Introduction
Generally, the ectopic overaccumulation of lipids
can trigger cellular dysregulation and functional tissue
impairment, a process referred to as lipotoxicity (1). The investigation of the molecular
mechanisms involved in lipotoxicity strongly indicates that excess
lipids impair cell functions, leading to metabolically relevant
cellular dysfunction, inflammation and oxidative stress (2,3).
The prolonged exposure of β cells to increased levels of fatty
acids, a major characteristic of lipotoxicity, elicits decreased
insulin secretion, compromised insulin gene expression and β cell
apoptosis (2). Glucagon-like
peptide-1 (GLP-1) is a gastrointestinal hormone primarily secreted
by L cells in the intestine in response to food intake (4). Intriguingly, GLP-1 exhibits
beneficial pleiotropic effects on β cells by binding to a specific
receptor (GLP-1R) and enhancing β cell proliferation and survival
(5,6). GLP-1 has also been shown to protect
pancreatic β cells against lipotoxicity-induced apoptosis and
promotes insulin gene expression (7–10).
Moreover, the GLP-1R agonist, liraglutide, has been shown to
suppress oxidative stress in rats with streptozotocin-induced
diabetes (11). Importantly,
intra-islet GLP-1 functions as a paracrine signal for damaged β
cells and is involved in protecting and regenerating them (12). In addition, the GLP-1R agonist,
exendin-4, inhibits human islet inflammation (13).
Recently, interest in GLP-1 has intensified due to
numerous important discoveries revealing that a functional GLP-1
system resides in α cells and is responsive to β cell stress and
injury (14,15). Under normal conditions, pancreatic
α cells secrete proglucagon, which is cleaved into glucagon by
prohormone convertase 2 (PC2). However, under β cell stress
conditions, α cell hyperplasia, which is a hallmark of β cell
injury, develops (16). Notably,
the dedifferentiation of hyperplastic α cells in adult islets seems
to be preserved as an immature, pro-α phenotype attributable to the
expression of prohormone convertase 1/3 (PC1/3) (15), which is the key enzyme in the
processing of proglucagon into GLP-1 peptides in α cells (17–20). Thus, we hypothesized that the
activation of the intra-islet GLP-1 system may be a protective
measure for enhancing cellular survival.
We thus hypothesized that an intra-islet GLP-1
system may be the direct target of signals and self-preservation
mechanisms that enhance β cell survival against lipotoxicity, in
which oxidative stress plays a critical role. Our findings suggest
that an elevated number of immature pro-α cells and the generation
of GLP-1 are an advantage to β cells during conditions of high
metabolic demand or stress, and that this ‘self-defense’ behavior
facilitates β cell survival by reshaping the oxidative balance and
inhibiting inflammation.
Materials and methods
Animals
Male wild-type C57BL/6J mice were used for all the
islet experiments. The mice were maintained under standard light
conditions (12/12-h light/dark cycle) and were allowed free access
to food and water. The male C57BL/6 mice and their food were
purchased from Beijing HFK Bio-Technology Co., Ltd. (Beijing,
China). The care and experimental treatment of the animals were
approved by the Animal Research Committee of Tongji Medical
College, Huazhong University of Science and Technology, Wuhan,
China. Six-week-old mice, weighing 21±2 g, were randomized into
groups and fed either a high-fat diet (HFD; 20% protein, 20%
carbohydrate and 60% fat), or a standard rodent chow diet [low-fat
diet (LFD); 20% protein, 70% carbohydrate and 10% fat]. After 4
weeks on the HFD or LFD, the mice were injected with liraglutide
(200 mg/kg body weight; Novo Nordisk, Princeton, NJ, USA) or a
placebo [phosphate-buffered saline (PBS)] daily for 4 weeks. Body
weight and food intake were measured weekly. To exclude the
differences induced by food intake, all the mice fed the HFD were
pair-fed. After the 8 weeks of feeding and drug administration, the
mice were anesthetized with an intraperitoneal injection of
pentobarbital sodium (0.6 mg/kg body weight).
Metabolic measurements
For the measurement of fasting blood glucagon and
insulin levels, the mice were fasted 6 h. The insulin and glucagon
concentrations were determined using the the insulin enzyme-linked
immunosorbent assay (ELISA) kit (Millipore, Boston, MA, USA). The
plasma GLP-1 concentrations were measured using an ELISA kit (Linco
Research, St. Charles, MO, USA).
