Introduction
Cigarette smoke has long been recognized as a risk
factor associated with lung cancer. However, the underlying
molecular mechanisms through which cigarette smoke induces lung
carcinogenesis remain unclear.
Increasing evidence indicates that tumor suppressor
genes play important roles in cigarette carcinogenesis. Nuclear
receptor subfamily 1, group D member 1 (Nr1d1), also known as
Rev-erb-α, a candidate tumor suppressor controlling cell
proliferation, lipid metabolism and inflammation, has been found to
be downregulated by cigarette smoke (1). Glypican 3, a
glycosylphosphatidylinositol-linked heparan sulfate proteoglycan,
functioning as a tumor suppressor by inhibiting cell growth and
inducing apoptosis, is decreased in lung adenocarcinoma and in the
normal lungs of smokers compared with the normal lungs of
non-smokers (2). A number of the
tumor suppressor genes are hypermethylated in the malignant lung
tissues of smokers. Deleted in liver cancer 1 (DLC-1), homologous
to RhoGAP, has been described as a novel tumor suppressor gene. A
previous study demonstrated a significant association between DLC-1
methylation and cigarette smoke (3). The aberrant methylation of other
tumor suppressor genes, such as p16 and fragile histidine triad
(FHIT), is also found in smokers (4,5).
Activating enhancer-binding protein 2 (AP-2), a
eukaryotic transcriptional factor, plays a pivotal role in normal
development and morphogenesis during embryogenesis. The AP-2 family
consists of 5 different isoforms, namely AP-2α, AP-2β, AP-2γ, AP-2δ
and AP-2ɛ (6). All AP-2 family
members share a highly conserved helix-span-helix dimerization
motif, a central basic region and a less conserved proline-rich
domain. The AP-2 proteins can form homo- or heterodimers and
transactivate target DNA. AP-2α can regulate a n umber of
tumor-related genes, such as p21 (7) human epidermal growth factor receptor
2 (HER-2) (8) and Bcl-2 (9). AP-2α expression is reduced in many
types of tumor tissue. The decreased expression of AP-2α has been
shown to significantly correlate with increased tumorigenic
potential and poor prognosis (10,11), suggesting that AP-2α is a
candidate tumor suppressor.
Previous studies have identified AP-2 as an
important transcriptional regulator of CYP11A1, a member of the
cytochrome P450 family, which is an important metabolizing enzyme
of cigarette smoke (12,13). Nicotine can suppress AP-2α
expression and its target DNA binding activity through the
activation of peroxisome proliferator-activated receptor (PPAR), a
key regulator of cancer cell proliferation (14). These findings prompted us to study
the association between AP-2α and cigarette smoke.
In the present study, we examined the role of AP-2α
in cigarette smoke and found that the presence of cigarette smoke
condensate (CSC) suppressed AP-2α expression in human lung cancer
cell lines. AP-2α promoter activity was markedly decreased in the
presence of CSC. Furthermore, the overexpression or silencing of
cellular p53 substantially affected the CSC-induced suppression of
AP-2α.
Materials and methods
Cell culture and transfection
NCI-H1299, NCI-H446 and A549 human lung cancer cell
lines were routinely cultured in complete medium containing
RPMI-1640 + 10% fetal bovine serum (FBS). One day prior to
transfection, the cells were seeded in 60-mm culture disks to reach
80–90% confluence at transfection. Cell transfection was performed
using Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The
transfection medium was substituted by complete medium 5 h after
transfection.
CSC preparation and treatment
CSC was prepared as previously described (15). Briefly, cigarette smoke (from a
popular type of cigarette, Honghe, 15 mg tar, 1.2 mg nicotine) was
condensed in a 2-litre flask submerged in liquid nitrogen. The
weight increase of the flask was representative of the amount of
the smoke condensate. The condensate was dissolved in dimethyl
sulfoxide (DMSO) up to 20 mg/ml, which was aliquoted and stored at
−80°C. When used for the experiments, CSC was diluted in complete
medium to the desired concentration. Control treatment was
performed with complete medium containing an equivalent amount of
DMSO.
Plasmids and siRNA
The promoter of AP-2α spanning nucleotides −1728 to
+286 was cloned from genomic DNA, as previously described (16). The p53-SN3 plasmid, which
expresses wild-type p53 driven by a cytomegalovirus (CMV) promoter,
was a kind gift from B. Vogelstein (Ohio State University,
Columbus, OH, USA). AP-2-LUC, a luciferase reporter downstream of 3
repeats of AP-2 binding sites, was kindly provided by Carlo M.
Croce (Ohio State University). p53-specific siRNA or scrambled
siRNA were purchased from Santa Cruz Biotechnology (Santa Cruz, CA,
USA).
RNA extraction and quantitative reverse
transcription PCR (RT-qPCR)
Total RNA was extracted using TRIzol reagent
(Invitrogen) according to the manufacturer’s instructions.
Following quantification, 5 μg of total RNA were
reverse-transcribed into cDNA using the SuperScript™ First-Strand
Synthesis System (Invitrogen). Quantitative PCR (qPCR) was
performed using ABI PRISM 7000 (Applied Biosystems, Foster City,
CA, USA) and the SYBR® Premix Ex Taq™ II kit (Takara,
Dalian, China). The detailed procedure followed the manufacturer’s
manual. The specific primer pairs used for qPCR were as follows:
5′-GATCCTCGCAGGGACTACAG-3′ (sense) and 5′-TACCCGGGTCTTCTACATGC-3′
for AP-2α; 5′-TGGTCACCAGGGCTGCTT-3′ (sense) and 5′-AGCTT
CCCGTTCTCAGCCTT-3′ for GAPDH. GAPDH was used as an internal
control. The AP-2α relative expression levels were calculated using
the ΔΔCT method. Changes in AP-2α expression levels were expressed
as a percentage change compared to the corresponding control
(defined as 100%). All experiments were performed in triplicate and
repeated thrice.
