Introduction
The endoplasmic reticulum (ER) is the cellular
organelle in which membrane and secretory proteins are synthesized,
glycosylated and acquire their correct conformation. While the
properly folded proteins can leave the ER and traffick to their
final destination along the secretary pathway, proteins that fail
to fold are retrotranslocated to the cytoplasm for degradation, a
process that is referred to as ER-associated degradation (ERAD)
(1). ERAD is initiated by
substrate recognition in the ER, followed by retrotranslocation
into the cytoplasm for ubiquitin-mediated proteasomal degradation.
For ERAD to occur properly, a machinery that can recognize
misfolded proteins is required. While ER degradation-enhancing
α-mannosidase-like proteins (EDEMs) were initially considered
lectins (2,3), recent studies have revealed that
EDEMs can function as mannosidases (4,5)
and molecular chaperones (6). We
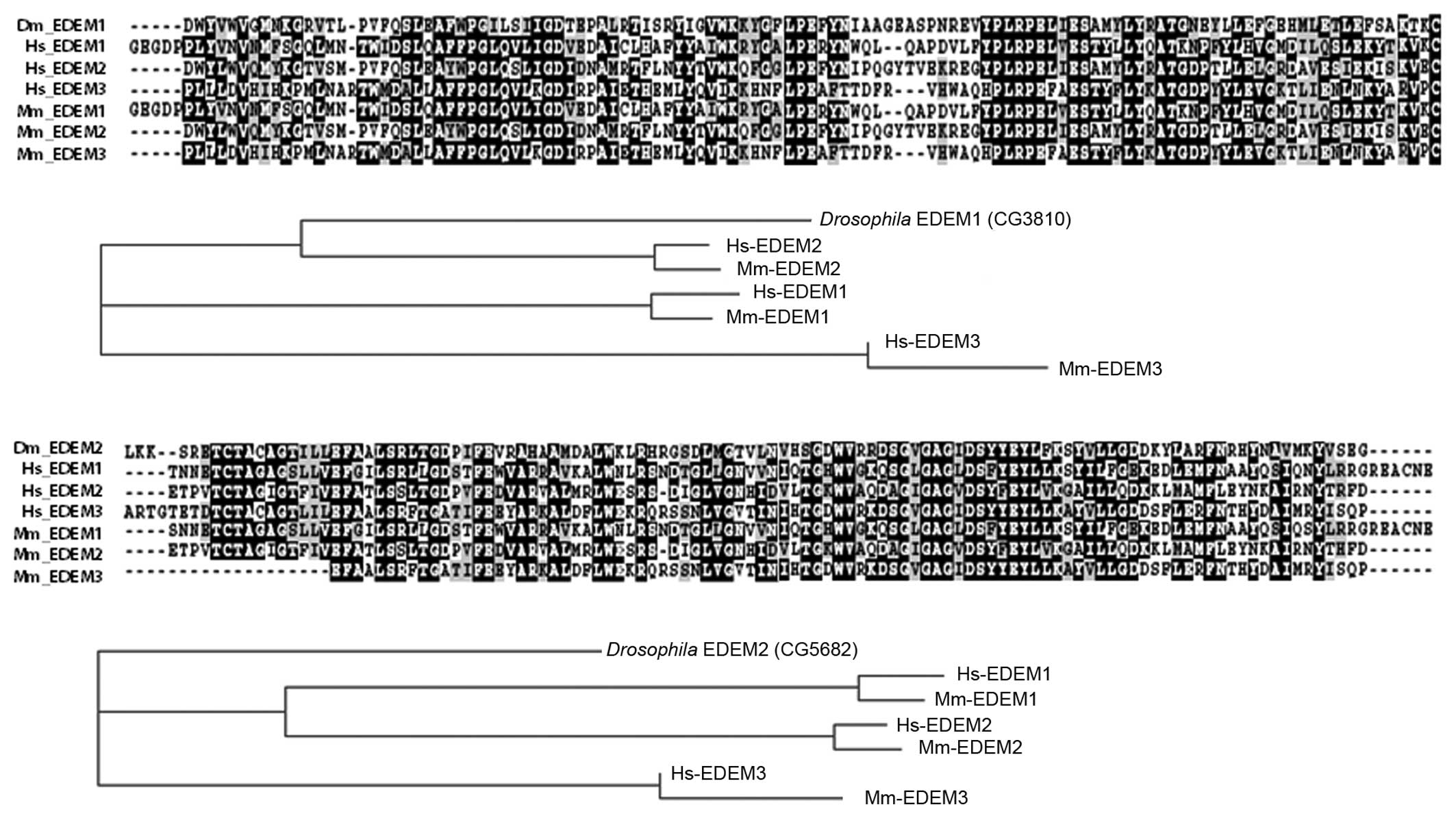
have previously reported that the Drosophila genome encodes
2 EDEM homologs, EDEM1 (CG3810) and EDEM2 (CG5682) (14). Sequence analysis indicated that
whereas Drosophila EDEM1 was similar to human EDEM2,
Drosophila EDEM2 showed a closer resemblance to EDEM3 in
mammals (Fig. 1).
Proteins that misfold in the ER underlie a number of
conformational diseases in humans. Among these diseases is
alpha-1-antitrypsin (A1AT) deficiency, which is caused by mutations
in the A1AT gene that impairs its protein folding properties during
biogenesis. The classical form of mutant A1AT protein is
alpha-1-antitrypsin Z (ATZ) that results from a Glu342Lys
substitution, rendering it prone to polymerization and aggregation
(7). Misfolded ATZ is rapidly
cleared from cells, through a combination of ERAD (8–10)
and autophagy (11–13).
