1. Introduction
Cardiovascular disease (CVD) of atherosclerotic
origin remain the leading cause of mortality worldwide, accounting
for approximately 27% of deaths among males and 32% among females.
The initiation and progression of atherosclerosis (AS) involves the
chronic expansion of arterial intima with diverse types of cells,
including macrophages, dendric cells (DCs), vascular smooth muscle
cells (VSMCs) and endothelial cells (ECs), as well as various sets
of pro-inflammatory substances, such as adhesion molecules,
cytokines, chemotactic factors and extracellular matrix (ECM)
proteins (1).
VSMCs are the most abundant cell type in the
arterial wall and are involved in the progression of AS. VSMCs
exist in many phenotypes (2,3).
Under normal conditions, the predominant phenotype is
quiescent/contractile with non-migratory and non-proliferative
VSMCs. The loss of ECs due to mechanical forces, including
hemodynamic forces and stenting or cell apoptosis, can lead to VSMC
migration from the media to the intima, where VSMCs can proliferate
to form the neointima and plaque (4–6).
VSMCs contribute to the development of AS through a variety of
functions, including migration, proliferation, ECM synthesis and
apoptosis, as well as inflammation and foam cell formation through
cholesterol uptake (7).
MicroRNAs (miRNAs, miRs) are small, non-coding RNAs
that can control gene expression by binding to their target
messenger RNAs (mRNAs) through translational repression or mRNA
decay (8). By binding to target
genes and directly degradating mRNA or repressing translation, miRs
are able to regulate the expression of hundreds or thousands of
genes. The regulation of miRs is involved in a number of
physiological and pathological conditions, such as metabolism,
differentiation, development, apoptosis, proliferation and
oncogenesis (9).
Recent studies have found that miRs play an
important role in vascular pathophysiology and AS (10–12). Abnormal miR expression profiles
have been reported to be related to CVD in over 400 studies (e.g.,
13). Moreover, accumulating
evidence suggests a close association between miRs and the
functions of VSMCs (14). The
identification of the role of miRs in the regulation of VSMCs may
lead to an improved in the understanding of the pathophysiology of
AS and is likely to provide novel diagnostic and therapeutic
targets.
In this review, we summarize the current knowledge
on the role of miRs in the regulation of VSMC functions, including
differentiation, proliferation, ECM synthesis, apoptosis and
calcification. In particular, we discuss the mechanisms of action
of miRs in the regulation of VSMC functions and their possible
implications in AS. In addition, we discuss the potential
applicability of miRs in the diagnosis and treatment of AS.
2. miRNAs regulate VSMC differentiation and
phenotype
VSMCs can switch between a differentiated
(contractile) state and a dedifferentiated (synthetic) phenotype in
response to extracellular stimulation. Spindle-shaped
differentiated VSMCs have low rates of proliferation, migration and
production of ECM, but high levels of contractile genes, while
rhomboid-shaped dedifferentiated VSMCs have increased rates of
proliferation, migration and production of ECM, as well as low
levels of contractile genes (15). Under pathological conditions,
VSMCs can undergo the phenotypic change from the normal contractile
state to synthetic, proliferative cells, which have an increased
ability to migrate, proliferate and produce ECM (16). In atherosclerotic lesions and
neointimal tissue, synthetic VSMC phenotypes are the major cellular
types in vascular repair following various injuries. The phenotypic
switching of VSMCs depends on numerous local signals, such as
cytokines, cell-cell contact, cell adhesion, ECM interactions,
injury stimuli, among which platelet-derived growth factor
(PDGF)-BB and transforming growth factor-β (TGF-β)/bone
morphogenetic protein (BMP) are the key mediators of VSMC
phenotypic switching (17).
PDGF-BB significantly promotes the VSMC phenotype switch from a
differentiated, contractile state into a dedifferentiated,
synthetic state, thus promoting VSMC proliferation and migration,
while TGF-β and BMP help to maintain the VSMC contractile phenotype
(18,19). Abnormal regulation of this
phenotype switching leads to the development and progression of AS
and restenosis (20). By
preventing the VSMC phenotypic modulation from a contractile state
to a synthetic state, neointimal progression in AS may be
controlled. Previous studies have identified many miRs in the
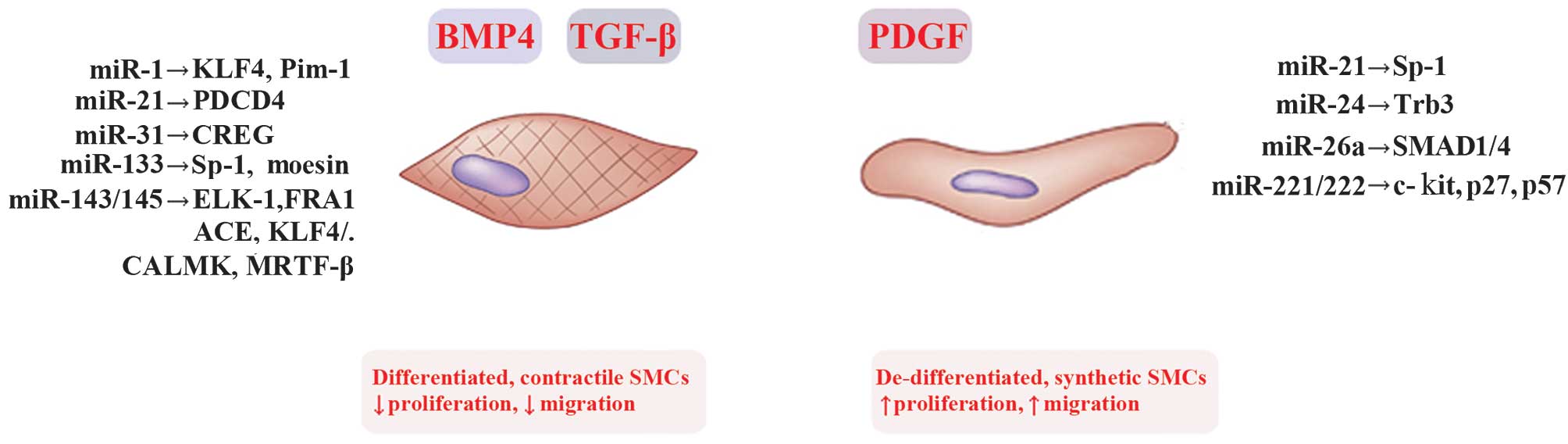
regulation of VSMCs phenotypic modulation (Table I and Fig. 1).
 | Table ImiRs involved in the regulation of
the VSMC phenotype. |
Table I
miRs involved in the regulation of
the VSMC phenotype.
| Function | miRs | Targets | (Refs.) |
|---|
| Promote contractile
phenotype | miR-1 | KLF4, Pim-1 | (19–21) |
| miR-31 | CREG | (29) |
| miR-133 | Sp-1, moesin | (3) |
| miR-143/145 | ELK1,
FRA1
ACE, KLF4/5, CALMK, MRTF-B | (35,40) |
| Promote synthetic
phenotype | miR-24 | Trb3 | (41) |
| miR-26a | SMAD1/4 | (43) |
| miR-221/222 | c-kit, p27
(Kip1)
p57 (Kip2) | (46) |
| Dual effect | miR-21 | PDCD4, Sp-1 | (50–52) |
3. miRNAs and pro-contractile phenotype
miR-1 levels are significantly higher in more
differentiated and less proliferative VSMCs and downregulated
during neointima formation (21).
miR-1 has been proven to regulate the differentiation of VSMCs by
targeting Krüppel-like factor 4 (KLF4) and proviral integration
site for Moloney murine leukemia virus 1 (Pim-1) (21–23). KLFs are a subclass of
evolutionarily conserved transcription factors (24), and KLF4 has been demonstrated to
significantly repress the expression of multiple genes in VSMCs
during the phenotypic switching of SMCs in response to vascular
injury, TGF-β or PDGF-BB (25–27). For instance, KLF4 can downregulate
the dedifferentiation marker gene, non-muscle myosin heavy chain B
(Smemb) (28). Pim-1, an
oncogenic serine/threonine kinase, has been reported to induce
neointimal hyperplasia formation and promote the proliferation of
cultured VSMCs (29).