Protein preparation and western blot
analysis
Total proteins were extracted using modified RIPA
buffer and quantified using Bradford assay (Bio-Rad Laboratories,
Hercules, CA, USA). Total proteins (40 μg) were loaded and run on a
10% SDS-PAGE gel. Following electrophoresis, the proteins were
semi-dry transferred onto nitrocellulose membranes. After blocking
in 5% non-fat milk and washing with PBS with 0.1% Tween-20, the
membranes were incubated with the appropriately diluted primary
antibodies against AP-2α or β-actin (all from Santa Cruz
Biotechnology) at 4°C overnight. Following incubation with
HRP-conjugated secondary antibody, the HRP signal was detected
using Super ECL Plus Detection reagent (Applygen Technologies Inc.,
Beijing, China) and exposed to X-ray film.
Luciferase reporter assay
The Dual-Luciferase® Reporter Assay
System (Promega, Madison, WI, USA) was used to detect AP-2α
promoter activity. Briefly, the cells were co-transfected with the
test plasmids or the empty pGL3-Basic vector and pRL-SV40 as
controls (Promega). Following transfection and CSC treatment, the
cells were washed in PBS and harvested using passive lysis buffer.
The enzyme activities of firefly luciferase and Renilla
luciferase were measured sequentially using the
Dual-Luciferase® Reporter Assay System (Promega). The
ratio of firefly luciferase levels to Renilla luciferase
levels was used as the relative luciferase activity. Changes in
luciferase activity were expressed as percentage change compared to
the corresponding control (defined as 100%). All experiments were
performed in triplicate and repeated thrice.
Statistical analysis
All qPCR results are presented as the means ±
standard error. The Student’s t test was used to identify
significant differences between groups. Statistical significance
was set at P<0.05.
Results
CSC inhibits AP-2α expression in lung
cancer cell lines
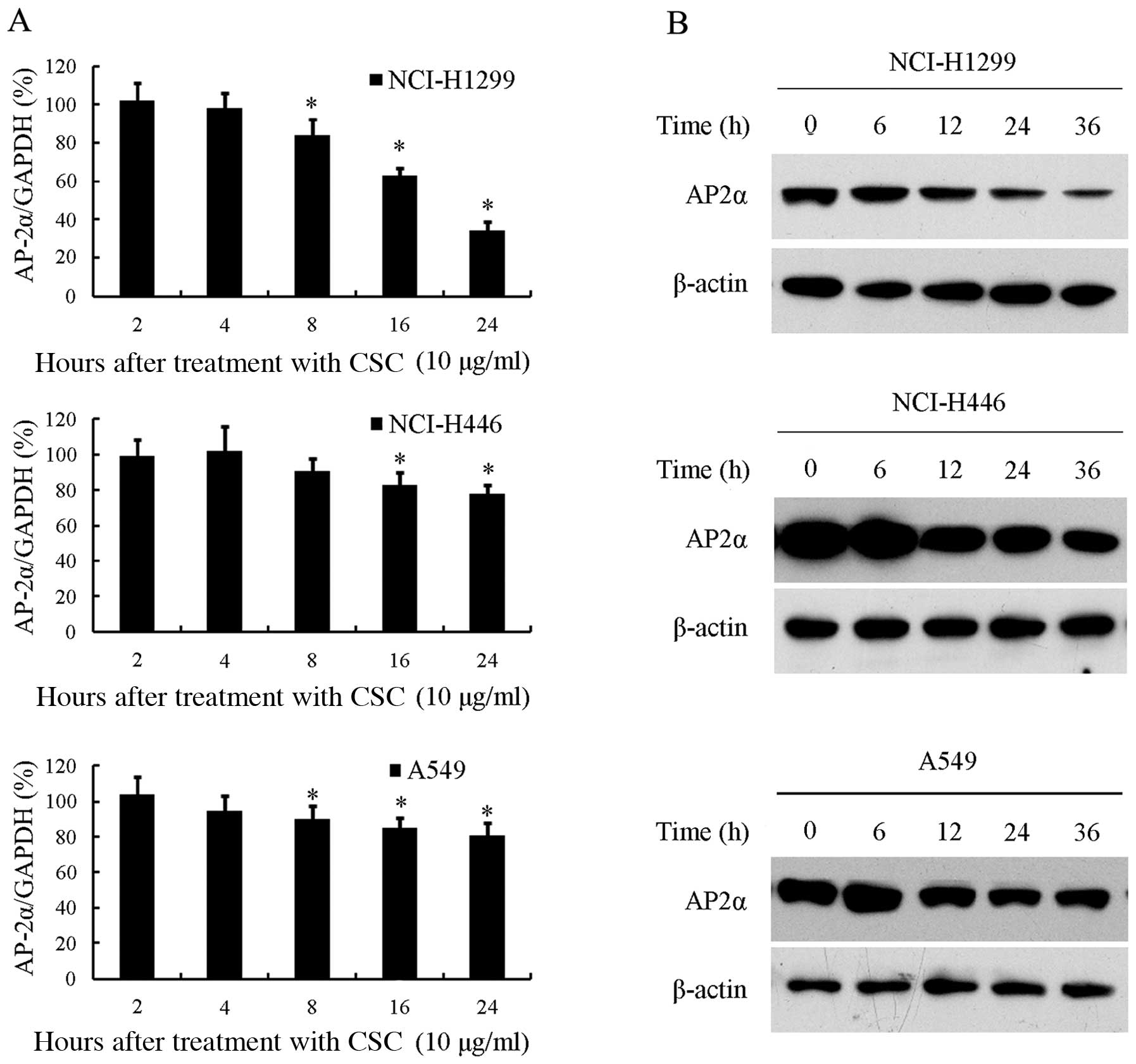
We first investigated the effects of CSC on AP-2α
mRNA expression. Three human lung cancer cell lines NCI-H1299,
NCI-H446 and A549, were cultured to 70% confluence and then
incubated with 10 μg/ml CSC or DMSO for different periods of time.
