Introduction
Ischemic brain injury is an important disorder that
threatens human health and life (1). Ischemic brain injury can cause high
mortality and is the third leading cause of mortality among
Americans (2). In addition to
high mortality, ischemic brain injury can also lead to long-term
disability (3). Cerebral
ischemia/reperfusion (I/R) can induce neuronal death, particularly
in the hippocampal formation (HF) (4). Molecular genetic studies have
suggested that the activities of the transcription factor,
hypoxia-inducible factor-1α (HIF-1α), are closely linked to
ischemia-induced neuronal death (5,6).
Cerebral ischemia results in low oxygen delivery and a decrease in
the adenosine triphosphate (ATP) concentration (7), and the lack of oxygen can lead to
the activation of HIF-1. The HIF-1 transcriptional complex plays an
important role in the regulation of oxygen homeostasis in mammalian
cells (8–10). HIF-1 is a member of the basic
helix-loop-helix-Per-Arnt-Sim superfamily, and is composed of an
HIF-1α and an HIF-1β subunit (11). HIF-1α is regulated by oxygen
levels and determines the level of HIF-1 activity, whereas HIF-1β
is constitutively expressed, and its activity is not affected by
hypoxia (12,13). HIF-1 can affect many cellular
processes, such as energy metabolism, tumor invasion,
erythropoiesis, cell migration, angiogenesis and pH regulation, by
controling the transcription of hundreds of HIF-mediated cells
(14). The constitutive
activation of HIF-1 inhibits mitochondrial biogenesis and cellular
respiration by the repression of c-Myc activity in von
Hippel-Lindau protein-deficient renal carcinoma cells (15). In addition, HIF-1 can affect tumor
cell apoptosis (16). Zhang et
al indicated that HIF-1 increased hypoxic cell survival under
hypoxic conditions (17) and also
suggested that HIF-1 activity may be involved in the protection of
neuronal cells through the regulation of mitochondrial autophagy
(17).
The mitochondria are replaced every 2–4 weeks in the
rat liver, kidneys, heart and brain (18). Mitochondrial degradation has been
implicated in the irreversible cell damage that may occur during
cerebral ischemia and reperfusion (19). The destruction of the mitochondria
may be the result of the process of autophagy, in which parts of
the cytoplasm are sequestered in autophagosomes. A number of
studies have suggested that autophagy, which is induced in the
heart under hypoxic or ischemic conditions, plays either a
protective or pathogenic role in heart disease (20–22). Moreover, autophagy may induce
changes in mitochondrial mass. Mitochondrial mass can change either
due to mitochondrial degradation or due to mitochondrial
amplification, and it has been suggested that autophagy results in
decreased mitochondrial mass through the accelerated mitochondrial
degradation (23).
To the best of our knowledge, the present study
provides the first direct evidence that the process of
mitochondrial autophagy is dependent on the expression of HIF-1
under conditions of cerebral I/R. We demonstrate that mitochondrial
autophagy protects neuronal cells, and the effects of HIF-1 on
mitochondrial autophagy involve the inhibition of the mTOR
signaling pathway.
Materials and methods
Animals
All procedures were carried out according to the
protocols approved by the Ethics Committee for Animal
Experimentation of the General Hospital of People’s Liberation Army
Chengdu Military Region (Chengdu, China). The Sprague-Dawley rats
(210–240 g) that were used in this study were obtained from the
Animal Center of the Institute of Field Surgery of the Third
Military Medical University in China.
Primary cortical neuronal culture
Mouse cortical neurons were cultured according to a
previously described method (24). Newborn (1–3 days old)
Sprague-Dawley rats were anesthetized with halothane and sacrificed
by cervical dislocation. The cortices obtained from the
Sprague-Dawley rats were suspended in cold D-Hank’s (HyClone, Salt
Lake City, UT, USA) solution and dissected free of meninges and
blood vessels. The cerebral tissues were digested in 0.125% trypsin
for 30 min at 37°C. The cell suspension was centrifuged at 3,000 ×
g for 10 min at 4°C, and the precipitate was re-suspended in
DMEM/F12 medium (HyClone) supplemented with 20% FBS (HyClone), 100
mg/l streptomycin and 100 kU/l benzylpenicillin. The cells were
plated at 1×106/ml on 96-well plates coated with 10 mg/l
poly-L-lysine. After 72 h, 5 μg/ml arabinosylcytosine were added to
the cells to prevent the growth of non-neuronal cells.
Subsequently, the culture medium was replaced with normal medium
after 24 h and was refreshed every 2–3 days (25). The cultures typically contained
>95% neurons under these conditions.
In vitro simulation of I/R
Oxygen-glucose-deprivation (OGD) was induced
according to a previously described method with minor modifications
(24,26). Briefly, the cells were rinsed
twice with PBS and glucose-free Earl’s solution [116.4 mmol/l
NaCl, 5.4 mmol/l KCl, 1.8 mmol/l CaCl2, 0.8
mmol/l MgSO4, 2.6 mmol/l NaH2PO4,
26.2 mmol/l NaHCO3 and 20.1 mmol/l HEPES (pH 7.4)] was
added and the cells were cultured at 37°C in an incubator in an
atmosphere of 5% CO2 and 95% N2 (OGD) for 2
h. OGD was terminated by the replacement of stored medium (Earl’s
solution, with the addition of 5.6 mmol/l glucose into the
glucose-free Earl’s solution) and by returning the cultures to an
atmosphere of 5% CO2 and 95% O2 for another
12 h.
Construction of clone and vector
expressing HIF-1α
The Ad-CMV-HIF-1α vector was constructed as
described in a previous study (27). Briefly, total RNA was extracted
from the cultured OGD-treated neurons using TRIzol reagent
(Invitrogen Life Technologies, Carlsbad, CA, USA) according to the
manufacturer’s instructions. Approximately 5 μg of total RNA was
reverse transcribed into cDNA using a PrimeScript RT reagent kit
(Takara Bio, Inc., Shiga, Japan). The cDNAs were used as templates
for the amplification of HIF-1α using 2 primers: forward,
5′-CGGTACCATGGAGGGCGCCGGCG GCGCG-3′ and reverse,
5′-CGCGGCCGCTCAGTTAACTTG ATCCAAAGC-3′. The obtained sequences were
fully sequenced (Shanghai Sangon Biotech Co., Ltd., Shanghai,
China). The cloned HIF-1α was inserted into a pENTR11 vector with
KpnI and NotI restriction sites. Ad-CMV-HIF-1α was
generated using the ViraPower™ Adenoviral Expression System
(Invitrogen Life Technologies) according to the operating
protocols. Cells were seeded in 6-well plates (1.5×105
cells/well) and 100 μl (5×1010 IU/ml) Ad-CMV-HIF-1α were
added and the cells were incubated at 37°C with 5% CO2
for the indicated periods of time.