miR-31 is upregulated in contractile VSMCs and
downregulated in proliferative VSMCs, the proliferation of which
was induced by PDGF (58). In
addition, miR-31 levels have been shown to be higher in the serum
of patients with coronary artery disease (CAD) with restenosis
compared to patients with CAD without restenosis (30). miR-31 has been proven to promote
the VSMC contractile phenotype by repressing cellular repressor of
E1A-stimulated genes (CREG) expression. It has been reported that
the overexpression of CREG maintains the differentiated phenotype
in cultured VSMCs, whereas shRNA-mediated CREG knockdown promotes
VSMC dedifferentiation, implicating a critical role for CREG in
maintaining the phenotypic switching of VSMCs (31). However, the exact mechanism of
action of CREG in regulating VSMC phenotypic modulation remains
unclear.
miR-133 is downregulated in proliferating VSMCs but
upregulated when VSMCs are back to the quiescence phenotype
(32). Furthermore, the
circulating levels of miR-133 are also downregulated in humans with
CAD (33). miR-133 has been shown
to inhibit the VSMC switch to the synthetic phenotype by inhibiting
the tracription factor, specificity protein-1 (Sp-1) (32). Sp-1 is a transcription factor
activated by a phenotypic switch that promotes stimuli and is
upregulated in animal models of vascular injury (34,35). Activated Sp-1 in turn increases
the activity of KLF4, which can repress myocardin and therefore
downregulates the expression of most of the genes in VSMCs
(36).
miR-143/145, encoded by a bicistronic gene cluster,
are highly conserved and abundantly expressed in normal VSMCs,
regulating VSMC differentiation. miR-143/145 are downregulated in
animal models of AS (37,38). Furthermore, the circulating levels
of miR-143/145 are downregulated in humans with CAD (33). The transfer of miR-143 and miR-145
from ECs to VSMCs promotes the contractile VSMC phenotype and
attenuates AS (39). Aged
miR-143/145−/− mice develop spontaneous atherosclerotic
lesions in the femoral artery in the absence of
hypercholesterolemia, and the overexpression of miR-145 reduces AS
in Apoe−/− mice (40,41). A network of transcription factors,
such as KLF4, KLF5, ELK1, versican, several actin remodeling
proteins and angiotensin-converting enzyme (ACE), have been
identified as the targets of miR-143/145 (37,42). These targets function as
transcription and translation factors, receptors, phosphatases,
kinases, growth factors, RNA binding proteins, and so forth, and
are involved in different cellular processes in addition to
regulating VSMC plasticity. This represents an example of how a
single miRNA can regulate a number of related or unrelated cellular
processes.
4. miRNAs and pro-synthetic phenotype
In response to PDGF-BB, the expression of miR-24 is
upregulated and its target, Tribbles-like protein 3 (Trb3), is
downregulated (43). Trb3 has
been reported to interact with type-II BMP receptor (BMPRII) and
promote the degradation of Smad ubiquitin-regulatory factor-1
(Smurf1), a negative regulator of BMP and TGF-β Smad-dependent
signalling (44). Trb3 has been
proven to be essential for the PDGF-BB-mediated induction of the
VSMC synthetic phenotype. In addition, VSMC proliferation and the
repression of Trb3 coincides with the reduced expression of Smad
proteins and a decrease in BMP and TGF-β signalling, which promotes
a synthetic phenotype in VSMCs.
It has been demonstrated that miR-26a has multiple
roles in VSMCs, including the promotion of VSMC proliferation,
migration and the inhibition of apoptosis. miR-26a is significantly
upregulated in synthetic VSMCs (45). The overexpression of miR-26a
inhibits the differentiation of VSMCs and helps maintain the
balance of cells in the synthetic and contractile states and
governs phenotypic shifting by directly targeting SMAD1 and SMAD4
(45). SMAD1 and SMAD4 are two
TGF-β- and BMP-related pro-differentiation factors, in which SMAD-1
is primarily an element of BMP-responsive pathways, while SMAD-4 is
in the common pathway for BMP and TGF-β (45).
miR-221 and miR-222 are clustered on the X
chromosome and share a common seed sequence. Both are significantly
upregulated in vivo in VSMCs following the balloon injury of
the vessel (46). miR-221 is
upregulated in VSMCs upon the PDGF-BB-mediated VSMC phenotypic
switch into the synthetic phenotype (47). miR-221/222 can regulate multiple
functions of VSMCs, including the reduced expression of contractile
genes, and increased proliferation and migration. miR-221/222
regulate multiple functions through multiple targets, including p27
(Kip1), p57 (Kip2) and c-kit (48). p27 and p57 are cyclin-dependent
kinase inhibitors (CKIs), negatively regulating cell proliferation.
The overexpression of p27 or p57 inactivates the cyclin-CDK
complexes and leads to cell cycle arrest, inversely correlates with
VSMC proliferation (49). Other
than the cell cycle control, p57 has been reported to be involved
in cell differentiation in chondrocytes (50). c-kit, a proto-oncogene, is
involved in multiple cellular functions, such as cell survival,
proliferation, differentiation, adhesion, homing and migration
(51) However, to the best of our
knowledge, the role of p27, p57 and c-kit in VSMC differentiation
has not been investigated to date.
miR-21 regulates multiple functions of VSMCs,
including phenotypic changes, proliferation and apoptosis. However,
its role in the phenotypic modulation and differentiation of VSMCs
remains controversial. In some studies, miR-21 has been proven to
inhibit VSMC differentiation, promote proliferation and reduce
apoptosis through Sp-1 (52,53). However, another study demonstrated
that miR-21 increases the biosynthesis of contractile protein in
VSMCs by targeting programmed cell death protein 4 (PDCD4)
(54). PDCD4, a tumor suppressor
gene, has been reported to promote the differentiation of acute
myeloid leukemia (AML) cells and female germline stem cells
(55,56). However, to our knowledge, the role
of PDCD4 in VSMC differentiation has not been investigated to date.
The dual effect of miR-21 may be due to the diverse target genes of
miR-21 in regulating VSMC differentiation. Further studies are
required to elucidate the exact role of miR-21 in the phenotypic
regulation of VSMCs.
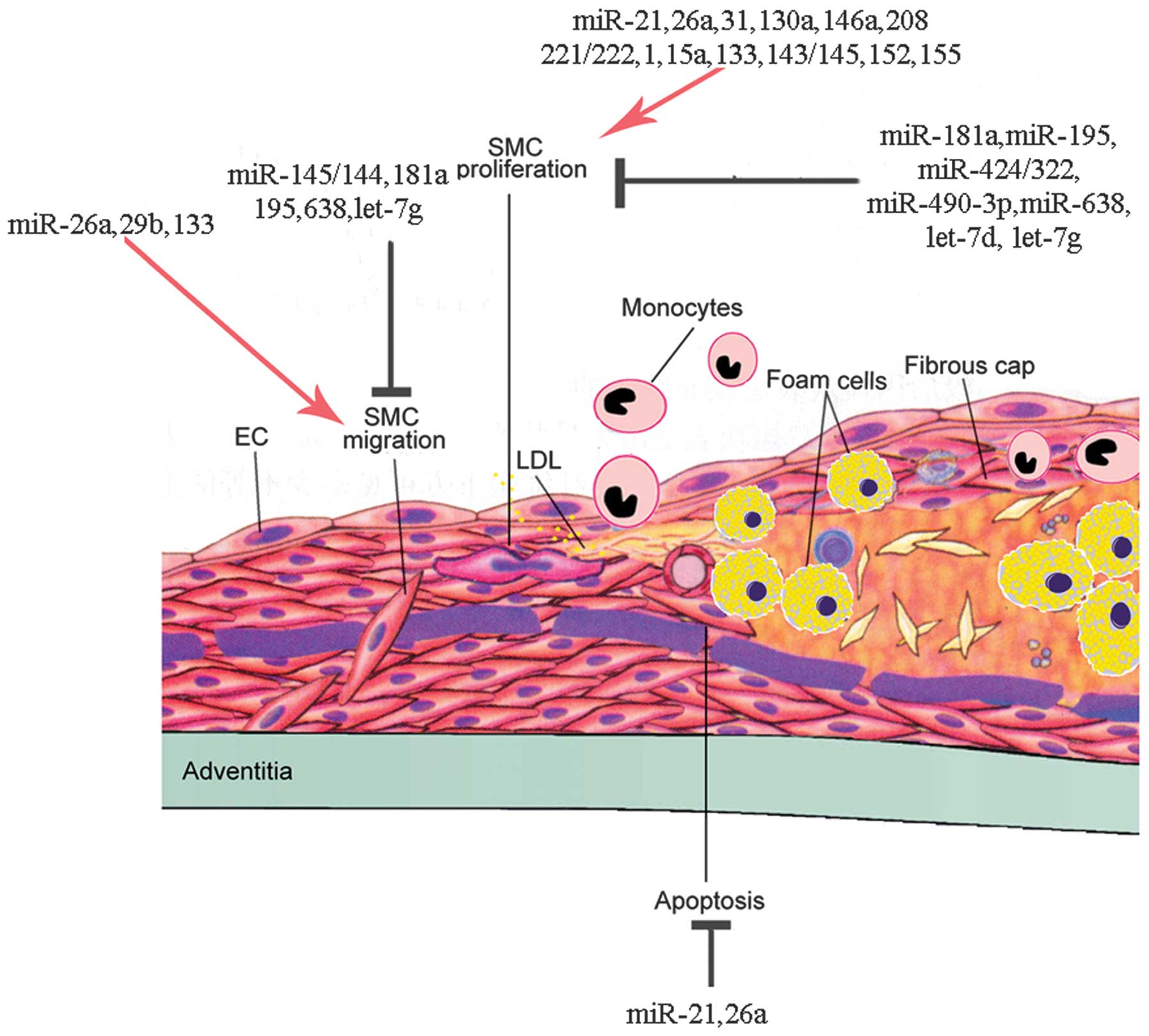
5. miRNAs regulate VSMC proliferation
Aberrant VSMC proliferation significantly
contributes in neointimal plaque formation during the progression
of AS. VSMC proliferation has been proven to be solely responsible
for in-stent restenosis (57). As
mentioned above, VSMCs can switch between two phenotypes, a
differentiated/contractile phenotype and a
dedifferentiated/synthetic phenotype. VSMCs with the
dedifferentiated/synthetic phenotype have an increased ability to
migrate, proliferate and produce ECM, which contributes to the
development of AS. Thus, the proliferation of VSMCs is closely
associated with the phenotypic regulation of VSMCs. Accumulating
evidence has demonstrated that miRs play an important role in the
mediation of VSMC proliferation through the regulation of the
post-transcriptional expression of several genes (Table II). Some miRs have been found to
promote the proliferation of VSMCs, while others have been reported
to inhibit the proliferation of VSMCs. By preventing VSMC
proliferation, neointimal progression in AS may be controlled.