Previously, we established a Drosophila model
to study how A1AT is degraded through ERAD (14). In that previous study, we had
focused on the rare null Hong Kong (NHK) allele (2,3,15),
and we had shown that the overexpression of Drosophila EDEM2
promotes ERAD of NHK. In this study, we performed a more in-depth
investigation of Drosophila EDEMs, focusing on the
predominant disease allele Z. Specifically, we demonstrate that the
two Drosophila EDEMs play redundant roles in the degradation
of the Z allele. We also demonstrate that the knockdown of these
two genes leads to the accumulation of glycosylated ATZ proteins,
while the overexpression of EDEMs promotes the degradation of ATZ.
In addition, we provide evidence of A1AT ubiquitination, using
cell-based ubiquitination assays.
Materials and methods
Plasmids and fly stocks
Genes were expressed in Drosophila through
the standard GAL4/UAS system (16). The following flies and DNA have
been described previously: armadillo-GAL4 (17), uas-myc-EDEM1 (14), uas-myc-EDEM2 (14) and uas-NHK (14). The DNA encoding ATZ (18) was subcloned into a pUAST
plasmid.
Cell culture and RNAi treatment
Drosophila Schneider 2 (S2) cells were grown
in Schneider’s medium supplemented with 10% fetal bovine serum and
1% penicillin/streptomycin (Invitrogen, Grand Island, NY, USA).
Treatment with double-stranded RNA (dsRNA) was performed as
described in a previous study (19). Briefly, the cells were plated into
6-well plates at a density of 1×106 cells/well before
treatment with dsRNA (day 1). After 24 h, 20 μg of EGFP,
EDEM1 or EDEM2 dsRNA were added to each well following another
boost with 20 μg dsRNA on day 4. The cells were then
transiently transfected with either NHK or ATZ using Effectene™
(Qiagen, Valencia, CA, USA) on day 5. The cells were split on day 8
and lysed to examine the level of NHK or ATZ on day 9. The EDEM1
dsRNA consisted of a 516-nt region (Amplicon ID: DRSC18573), as
described by the Drosophila RNAi Screening Center
(http://www.flyrnai.org). The following primers
were used to amplify this sequence from an embryo cDNA library: ‘R’
primer, CAATGTTGTCACCCACGAAA; ‘S’ primer, TCGAAGTT GCTTACTAACAGA.
This amplicon has no predicted off-targets. The EDEM2 dsRNA
consisted of a 520-nucleotide region (amplicon ID: DRSC02877). The
following primers were used to amplify this sequence from an embryo
cDNA library: ‘R’ primer, 5′-CATGCGCGGGTTAAT CTC-3′; ‘S’ primer,
5′-GATAGAGCATCTCGTGTGTC-3′. To induce ER stress, Drosophila
S2 cells were treated with dithiothreitol (DTT; Cat. no. 43815;
Sigma, St. Louis, MO, USA) or thapsigargin (Tg; Cat. no. T9033;
Sigma) for the indicated periods of time.
Immunohistochemistry
All fluorescent images were captured under a Zeiss
LSM 510 confocal microscope, using x100 objective lenses. The
following antibodies were used: guinea pig anti-Hsc3 antibody
(1:500) as previously described (17), mouse anti-myc (1:1,000 for
immunohistochemistry; 9E10; Cat. no. 11667149001; Roche Diagnostics
GmbH, Mannheim, Germany), rhodamine red anti-mouse secondary
antibody (Cat. no. 715-295-150; 1:500) and FITC anti-guinea pig
secondary antibody (Cat. no. 706-095-148; 1:500) (both from Jackson
ImmunoResearch, West Grove, PA, USA).
Western blot analysis and
immunoprecipitation
For western blot analyses, Drosophila S2
cells were extracted with 1% SDS lysis buffer (10 mM Tris pH 7.5, 1
mM EDTA, 150 mM NaCl and 1% SDS; Sigma). Following centrifugation
at 16,100 x g for 10 min, the supernatants were fractionated by
SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF)
membranes (Millipore, Billerica, MA, USA). The membranes were then
probed with the indicated antibodies: polyclonal rabbit anti-A1AT
(1:5,000 for western blot analysis; Cat. no. A0012; Dako, Glostrup,
Denmark), mouse anti-profilin (1:2,000 for western blot analysis;
Developmental Studies Hybridoma Bank, chi 1J, University of Iowa,
Iowa City, IA, USA), rat anti-HA antibody (Cat. no. 11867423001;
1:1,000; Roche Diagnostics GmbH) and rabbit anti-GFP antibody
(1:5,000; Cat. no. A6455; Molecular Probes, Eugene, OR, USA). The
fractionation of the Drosophila S2 cells was performed as
previously described (20). For
immunoprecipitation, the Drosophila S2 cells were extracted
with 1% Triton X-100 lysis buffer (50 mM Tris pH 8.0, 150 mM NaCl,
digitonin and 1% Triton X-100; Sigma) for 20 min on ice, and
centrifuged at 16,100 × g. The supernatant was quantified by
Bradford assay, and used for immunoprecipitation.
Immunoprecipitation was performed with anti-A1AT antibody and
protein-G-agarose beads (Roche Diagnostics GmbH). The beads were
washed 3 times in low-ionic-strength buffer (50 mM Tris-Cl pH 8.0,
100 mM NaCl and 1% Triton X-100; Sigma). Rat anti-HA antibody was
used to detect the ubiquitination of A1AT. For quantification of
western bands, we used ImageJ software (http://rsbweb.nih.gov/ij). The intensity of the band
of interest was normalized with an anti-profilin band.