RT-qPCR was used to detect the mRNA levels of AP-2α. As shown in
Fig. 1A, CSC significantly
decreased AP-2α mRNA expression in the NCI-H1299 cells in a
time-dependent manner. AP-2α mRNA expression slightly decreased
after 8 h of exposure to CSC and then remained decreased until it
reached its lowest level at 24 h, at which time the AP-2α level was
decreased to 34% compared to the untreated controls (P<0.05).
AP-2α mRNA expression was also inhibited by exposure to CSC in the
NCI-H446 and A549 cells (P<0.05), although to a much lesser
extent. DMSO treatment did not alter the AP-2α mRNA expression
levels (data not shown).
Subsequently, we examined AP-2α protein expression
following incubation with CSC. In parallel to the mRNA results, we
also found that AP-2α protein epxression was markedly inhibited by
CSC, but not by DMSO (data not shown), in the NCI-H1299 cells, and
to a much lesser extent in the NCI-H446 and A549 cells (Fig. 1B).
CSC inhibits the transactivating ability
of AP-2α, but does not affect its subcellular localization
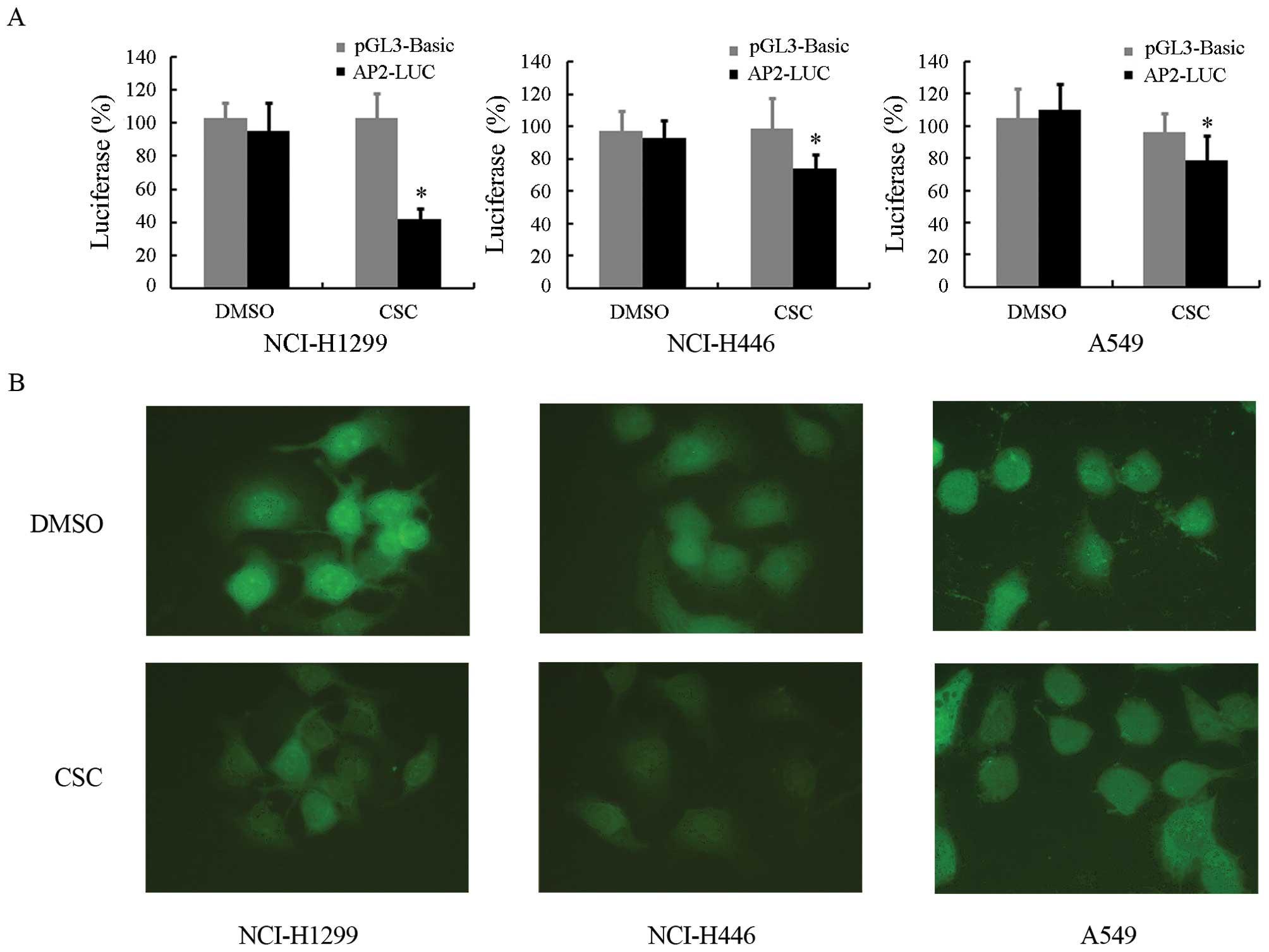
To evaluate the functional changes of AP-2α which
occur due to smoke, we examined the transactivating ability of
AP-2α following exposure to CSC. The NCI-H1299, NCI-H446 and A549
cells were transfected with the empty vector, pGL3-basic, or
AP-2-LUC, a luciferase reporter downstream of 3 repeats of AP-2
binding sites. At 24 h after transfection, the cells were incubated
with 10 μg/ml CSC or DMSO. After a further 24 h of incubation,
luciferase activity was detected. As shown in Fig. 2A, the luciferase activity of
AP-2-LUC in the NCI-H1299 cells was reduced by CSC, but not by
DMSO, while no significant changes in pGL3-basic luciferase
activity were observed in the presence of CSC or DMSO. Similar
results were also observed in the NCI-H446 and A549 cells, but not
as profoundly as in the NCI-H1299 cells. The results coincided with
the observations that CSC downregulated the expression of
AP-2α.