qRT-PCR
The transcription of HIF-1α was detected using the
RT-PCR method as previously described with minor modifications
(28). The total RNA of primary
neuronal cultures was isolated using TRIzol reagent according to
the manufacturer’s instructions (Biostar, Shanghai, China) and was
then purified using the RNeasy mini kit (Qiagen, Palo Alto, CA,
USA). Approximately 2 μg of total RNA was reverse transcribed into
first-strand cDNA using a QuantiTect® reverse
transcription (RT) kit (Qiagen, Valencia, CA, USA) according to the
manufacturer’s instructions. qRT-PCR was performed to analyze the
levels of mRNA transcripts using specific primers, using the
QuantiTect™ SYBR®-Green PCR kit (Qiagen) and a Smart
Cycler® 1.2f Detection System (Cepheid, Sunnyvale, CA,
USA). The primer sequences used for qRT-PCR were as follows: HIF-1α
sense, 5′-CCAGCAGACTCAAATACAAGAACC-3′ and antisense,
5′-TGTATGTGGGTAGGAGATGGAGAT-3′; β-actin sense,
5′-AAGCAGGAGTATGACGAGTCCG-3′ and antisense,
5′-GCCTTCATACATCTCAAGTTGG-3′. These primers were all synthesized by
Shanghai Sangon Biotech Co., Ltd. Cycling conditions were as
follows: pre-incubation at 95°C, 15 min; PCR: 95°C, 15 sec and
56°C, 30 sec, 40 cycles; final elongation: 72°C, 10 min. β-actin
was used as an internal control and the expression levels of the
relative genes were calculated using the 2−ΔΔCT method,
as previously described (29).
Western blot analysis
The total protein of the OGD-treated neurons was
extracted using RIPA lysis buffer (Beyotime, Nantong, China)
according to the manufacturer’s instructions. A total of 40 μg of
protein per lane was subjected to 10% SDS-PAGE and electroblotted
onto nitrocellulose membranes (Amersham Pharmacia Biotech,
Freiburg, Germany). The immunodetection of HIF-1α, LC3, p70S6
kinase and β-actin was carried out using HIF-1-α antibody (Abcam,
Cambridge, MA, USA), LC3 antibody (Abcam), phospho-p70S6 kinase
antibody (Thr-389; Cell Signaling Technology, Inc., Danvers, MA,
USA) and β-actin antibody (Abcam). Goat anti-rabbit IgG (Abcam) was
used as the secondary antibody. The bound antibodies were
visualized using LumiGLO® reagent (Pierce Biotechnology,
Inc., Rockford, IL, USA). All experiments were repeated 3
times.
Fluorescence-activated cell sorting
(FACS) analysis
The analysis of mitochondrial mass was performed
using a FACScan flow cytometer (BD Biosciences, San Jose, CA, USA)
according to a previously described method with minor
modifications. Briefly, the cells were stained with 1 μM
dichlorodihydrofluorescein diacetate, 10 nM nonyl acridine orange
(NAO), or 1 μM ER-tracker™ Green dye, and then cultured in PBS
solution supplemented with 5% fetal bovine serum at 37°C for 15
min. The stained cells were analyzed using a FACScan flow
cytometer. Apoptosis was measured by flow cytometry using an
Annexin V-FITC/PI kit (BD Pharmingen, San Diego, CA, USA) according
to the manufacturer’s instructions.
MTT cell proliferation assay
Cortical neuronal proliferation was measured by MTT
assay as previously described (30). Briefly, the cortical neurons were
seeded into 96-well culture plates for 24, 48 and 72 h, and then 5
mg/ml MTT (20 μl) were added to the cells at 37°C for 4 h. A total
of 200 μl of DMSO was added to solubilize the crystals. The OD
value was measured at a wavelength of 490 nm using a
spectrophotometer (Multiskan MK3; Thermo Scientific, Waltham, MA,
USA). Data are expressed as the means ± standard error of the mean
(SEM). Differences between the treatment groups were analyzed using
the Student’s t-test and a value of P≤0.05 was considered to
indicate a statistically significant difference.
Measurement of ATP levels
ATP levels in the neurons were determined using
recombinant firefly luciferase and its substrate, D-luciferin
(Invitrogen Life Technologies) according to the manufacturer’s
instructions and as previously described (31). ATP levels were quantitatively
detected, and luminescence was measured using a luminometer
(emission maximum at 560 nm).
Transfection with small interfering RNA
(siRNA)
The neuronal cells expressing the HIF-1α protein
were transfected with HIF-1α siRNA (siHIF-1α) or scrambled siRNA
(siMock) using Lipofectamine 2000 (Invitrogen Life Technologies)
according to the manufacturer’s instructions. For the siRNA
experiments, the HIF-1α overexpressing cells and the wild-type
control cells were seeded on 60-mm plates. The cells were
transfected and, after 20 h, were divided into 24-well plates. The
cells were analyzed 3 days after transfection. The siRNA oligo
sequences for HIF-1α were as previously described (32,33). The siHIF-1α target sequences were:
sense, 5′-CUGAUGA CCAGCAACUUGAdTdT-3′ and antisense, 5′-UCAAGUUGC
UGGUCAUCAGdTdT-3′.
Statistical analysis
All values are expressed as the means ± SEM.
Statistical analysis was performed using the Student’s t-test. A
value of P<0.05 was considered to indicate a statistically
significant difference. All experiments were repeated at least 3
times.