Measurement of RNA levels
Total RNA was isolated using TRIzol reagent, and
reverse transcription was performed from 200 ng of total RNA using
the SuperScript First-Strand Synthesis kit (both from Invitrogen).
The following primer sequences were used for amplification and
quantification: dEDEM1-F, ACGCCTACGATGGTTACCTG; dEDEM1-R,
ACACGTTGATGTCCCTGTCA; dEDEM2-F, CTTAGCACCGAAACCACCAT; dEDEM2-R,
ACTCCTCGGTACCGTCCTTT.
Results
Characterization of Drosophila EDEMs
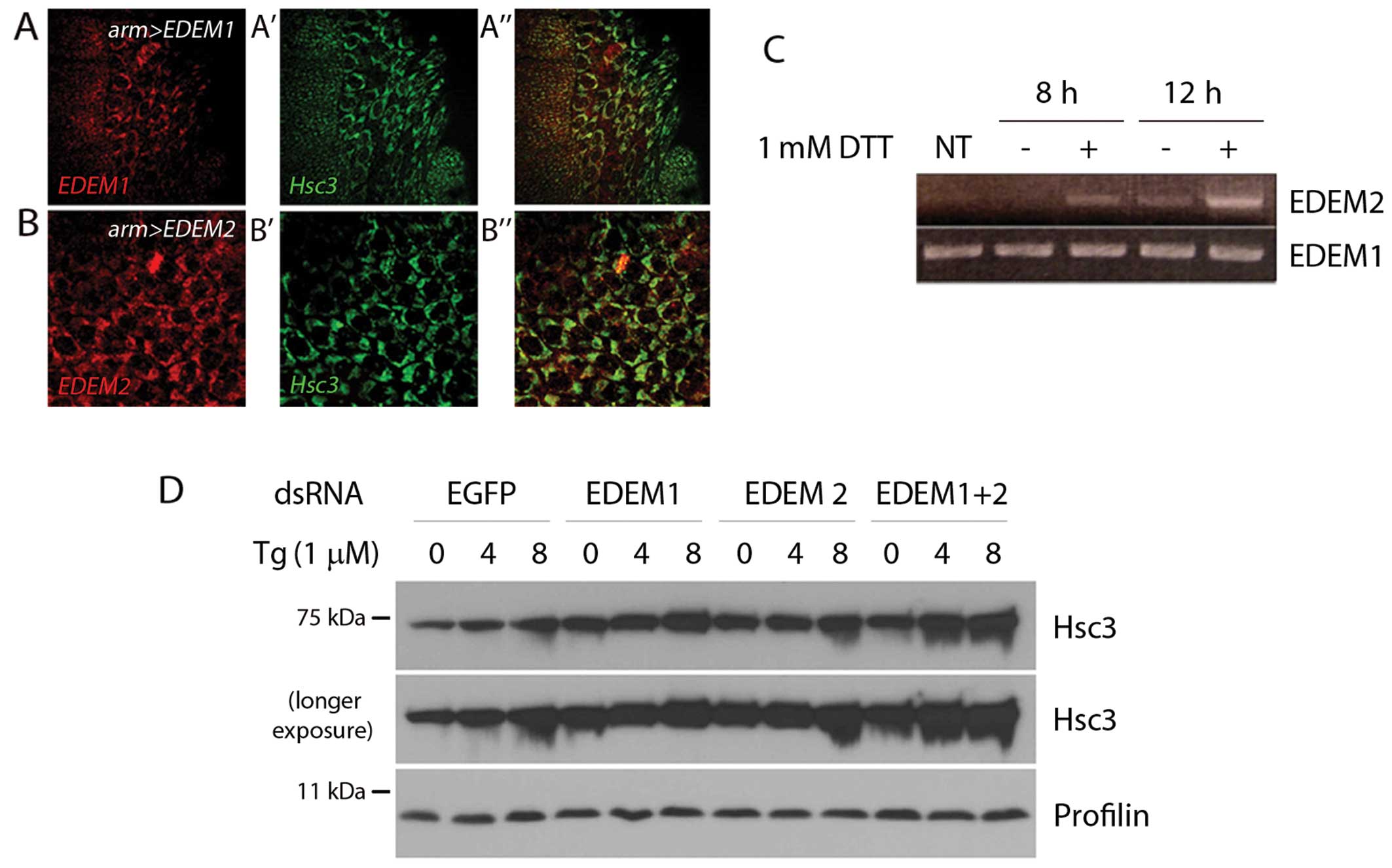
To determine the subcellular distribution of EDEMs
in Drosophila, we generated transgenic EDEM lines with
epitope tags and expressed them in Drosophila embryo
amnioserosa cells. Immunolabeling with anti-myc antibody revealed
that the Drosophila EDEMs co-localized with Hsc3, the
Drosophila orthologue of mammalian BIP. This result is
consistent with the hypothesis that Drosophila EDEMs reside
in the ER (Fig. 2A and B).
Mammalian EDEMs are stress-regulated proteins that
are induced by ER stress (21,22). To determine whether the
Drosophila EDEM homologs are similarly regulated, we treated
the Drosophila S2 cells with dithiothreitol (DTT), which
imposes ER stress by reducing disulfide bonds. Under these
conditions, Drosophila EDEM2 expression was
transcriptionally induced (Fig.