To exclude the possibility that the decreased
transactivating ability of AP-2α induced by CSC may result from the
changes in its subcellular localization, we examined the
subcellular localization of AP-2α following exposure of the cells
to CSC by immunofluorescence. As shown in Fig. 2B, AP-2α was mainly localized in
the nucleus under normal growth conditions. Treatment with 24 h
with 10 μg/ml CSC did not alter its subcellular localization;
however, its expression levels were decreased, which is in
agreement with the above results.
CSC suppresses the promoter activity of
AP-2α
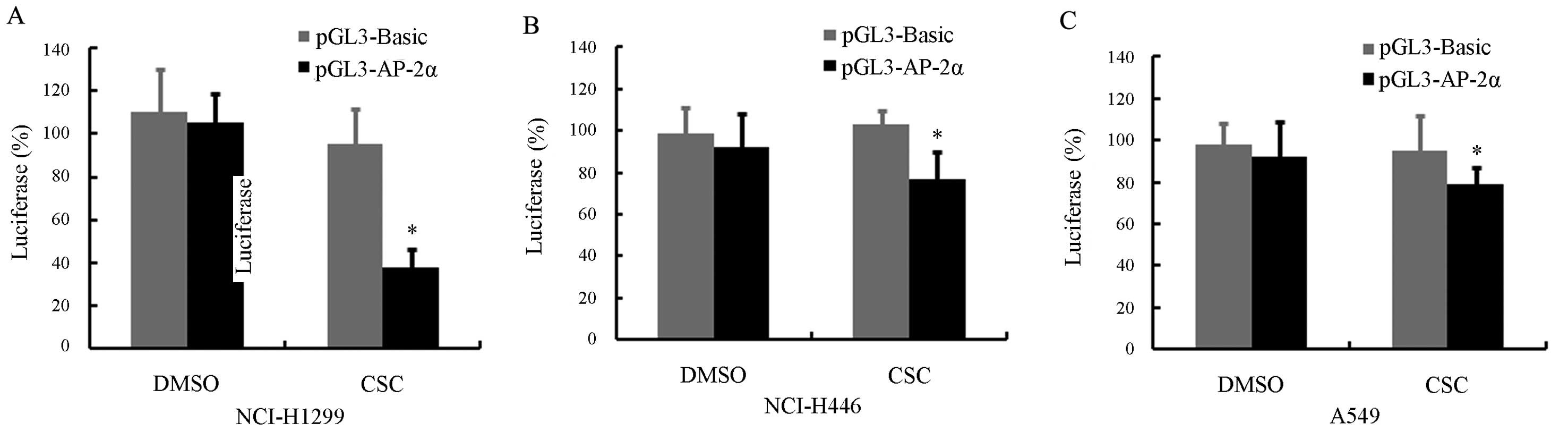
To determine whether the CSC-induced downregulation
of AP-2α occurs at its transcriptional level, we constructed a
luciferase reporter plasmid, pGL3-AP-2α, containing a putative
AP-2α promoter spanning from position −1728 to +286. The NCI-H1299
cells were transfected with pGL3-AP-2α or the control plasmid,
pGL3-basic, and 24 h later the cells were treated with 10 μg/ml CSC
or DMSO for 24 h. Unsurprisingly, CSC treatment significantly
reduced the ability of the AP-2α promoter to drive the luciferase
expression in the NCI-H1299 cells (P<0.05) (Fig. 3A). However, the inhibitory effect
was not pronounced, but statistically significant, in the NCI-H446
and A549 cells (Fig. 3B and
C).
Transfection of p53 inhibits the
CSC-induced downregulation of AP-2α by rescuing its promoter
activity in NCI-H1299 cells
The above-mentioned results showed a differential
effect of CSC on different human lung cancer cell lines. The
NCI-H1299 cell line was more sensitive to the CSC-induced
inhibition of AP-2α expression. As is already known, NCI-H1299 is a
p53-deficient cell line, whereas NCI-H446 and A549 cells harbor
wild-type p53. This prompted us to examine whether p53 plays a role
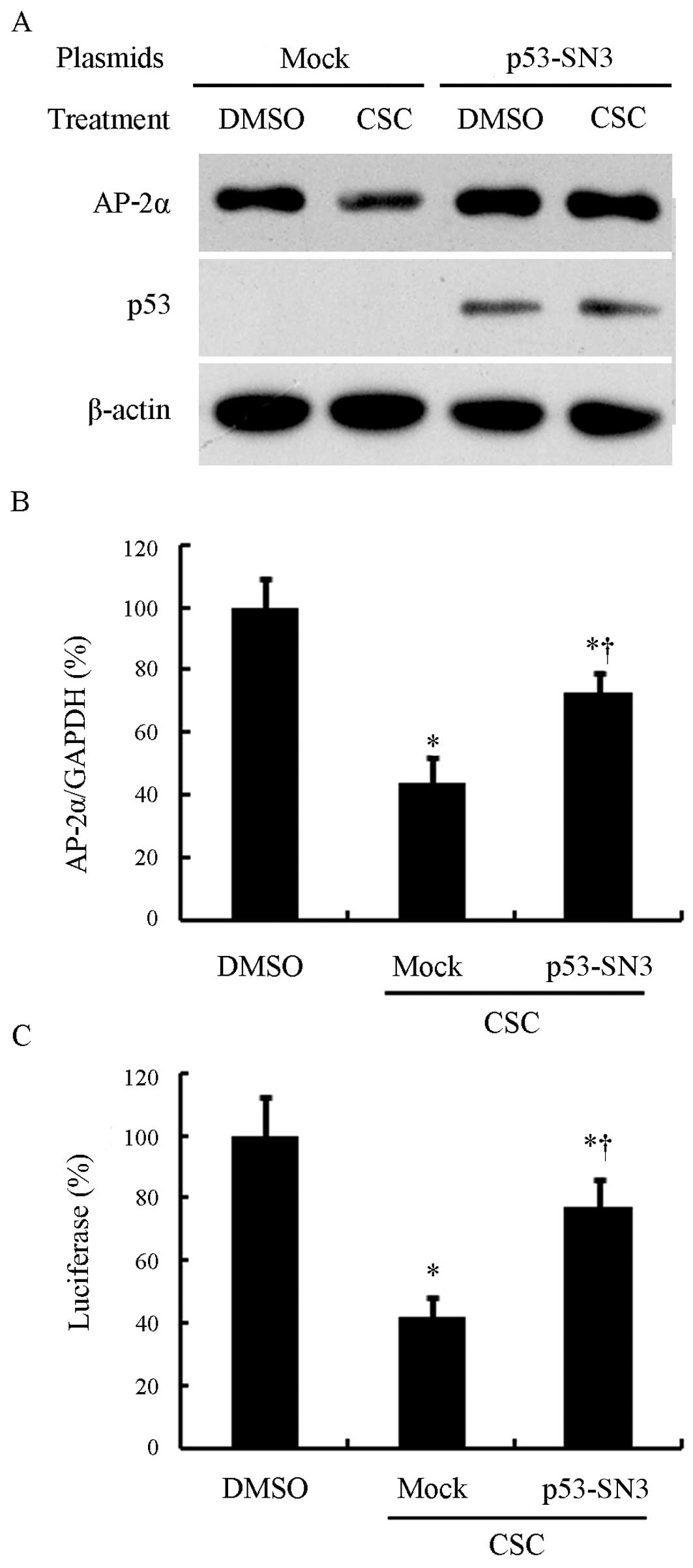
in the CSC-mediated downregulation of AP-2α. Therefore, we restored
p53 expression in the NCI-H1299 cells and investigated its effects
on the CSC-induced downregulation of AP-2α. The wild-type p53
plasmid, p53-SN3, or the empty vector, pCMV, was transiently
transfected into the NCI-H1299 cells. After 24 h, the cells were
incubated with 10 μg/ml CSC or DMSO for 24 h. As shown in Fig. 4A, in the mock-transfected cells
(emtpy vector), we did not detect any p53 expression, while p53
expression was clearly detected following transfection with
p53-SN3. As expected, p53 transfection attenuated the loss of AP-2α
induced by exposure to CSC. We further analyzed AP-2α mRNA levels.