Results
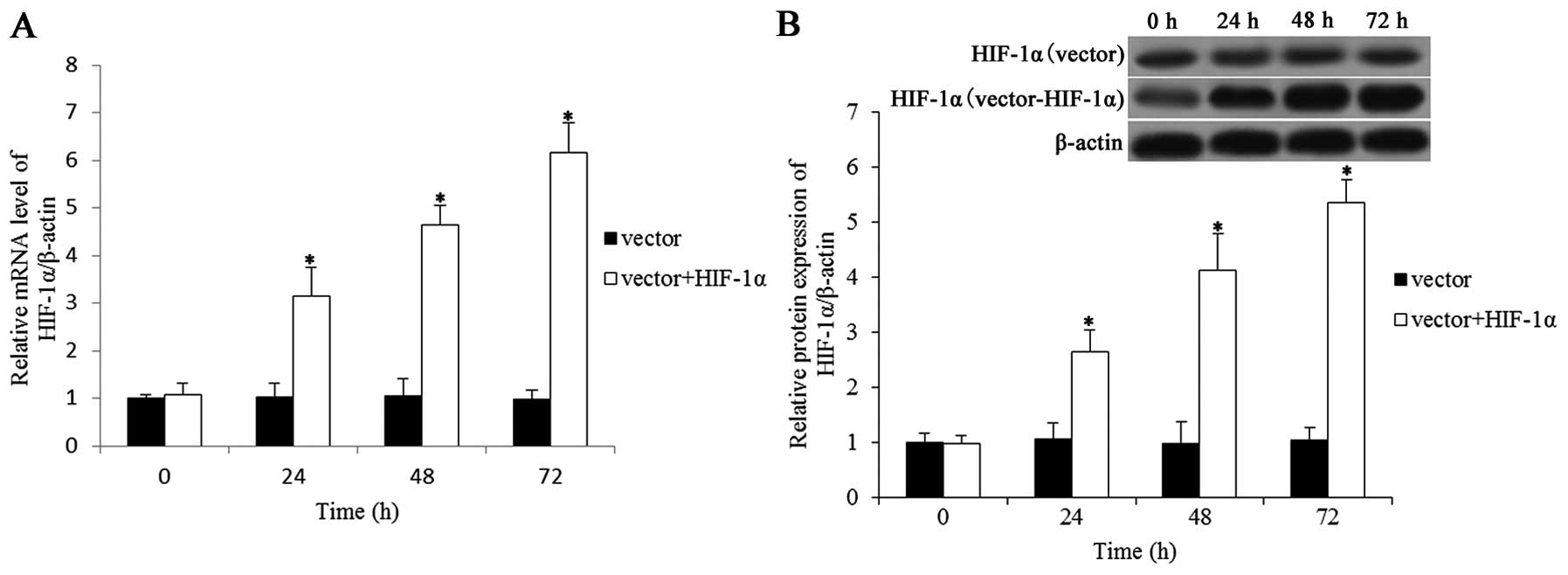
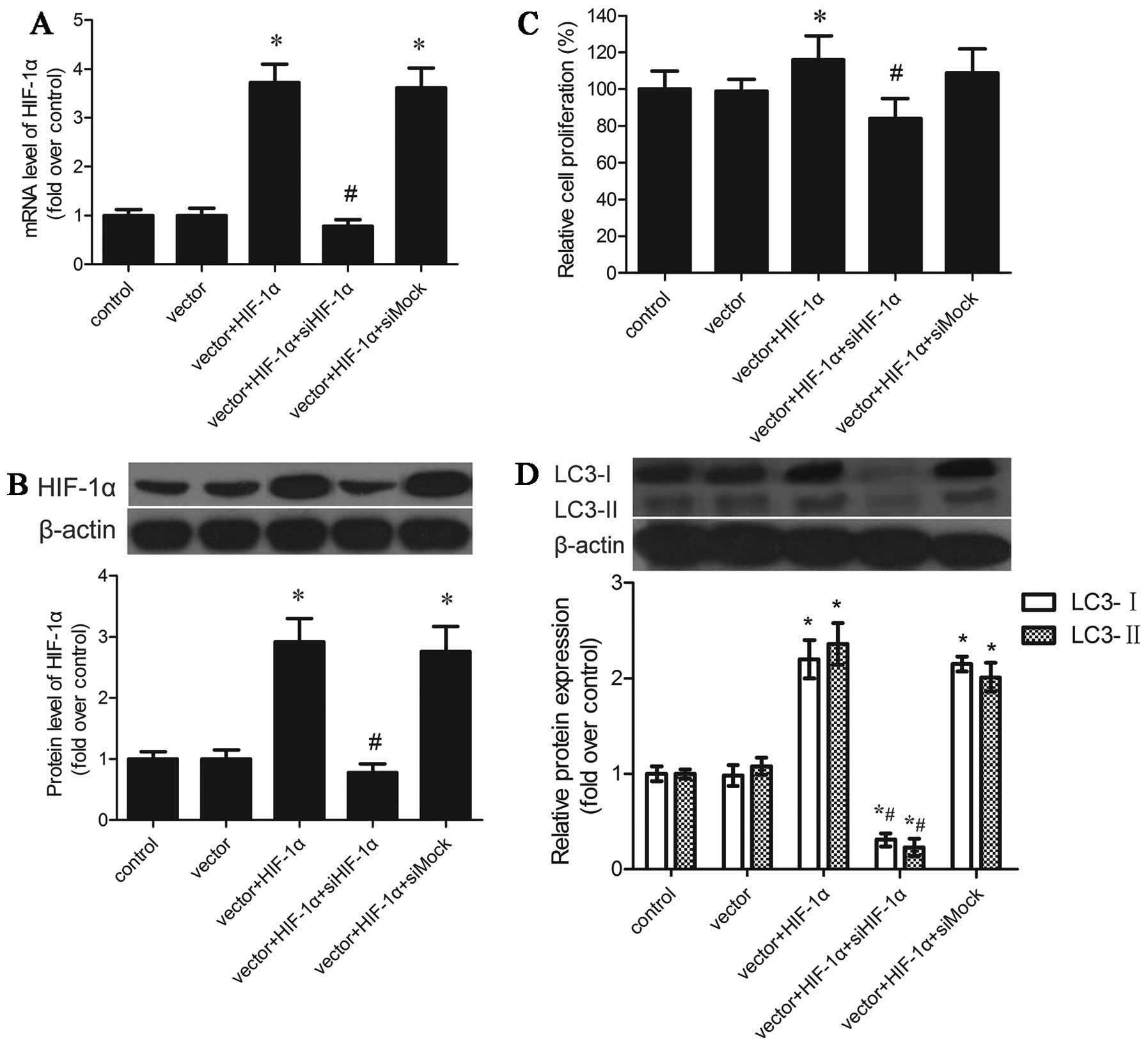
HIF-1α levels were increased in neurons
transfected with Ad-CMV-HIF-1α
In order to induce the overexpression of HIF-1α in
primary cortical neurons, we created an adenoviral vector that
encodes HIF-1α under normoxic conditions. The expression of HIF-1α
in the cortical neurons was determined by qRT-PCR and western blot
analysis. The qRT-PCR results (Fig.
1A) showed that the HIF-1α mRNA levels in the neurons
transfected with Ad-CMV-HIF-1α were markedly higher than those of
the control cells 24 h later. The protein expression was also
determined and the results revealed that the HIF-1α protein levels
were upregulated in the neurons transfected with Ad-CMV-HIF-1α
(Fig. 1B).
