyrosine kinases.
Cell. 80:661–670. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schneider C, King RM and Philipson L:
Genes specifically expressed at growth arrest of mammalian cells.
Cell. 54:787–793. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Melaragno MG, Cavet ME, Yan C, et al: Gas6
inhibits apoptosis in vascular smooth muscle: role of Axl kinase
and Akt. J Mol Cell Cardiol. 37:881–887. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Stenhoff J, Dahlback B and Hafizi S:
Vitamin K-dependent Gas6 activates ERK kinase and stimulates growth
of cardiac fibroblasts. Biochem Biophys Res Commun. 319:871–878.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McCloskey P, Fridell YW, Attar E, et al:
GAS6 mediates adhesion of cells expressing the receptor tyrosine
kinase Axl. J Biol Chem. 272:23285–23291. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nakano T, Higashino K, Kikuchi N, et al:
Vascular smooth muscle cell-derived, Gla-containing
growth-potentiating factor for Ca(2+)-mobilizing growth factors. J
Biol Chem. 270:5702–5705. 1995.PubMed/NCBI
|
|
12
|
Melaragno MG, Wuthrich DA, Poppa V, et al:
Increased expression of Axl tyrosine kinase after vascular injury
and regulation by G protein-coupled receptor agonists in rats. Circ
Res. 83:697–704. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
O’Donnell K, Harkes IC, Dougherty L and
Wicks IP: Expression of receptor tyrosine kinase Axl and its ligand
Gas6 in rheumatoid arthritis: evidence for a novel endothelial cell
survival pathway. Am J Pathol. 154:1171–1180. 1999.PubMed/NCBI
|
|
14
|
Hasanbasic I, Cuerquis J, Varnum B and
Blostein MD: Intracellular signaling pathways involved in
Gas6-Axl-mediated survival of endothelial cells. Am J Physiol Heart
Circ Physiol. 287:H1207–H1213. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
D’Arcangelo D, Gaetano C and Capogrossi
MC: Acidification prevents endothelial cell apoptosis by Axl
activation. Circ Res. 91:e4–e12. 2002.PubMed/NCBI
|
|
16
|
Hasanbasic I, Rajotte I and Blostein M:
The role of gamma-carboxylation in the anti-apoptotic function of
gas6. J Thromb Haemost. 3:2790–2797. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ruan GX and Kazlauskas A: Axl is essential
for VEGF-A-dependent activation of PI3K/Akt. Embo J. 31:1692–1703.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Alciato F, Sainaghi PP, Sola D, Castello L
and Avanzi GC: TNF-alpha, IL-6, and IL-1 expression is inhibited by
GAS6 in monocytes/macrophages. J Leukoc Biol. 87:869–875. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ganopolsky JG, Abid MR, Aird WC and
Blostein MD: GAS6-induced signaling in human endothelial cells is
mediated by FOXO1a. J Thromb Haemost. 6:1804–1811. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kawasaki K, Watabe T, Sase H, et al: Ras
signaling directs endothelial specification of VEGFR2(+) vascular
progenitor cells. J Cell Biol. 181:131–141. 2008.PubMed/NCBI
|
|
21
|
Xu J, Liu X, Jiang Y, et al: MAPK/ERK
signalling mediates VEGF-induced bone marrow stem cell
differentiation into endothelial cell. J Cell Mol Med.
12:2395–2406. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yan X, Cai S, Xiong X, et al: Chemokine
receptor CXCR7 mediates human endothelial progenitor cells
survival, angiogenesis, but not proliferation. J Cell Biochem.
113:1437–1446. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang F, Wang Y, Zhang L and Zou L: Gene
modification with integrin-linked kinase improves function of
endothelial progenitor cells in pre-eclampsia in vitro. J Cell
Biochem. 112:3103–3111. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kalka C, Masuda H, Takahashi T, et al:
Transplantation of ex vivo expanded endothelial progenitor cells
for therapeutic neovascularization. Proc Natl Acad Sci USA.