As observed in the above-mentioned experiments, AP-2α mRNA
expression was markedly inhibited by exposure to CSC in both the
mock-transfected and p53-SN3-transfected cells (P<0.05).
However, the p53-transfected cells showed a much higher AP-2α
expression compared with the mock-transfected cells (P<0.05)
(Fig. 4B).
Subsequently, we investigated the effects of p53
transfection on AP-2α promoter activity following exposure to CSC.
pGL3-AP-2α and the control plasmid, phRL-SV40, were co-transfected
into the NCI-H1299 cells with p53-SN3 or the empty vector, pCMV.
After 24 h, the cells were incubated with 10 μg/ml CSC or DMSO for
24 h and the luciferase activity was determined. As expected, p53
partially reversed the suppression of the AP-2α promoter induced by
CSC, while the mock-transfected cells still showed a significant
loss of AP-2α following treatment with CSC (Fig. 4C).
p53 silencing enhances the CSC-induced
downregulation of AP-2α in A549 cells
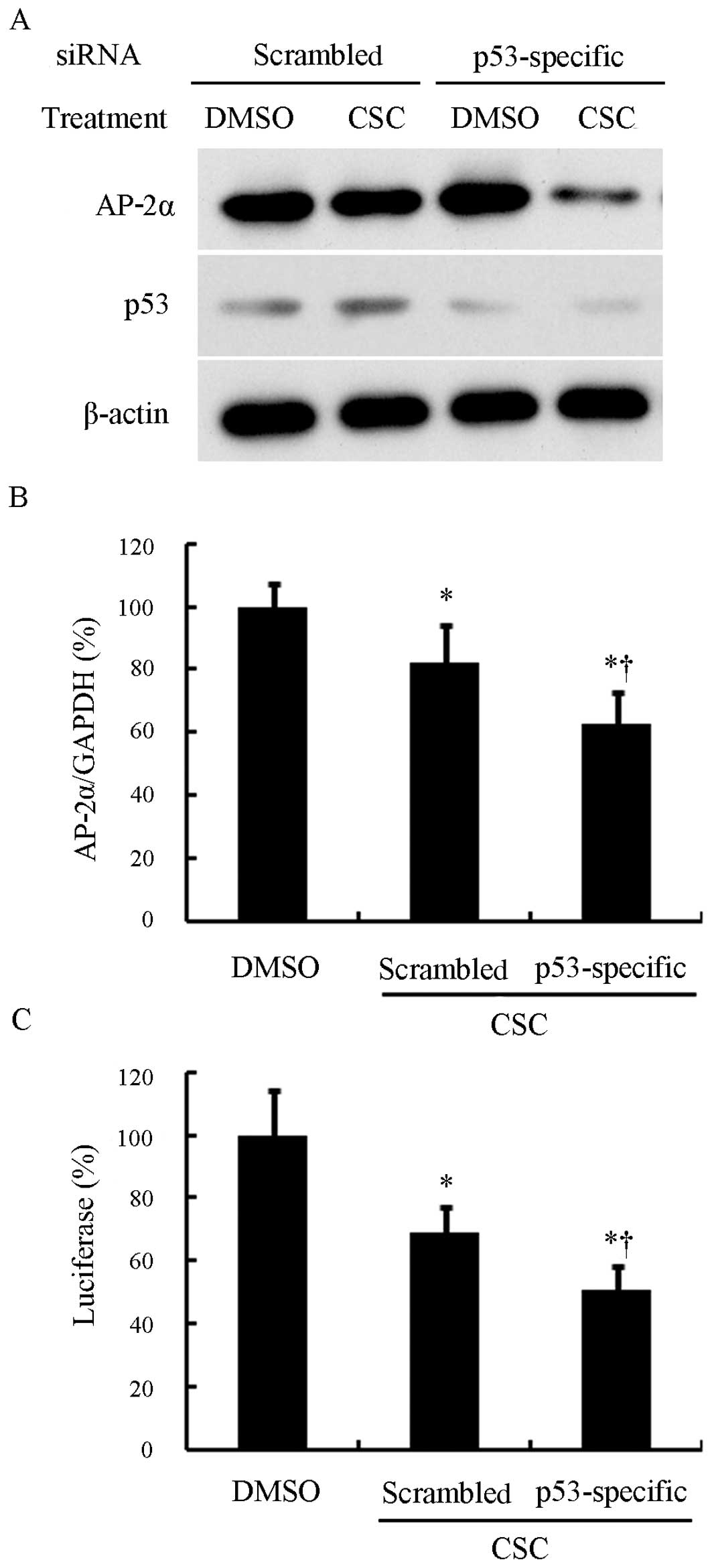
To further confirm the involvement of p53 in the
downregulation of AP-2α by CSC, we silenced p53 in the A549 lung
cancer cells using p53-specific siRNA (Fig. 5A). As expected, following
treatment with CSC, the p53-specific siRNA-transfected cells and
scrambled siRNA-transfected cells both showed a decrease in the
AP-2α mRNA expression level (P<0.05) (Fig. 5B). However, the p53-specific
siRNA-transfected cells expressed much lower levels of AP-2α than
the scrambled siRNA-transfected cells following treatment with CSC
(P<0.05) (Fig. 5B). In
addition, the silencing of p53 intensified the inhibition of the
AP-2α promoter induced by CSC (Fig.
5C). These results suggest a counteracting role of p53 in the
CSC-induced AP-2α downregulation.
Discussion
Cigarette smoke is a complex mixture of which more
than 4,800 compounds have been identified (17), over 55 of which have been
implicated in lung cancer, such as
4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK),
benzo[a]pyrene (B[a]P) and acrolein (18,19). Polycyclic aromatic hydrocarbons
(PAHs), one of the most studied tobacco toxins, is present in the
combustion products of cigarette smoke and have long been
considered to be an important carcinogen responsible for the
initiation and development of lung cancer (20). PAHs can be activated by cytochrome
P450 enzymes to form its metabolite, diol epoxides, which induce
DNA mutation by forming covalent adducts with DNA (21).
Studies using DNA microarray have revealed that
cigarette smoke can induce significant changes in the