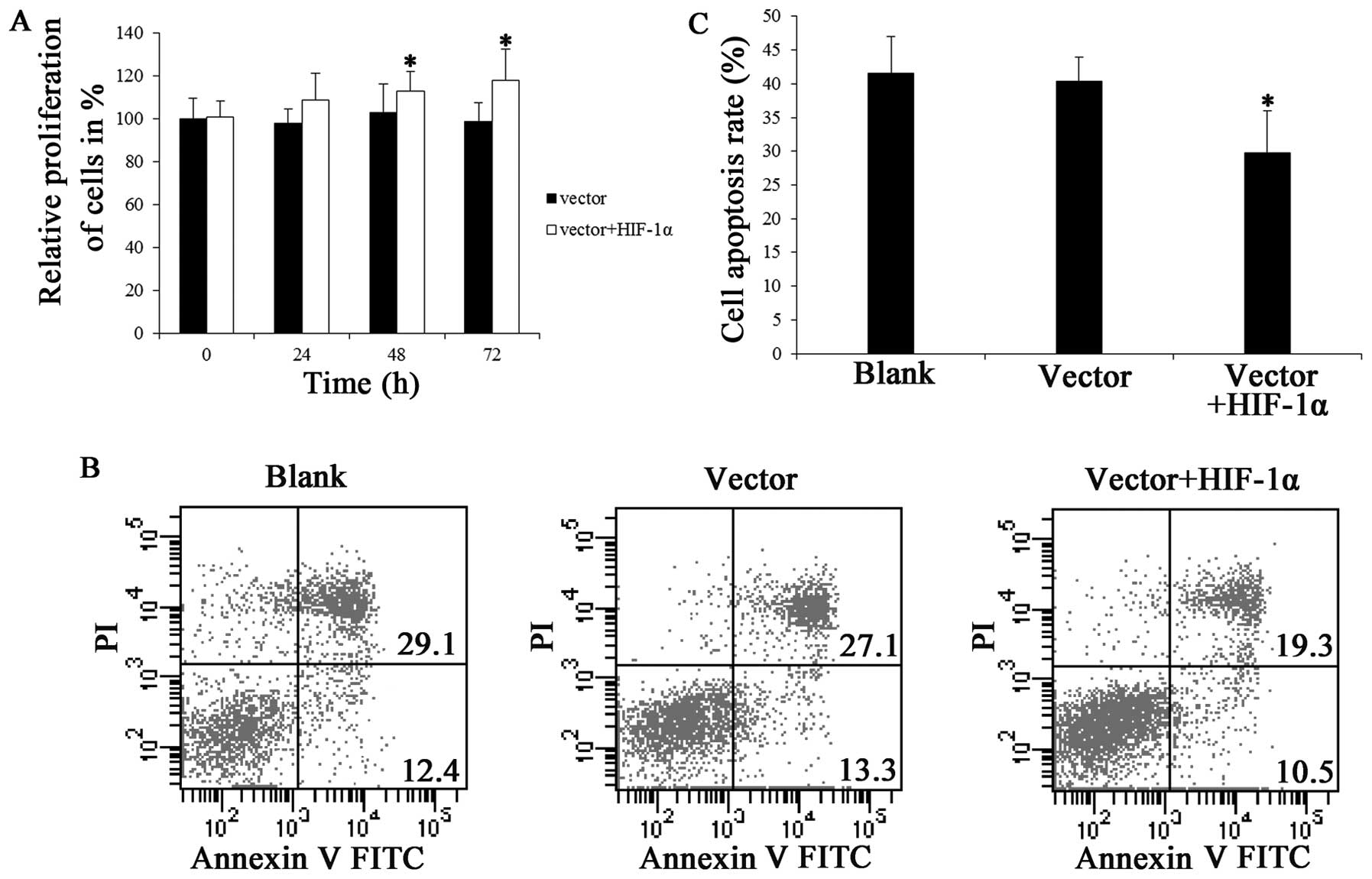
Effect of HIF-1α overexpression on
neuronal cell proliferation and apoptosis
The effect of HIF-1α overexpression on neuronal cell
proliferation was determined by MTT assay. The results indicated
that there was an increase in the proliferation of cortical neurons
which overexpressed HIF-1α and were subjected to OGD, and the
difference was significant following transfection with HIF-1α for
48 h; however, there was no significant difference at 24 h
(Fig. 2A). We found that HIF-1α
overexpression led to an almost 18% increase in the number of
cells. This indicated that HIF-1α exerts positive effects by
increasing the number of cells.
It has recently been demonstrated that HIF-1 exerts
an anti-apoptotic effect on p53-mediated apoptosis through a
secreted neuronal tyrosinase (34). Moreover, HIF-1 can induce
erythropoietin production under hypoxic and hypoglycemic
conditions, and erythropoietin can prevent neuronal apoptosis
following cerebral ischemia and metabolic stress (35,36). Thus, we examined whether the cell
increase in our experiments was mediated by apoptosis. Apoptosis
was first examined by flow cytometric analysis of the cells stained
with Annexin V and PI. The results revealed that HIF-1α
overexpression prevented cell apoptosis, and that the apoptotic
rate decreased to 29.8% when the cells were transfected with
Ad-CMV-HIF-1α for 72 h (Fig. 2B and
C).