97:3422–3427. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kawamoto A, Tkebuchava T, Yamaguchi J, et
al: Intramyocardial transplantation of autologous endothelial
progenitor cells for therapeutic neovascularization of myocardial
ischemia. Circulation. 107:461–468. 2003. View Article : Google Scholar
|
|
26
|
Nakano T, Kawamoto K, Kishino J, Nomura K,
Higashino K and Arita H: Requirement of gamma-carboxyglutamic acid
residues for the biological activity of Gas6: contribution of
endogenous Gas6 to the proliferation of vascular smooth muscle
cells. Biochem J. 323:387–392. 1997.PubMed/NCBI
|
|
27
|
Yanagita M, Arai H, Nakano T, et al: Gas6
induces mesangial cell proliferation via latent transcription
factor STAT3. J Biol Chem. 276:42364–42369. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Bellosta P, Zhang Q, Goff SP and Basilico
C: Signaling through the ARK tyrosine kinase receptor protects from
apoptosis in the absence of growth stimulation. Oncogene.
15:2387–2397. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Goruppi S, Ruaro E, Varnum B and Schneider
C: Gas6-mediated survival in NIH3T3 cells activates stress
signalling cascade and is independent of Ras. Oncogene.
18:4224–4236. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Allen MP, Linseman DA, Udo H, et al: Novel
mechanism for gonadotropin-releasing hormone neuronal migration
involving Gas6/Ark signaling to p38 mitogen-activated protein
kinase. Mol Cell Biol. 22:599–613. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fridell YW, Villa JJ, Attar EC and Liu ET:
GAS6 induces Axl-mediated chemotaxis of vascular smooth muscle
cells. J Biol Chem. 273:7123–7126. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Angelillo-Scherrer A, de Frutos P,
Aparicio C, et al: Deficiency or inhibition of Gas6 causes platelet
dysfunction and protects mice against thrombosis. Nat Med.
7:215–221. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Jiang L, Liu CY, Yang QF, Wang P and Zhang
W: Plasma level of growth arrest-specific 6 (GAS6) protein and
genetic variations in the GAS6 gene in patients with acute coronary
syndrome. Am J Clin Pathol. 131:738–743. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Holland SJ, Powell MJ, Franci C, et al:
Multiple roles for the receptor tyrosine kinase axl in tumor
formation. Cancer Res. 65:9294–9303. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gallicchio M, Mitola S, Valdembri D, et
al: Inhibition of vascular endothelial growth factor receptor
2-mediated endothelial cell activation by Axl tyrosine kinase
receptor. Blood. 105:1970–1976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Li Y, Ye X, Tan C, et al: Axl as a
potential therapeutic target in cancer: role of Axl in tumor
growth, metastasis and angiogenesis. Oncogene. 28:3442–3455. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ruan GX and Kazlauskas A: Lactate engages
receptor tyrosine kinases axl, tie2, and vascular endothelial
growth factor receptor 2 to activate phosphoinositide 3-kinase/akt
and promote angiogenesis. J Biol Chem. 288:21161–21172. 2013.
View Article : Google Scholar
|
|
38
|
Allen MP, Zeng C, Schneider K, et al:
Growth arrest-specific gene 6 (Gas6)/adhesion related kinase (Ark)
signaling promotes gonadotropin-releasing hormone neuronal survival
via extracellular signal-regulated kinase (ERK) and Akt. Mol
Endocrinol. 13:191–201. 1999. View Article : Google Scholar
|
|
39
|
Shankar SL, O’Guin K, Cammer M, et al: The
growth arrest-specific gene product Gas6 promotes the survival of
human oligodendrocytes via a phosphatidylinositol
3-kinase-dependent pathway. J Neurosci. 23:4208–4218.
2003.PubMed/NCBI
|
|
40
|
Wang JY, Lee YT, Chang PF and Chau LY:
Hemin promotes proliferation and differentiation of endothelial
progenitor cells via activation of AKT and ERK. J Cell Physiol.
219:617–625. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Munoz-Chapuli R, Quesada AR and Angel MM:
Angiogenesis and signal transduction in endothelial cells. Cell Mol
Life Sci. 61:2224–2243. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Park IK, Giovenzana C, Hughes TL, Yu J,
Trotta R and Caligiuri MA: The Axl/Gas6 pathway is required for
optimal cytokine signaling during human natural killer cell
development. Blood. 113:2470–2477. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Angelillo-Scherrer A, Burnier L,
Lambrechts D, et al: Role of Gas6 in erythropoiesis and anemia in
mice. J Clin Invest. 118:583–596. 2008.PubMed/NCBI
|
|
44
|
Chao H and Hirschi KK: Hemato-vascular
origins of endothelial progenitor cells? Microvasc Res. 79:169–173.
2010. View Article : Google Scholar : PubMed/NCBI
|