transcriptome. Spira et al (22), using high-density gene expression
arrays, found that the normal airway transcriptome consists of more
than 7,000 measurable genes, while smoking alters the expression of
numerous genes. Although the levels of most of the susceptible
genes return to normal after smoking cessation, several potential
oncogenes (such as CEACAM6 and HN1) are permanently increased and
tumor suppressor genes (such as TU3A and CX3CL1) are permanently
decreased. Another study, using serial analysis of gene expression
(SAGE) also verified many reversible and irreversible gene
expression changes upon the cessation of smoking and the
irreversible changes may lead to persistent lung cancer risk
despite smoking cessation (23).
In the present study, we demonstrated that AP-2α, one of the
putative tumor suppressors, was downregulated by CSC in lung cancer
cell lines, suggesting the involvement of the loss of AP-2α in
smoking-related lung cancer development.
It has long been suspected that the loss of AP-2 may
be associated with tumorigenesis. Early investigations on primary
invasive breast cancers demonstrated that AP-2 expression is
reduced in the majority of tumors, suggesting that AP-2 acts as a
tumor suppressor during breast carcinogenesis (24). Similar findings have also been
reported for prostate and colon cancers. The enforced AP-2
overexpression in melanoma and prostate cancer cell lines has been
shown to lead to reduced tumorigenicity (25,26). Functional studies have indicated
that AP-2α can bind as a homo- or heterodimer to the motif
‘GCCNNNGGC’ in the regulatory regions of its target genes. After
binding, AP-2α can activate the transcription of target genes
(27). In melanoma, AP-2α may
function as a tumor suppressor by directly activating the
transcription of downstream genes, such as c-KIT, E-cadherin,
protease activated receptor 1 (PAR-1), matrix metalloproteinase-2
(MMP-2) and vascular endothelial growth factor (VEGF) (28). AP-2α can also act as
transcriptional repressor. A previous study demonstrated that AP-2α
competes with SP1 for binding to the CYP17 gene promoter to repress
its transcription (29).
Epigenetic studies have revealed that AP-2α can recruit histone
deacetylase (HDAC) to the promoter regions of target genes, thus
silencing these genes (30).
Another study demonstrated that frequent methylation regions in
acute lymphoblastic leukemia contain multiple AP-2α binding sites,
suggesting a potential role of AP-2α in epigenetic regulation
(31).