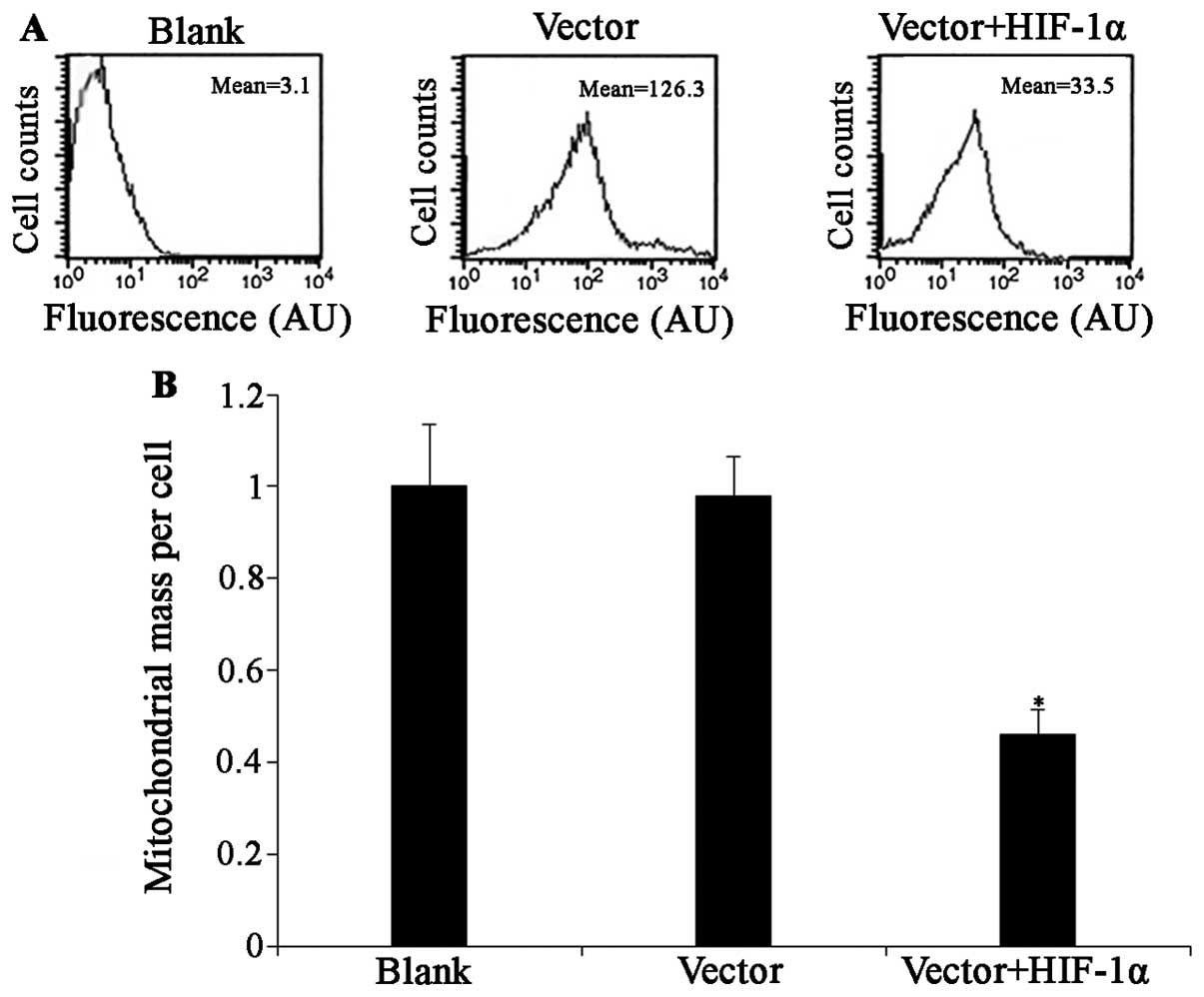
Effect of HIF-1α overexpression on
mitochondrial mass in cortical neurons
The above-mentioned results suggested that HIF-1α
overexpression protected neurons against apoptosis; however, the
mechanisms involved remain unclear. The constitutive activation of
HIF-1 inhibits mitochondrial biogenesis by repressing c-Myc
activity in von Hippel-Lindau protein-deficient renal carcinoma
cells (15). In addition, Zhang
et al also suggested that HIF-1 activity may be involved in
protecting neuron cells through the regulation of mitochondrial
autophagy (17). We first
evaluated mitochondrial mass using FACS analysis. Living cells were
stained with MitoTracker Green (MTG), a fluorescent vital stain
specific for mitochondria (23).
The cells were transfected using the HIF-1α adenoviral construct
and analyzed at several time points following transfection. We
found that the overexpression of HIF-1α for 24 h or longer caused a
significant decrease in mitochondrial mass compared with the
control group (Fig. 3).
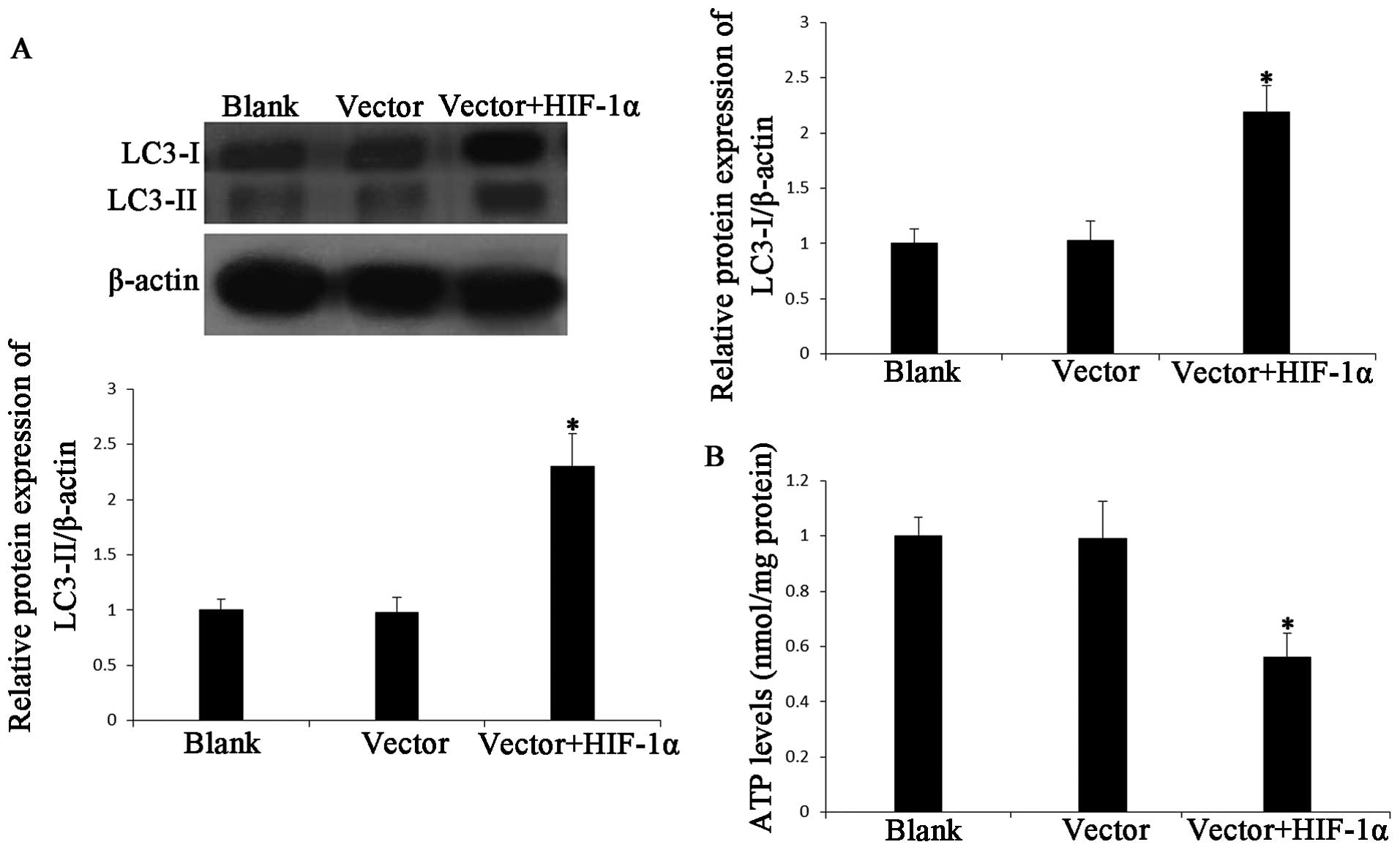
Effect of HIF-1α overexpression on
mitochondrial autophagy
Mitochondrial mass can change due to mitochondrial
degradation, and autophagy results in a decrease in mitochondrial
mass through accelerated mitochondrial degradation (23). We subsequently detected the effect
of HIF-1α overexpression on mitochondrial autophagy. Certain
studies have demonstrated that LC3 (ATG8 in yeast) plays a central
role in the autophagy pathway and promotes the formation of
autophagosomes (37,38). LC3 is known to exist in a soluble
form, termed LC3-I and a lipidated form termed, LC3-II that is
associated with autophagosomal membranes (39). LC3 levels in the cortical neurons
following the induction of HIF-1α overexpression were detected by
western blot analysis. The levels of LC3-I and LC3-II were
significantly increased in the cortical neurons following the
induction of the overexpression of HIF-1α for 24 h (data not
shown), and maximally at 72 h (Fig.