| Table IImiRs involved in the regulation of
VSMC proliferation. |
Table II
miRs involved in the regulation of
VSMC proliferation.
| Function | miRs | Targets | (Refs.) |
|---|
| Promote
proliferation | miR-21 | PTEN, Bcl-2 | (51) |
| miR-26a | SMAD-1/4 | (43) |
| miR-31 | LATS2 | (56) |
| miR-130a | GAX | (58) |
| miR-146a | KLF4 | (66) |
| miR-208 | p21 | (67) |
| miR-221/222 | PTEN, Bcl-2, p27,
p57 | (44,45) |
| Inhibit
proliferation | miR-1 | Pim-1 | (13) |
| miR-15a | ? | (39) |
| miR-133 | Sp-1 | (17) |
| miR-143/145 | KLF5, ELK1,
myocardin | (19) |
| miR-152 | DNMT1 | (42) |
| miR-155 | - | (44) |
| miR-181a | - | (45) |
| miR-195 | Cdc42, CCND1,
FGF1 | (46) |
| miR-424/322 | Cyclin D1,
calumenin | (47) |
| miR-490-3p | PAPP-A | (48) |
| miR-638 | NDR1 | (49) |
| let-7d | KRAS | (50) |
| let-7g | LOX-1 | (51) |
6. miRNAs promote the proliferation of
VSMCs
miR-21 is one of the most upregulated miRs in the
vascular wall following balloon injury. The inhibition of miR-21
can decrease the proliferation of cultured VSMCs in injured rat
carotid arteries (53).
In addtion to phenotypic regulation, miR-26a can
also promote VSMC proliferation and aortic SMCs deficient in
miRNA-26a show a significant reduction in proliferation (45).
As mentioned above, miR-31 can modulate the VSMC
phenotype by targeting CREG (30). It can also regulate the
proliferation of VSMCs. The expression of miR-31 is significantly
increased in proliferative VSMCs. miR-31 exerts a pro-proliferative
effect on VSMCs by targeting large tumor suppressor homolog 2
(LATS2) (58). LATS2 has been
proven to play a major role in cell proliferation and apoptosis and
is an important regulator of tissue and organ development (59).
miR-130a is upregulated in the remodeled aorta and
arteries with hypertension. The overexpression of miR-130a has been
shown to significantly promote the proliferation of VSMCs by
inhibiting the expression of growth arrest-specific homeobox (GAX)
(60). GAX has been proven to be
downregulated in some vascular diseases, such as balloon injury
(61), pulmonary hypertension
(62), or by certain growth
factors including Ang-II (63)
and PDGF (64). GAX can
significantly inhibit the proliferation, differentiation and
migration of VSMCs (65,66).
The expression of miR-146a is significantly
upregulated in atherosclerotic arteries compared with
non-atherosclerotic arteries (38). miR-146a promotes VSMC
proliferation in cultured VSMCs by targeting KLF4. KLF4 exerts an
anti-proliferative effect on VSMCs through the upregulation of p21,
which is a member of the cyclin-dependent kinase (CDK)-inhibitory
protein family (67). The
transfection of antisense miR-146a oligonucleotide into
balloon-injured rat carotid arteries has been shown to markedly
decrease neointimal hyperplasia (68).
miR-208 regulates insulin-induced VSMC proliferation
through the downregulation of its potential target, p21. However,
as previously demonstrated, an miR-208 inhibitor alone had no
effect on VSMC proliferation (69).
As mentioned above, miR-221/222 regulates multiple
functions of VSMCs, such as reducing the expression of contractile
genes and increasing proliferation and migration. The expression of
miR-221/222 is significantly increased in proliferative VSMCs. The
knockdown of miR-221/222 has been shown to decrease the
proliferation of cultured VSMCs and neointima formation in
balloon-injured rat arteries (46). miR-221 and miR-222 have been
proven to regulate VSMC proliferation by targeting p27 and p57
(46,47). p27 and p57 are CKIs, negatively
regulating cell proliferation (Fig.
2).
 | Figure 2Regulation of vascular smooth muscle
cell (VSMC) phenotypic switch by microRNAs (miRs) and their
targets. VSMCs switch between the contractile phenotype and the
synthetic phenotuype in response to several external stimuli.
Deregulation of switching between these phenotypes is associated
with vascular disorders and atherosclerosis (AS). Under normal
conditions, platelet-derived growth factor (PDGF) induces multiple
aspects of the synthetic VSMC phenotype, characterized by increased
rates of proliferation and migration. Inversely, transforming
growth factor-β (TGF-β) and the related factor bone morphogenetic
protein 4 (BMP 4) promote the contractile VSMC phenotype,
characterized by decreased rates of proliferation and migration.
miRNAs reported to modulate the VSMC phenotype switching are shown.
KLF4/5, Krüppel-like factor 4/5; Pim-1, proviral integration site
1; CREG, cellular repressor of E1A-stimulated gene; SP-1,
specificity protein-1 transcription factor; ELK1, ELK1 members of
ETS oncogene family; FRA1, Fos-related antigene 1; ACE,
angiotensin-converting enzyme; CALMK, calmodulin K; MRTF-B,
myocardin related transcription factor B; Trb3, Trbbles-like
protein-3; p27, cyclin-dependent kinase inhibitor 1C (CDKN1C);
PDCD4, programmed cell death protein 4. |
7. miRNAs inhibit the proliferation of
VSMCs
As mentioned above, miR-1 expression is
significantly higher in more differentiated and less proliferative
VSMCs and is downregulated during neointima formation. miR-1
expression is higer in VSMCs treated with myocardin, which can
strongly inhibit the proliferation of VSMCs. The introduction of
miR-1 into VSMCs inhibits their proliferation by directly targeting
Pim-1 (21). Pim-1 has been
proven to encode an oncogenic serine/threonine kinase, which is
required for injury-induced neointima formation and SMC
proliferation (29). miR-15a is
upregulated in VSMCs treated with KLF4, which can strongly inhibit
the proliferation of VSMCs. Therefore, miR-15a contributes to the
anti-proliferative behaviors of KLF4, which has a strong inhibitory
effect on the proliferation of VSMCs (70).