Islet isolation and cell culture
Non-diabetic mouse islets were isolated from the
pancreata according to a previously described method (21). Specifically, the pancreas was
perfused through the common bile duct with 1.5 mg/ml collagenase P
(Roche Applied Science, Indianapolis, IN, USA), incubated at room
temperature, and then further separated from the acinar tissue
using a Histopaque 1077 gradient. The islets were isolated by hand
and cultured for 24–72 h in RPMI-1640 medium in a 5% CO2
incubator. Subsequently, the islets were examined according to the
steps outlined below for the functional analysis.
Functional analysis
The non-diabetic islets were seeded into 24-well
plates; 25 islets were added to each well. The palmitate solution
was prepared as previously described (22). Following incubation in
Krebs-Ringer Bicarbonate (KRB) buffer with 5.6 mmol/l glucose for 1
h, the islets were incubated under the following conditions: 0.5%
BSA (as a control) or 0.5 mmol/l palmitate bound to 0.5% BSA for
24, 48 or 72 h. We examined the role of oxidative stress in the
detrimental effects of palmitate by treating the islets with the
antioxidant, N-acetylcysteine (NAC) (5 mmol/l; Sigma-Aldrich, St.
Louis, MO, USA). Finally, to examine the bioactivity of
islet-released GLP-1, experiments were performed in the absence or
presence of the GLP-1R antagonist, exendin-(9-39)
(0.5 µmol/l; Sigma-Aldrich) (14). The islets were pre-treated with
100 nmol/l liraglutide, 5 mmol/l NAC or 0.5 µmol/l
exendin-(9-39) for 2 h followed by exposure to 0.5
mmol/l palmitate. The active GLP-1 content in the cell culture
medium and cell lysates was measured using an ELISA kit (Linco
Research).
MTT assay
After functional analysis,
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay was used to determine the proportion of viable cells in the
treated group compared to the control group, as previously
described (23). The islets were
incubated with 500 µg/ml MTT in RPMI-1640 medium for 3 h at
37°C. At the end of the incubation period, the medium containing
MTT was removed, and the islets or cells were dissolved in dimethyl
sulfoxide (DMSO). The absorbance was measured at 540 and 690 nm
using a microplate reader (Perkin Elmer EnSpire; Perkin Elmer,
Waltham, MA, USA).
Apoptosis assay
DNA fragmentation activity in the islets was
quantified using a Cell Death Detection ELISA Plus kit (Roche
Diagnostics GmbH, Mannheim, Germany) according to the
manufacturer\s instructions. Following treatment, the cells were
washed twice with PBS and incubated with lysis buffer for 20 min at
room temperature. Following centrifugation to remove the nuclei and
cellular debris, the supernatants were diluted 1:5 with lysis
buffer, and each sample was analyzed using ELISA.
Mmeasurement of intracellular levels of
reactive oxygen species (ROS)
The quantification of intracellular ROS levels was
carried out using the fluorescent probe, 2,7-dichlorofluorescein
diacetate (DCFH-DA), as described in our previous study (24). The cells were washed twice with
PBS and incubated in culture medium containing 20 µmol/l
DCFH-DA (Beyotime Institute of Biotechnology, Beijing, China) for
20 min at 37°C in the dark. Subsequently, the cells were lysed with
lysis buffer, and the carboxy-DCF fluorescence in the cell lysates
was measured using a multimode microplate reader (Bio-Rad,
Hercules, CA, USA) and excitation and emission wavelengths of 488
and 530 nm, respectively.
Immunoblot analysis
The islets were seeded into 24-well plates; 50
islets were added to each well. Following treatment, the islets
were washed with PBS prior to the addition of cell lysis buffer
containing protease inhibitor cocktail and PhoshopStop tablets
(Roche Applied Science). The primary antibodies used were PC1/3
(1:1,000; AB10553; Millipore) and pancreatic duodenal homeobox 1
(PDX1) (1:1,000; 5679; Cell Signaling Technology, Boston, MA, USA).
An antibody against mouse β-actin (A1978) was obtained from
Sigma-Aldrich. Densitometric analysis was performed using ImageJ
software.