2C). Similar results were obtained with independent ER
stress-causing chemicals; tunicamycin (10 μg/ml), which
inhibits the glycosylation of proteins in the ER and thapsigargin
(Tg; 1 μM), which perturbs ER-calcium homeostasis (data not
shown). On the other hand, we were not able to detect the induction
of Drosophila EDEM1 under these conditions.
The degree of ER stress in cells can be indirectly
assessed by the extent of the transcriptional response that induces
ER chaperones and ERAD genes. To examine the protective role of
Drosophila EDEMs under ER stress conditions, we treated the
Drosophila S2 cells with dsRNAs that target either EGFP (as
a control), EDEM1 or EDEM2, or both EDEM1 and EDEM2 and
subsequently exposed them to Tg. The level of Hsc3 increased after
4 h, and this increase was even more pronounced when both EDEM1 and
EDEM2 were knocked down (Fig.
2D). These results suggest that Drosophila EDEMs play
protective roles against ER stress.
The ER stress reporter is activated by
NHK, but not by the ATZ variant
Excessive protein misfolding in the ER triggers the
activation of signaling pathways referred to as the unfolded
protein response (UPR). One such pathway involves the mRNA splicing
of X-box binding protein 1 (XBP1), which causes a shift in the
reading frame of the downstream sequences and the generation of a
distinct protein isoform (17,23). We have previously exploited this
property to develop a UPR sensor, XBP1-EGFP, in which EGFP is
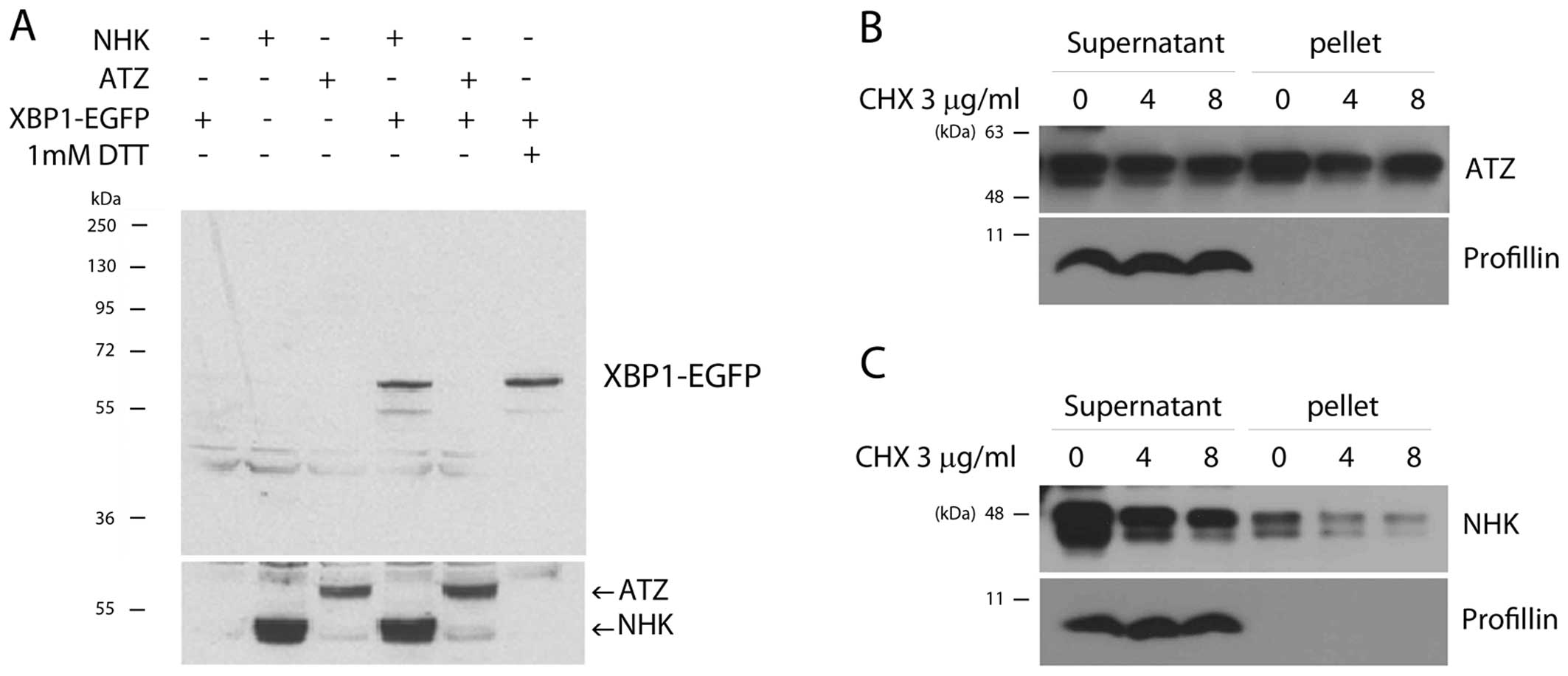
expressed in frame when UPR is activated (17). We thus employed this UPR sensor to
assess whether the mutant variants of A1AT cause ER stress in
Drosophila. As we have reported previously (14), this UPR marker was activated by
NHK expression (Fig. 3A, lane 4)
to a similar extent as that induced by DTT treatment (Fig. 3A, lane 6). Intriguingly, ATZ
expression in the Drosophila S2 cells did not trigger XBP1
mRNA splicing (Fig. 3A, lane 5).