In addition to the phenotypic regulation of VSMCs
mentioned earlier, miR-133 expression is decreased in proliferating
VSMCs and following vascular injury. The overexpression of miR-133
decreases VSMC proliferation and migration, while anti-miR-133 can
lead to an increased proliferation and migration by specifically
suppressing the expression of the transcription factor, Sp-1
(32).
As mentioned earlier, miR-143/145 expression is
downregulated following both vascular injury and PDGF-BB treatment.
The restoration of the expression of miR-143/145 inibits neointima
formation in the rat carotid artery following balloon injury
(37,71,72). The adenovirus-mediated gene
transfer of miR-145 in rat balloon-injured carotid arteries has
been proven to inhibit neointimal lesion formation (71).
miR-152 is downregulated in lipopolysaccharide
(LPS)-treated VSMCs. The overexpression of miR-152 decreases cell
proliferation in LPS-treated VSMCs. miR-152 exerts its effects by
downregulating DNA methyltransferase 1 (DNMT1) and subsequently
decreasing the methylation of ERα gene promoter region (73). DNMT1 is the most abundant DNA
methyltransferase in mammalian cells and is the key maintenance
enzyme for hemimethylated DNA during DNA replication of various
types of cancer (74).
miR-155 is specifically expressed in atherosclerotic
plaques, where it is induced by oxidized low-density lipoprotein
(ox-LDL) (75). However, the
circulating levels of miR-155 have been shown to be significantly
downregulated in patients with CAD compared to the controls
(33). Therefore, in vivo
studies on the role of miR-155 in AS are conflicting. miR-155 is
considered to contribute to the suppression of certain members of
the matrix metalloproteinase (MMP) family (76). MMPs are involved in the
atherosclerotic plaque formation by participating in ECM production
and cell migration. In particular, MMPs can destruct the existing
collagen that lends strength to the protective fibrous cap of
atherosclerotic plaques, which may lead to lethal thrombosis and
stenosis in AS.
miR-181a is downregulated by Ang-II, which plays a
central role in the pathogenesis of AS. miR-181a inhibits the
adhesion of VSMCs to collagen in response to Ang-II, which is
essential for migration and proliferation (77). The overexpression of miR-181a
inhibits Ang-II-mediated osteopontin (OPN) expression, which is a
pro-inflammatory molecule and is found in abundance in
atherosclerotic plaques.
miR-195 expression is altered in ox-LDL-treated
VSMCs. miR-195 inhibits ox-LDL-induced VSMC proliferation,
migration and synthesis the of IL-1β, IL-6, and IL-8. In addition,
the administration of miR-195 has been shown to substantially
reduce neointima formation, and thus plays a potential therapeutic
role (78). miR-195 can repress
the expression of the Cdc42, cyclin D1 (CCND1) and fibroblast
growth factor 1 (FGF1) genes, which are functionally related in the
same signalling pathway and are involved in cell cycle control,
migration and proliferation. Cdc42 is a downstream effector of
phosphoinositide 3-kinase (PI3K) and can prommote CCND1 (78) and FGF1 (80) expression. FGF1 has been reported
to be an important angiogenic factor that can induce VSMC
proliferation and migration (81,82).
miR-424, the ortholog of rat miR-322, is upregulated
in proliferative VSMCs and after vascular injury. However,
increased levels of miR-424/322 inhibit VSMC proliferation in
vitro and in injury-induced remodeling in vivo (83). In addition, the early and ample
introduction of miR-424/322 in vivo in balloon-injured
vessels has been shown to markedly reduce neointima formation in a
rat model of restenosis. Cyclin D1 and calumenin have been
confirmed to be the direct target genes of miR-424/322. Cyclin D1
has been proven to be a direct target of miR-424 in other tissues
(84). It has been demonstrated
that calumenin, a Ca2+-binding protein, is a regulator
of SERCA2a, which is able to inhibit its activity in cardiac
myocytes (85). miR-424/322 can
inhibit calumenin protein expression. This decrease can improve
SERCA2a activity and allow better Ca2+ cycling in
VSMCs.
miR-490-3p is significantly downregulated in in
ox-LDL-treated VSMCs and human AS plaques. miR-490-3p inhibits the
proliferation of VSMCs induced by ox-LDL by targeting
pregnancy-associated plasma protein A (PAPP-A) (86). PAPP-A, a metalloproteinase, is
abundantly expressed in VSMCs. PAPP-A can activate local
insulin-like growth factor (IGF). However, whether PAPP-A is pro-
or anti-atherosclerotic protein remains controversial since the
role of IGF in AS has not yet been fully elucidated.
miR-638, which is enriched in human aortic VSMCs, is
significantly downregulated in proliferative VSMCs. miR-638
inhibits PDGF-BB-induced VSMC proliferation and migration by
targeting neuron-derived orphan receptor 1 (NOR1) (87). NOR1 is a critical regulator
implicated in proliferative vascular diseases (88).
Let-7d is downregulated in proliferating VSMCs. The
overexpression of let-7d in VSMCs reduces VSMC growth by targeting
KRAS (89). KRAS, a member of the
small GTPase superfamily, regulates the pathway involved in
proliferation, differentiation and programmed cell death (90,91).
The human serum levels of let-7g are lower in
subjects with hypercholesterolemia compared with normal controls. A
lower level of let-7g has been observed in mice fed a high-fat diet
than in mice fed a normal chow diet. The tansfection of let-7g into
VSMCs has been shown to significantly inhibit VSMC proliferation
and migration induced by ox-LDL by targeting lectin-like
oxidized-low-density lipoprotein receptor-1 (LOX-1) (92). The role of LOX-1 in the
pathogenesis of vascular disorders has been well documented
(93). LOX-1 can be activated by
oxLDL and stimulates the expression of endothelial pro-inflammatory
genes and superoxide radical formation, which is believed to play
an active role in atherogenesis (94) (Fig.
2).
8. miRNAs regulate VSMC migration
In the native vessel, VSMCs are maintained in a
quiescent/contractile and non-migratory and non-proliferative
state, surrounded by a complex, highly structured ECM. In response
to vascular injury, such as platelet aggregation on the lining of
the vascular wall, or mechanical injury (ballooning with or without
stenting, VSMCs can migrate from the medial wall to the intimal
space, where they can proliferate and deposit ECM components,
converting fatty streak into mature fibrofatty atheroma. Therefore,
the migration of VSMCs plays a central role in the growth of AS
lesions. Similar to their proliferation, the migration of VSMCs is
regulated by growth promoters and inhibitors. PDGF-BB is one of the
most potent stimuli for the migration of VSMCs. VSMCs form a
structure known as podosomes when they migrate and invade. In fact,
the proliferation and migration of VSMCs are closely associated. An
increasing number of studies have found that miRs are involved in
this process (Table III and
Fig. 1).
| Table IIImiRs involved in the regulation of
VSMC migration. |
Table III
miRs involved in the regulation of
VSMC migration.
| Function | miRs | Targets | (Refs.) |
|---|
| Promote
migration | miR-26a | SMAD1/4 | (26) |
| miR-29b | DNMT3b | (52) |
| miR-133 | Sp-1 | (17) |
| Inhibit
migration | miR-143/145 | KLF5, ELK1,
myocardin | (41) |
| miR-181a | - | (45) |
| miR-195 | Cdc42, CCND1,
FGF1 | (46) |
| miR-638 | NDR1 | (49) |
| Let-7g | LOX-1 | (51) |
9. miRNAs promote the migration of
VSMCs
As mentioned earlier, miR-26a has multiple effects
on VSMC functions. We have discussed its role in phenotypic
regulation and proliferation. miR-26a can also promote the
migration of VSMCs. As previously shown, cells deficient in miR-26a
are less able to migrate towards a growth factor/serum gradient
compared with the control group (45).
The miR-29 family (i.e., miR-29a, miR-29b and
miR-29c) has been reported to be overexpressed in AS-related
diseases (95,96). More specifically, miR-29b
expression is upregulated in VSMCs treated with ox-LDL. miR-29b
promotes VSMC migration by directly targeting DNA methyltransferase
3b (DNMT3b) and epigenetically regulating the expression of
MMP-2/MMP-9 genes (97). The
MMP-2 and MMP-9 genes, which are involved in ECM remodeling, can
promote VSMC migration and contribute to neointima formation
(98).