To determine at which level AP-2α downregulation
occurs following exposure to CSC, we performed luciferase reporter
assays and revealed a reduced promoter activity of AP-2α by CSC
treatment. These findings suggest that the CSC-induced
downregulation of AP-2α occurs at the transcriptional level. To
date, the mechanisms underlying the transcriptional regulation of
AP-2 expression remain obscure. A previous study investigated the
transcriptional regulation of the AP-2α gene promoter and found
that many nuclear proteins can bind to the AP-2α promoter (32). The authors also found that BTEB-1
and AP-2rep are two transcriptional regulators of AP-2α. BTEB-1
acts as a positive regulator, while the function of AP-2rep depends
on its binding cis-elements. Another study identified an
enhancer in the AP-2α promoter, which contains an Ets-1 binding
site (16). Strikingly, AP-2α is
subjected to autoregulation since an AP-2α binding site exists in
the AP-2α promoter region (33).
AP-2 expression is also regulated at the post-transcriptional
level. A previous study demonstrated that the AP-2α protein is
unusually stable in HER-2-positive breast cancer cell lines, which
results from the insufficient proteasomal-degradation of AP-2α
(34).
As the changes in AP-2α levels were more pronounced
in p53-deficient NCI-H1299 cells than in NCI-H446 and A549 cells
which harbor wild-type p53, we transfected a wild-type p53
expression plasmid into the NCI-H1299 cells to examine the changes
in the levels of AP-2α following exposure of the cells to CSC. The
results revealed that the p53 restoration antagonized the effects
of the CSC-induced decrease in AP-2α expression. In addition, we
observed an enhanced ability of CSC to decrease AP-2α expression in
the A549 cells silenced by p53 siRNA. All these findings suggest
that p53 antagonizes CSC-induced AP-2α downregulation. It has
previously been demonstrated that AP-2α and AP-2γ are
transcriptionally regulated by p53 (35). p53 activates AP-2α and AP-2α
transcription by binding to and remodeling their promoters
(35). Although we did not
observe an obvious increase in the AP-2α level following p53
transfection, p53 restoration can counteract the CSC-induced AP-2α
downregulation. Other studies have reported that AP-2α can also
regulate the transcriptional activity of p53 through co-activation
and decreased stability (36).
The overexpression of AP-2α can repress p53-mediated p21 activation
(36). All these findings suggest
a reciprocal regulation between p53 and AP-2α.
Taken together, the results of the present study
provide new mechanistic information as to the mechanisms through
which cigarette smoke exerts cytotoxic and tumorigenic effects on
lung cells. However, given the complexity of cigarette smoke, it is
difficult to identify the specific AP-2α-killing culprits. Further
studies are required in order to screen potential cigarette
toxins.
Acknowledgements
We are thankful to B. Vogelstein and Carlo M. Croce
for providing the plasmids, p53-SN3 and AP-2-LUC, respectively.
This study was supported by the National Natural Science Foundation
of China (NSFC; 81100703) and the Fundamental Research Funds for
Jilin University (201103082).
References
|
1
|
Vasu VT, Cross CE and Gohil K: Nr1d1, an
important circadian pathway regulatory gene, is suppressed by
cigarette smoke in murine lungs. Integr Cancer Ther. 8:321–328.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim H, Xu GL, Borczuk AC, et al: The
heparan sulfate proteoglycan GPC3 is a potential lung tumor
suppressor. Am J Respir Cell Mol Biol. 29:694–701. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Yuan BZ, Jefferson AM, Baldwin KT,
Thorgeirsson SS, Popescu NC and Reynolds SH: DLC-1 operates as a
tumor suppressor gene in human non-small cell lung carcinomas.
Oncogene. 23:1405–1411. 2004.PubMed/NCBI
|
|
4
|
Georgiou E, Valeri R, Tzimagiorgis G, et
al: Aberrant p16 promoter methylation among Greek lung cancer
patients and smokers: correlation with smoking. Eur J Cancer Prev.
16:396–402. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim JS, Kim H, Shim YM, Han J, Park J and
Kim DH: Aberrant methylation of the FHIT gene in chronic smokers
with early stage squamous cell carcinoma of the lung.
Carcinogenesis. 25:2165–2171. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tummala R, Romano RA, Fuchs E and Sinha S:
Molecular cloning and characterization of AP-2 epsilon, a fifth
member of the AP-2 family. Gene. 321:93–102. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li H, Goswami PC and Domann FE: AP-2gamma
induces p21 expression, arrests cell cycle, and inhibits the tumor
growth of human carcinoma cells. Neoplasia. 8:568–577.
2006.PubMed/NCBI
|
|
8
|
Pellikainen J, Naukkarinen A, Ropponen K,
et al: Expression of HER2 and its association with AP-2 in breast
cancer. Eur J Cancer. 40:1485–1495. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wajapeyee N, Britto R, Ravishankar HM and
Somasundaram K: Apoptosis induction by activator protein 2alpha
involves transcriptional repression of Bcl-2. J Biol Chem.
281:16207–16219. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jean D, Gershenwald JE, Huang S, Luca M,
Hudson MJ, Tainsky MA and Bar-Eli M: Loss of AP-2 results in
up-regulation of MCAM/MUC18 and an increase in tumor growth and
metastasis of human melanoma cells. J Biol Chem. 273:16501–16508.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Karjalainen JM, Kellokoski JK, Eskelinen
MJ, Alhava EM and Kosma VM: Downregulation of transcription factor
AP-2 predicts poor survival in stage I cutaneous malignant
melanoma. J Clin Oncol. 16:3584–3591. 1998.PubMed/NCBI
|
|
12
|
Guo IC, Hu MC and Chung BC:
Transcriptional regulation of CYP11A1. J Biomed Sci. 10:593–598.
2003.PubMed/NCBI
|
|
13
|
Pena P, Reutens AT, Albanese C, et al:
Activator protein-2 mediates transcriptional activation of the
CYP11A1 gene by interaction with Sp1 rather than binding to DNA.