Previous studies on mammalian cells have also reported that, for
some reason, ATZ does not activate UPR; instead, ATZ expression
activates NF-κB signaling via ER overload response (EOR) and ERK
signaling (24–26). To determine whether this
difference is derived from the solubility of misfolded A1AT, we
simply fractionated the cell extracts as supernatants and pellets.
We found that the ratio of ATZ protein in the soluble versus the
insoluble fraction was roughly 1:1 (Fig. 3B). On the other hand, the majority
of NHK protein was found in the soluble fraction (Fig. 3C). These observations support the
hypothesis that the two disease alleles of A1AT have distinct
biochemical properties.
The degradation of mutant variants of
A1AT is regulated by Drosophila EDEMs
We have previously demonstrated that
Drosophila EDEM1 and EDEM2 are homologs of mammalian EDEM2
and EDEM3, respectively. Moreover, we demonstrated that the
overexpression of Drosophila EDEMs helps to lower the levels
of the A1AT NHK variant (14). In
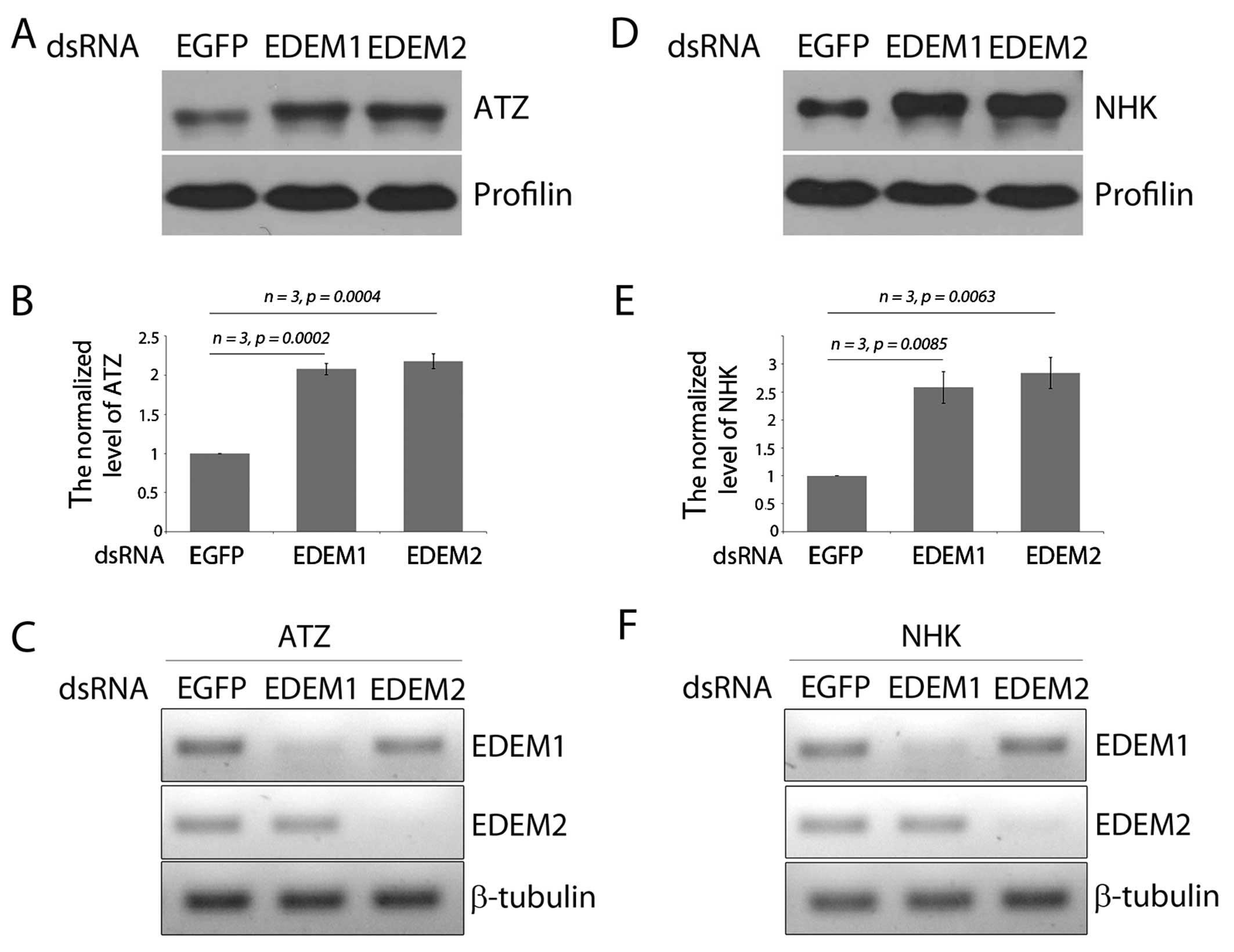
this study, to investigate whether Drosophila EDEMs also
show distinct specificity toward two misfolded A1AT variants, we
examined the effects of EDEM1 and EDEM2 on the degradation of the
misfolded ATZ and NHK variants. The level of ATZ increased by
approximately 2-fold after the knockdown of EDEM1 and EDEM2 by
dsRNA in Drosophila S2 cells (Fig. 4A and B). Similarly, the knockdown
of Drosophila EDEM1 and EDEM2 increased the levels of
another misfolded A1AT variant, NHK (Fig. 4D and E). Of note, a slightly
higher molecular weight band of ATZ was detected after the
knockdown of either Drosophila EDEM1 or EDEM2. As the
deglycosylation of ERAD substrates occur after being dislocated
back into the cytoplasm (27),
the slower migrating A1AT band suggests defective ERAD and the
accumulation of glycosylated ATZ species that accumulate in the
ER.