10. miRNAs inhibit the migration of
VSMCs
As mentioned earlier, the overexpression of miR-133
results in decreased VSMC proliferation and migration, while
anti-miR-133 can lead to an increased proliferation and migration
(32).
miR-143/145 inhibit podosome formation, which is an
important morphological feature of VSMC migration in vitro
(99,100). miR-143/145 are downregulated in
VSMCs treated with PDGF-BB, which has been proven to induce VSMC
migration through podosome formation (42,72).
As mentioned above, miR-181a inhibits the adhesion
of VSMCs to collagen in response to Ang-II, which is essential for
migration and proliferation (77). miR-195 can inhibit VSMC
proliferation, migration and the synthesis of IL-1β, IL-6 and IL-8
induced by ox-LDL (78). miR-638,
which is significantly downregulated in proliferative VSMCs,
inhibits VSMC migration in response to PDGF-BB by targeting the
NOR1/cyclin D1 pathway (87).
Let-7g can significantly inhibit the oxLDL-induced LOX-1 and OCT-1
expression in VSMCs, as well as their proliferation and migration
(101).
11. miRNAs regulate VSMC apoptosis
Apoptosis, or programmed cell death, is a part of
normal development, senescence and other biological processes. The
apoptosis of VSMCs in the fibrous cap causes the thinning of the
fibrous cap and the expansion of the underlying necrotic core.
Therefore, the apoptosis of VSMCs plays a critical role in
determining plaque stability and thus, the most important
consequence of AS, i.e., plaque rupture. The apoptosis of VSMCs is
a tightly regulated process that contributes to atherosclerotic
plaque rupture and restenosis (102). Myocardial infarction mainly
occurs due to the uneven thinning and rupture of the fibrous cap
and is often initiated at the shoulders of the lesion where cell
apoptosis may occur (103). The
rupture sites of the atherosclerotic plaque lack VSMCs and are rich
in macrophages and inflammatory cells (104). The apoptosis of VSMCs has been
detected in the shoulder regions of atherosclerotic plaques, which
are most likely to rupture (105). VSMC apoptosis may be a potential
critical target for the development of therapies for unstable AS.
However, studies on the role of of miRs in the regulation of VSMC
apoptosis in AS are relatively limited (Table IV).
| Table IVmiRs involved in the regulation of
VSMC apoptosis, ECM synthesis and calcification. |
Table IV
miRs involved in the regulation of
VSMC apoptosis, ECM synthesis and calcification.
| Function | miRs | Targets | (Refs.) |
|---|
| Inhibit
apoptosis | miR-21 | PDCD4, Sp-1 | (30–32) |
| miR-26a | SMAD1/4 | (26) |
| Inhibit ECM
synthesis | miR-145 | - | (60) |
| Inhibited
calcification | miR-29a/b | ADAMTS-7 | (62) |
| miR-125b | Sp-7 | (63) |
In addition to its role in regulating phenotypic
changes and the proliferation of VSMCs, miR-21 has an
anti-apoptotic effect in cultured VSMCs. The inhibition of miR-21
has been proven to increase apoptosis in cultured VSMCs, as well as
in vivo in injured rat carotid arteries (53). Phosphatase and tensin homolog
(PTEN) and Bcl-2 are targets of miR-21. PTEN has been proven to
regulate VSMC apoptosis through the inhibition of Akt
phosphorylation/activation (106). Bcl-2, an anti-apoptotic protein,
inhibits apoptosis by preventing the release of cytochrome c
from the mitochondria and plays a role in the initiation and
progression of vascular cell apoptosis (107).
In addition to the regulation of the phenotype
switch, and the proliferation and migration of VSMCs, miR-26a can
also regulate the apoptosis of VSMCs. Cells deficient in miR-26a
display significantly increased rates of apoptosis, indicating that
miR-26a may inhibit the apoptosis of VSMCs (45). miR-26a has several possible target
genes associated with apoptosis, incuding Bcl-2-antagonist/killer 1
(BAK1), p21 protein (Cdc42/Rac)-activated kinase 2 (PAK2) and
sulfatase 1 (SULF1). However, further studies are is required to
specify its target gene.
12. miRNAs regulate ECM synthesis by
VSMCs
Under pathological conditions, VSMCs may undergo the
phenotypic change from the normal contractile state to a synthetic
state, in which the cells an increased ability to migrate,
proliferate and produce ECM proteins. Synthetic VSMCs can
synthesize ECM conssting of fibrillar collagens and elastins and
matrix-modifying enzymes that remodel the ECM. ECM production and
remodeling are necessary to form the neointima (108). However, studies on the effects
of miRs on ECM synthesis by VSMCs are limited (Table IV).
Lysyl oxidase (Lox), which can crosslink adjacent
collagen triple helices, confer tensile strength to collagen
fibrils and contribute to neointimal growth through the chemotactic
activity of VSMCs and monocytes (109,110), is negatively regulated by
miR-145. ApoE, which can inhibit ECM gene expression, reduces the
levels of Lox by increasing the expression of miR-145 (111).
13. miRNAs regulate VSMC calcification
Vascular calcification is defined as the deposition
of calcium phosphate mineral in the vessel wall. The exact role of
vascular calcification in plaque stability remains unclear. VSMCs
have been proven to significantly contribute to vascular
calcification, which is a prominent feature of AS (112). miRs have been reported to be
involved in the regulation of VSMC calcification (Table IV).
In addition to the role of miR-29b in promoting VSMC
migration, miR-29a/b is downregulated in calcifying VSMCs.
miR-29a/b inhibits VSMC calcification by suppressing the expression
of a disintegrin and metalloproteinase with thrombospondin motifs-7
(ADAMTS-7) (113). In injured
vessels, ADAMTS-7 has been proven to mediate the degradation of
cartilage oligomeric matrix protein (COMP) (114,115), which can inhibit VSMC
calcification in vitro and in vivo by interfering
with BMP 2 expression and preventing the osteochondrogenic
transdifferentiation of VSMCs (116).
miR-125b is significantly downregulated in calcified
vessels. miR-125b expression may be increased during
atherosclerotic inflammation processes since an increased
expression of miR-125b has been observed in ApoE−/− mice
fed a high-fat diet (117).
Furthermore, the inhibition of endogenous miR-125b promotes the
transdifferentiation of VSMCs into osteoblast-like cells. miR-125b
has been proven to be involved in vascular calcification by
targeting SP7 (osterix) (118).
SP7 is a zinc finger transcription factor expressed by osteoblasts
is necessary for osteoblast differentiation in mice (119).
14. Implications in the management of
AS
The regulation of specific miRs during neointima
formation in AS and restenosis suggests that miRs are key
determinants of the changes in the function of VSMCs. Altered
circulating levels of miRs in patients with AS may provide a novel
approach for the detection of CAD. For example, as the circulating
levels of miR-145 are lower in the serum of patients with CAD
(33), it may serve as a
biomarker for CAD. However, large-scale studies are required to
confirm the use of miRs as biomarkers for the diagnosis of disease.
Furthermore, miRs may also serve as potential targets for
therapeutic strategies since their expression can be altered
through genetic approaches. Since the modifications of miRs are
reversible, it is possible to use specific miRs to reduce VSMC
proliferation, migration and apoptosis, in order to prevent
neointima formation in AS and restenosis. For example, the
adenovirus-mediated gene transfer of miR-145 in rat balloon-injured
carotid arteries has been proven to inhibit neointimal lesion
formation. The knockdown of miR-221 and −222 in vessels reduces
VSMC proliferation and intimal thickening in response to vascular
injury (46). However, in
vivo data on the role of miRs in the regulation of VSMC
functions in AS and restenosis are limited. Further studies are
required to elucidate the underlying mechanisms of action of miRs
and their contribution to VSMC functions and AS in vivo.
15. Conclusions
In this review, we summarized the roles of miRs in
the function of VSMCs and their contributions to AS. miRs
critically affect VSMC functions, including differentiation,
proliferation, migration, ECM synthesis, calcification and
apoptosis, all of which play critical roles in the pathogenesis of
AS and restenosis. We also discussed the changes in miR expression
patterns in humans and animal models associated of AS. We focused
on the different functions of VSMCs to elucidate the mechanisms
through which miRs regulate each specific cellular function. miRs
exert their functions by regulating a number of target genes, such
as KLF4, p27 and Sp-1. The discovery of miR interference provides a
promising insight into the understanding of the regulation of gene
expression.
References
|
1
|
Lusis AJ: Atherosclerosis. Nature.