4A). Certain studies have shown that mitochondrial autophagy is
associated with a change in the ATP levels (17,23). Thus, we also determined whether
HIF-1α regulates ATP levels. The overexpression of HIF-1α for 24,
48 (data not shown) and 72 h led to significantly decreased ATP
levels (Fig. 4B). These results
are consistent with the above-mentioned findings that HIF-1α leads
to mitochondrial degradation.
Specific inhibition of HIF-1α mRNA and
protein expression by HIF-1α-specific siRNA
The above-mentioned experiments were designed to
examine the effect of the overexpression of HIF-1α in cortical
neurons. We subsequently employed a complementary approach using
RNA interference technology and examined the effects of suppressing
the levels of HIF-1α in the HIF-1α-overexpressing cells. The cells
were transfected with siHIF-1α or control siRNA (siMock) and were
analyzed after 2 days of growth. Compared with the
siMock-transfected cells, the levels of HIF-1α in the cells
transfected with siHIF-1α were significantly decreased (Fig. 5A and B). We then determined the
effect of HIF-1α silencing on cell proliferation and mitochondrial
autophagy. As shown in Fig. 5C,
there was a decrease in the growth of the siHIF-1α-transfected
cells compared with the siMock-transfected cells. Moreover, the LC3
protein levels were also decreased in the siHIF-1α-transfected
cells (Fig. 5D). These results
indicated that the increase in cell growth and the activity of
mitochondrial autophagy in cortical neurons was associated with the
overexpression of HIF-1α.
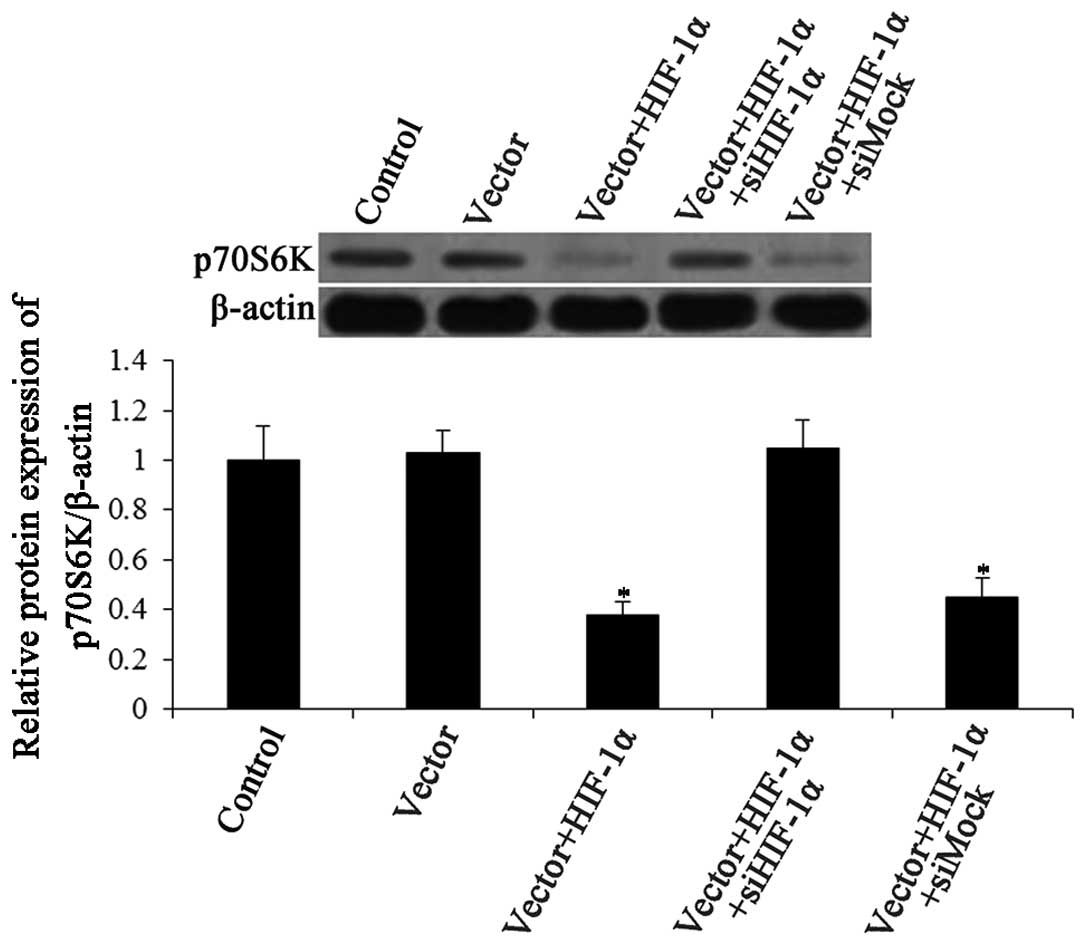
Overexpression of HIF-1α is directly
related to the inhibition of mTOR
There is evidence that the induction of autophagy is
mediated by the inhibition of the mTOR pathway (40). Since HIF-1α induces autophagy, we
examined whether it also inhibits mTOR signaling. The expression of
p70S6 kinase was detected by western blot analysis. p70S6 kinase
expression was significantly increased in the siHIF-1α-transfected
cells compared with siMock group (Fig. 6). These results demonstrated that
HIF-1α overexpression inhibited the mTOR pathway.