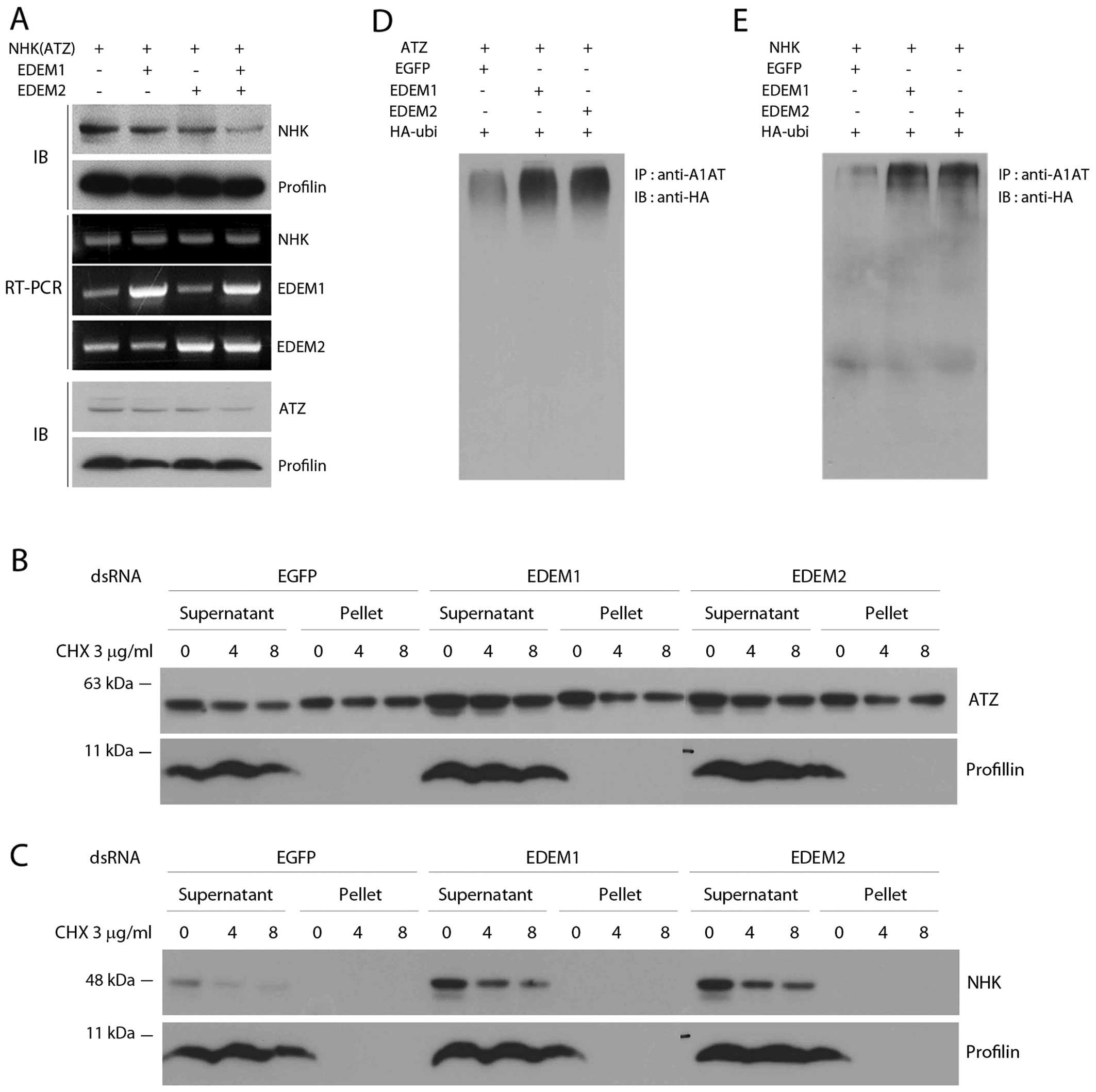
We also overexpressed EDEMs and found that mutant
A1AT degradation was accelerated by the overexpression of EDEMs.
Intriguingly, the two EDEMs from Drosophila had additive
effects on the degradation of both the NHK (Fig. 5A) and ATZ variants of A1AT
(Fig. 5A). Subsequently, we
wished to determine whether Drosophila EDEM1 or EDEM2
affects the solubility of ATZ or NHK. We did not observe any
significant change in the solubility of ATZ or NHK by knocking down
EDEM1 or EDEM2 (Fig. 5B and C).
These results indicate that Drosophila EDEMs regulate the
degradation of misfolded A1AT variants without affecting the
solubility of misfolded A1AT.
Drosophila EDEMs increase the level of
ubiquitinated misfolded A1AT variants
Previous studies have suggested that ATZ can be
degraded by either the ubiquitin-proteasomal pathway, or through
autophagy (13,28,29). In order to further confirm that
the Drosophila EDEMs act by stimulating the ubiquitin
proteasomal degradation of misfolded A1AT variants, we co-expressed
Drosophila EDEMs and ATZ with HA-tagged ubiquitin. The
co-expression of EDEM1 or EDEM2 with ATZ increased the level of
ubiquitinated ATZ (Fig. 5D). The
levels of ubiquitinated NHK also increased, albeit to a different
extent than that observed for ATZ (Fig. 5E). These results again suggest
that Drosophila EDEM1 and EDEM2 target misfolded A1AT
variants for proteasomal degradation.