407:233–241. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gittenberger-de Groot AC, DeRuiter MC,
Bergwerff M and Poelmann RE: Smooth muscle cell origin and its
relation to heterogeneity in development and disease. Arterioscler
Thromb Vasc Biol. 19:1589–1594. 1999.PubMed/NCBI
|
|
3
|
Frid MG, Dempsey EC, Durmowicz AG and
Stenmark KR: Smooth muscle cell heterogeneity in pulmonary and
systemic vessels. Importance in vascular disease. Arterioscler
Thromb Vasc Biol. 17:1203–1209. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Schachter M: Vascular smooth muscle cell
migration, atherosclerosis, and calcium channel blockers. Int J
Cardiol. 62(Suppl 2): S85–S90. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Libby P, Sukhova G, Lee RT and Liao JK:
Molecular biology of atherosclerosis. Int J Cardiol. 62(Suppl 2):
S23–S29. 1997. View Article : Google Scholar
|
|
6
|
Schwartz SM: Smooth muscle migration in
atherosclerosis and restenosis. J Clin Invest. 100(Suppl): S87–S89.
1997.PubMed/NCBI
|
|
7
|
Doran AC, Meller N and McNamara CA: Role
of smooth muscle cells in the initiation and early progression of
atherosclerosis. Arterioscler Thromb Vasc Biol. 28:812–819. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Huntzinger E and Izaurralde E: Gene
silencing by microRNAs: contributions of translational repression
and mRNA decay. Nat Rev Genet. 12:99–110. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cho WC: OncomiRs: the discovery and
progress of microRNAs in cancers. Mol Cancer. 6:602007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Small EM and Olson EN: Pervasive roles of
microRNAs in cardiovascular biology. Nature. 469:336–342. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Song Z and Li G: Role of specific
microRNAs in regulation of vascular smooth muscle cell
differentiation and the response to injury. J Cardiovasc Transl
Res. 3:246–250. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zernecke A: MicroRNAs in the regulation of
immune cell functions-implications for atherosclerotic vascular
disease. Thromb Haemost. 107:626–633. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aghabozorg Afjeh SS and Ghaderian SM: The
role of microRNAs in cardiovascular disease. Int J Mol Cell Med.
2:50–57. 2013.PubMed/NCBI
|
|
14
|
Robinson HC and Baker AH: How do microRNAs
affect vascular smooth muscle cell biology? Curr Opin Lipidol.
23:405–411. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Owens GK, Kumar MS and Wamhoff BR:
Molecular regulation of vascular smooth muscle cell differentiation
in development and disease. Physiol Rev. 84:767–801. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stegemann JP, Hong H and Nerem RM:
Mechanical, biochemical, and extracellular matrix effects on
vascular smooth muscle cell phenotype. J Appl Physiol (1985).
98:2321–2327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Tang Y, Yang X, Friesel RE, Vary CP and
Liaw L: Mechanisms of TGF-β-induced differentiation in human
vascular smooth muscle cells. J Vasc Res. 48:485–494. 2011.
|
|
18
|
Millette E, Rauch BH, Kenagy RD, Daum G
and Clowes AW: Platelet-derived growth factor-BB transactivates the
fibroblast growth factor receptor to induce proliferation in human
smooth muscle cells. Trends Cardiovasc Med. 16:25–28. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raines EW: PDGF and cardiovascular
disease. Cytokine Growth Factor Rev. 15:237–254. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ross R: The pathogenesis of
atherosclerosis: a perspective for the 1990s. Nature. 362:801–809.
1993. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Chen J, Yin H, Jiang Y, et al: Induction
of microRNA-1 by myocardin in smooth muscle cells inhibits cell
proliferation. Arterioscler Thromb Vasc Biol. 31:368–375. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xie C, Huang H, Sun X, et al: MicroRNA-1
regulates smooth muscle cell differentiation by repressing
Kruppel-like factor 4. Stem Cells Dev. 20:205–210. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang Y, Yin H and Zheng XL: MicroRNA-1
inhibits myocardin-induced contractility of human vascular smooth
muscle cells. J Cell Physiol. 225:506–511. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Suzuki T, Aizawa K, Matsumura T and Nagai
R: Vascular implications of the Kruppel-like family of
transcription factors. Arterioscler Thromb Vasc Biol. 25:1135–1141.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu Y, Sinha S, McDonald OG, Shang Y,
Hoofnagle MH and Owens GK: Kruppel-like factor 4 abrogates
myocardin-induced activation of smooth muscle gene expression. J
Biol Chem. 280:9719–9727. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yoshida T, Kaestner KH and Owens GK:
Conditional deletion of Kruppel-like factor 4 delays downregulation
of smooth muscle cell differentiation markers but accelerates
neointimal formation following vascular injury. Circ Res.
102:1548–1557. 2008. View Article : Google Scholar
|
|
27
|
King KE, Iyemere VP, Weissberg PL and
Shanahan CM: Kruppel-like factor 4 (KLF4/GKLF) is a target of bone
morphogenetic proteins and transforming growth factor beta 1 in the
regulation of vascular smooth muscle cell phenotype. J Biol Chem.
278:11661–11669. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang C, Han M, Zhao XM and Wen JK:
Kruppel-like factor 4 is required for the expression of vascular
smooth muscle cell differentiation marker genes induced by
all-trans retinoic acid. J Biochem. 144:313–321. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Katakami N, Kaneto H, Hao H, et al: Role
of pim-1 in smooth muscle cell proliferation. J Biol Chem.
279:54742–54749. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang J, Yan CH, Li Y, et al: MicroRNA-31
controls phenotypic modulation of human vascular smooth muscle
cells by regulating its target gene cellular repressor of
E1A-stimulated genes. Exp Cell Res. 319:1165–1175. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Han Y, Deng J, Guo L, et al: CREG promotes
a mature smooth muscle cell phenotype and reduces neointimal
formation in balloon-injured rat carotid artery. Cardiovasc Res.
78:597–604. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Torella D, Iaconetti C, Catalucci D, et
al: MicroRNA-133 controls vascular smooth muscle cell phenotypic
switch in vitro and vascular remodeling in vivo. Circ Res.
109:880–893. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fichtlscherer S, De Rosa S, Fox H, et al:
Circulating microRNAs in patients with coronary artery disease.
Circ Res. 107:677–684. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Miano JM: Serum response factor: toggling
between disparate programs of gene expression. J Mol Cell Cardiol.
35:577–593. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Madsen CS, Hershey JC, Hautmann MB, White
SL and Owens GK: Expression of the smooth muscle myosin heavy chain
gene is regulated by a negative-acting GC-rich element located
between two positive-acting serum response factor-binding elements.
J Biol Chem. 272:6332–6340. 1997. View Article : Google Scholar
|
|
36
|
Deaton RA, Gan Q and Owens GK:
Sp1-dependent activation of KLF4 is required for PDGF-BB-induced
phenotypic modulation of smooth muscle. Am J Physiol Heart Circ
Physiol. 296:H1027–H1037. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cordes KR, Sheehy NT, White MP, et al:
miR-145 and miR-143 regulate smooth muscle cell fate and
plasticity. Nature. 460:705–710. 2009.PubMed/NCBI
|
|
38
|
Raitoharju E, Lyytikäinen LP, Levula M, et
al: miR-21, miR-210, miR-34a, and miR-146a/b are upregulated in
human atherosclerotic plaques in the Tampere Vascular Study.
Atherosclerosis. 219:211–217. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Hergenreider E, Heydt S, Tréguer K, et al:
Atheroprotective communication between endothelial cells and smooth
muscle cells through miRNAs. Nat Cell Biol. 14:249–256. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lovren F, Pan Y, Quan A, et al:
MicroRNA-145 targeted therapy reduces atherosclerosis. Circulation.
126(Suppl 1): S81–S90. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Boettger T, Beetz N, Kostin S, et al:
Acquisition of the contractile phenotype by murine arterial smooth
muscle cells depends on the Mir143/145 gene cluster. J Clin Invest.
119:2634–2647. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rangrez AY, Massy ZA, Metzinger-Le Meuth V
and Metzinger L: miR-143 and miR-145: molecular keys to switch the
phenotype of vascular smooth muscle cells. Circ Cardiovasc Genet.
4:197–205. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chan MC, Hilyard AC, Wu C, et al:
Molecular basis for antagonism between PDGF and the TGFbeta family
of signalling pathways by control of miR-24 expression. EMBO J.