Discussion
The main findings of the present study were the
following: i) the overexpression of HIF-1α improves cell survival
in the OGD/RP model using cultured neonatal rat cortical neurons;
ii) the overexpression of HIF-1α significantly induces
mitochondrial autophagy and degradation in the OGD/RP model using
cultured neonatal rat cortical neurons; iii) the inhibition of
HIF-1α markedly suppresses mitochondrial autophagy in rat cortical
neurons; and iv) the overexpression of HIF-1α induced mitochondrial
autophagy and this was accompanied by the inhibition of the mTOR
pathway. To the best of our knowledge, these data demonstrate for
the first time that the neuroprotective effects of the
overexpression of HIF-1α in a model of OGD/RP using cultured
neonatal rat cortical neurons involve an increase in mitochondrial
autophagy. Thus, the results provide insight into the role of
HIF-1α in cerebral I/R injury.
In the present study, we analyzed the effects of
HIF-1α overexpression. HIF-1 is a major regulator of oxygen
homeostasis (10). It is composed
of 2 subunits, an α and a β subunit, but the regulation of HIF-1
activity mostly depends on the α subunit. The upregulation of HIF-1
and the increased HIF-1 binding activity have been shown to protect
astrocytes from ischemic injury during 6 h of hyperthermia (38 or
40°C) (41). Moreover, the
upregulation of HIF-1α and the increased binding activity of HIF-1
are associated with the neuroprotective effects induced by
hypoxia/ischemia pre-conditioning (42). The present study demonstrates that
the overexpression of HIF-1α improves cell survival in a model of
OGD/RP using cultured neonatal rat cortical neurons. We found that
a high level of HIF-1α is associated with an increase in cell
proliferation and a decrease in cell apoptosis. These data suggest
that the upregulation of the level of HIF-1α protects cortical
neurons from ischemia-reperfusion injury.
The present study also demonstrates that the
overexpression of HIF-1α significantly induces changes in
mitochondrial mass in cortical neurons. Changes in mitochondrial
mass may be due to mitochondrial degradation, and autophagy results
in a decrease in the mitochondrial mass through accelerated
mitochondrial degradation (23).
Thus, we also examined the effect of HIF-1α overexpression on
mitochondrial autophagy. LC3 (ATG8 in yeast) is a well-accepted
marker of increased autophagy, plays a central role in the
autophagy pathway and can promote the formation of autophagosomes
(37,38). Our study demonstrated that HIF-1α
overexpression is followed by increased LC3 protein levels,
particularly LC3-II. Our data indicated that HIF-1α overexpression
induced mitochondrial autophagy. The present study also
demonstrated that the inhibition of HIF-1α expression markedly
suppressed cortical neuron survival and mitochondrial autophagy. A
previous study demonstrated that mitochondrial autophagy is
dependent on the level of HIF-1 (17). We found that siHIF-1α suppressed
cell survival and increased the apoptotic rate. Moreover, we
demonstrated that HIF-1α silencing also inhibited mitochondrial
autophagy. In addition, we found that HIF-1α induced mitochondrial
autophagy through the inhibition of the mTOR pathway.
In conclusion, the present results indicate that
HIF-1α overexpression increases cell survival in a model of OGD/RP
using cultured neonatal rat cortical neurons. Moreover, HIF-1α
overexpression increased mitochondrial autophagy. The inhibition of
the expression of HIF-1α suppressed cell survival and decreased
mitochondrial autophagy. It is, therefore, conceivable that HIF-1α
targeting may have detrimental effects on cortical neurons
subjected to OGD/RP. More importantly, our findings raise the
possibility that HIF-1α may serve as a novel regulator of cerebral
I/R injury.
Acknowledgements
This study was supported by the Project of General
Hospital of the People’s Liberation Army Chengdu Military Region
(2013YG-A010).
Abbreviations:
|
I/R
|
cerebral ischemia/reperfusion
|
|
HIF-1α
|
hypoxia- inducible factor-1α
|
|
OGD
|
oxygen-glucose-deprivation
|
|
HF
|
hippocampal formation
|
|
siRNA
|
small interference RNA
|
References
|
1
|
Rosamond W, Flegal K, Furie K, et al:
Heart disease and stroke statistics--2008 update: a report from the
American Heart Association Statistics Committee and Stroke
Statistics Subcommittee. Circulation. 117:e25–e146. 2008.
View Article : Google Scholar
|
|
2
|
Wass CT and Lanier WL: Glucose modulation
of ischemic brain injury: review and clinical recommendations. Mayo
Clin Proc. 801–812. 1996.
|
|
3
|
Hackett ML, Duncan JR, Anderson CS, Broad
JB and Bonita R: Health-related quality of life among long-term
survivors of stroke: results from the Auckland Stroke Study,
1991–1992. Stroke. 31:440–447. 2000.PubMed/NCBI
|
|
4
|
Nitatori T, Sato N, Waguri S, et al:
Delayed neuronal death in the CA1 pyramidal cell layer of the
gerbil hippocampus following transient ischemia is apoptosis. J
Neurosci. 15:1001–1011. 1995.PubMed/NCBI
|
|
5
|
Halterman MW and Federoff HJ: HIF-1α and
p53 promote hypoxia-induced delayed neuronal death in models of CNS
ischemia. Exp Neurol. 159:65–72. 1999.
|
|
6
|
Halterman MW, Miller CC and Federoff HJ:
Hypoxia-inducible factor-1alpha mediates hypoxia-induced delayed
neuronal death that involves p53. J Neurosci. 19:6818–6824.
1999.PubMed/NCBI
|
|
7
|
Chen CJ, Cheng FC, Liao SL, Chen WY, Lin
NN and Kuo JS: Effects of naloxone on lactate, pyruvate metabolism
and antioxidant enzyme activity in rat cerebral
ischemia/reperfusion. Neurosci Lett. 287:113–116. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Semenza GL: HIF-1: mediator of
physiological and pathophysiological responses to hypoxia. J Appl
Physiol. 88:1474–1480. 2000.PubMed/NCBI
|
|
9
|
Melillo G: HIF-1: a target for cancer,
ischemia and inflammation--too good to be true? Cell Cycle.
3:149–150. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Brahimi-Horn MC and Pouysségur J: Oxygen,
a source of life and stress. FEBS Lett. 581:3582–3591.
2007.PubMed/NCBI
|
|
11
|
Wang GL, Jiang B-H and Semenza GL: Effect
of protein kinase and phosphatase inhibitors on expression of
hypoxia inducible factor 1. Biochem Biophys Res Commun.
216:669–675. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang LE, Gu J, Schau M and Bunn HF:
Regulation of hypoxia-inducible factor 1alpha is mediated by an
O2-dependent degradation domain via the
ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 95:79871998.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang GL, Jiang B-H, Rue EA and Semenza GL:
Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS
heterodimer regulated by cellular O2 tension. Proc Natl
Acad Sci USA. 92:5510–5514. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Mazure NM and Pouysségur J:
Hypoxia-induced autophagy: cell death or cell survival? Current
opinion in cell biology. 22:177–180. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhang H, Gao P, Fukuda R, et al: HIF-1
inhibits mitochondrial biogenesis and cellular respiration in
VHL-deficient renal cell carcinoma by repression of C-MYC activity.