Discussion
In the present study, we report the use of a
Drosophila model for the study of the mechanisms of
misfolded A1AT degradation that underly A1AT deficiency (30–32). Specifically, we focused on EDEMs,
which are ER resident proteins with mannosidase-like domains.
Similar to the mammalian EDEMs, we find that the Drosophila
EDEM2 mRNA level increases in response to ER stress. We did not
observe a similar induction with the EDEM1 mRNA level. The
examination of the temporal and tissue-specific expression of
Drosophila EDEM2 indicated that the tissues with the highest
levels of Drosophila EDEM2 transcripts include the larval
salivary gland, the adult intestine and the fat body, all of which
have a high protein secretion load (33). Of note, these three organs are all
characterized by high levels of IRE1/XBP1 activity (17,34). As mammalian EDEMs are regulated by
IRE1/XBP1 signaling, it is likely that the Drosophila
IRE1/XBP1 pathway contributes to the induction of EDEM2 during
specific developmental stages, as well as upon ER stress.
EDEMs are involved in one of the early steps of ERAD
substrate recognition. The tight regulation of ERAD is important as
the inefficient detection of misfolded or unfolded proteins causes
their accumulation in the ER, and leads to ER stress. On the other
hand, overactive ERAD can degrade ER resident proteins or folding
intermediates. Although the expression of the majority of ERAD
components is upregulated by ER stress, we observed a significantly
high level of Drosophila EDEM1 transcripts even under
conditions of no stress (Fig.
2D). The mechanisms through which EDEM1 distinguishes
terminally misfolded proteins versus folding intermediates remains
to be explored.
In conclusion, the results from the present study
indicate that the Drosophila EDEMs, EDEM1 and EDEM2, are
resident in the ER, and that the expression of Drosophila
EDEM2 is upregulated by ER stress. Both EDEM1 and EDEM2 in
Drosophila promote the degradation of misfolded A1AT
variants by increasing the ubiquitination of its substrates. Given
the striking similarity between Drosophila and humans in
terms of this process, the present study provides a novel approach
for the study of A1AT deficiency.
Acknowledgments
We would like to thank the Kang laboratory for their
helpful comments on the manuscript. This study was supported by
grants from the Korean Health Technology R&D Project, Ministry
of Health and Welfare, Republic of Korea (HI13C1821), the National
Research Foundation of Korea (NRF-2013R1A1A1009437), the Korean
Government (MSIP) (MRC grant 2008-0062286) to M.-J.K. and the
National Institutes of Health grant R01 EY020866 to H.D.R.
References
|
1
|
Vembar SS and Brodsky JL: One step at a
time: Endoplasmic reticulum-associated degradation. Nat Rev Mol
Cell Biol. 9:944–957. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Molinari M, Calanca V, Galli C, Lucca P
and Paganetti P: Role of EDEM in the release of misfolded
glycoproteins from the calnexin cycle. Science. 299:1397–1400.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oda Y, Hosokawa N, Wada I and Nagata K:
EDEM as an acceptor of terminally misfolded glycoproteins released
from calnexin. Science. 299:1394–1397. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Clerc S, Hirsch C, Oggier DM, Deprez P,
Jakob C, Sommer T and Aebi M: Htm1 protein generates the N-glycan
signal for glycoprotein degradation in the endoplasmic reticulum. J
Cell Biol. 184:159–172. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hosokawa N, Tremblay LO, Sleno B, Kamiya
Y, Wada I, Nagata K, Kato K and Herscovics A: EDEM1 accelerates the
trimming of alpha1,2-linked mannose on the C branch of N-glycans.
Glycobiology. 20:567–575. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hebert DN and Molinari M: Flagging and
docking: Dual roles for N-glycans in protein quality control and
cellular proteostasis. Trends Biochem Sci. 37:404–410. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lomas DA, Evans DL, Finch JT and Carrell
RW: The mechanism of Z alpha 1-antitrypsin accumulation in the
liver. Nature. 357:605–607. 1992. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Termine DJ, Moremen KW and Sifers RN: The
mammalian UPR boosts glycoprotein ERAD by suppressing the
proteolytic down-regulation of ER mannosidase I. J Cell Sci.
122:976–984. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wu Y, Swulius MT, Moremen KW and Sifers
RN: Elucidation of the molecular logic by which misfolded alpha
1-antitrypsin is preferentially selected for degradation. Proc Natl
Acad Sci USA. 100:8229–8234. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mast SW, Diekman K, Karaveg K, Davis A,
Sifers RN and Moremen KW: Human EDEM2, a novel homolog of family 47
glycosidases, is involved in ER-associated degradation of
glycoproteins. Glycobiology. 15:421–436. 2005. View Article : Google Scholar
|
|
11
|
Teckman JH and Perlmutter DH: Retention of
mutant alpha(1)-antitrypsin Z in endoplasmic reticulum is
associated with an autophagic response. Am J Physiol Gastrointest
Liver Physiol. 279:G961–G974. 2000.PubMed/NCBI
|
|
12
|
Kamimoto T, Shoji S, Hidvegi T, Mizushima
N, Umebayashi K, Perlmutter DH and Yoshimori T: Intracellular
inclusions containing mutant alpha1-antitrypsin Z are propagated in
the absence of autophagic activity. J Biol Chem. 281:4467–4476.