29:559–573. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Chan MC, Nguyen PH, Davis BN, et al: A
novel regulatory mechanism of the bone morphogenetic protein (BMP)
signaling pathway involving the carboxyl-terminal tail domain of
BMP type II receptor. Mol Cell Biol. 27:5776–5789. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Leeper NJ, Raiesdana A, Kojima Y, et al:
MicroRNA-26a is a novel regulator of vascular smooth muscle cell
function. J Cell Physiol. 226:1035–1043. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu X, Cheng Y, Zhang S, Lin Y, Yang J and
Zhang C: A necessary role of miR-221 and miR-222 in vascular smooth
muscle cell proliferation and neointimal hyperplasia. Circ Res.
104:476–487. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Davis BN, Hilyard AC, Nguyen PH, Lagna G
and Hata A: Induction of microRNA-221 by platelet-derived growth
factor signaling is critical for modulation of vascular smooth
muscle phenotype. J Biol Chem. 284:3728–3738. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu X, Cheng Y, Yang J, Xu L and Zhang C:
Cell-specific effects of miR-221/222 in vessels: molecular
mechanism and therapeutic application. J Mol Cell Cardiol.
52:245–255. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Tanner FC, Yang ZY, Duckers E, Gordon D,
Nabel GJ and Nabel EG: Expression of cyclin-dependent kinase
inhibitors in vascular disease. Circ Res. 82:396–403. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Stewart MC, Kadlcek RM, Robbins PD,
MacLeod JN and Ballock RT: Expression and activity of the CDK
inhibitor p57Kip2 in chondrocytes undergoing hypertrophic
differentiation. J Bone Miner Res. 19:123–132. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Ashman LK: The biology of stem cell factor
and its receptor c-kit. Int J Biochem Cell Biol. 31:1037–1051.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Yang G, Pei Y, Cao Q and Wang R:
MicroRNA-21 represses human cystathionine gamma-lyase expression by
targeting at specificity protein-1 in smooth muscle cells. J Cell
Physiol. 227:3192–3200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ji R, Cheng Y, Yue J, et al: MicroRNA
expression signature and antisense-mediated depletion reveal an
essential role of microRNA in vascular neointimal lesion formation.
Circ Res. 100:1579–1588. 2007. View Article : Google Scholar
|
|
54
|
Davis BN, Hilyard AC, Lagna G and Hata A:
SMAD proteins control DROSHA-mediated microRNA maturation. Nature.
454:56–61. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Ozpolat B, Akar U, Steiner M, et al:
Programmed cell death-4 tumor suppressor protein contributes to
retinoic acid-induced terminal granulocytic differentiation of
human myeloid leukemia cells. Mol Cancer Res. 5:95–108. 2007.
View Article : Google Scholar
|
|
56
|
Cash AC and Andrews J: Fine scale analysis
of gene expression in Drosophila melanogaster gonads reveals
programmed cell death 4 promotes the differentiation of female
germline stem cells. BMC Dev Biol. 12:42012.
|
|
57
|
Curcio A, Torella D and Indolfi C:
Mechanisms of smooth muscle cell proliferation and endothelial
regeneration after vascular injury and stenting: approach to
therapy. Circ J. 75:1287–1296. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu X, Cheng Y, Chen X, Yang J, Xu L and
Zhang C: MicroRNA-31 regulated by the extracellular regulated
kinase is involved in vascular smooth muscle cell growth via large
tumor suppressor homolog 2. J Biol Chem. 286:42371–42380. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
An Y, Kang Q, Zhao Y, Hu X and Li N: Lats2
modulates adipocyte proliferation and differentiation via hippo
signaling. PloS One. 8:e720422013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Wu WH, Hu CP, Chen XP, et al:
MicroRNA-130a mediates proliferation of vascular smooth muscle
cells in hypertension. Am J Hypertens. 24:1087–1093. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Weir L, Chen D, Pastore C, Isner JM and
Walsh K: Expression of gax, a growth arrest homeobox gene, is
rapidly downregulated in the rat carotid artery during the
proliferative response to balloon injury. J Biol Chem.
270:5457–5461. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Xia S, Tai X, Wang Y, et al: Involvement
of Gax gene in hypoxia-induced pulmonary hypertension,
proliferation, and apoptosis of arterial smooth muscle cells. Am J
Respir Cell Mol Biol. 44:66–73. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Saito T, Itoh H, Yamashita J, et al:
Angiotensin II suppresses growth arrest specific homeobox (Gax)
expression via redox-sensitive mitogen-activated protein kinase
(MAPK). Regul Pept. 127:159–167. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Gorski DH, LePage DF, Patel CV, Copeland
NG, Jenkins NA and Walsh K: Molecular cloning of a diverged
homeobox gene that is rapidly downregulated during the G0/G1
transition in vascular smooth muscle cells. Mol Cell Biol.
13:3722–3733. 1993.PubMed/NCBI
|
|
65
|
Yamashita J, Itoh H, Ogawa Y, et al:
Opposite regulation of Gax homeobox expression by angiotensin II
and C-type natriuretic peptide. Hypertension. 29:381–387. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Witzenbichler B, Kureishi Y, Luo Z, Le
Roux A, Branellec D and Walsh K: Regulation of smooth muscle cell
migration and integrin expression by the Gax transcription factor.
J Clin Invest. 104:1469–1480. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Wassmann S, Wassmann K, Jung A, et al:
Induction of p53 by GKLF is essential for inhibition of
proliferation of vascular smooth muscle cells. J Mol Cell Cardiol.
43:301–307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Sun SG, Zheng B, Han M, et al: miR-146a
and Krüppel-like factor 4 form a feedback loop to participate in
vascular smooth muscle cell proliferation. EMBO Rep. 12:56–62.
2011.
|
|
69
|
Zhang Y, Wang Y, Wang X, et al: Insulin
promotes vascular smooth muscle cell proliferation via
microRNA-208-mediated downregulation of p21. J Hypertens.
29:1560–1568. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Zheng X, Li A, Zhao L, et al: Key role of
microRNA-15a in the KLF4 suppressions of proliferation and
angiogenesis in endothelial and vascular smooth muscle cells.
Biochem Biophys Res Commun. 437:625–631. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Cheng Y, Liu X, Yang J, et al:
MicroRNA-145, a novel smooth muscle cell phenotypic marker and
modulator, controls vascular neointimal lesion formation. Circ Res.
105:158–166. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Quintavalle M, Elia L, Condorelli G and
Courtneidge SA: MicroRNA control of podosome formation in vascular
smooth muscle cells in vivo and in vitro. J Cell Biol. 189:13–22.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Wang YS, Chou WW, Chen KC, Cheng HY, Lin
RT and Juo SH: MicroRNA-152 mediates DNMT1-regulated DNA
methylation in the estrogen receptor α gene. PloS One.
7:e306352012.PubMed/NCBI
|
|
74
|
Li E, Bestor TH and Jaenisch R: Targeted
mutation of the DNA methyltransferase gene results in embryonic
lethality. Cell. 69:915–926. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Huang RS, Hu GQ, Lin B, Lin ZY and Sun CC:
MicroRNA-155 silencing enhances inflammatory response and lipid
uptake in oxidized low-density lipoprotein-stimulated human THP-1
macrophages. J Investig Med. 58:961–967. 2010.PubMed/NCBI
|
|
76
|
Ma X, Ma C and Zheng X: MicroRNA-155 in
the pathogenesis of atherosclerosis: a conflicting role? Heart Lung
Circ. 22:811–818. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Remus EW, Lyle AN, Weiss D, et al: miR181a
protects against angiotensin II-induced osteopontin expression in
vascular smooth muscle cells. Atherosclerosis. 228:168–174. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Wang YS, Wang HY, Liao YC, et al:
MicroRNA-195 regulates vascular smooth muscle cell phenotype and
prevents neointimal formation. Cardiovasc Res. 95:517–526. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Ammit AJ and Panettieri RA Jr: Invited
review: the circle of life: cell cycle regulation in airway smooth
muscle. J Appl Physiol (1985). 91:1431–1437. 2001.PubMed/NCBI
|
|
80
|
Chotani MA, Touhalisky K and Chiu IM: The
small GTPases Ras, Rac, and Cdc42 transcriptionally regulate
expression of human fibroblast growth factor 1. J Biol Chem.