Cancer Cell. 11:407–420. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sowter HM, Ratcliffe PJ, Watson P,
Greenberg AH and Harris AL: HIF-1-dependent regulation of hypoxic
induction of the cell death factors BNIP3 and NIX in human tumors.
Cancer Res. 61:6669–6673. 2001.PubMed/NCBI
|
|
17
|
Zhang H, Bosch-Marce M, Shimoda LA, et al:
Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic
response to hypoxia. J Biological Chem. 283:10892–10903. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Menzies RA and Gold PH: The turnover of
mitochondria in a variety of tissues of young adult and aged rats.
J Biol Chem. 246:2425–2429. 1971.PubMed/NCBI
|
|
19
|
Rosenthal RE, Hamud F, Fiskum G, Varghese
PJ and Sharpe S: Cerebral ischemia and reperfusion: prevention of
brain mitochondrial injury by lidoflazine. J Cereb Blood Flow
Metab. 7:752–758. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mei Y, Thompson MD, Cohen RA and Tong X:
Autophagy and oxidative stress in cardiovascular diseases. Biochim
Biophys Acta. May 13–2014.(Epub ahead of print).
|
|
21
|
Ma S, Wang Y, Chen Y and Cao F: The role
of the autophagy in myocardial ischemia/reperfusion injury. Biochim
Biophys Acta. May 21–2014.(Epub ahead of print).
|
|
22
|
Ghavami S, Gupta S, Ambrose EC, Hnatowich
M, Freed DH and Dixon IM: Autophagy and Heart Disease: Implications
for Cardiac Ischemia-Reperfusion Damage. Curr Mol Med. June
2–2014.(Epub ahead of print).
|
|
23
|
Ermak G, Sojitra S, Yin F, Cadenas E,
Cuervo AM and Davies KJ: Chronic expression of RCAN1-1L protein
induces mitochondrial autophagy and metabolic shift from oxidative
phosphorylation to glycolysis in neuronal cells. J Biol Chem.
287:14088–14098. 2012. View Article : Google Scholar
|
|
24
|
Gong QH, Wang Q, Shi JS, Huang X-n, Liu Q
and Ma H: Inhibition of caspases and intracellular free Ca2+
concentrations are involved in resveratrol protection against
apoptosis in rat primary neuron cultures. Acta Pharmacol Sin.
28:1724–1730. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Murphy TH and Baraban JM: Glutamate
toxicity in immature cortical neurons precedes development of
glutamate receptor currents. Brain Res Dev Brain Res. 57:146–150.
1990. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Choi DW, Maulucci-Gedde M and Kriegstein
AR: Glutamate neurotoxicity in cortical cell culture. J Neurosci.
7:357–368. 1987.PubMed/NCBI
|
|
27
|
Wei L, Lu J, Feng L, Li S, Shan J and Li
Y: Construction of recombinant adenovirus vector containing a
modified gene that codes for human hypoxia-inducible factor-1α
without oxygen-dependent degradation domain. Plasmid. 63:20–26.
2010.PubMed/NCBI
|
|
28
|
Bryant CS, Munkarah AR, Kumar S, et al:
Reduction of hypoxia-induced angiogenesis in ovarian cancer cells
by inhibition of HIF-1 alpha gene expression. Arch Gynecol Obstet.
282:677–683. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Livak K and Schmittgen T: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
30
|
Song YX, Yue ZY, Wang ZN, et al:
MicroRNA-148b is frequently down-regulated in gastric cancer and
acts as a tumor suppressor by inhibiting cell proliferation. Mol
Cancer. 10:12011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zadran S, Sanchez D, Zadran H, Amighi A,
Otiniano E and Wong K: Enhanced-acceptor fluorescence-based single
cell ATP biosensor monitors ATP in heterogeneous cancer populations
in real time. Biotechnol Lett. 35:175–180. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sowter HM, Raval R, Moore J, Ratcliffe PJ
and Harris AL: Predominant role of hypoxia-inducible transcription
factor (Hif)-1α versus Hif-2α in regulation of the transcriptional
response to hypoxia. Cancer Res. 63:6130–6134. 2003.
|
|
33
|
Esteban MA, Tran MG, Harten SK, et al:
Regulation of E-cadherin expression by VHL and hypoxia-inducible
factor. Cancer Res. 66:3567–3575. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sendoel A, Kohler I, Fellmann C, Lowe SW
and Hengartner MO: HIF-1 antagonizes p53-mediated apoptosis through
a secreted neuronal tyrosinase. Nature. 465:577–583. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sirén AL, Fratelli M, Brines M, et al:
Erythropoietin prevents neuronal apoptosis after cerebral ischemia
and metabolic stress. Proc Natl Acad Sci USA. 98:4044–4049.
2001.PubMed/NCBI
|
|
36
|
Chandel N, Maltepe E, Goldwasser E,
Mathieu C, Simon M and Schumacker P: Mitochondrial reactive oxygen
species trigger hypoxia-induced transcription. Proc Natl Acad Sci
USA. 95:11715–11720. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Levine B, Mizushima N and Virgin HW:
Autophagy in immunity and inflammation. Nature. 469:323–335. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Geng J and Klionsky DJ: The Atg8 and Atg12
ubiquitin-like conjugation systems in macroautophagy. EMBO Rep.
9:859–864. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kraft LJ and Kenworthy AK: Imaging protein
complex formation in the autophagy pathway: analysis of the
interaction of LC3 and Atg4BC74A in live cells using Förster
resonance energy transfer and fluorescence recovery after
photobleaching. J Biomed Opt. 17:0110081–01100812. 2012.PubMed/NCBI
|
|
40
|
Mijaljica D, Prescott M and Devenish RJ:
Different fates of mitochondria: alternative ways for degradation?
Autophagy. 3:4–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Du F, Zhu L, Qian ZM, Wu XM, Yung WH and
Ke Y: Hyperthermic preconditioning protects astrocytes from
ischemia/reperfusion injury by up-regulation of HIF-1 alpha
expression and binding activity. Biochim Biophys Acta.
1802:1048–1053. 2010. View Article : Google Scholar
|
|
42
|
Du F, Wu XM, Gong Q, He X and Ke Y:
Hyperthermia conditioned astrocyte-cultured medium protects neurons
from ischemic injury by the up-regulation of HIF-1 alpha and the
increased anti-apoptotic ability. Eur J Pharmacol. 666:19–25. 2011.
View Article : Google Scholar
|