2006. View Article : Google Scholar
|
|
13
|
Kroeger H, Miranda E, MacLeod I, Pérez J,
Crowther DC, Marciniak SJ and Lomas DA: Endoplasmic
reticulum-associated degradation (ERAD) and autophagy cooperate to
degrade polymerogenic mutant serpins. J Biol Chem. 284:22793–22802.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kang MJ and Ryoo HD: Suppression of
retinal degeneration in Drosophila by stimulation of ER-associated
degradation. Proc Natl Acad Sci USA. 106:17043–17048. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Hosokawa N, Tremblay LO, You Z, Herscovics
A, Wada I and Nagata K: Enhancement of endoplasmic reticulum (ER)
degradation of misfolded Null Hong Kong alpha1-antitrypsin by human
ER mannosidase I. J Biol Chem. 278:26287–26294. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Brand AH and Perrimon N: Targeted gene
expression as a means of altering cell fates and generating
dominant phenotypes. Development. 118:401–415. 1993.PubMed/NCBI
|
|
17
|
Ryoo HD, Domingos PM, Kang MJ and Steller
H: Unfolded protein response in a Drosophila model for retinal
degeneration. EMBO J. 26:242–252. 2007. View Article : Google Scholar
|
|
18
|
Wu Y, Whitman I, Molmenti E, Moore K,
Hippenmeyer P and Perlmutter DH: A lag in intracellular degradation
of mutant alpha 1-antitrypsin correlates with the liver disease
phenotype in homozygous PiZZ alpha 1-antitrypsin deficiency. Proc
Natl Acad Sci USA. 91:9014–9018. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Armknecht S, Boutros M, Kiger A, Nybakken
K, Mathey-Prevot B and Perrimon N: High-throughput RNA interference
screens in Drosophila tissue culture cells. Methods Enzymol.
392:55–73. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shen Y, Ballar P and Fang S: Ubiquitin
ligase gp78 increases solubility and facilitates degradation of the
Z variant of alpha-1-anti-trypsin. Biochem Biophys Res Commun.
349:1285–1293. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Olivari S, Galli C, Alanen H, Ruddock L
and Molinari M: A novel stress-induced EDEM variant regulating
endoplasmic reticulum-associated glycoprotein degradation. J Biol
Chem. 280:2424–2428. 2005. View Article : Google Scholar
|
|
22
|
Hosokawa N, Wada I, Hasegawa K, Yorihuzi
T, Tremblay LO, Herscovics A and Nagata K: A novel ER
alpha-mannosidase-like protein accelerates ER-associated
degradation. EMBO Rep. 2:415–422. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Coelho DS, Cairrão F, Zeng X, Pires E,
Coelho AV, Ron D, Ryoo HD and Domingos PM: Xbp1-independent Ire1
signaling is required for photoreceptor differentiation and
rhabdomere morphogenesis in Drosophila. Cell Rep. 5:791–801. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hidvegi T, Schmidt BZ, Hale P and
Perlmutter DH: Accumulation of mutant alpha1-antitrypsin Z in the
endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and
BAP31 but not the unfolded protein response. J Biol Chem.
280:39002–39015. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lawless MW, Greene CM, Mulgrew A, Taggart
CC, O’Neill SJ and McElvaney NG: Activation of endoplasmic
reticulum-specific stress responses associated with the
conformational disease Z alpha 1-antitrypsin deficiency. J Immunol.
172:5722–5726. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
van ’t Wout EF, Dickens JA, van Schadewijk
A, et al: Increased ERK signalling promotes inflammatory signalling
in primary airway epithelial cells expressing Z α1-antitrypsin. Hum
Mol Genet. 23:929–941. 2014. View Article : Google Scholar
|
|
27
|
Hirsch C, Blom D and Ploegh HL: A role for
N-glycanase in the cytosolic turnover of glycoproteins. EMBO J.
22:1036–1046. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Y, Choudhury P, Cabral CM and Sifers
RN: Oligosaccharide modification in the early secretory pathway
directs the selection of a misfolded glycoprotein for degradation
by the proteasome. J Biol Chem. 274:5861–5867. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Teckman JH, Gilmore R and Perlmutter DH:
Role of ubiquitin in proteasomal degradation of mutant
alpha(1)-antitrypsin Z in the endoplasmic reticulum. Am J Physiol
Gastrointest Liver Physiol. 278:G39–G48. 2000.PubMed/NCBI
|
|
30
|
Perlmutter DH: Liver injury in
alpha1-antitrypsin deficiency: An aggregated protein induces
mitochondrial injury. J Clin Invest. 110:1579–1583. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lin L, Schmidt B, Teckman J and Perlmutter
DH: A naturally occurring nonpolymerogenic mutant of alpha
1-antitrypsin characterized by prolonged retention in the
endoplasmic reticulum. J Biol Chem. 276:33893–33898. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Parfrey H, Mahadeva R and Lomas DA:
Alpha(1)-antitrypsin deficiency, liver disease and emphysema. Int J
Biochem Cell Biol. 35:1009–1014. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Gelbart WM and Emmert DB: FlyBase high
throughput expression pattern data. http://flybase.org/reports/FBrf0221009.htmlurisimpleflybase.org/reports/FBrf0221009.html.
2013
|
|
34
|
Sone M, Zeng X, Larese J and Ryoo HD: A
modified UPR stress sensing system reveals a novel tissue
distribution of IRE1/XBP1 activity during normal Drosophila
development. Cell Stress Chaperones. 18:307–319. 2013. View Article : Google Scholar :
|