275:30432–30438. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Lindner V and Reidy MA: Proliferation of
smooth muscle cells after vascular injury is inhibited by an
antibody against basic fibroblast growth factor. Proc Natl Acad Sci
USA. 88:3739–3743. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Hanna AK, Fox JC, Neschis DG, Safford SD,
Swain JL and Golden MA: Antisense basic fibroblast growth factor
gene transfer reduces neointimal thickening after arterial injury.
J Vasc Surg. 25:320–325. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Merlet E, Atassi F, Motiani RK, et al:
miR-424/322 regulates vascular smooth muscle cell phenotype and
neointimal formation in the rat. Cardiovasc Res. 98:458–468. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Liu Q, Fu H, Sun F, et al: miR-16 family
induces cell cycle arrest by regulating multiple cell cycle genes.
Nucleic acids Res. 36:5391–5404. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Sahoo SK, Kim T, Kang GB, Lee JG, Eom SH
and Kim do H: Characterization of calumenin-SERCA2 interaction in
mouse cardiac sarcoplasmic reticulum. J Biol Chem. 284:31109–31121.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Sun Y, Chen D, Cao L, et al: miR-490-3p
modulates the proliferation of vascular smooth muscle cells induced
by ox-LDL through targeting PAPP-A. Cardiovasc Res. 100:272–279.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Li P, Liu Y, Yi B, et al: MicroRNA-638 is
highly expressed in human vascular smooth muscle cells and inhibits
PDGF-BB-induced cell proliferation and migration through targeting
orphan nuclear receptor NOR1. Cardiovasc Res. 99:185–193. 2013.
View Article : Google Scholar
|
|
88
|
Bonta PI, Pols TW and de Vries CJ: NR4A
nuclear receptors in atherosclerosis and vein-graft disease. Trends
Cardiovasc Med. 17:105–111. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Yu ML, Wang JF, Wang GK, et al: Vascular
smooth muscle cell proliferation is influenced by let-7d microRNA
and its interaction with KRAS. Circ J. 75:703–709. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Doe Z, Fukumoto Y, Takaki A, et al:
Evidence for Rho-kinase activation in patients with pulmonary
arterial hypertension. Circ J. 73:1731–1739. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zhou YC and Waxman DJ: Cross-talk between
janus kinase-signal transducer and activator of transcription
(JAK-STAT) and peroxisome proliferator-activated receptor-alpha
(PPARalpha) signaling pathways. Growth hormone inhibition of
pparalpha transcriptional activity mediated by stat5b. J Biol Chem.
274:2672–2681. 1999.
|
|
92
|
Chen KC, Hsieh IC, Hsi E, et al: Negative
feedback regulation between microRNA let-7g and the oxLDL receptor
LOX-1. J Cell Sci. 124:4115–4124. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Mehta JL, Chen J, Hermonat PL, Romeo F and
Novelli G: Lectin-like, oxidized low-density lipoprotein receptor-1
(LOX-1): a critical player in the development of atherosclerosis
and related disorders. Cardiovasc Res. 69:36–45. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Mehta JL and Li DY: Identification and
autoregulation of receptor for OX-LDL in cultured human coronary
artery endothelial cells. Biochem Biophys Res Commun. 248:511–514.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Tan KS, Armugam A, Sepramaniam S, et al:
Expression profile of MicroRNAs in young stroke patients. PloS One.
4:e76892009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Urbich C, Kuehbacher A and Dimmeler S:
Role of microRNAs in vascular diseases, inflammation, and
angiogenesis. Cardiovasc Res. 79:581–588. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Chen KC, Wang YS, Hu CY, et al: OxLDL
upregulates microRNA-29b, leading to epigenetic modifications of
MMP-2/MMP-9 genes: a novel mechanism for cardiovascular diseases.
FASEB J. 25:1718–1728. 2011. View Article : Google Scholar
|
|
98
|
Aoyagi M, Yamamoto M, Azuma H, et al:
Immunolocalization of matrix metalloproteinases in rabbit carotid
arteries after balloon denudation. Histochem Cell Biol. 109:97–102.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Gimona M, Kaverina I, Resch GP, Vignal E
and Burgstaller G: Calponin repeats regulate actin filament
stability and formation of podosomes in smooth muscle cells. Mol
Biol Cell. 14:2482–2491. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Linder S and Aepfelbacher M: Podosomes:
adhesion hot-spots of invasive cells. Trends Cell Biol. 13:376–385.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Kunugi S, Iwabuchi S, Matsuyama D, Okajima
T and Kawahara K: Negative-feedback regulation of ATP release: ATP
release from cardiomyocytes is strictly regulated during ischemia.
Biochem Biophys Res Commun. 416:409–415. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Bennett MR: Apoptosis of vascular smooth
muscle cells in vascular remodelling and atherosclerotic plaque
rupture. Cardiovasc Res. 41:361–368. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Fuster V: Lewis A. Conner Memorial
Lecture. Mechanisms leading to myocardial infarction: insights from
studies of vascular biology. Circulation. 90:2126–2146.
1994.PubMed/NCBI
|
|
104
|
Davies MJ, Richardson PD, Woolf N, Katz DR
and Mann J: Risk of thrombosis in human atherosclerotic plaques:
role of extracellular lipid, macrophage, and smooth muscle cell
content. Br Heart J. 69:377–381. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Geng YJ and Libby P: Evidence for
apoptosis in advanced human atheroma. Colocalization with
interleukin-1 beta-converting enzyme. Am J Pathol. 147:251–266.
1995.PubMed/NCBI
|
|
106
|
Sedding DG, Widmer-Teske R, Mueller A, et
al: Role of the phosphatase PTEN in early vascular remodeling. PloS
One. 8:e554452013. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Geng YJ: Molecular signal transduction in
vascular cell apoptosis. Cell Res. 11:253–264. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Clowes AW, Clowes MM, Fingerle J and Reidy
MA: Regulation of smooth muscle cell growth in injured artery. J
Cardiovasc Pharmacol. 14(Suppl 6): S12–S15. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Li W, Liu G, Chou IN and Kagan HM:
Hydrogen peroxide-mediated, lysyl oxidase-dependent chemotaxis of
vascular smooth muscle cells. J Cell Biochem. 78:550–557. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Lazarus HM, Cruikshank WW, Narasimhan N,
Kagan HM and Center DM: Induction of human monocyte motility by
lysyl oxidase. Matrix Biol. 14:727–731. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Kothapalli D, Liu SL, Bae YH, et al:
Cardiovascular protection by ApoE and ApoE-HDL linked to
suppression of ECM gene expression and arterial stiffening. Cell
Rep. 2:1259–1271. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Iyemere VP, Proudfoot D, Weissberg PL and
Shanahan CM: Vascular smooth muscle cell phenotypic plasticity and
the regulation of vascular calcification. J Intern Med.
260:192–210. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Du Y, Gao C, Liu Z, et al: Upregulation of
a disintegrin and metalloproteinase with thrombospondin motifs-7 by
miR-29 repression mediates vascular smooth muscle calcification.
Arterioscler Thromb Vasc Biol. 32:2580–2588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Wang L, Zheng J, Bai X, et al: ADAMTS-7
mediates vascular smooth muscle cell migration and neointima
formation in balloon-injured rat arteries. Circ Res. 104:688–698.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Liu CJ, Kong W, Ilalov K, et al: ADAMTS-7:
a metalloproteinase that directly binds to and degrades cartilage
oligomeric matrix protein. FASEB J. 20:988–990. 2006. View Article : Google Scholar
|
|
116
|
Du Y, Wang Y, Wang L, et al: Cartilage
oligomeric matrix protein inhibits vascular smooth muscle
calcification by interacting with bone morphogenetic protein-2.
Circ Res. 108:917–928. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Villeneuve LM, Kato M, Reddy MA, Wang M,
Lanting L and Natarajan R: Enhanced levels of microRNA-125b in
vascular smooth muscle cells of diabetic db/db mice lead to
increased inflammatory gene expression by targeting the histone
methyltransferase Suv39h1. Diabetes. 59:2904–2915. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Goettsch C, Rauner M, Pacyna N, Hempel U,
Bornstein SR and Hofbauer LC: miR-125b regulates calcification of
vascular smooth muscle cells. Ame J Pathol. 179:1594–1600. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Zhu F, Friedman MS, Luo W, Woolf P and
Hankenson KD: The transcription factor osterix (SP7) regulates
BMP6-induced human osteoblast differentiation. J Cell Physiol.
227:2677–2685. 2012. View Article : Google Scholar : PubMed/NCBI
|