Mol Endocrinol. 13:1402–1416. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Sun X, Ritzenthaler JD, Zhong X, Zheng Y,
Roman J and Han S: Nicotine stimulates PPARbeta/delta expression in
human lung carcinoma cells through activation of PI3K/mTOR and
suppression of AP-2alpha. Cancer Res. 69:6445–6453. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yuan J, Ma J, Zheng H, et al:
Overexpression of OLC1, cigarette smoke, and human lung
tumorigenesis. J Natl Cancer Inst. 100:1592–1605. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cheng YH and Handwerger S: Identification
of an enhancer of the human activating protein-2alpha gene that
contains a critical Ets1 binding site. J Clin Endocrinol Metab.
88:3305–3311. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hoffmann D, Hoffmann I and El-Bayoumy K:
The less harmful cigarette: a controversial issue. a tribute to
Ernst L Wynder. Chem Res Toxicol. 14:767–790. 2001.PubMed/NCBI
|
|
18
|
Feng Z, Hu W, Hu Y and Tang MS: Acrolein
is a major cigarette-related lung cancer agent: preferential
binding at p53 mutational hotspots and inhibition of DNA repair.
Proc Natl Acad Sci USA. 103:15404–15409. 2006. View Article : Google Scholar
|
|
19
|
Pfeifer GP, Denissenko MF and Tang M: p53
mutations, benzo[a]pyrene and lung cancer: a reply. Mutagenesis.
13:537–538. 1998.
|
|
20
|
Hecht SS, Carmella SG, Murphy SE, Foiles
PG and Chung FL: Carcinogen biomarkers related to smoking and upper
aerodigestive tract cancer. J Cell Biochem Suppl. 17F:27–35. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Smith LE, Denissenko MF, Bennett WP, Li H,
Amin S, Tang M and Pfeifer GP: Targeting of lung cancer mutational
hotspots by polycyclic aromatic hydrocarbons. J Natl Cancer Inst.
92:803–811. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Spira A, Beane J, Shah V, et al: Effects
of cigarette smoke on the human airway epithelial cell
transcriptome. Proc Natl Acad Sci USA. 101:10143–10148. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chari R, Lonergan KM, Ng RT, MacAulay C,
Lam WL and Lam S: Effect of active smoking on the human bronchial
epithelium transcriptome. BMC Genomics. 8:2972009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Gee JM, Robertson JF, Ellis IO, Nicholson
RI and Hurst HC: Immunohistochemical analysis reveals a tumour
suppressor-like role for the transcription factor AP-2 in invasive
breast cancer. J Pathol. 189:514–520. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nyormoi O and Bar-Eli M: Transcriptional
regulation of metastasis-related genes in human melanoma. Clin Exp
Metastasis. 20:251–263. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ruiz M, Pettaway C, Song R, Stoeltzing O,
Ellis L and Bar-Eli M: Activator protein 2alpha inhibits
tumorigenicity and represses vascular endothelial growth factor
transcription in prostate cancer cells. Cancer Res. 64:631–638.
2004. View Article : Google Scholar
|
|
27
|
Liu R, Zhou A, Ren D, et al: Transcription
factor specificity protein 1 (SP1) and activating protein 2alpha
(AP-2alpha) regulate expression of human KCTD10 gene by binding to
proximal region of promoter. FEBS J. 276:1114–1124. 2009.
View Article : Google Scholar
|
|
28
|
Hilger-Eversheim K, Moser M, Schorle H and
Buettner R: Regulatory roles of AP-2 transcription factors in
vertebrate development, apoptosis and cell-cycle control. Gene.
260:1–12. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang G and Veldhuis JD: Requirement for
proximal putative Sp1 and AP-2 cis-deoxyribonucleic acid elements
in mediating basal and luteinizing hormone- and insulin-dependent
in vitro transcriptional activation of the CYP17 gene in porcine
theca cells. Endocrinology. 145:2760–2766. 2004. View Article : Google Scholar
|
|
30
|
Bennett KL, Romigh T and Eng C: AP-2alpha
induces epigenetic silencing of tumor suppressive genes and
microsatellite instability in head and neck squamous cell
carcinoma. PLoS One. 4:e69312009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Taylor KH, Pena-Hernandez KE, Davis JW, et
al: Large-scale CpG methylation analysis identifies novel candidate
genes and reveals methylation hotspots in acute lymphoblastic
leukemia. Cancer Res. 67:2617–2625. 2007. View Article : Google Scholar
|
|
32
|
Imhof A, Schuierer M, Werner O, Moser M,
Roth C, Bauer R and Buettner R: Transcriptional regulation of the
AP-2alpha promoter by BTEB-1 and AP-2rep, a novel wt-1/egr-related
zinc finger repressor. Mol Cell Biol. 19:194–204. 1999.PubMed/NCBI
|
|
33
|
Bauer R, Imhof A, Pscherer A, et al: The
genomic structure of the human AP-2 transcription factor. Nucleic
Acids Res. 22:1413–1420. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li M, Wang Y, Hung MC and Kannan P:
Inefficient proteasomal-degradation pathway stabilizes AP-2alpha
and activates HER-2/neu gene in breast cancer. Int J Cancer.
118:802–811. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Li H, Watts GS, Oshiro MM, Futscher BW and
Domann FE: AP-2alpha and AP-2gamma are transcriptional targets of
p53 in human breast carcinoma cells. Oncogene. 25:5405–5415. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Stabach PR, Thiyagarajan MM, Woodfield GW
and Weigel RJ: AP2alpha alters the transcriptional activity and
stability of p53. Oncogene. 25:2148–2159. 2006. View Article : Google Scholar : PubMed/NCBI
|