Immunodetection
After the mice were euthanized, and their pancreata
were removed, the pancreatic tissue was harvested and fixed in 4%
formaldehyde overnight and stored in 70% ethanol. Fixed sections of
pancreatic tissues were embedded in paraffin and cut into
5-µm-thick sections. Paraffin-embedded sections were
rehydrated, and antigen retrieval was performed using a PickCell
pressure cooker. The primary antibodies used were guinea pig
anti-insulin (1:150; ab7842; Abcam, Cambridge, UK), rabbit
anti-PC1/3 (1:200; Millipore), rabbit anti-glucagon (1:200; 8233;
Cell Signaling Technology) and rabbit anti-NF-κB p65 (1:200,
ab16502; Abcam). The secondary antibodies were conjugated to Alexa
Fluor 488 [1:200; AffiniPure goat anti-guinea pig IgG (H+L); Cat.
no. 106-545-003; Jackson ImmunoResearch Laboratories, West Grove,
PA, USA] or DyLight 549 [1:200; goat anti-rabbit IgG (H+L); Cat.
no. A23320; Abbkine Inc., Redlands, CA, USA]. The nuclear
counterstain, 4′6′-diamidino-2-phenylindole (DAPI; Invitrogen,
Carlsbad, CA, USA), was also used. All the digital images were
acquired using a fluorescence microscope equipped with a DC200
digital camera (C-1/TE200U; Nikon, Tokyo, Japan) and were
subsequently analyzed using Image-Pro Plus version 5.0 image
analysis software (Media Cybernetics, Rockville, MD, USA). The
density threshold selection tool was used to select the pancreatic
islet areas marked with insulin and glucagon, which was depicted as
a percentage of the mean islet cross-sectional area
(immuno-density), as previoulsy descibed (25).
Analyses of mRNA expression by
quantitative polymerase chain reaction (qPCR)
Total RNA from the isolated islets and mouse
pancreatic tissue samples was extracted using TRIzol reagent
(Invitrogen). First-strand cDNA synthesis was performed using a
cDNA synthesis kit (Takara Shuzo Co., Ltd., Kyoto, Japan) and qPCR
was performed using a LightCycler (Roche Diagnostics GmbH). The
primers used in this study are listed in Table I. The relative transcript levels
were normalized to 36B4 and calculated using the 2−ΔΔCT
statistical method.
 | Table IList of primers used for qPCR using
SYBR-Green. |
Table I
List of primers used for qPCR using
SYBR-Green.
| Gene | Gene ID | Forward sequence
(5′→3′) | Reverse sequence
(5′→3′) | Length |
|---|
| Nkx6.1 | NM_144955.2 |
ACTTGGCAGGACCAGAGAG |
GCGTGCTTCTTTCTCCACTT | 109 |
| PDX1 | NM_008814.3 |
TGAACTTGACCGAGAGACACAT |
GGTCCCGCTACTACGTTTCTTA | 92 |
| PC1/3 | NM_013628.2 |
ACATGGGGAGAGAATCCTGTAGGCA |
CATGGCCTTTGAAGGAGTTCCTTGT | 220 |
| Insulin-1 | NM_008386.3 |
GGACCCACAAGTGGAACAAC |
GCTGGTAGAGGGAGCAAATG | 130 |
| GLUT2 | NM_031197.2 |
GCCAAGTAGGATGTGCCAAT |
CCCTGGGTACTCTTCACCAA | 110 |
| NOX4 | NM_015760.5 |
ATTTGGATAGGCTCCAGGCAAAC |
CACATGGGTATAAGCTTTGTGAGC | 155 |
| p22phox | NM_001301284.1 |
GGCACCATCAAGCAACCACC |
CTCATCTGTCACTGGCATTGGG | 135 |
| gp91phox | NM_007807.5 |
TCCGTATTGTGGGAGACTGGACG |
AATGGAGGCAAAGGGCGTGAC | 194 |
| SOD2 | NM_013671.3 |
CAGACCTGCCTTACGACTATGG |
CTCGGTGGCGTTGAGATTGTT | 113 |
| GPx-1 | NM_008160.6 |
CCTCAAGTACGTCCGACCTG |
CAATGTCGTTGCGGCACACC | 197 |
| IL-1β | NM_008361.3 |
GCACACCCACCCTGCA |
ACCGCTTTTCCATCTTCTTCTT | 69 |
| IL-6 | NM_031168.1 |
TCCAGAAACCGCTATGAAGTTC |
CACCAGCATCAGTCCCAAGA | 73 |
| TNF-α | NM_013693.3 |
CTCCAGGCGGTGCCTATG |
GGGCCATAGAACTGATGAGAGG | 149 |
| 36B4 | NM_007475.5 |
CAGCAAGTGGGAAGGTGTAATCC |
CCCATTCTATCATCAACGGGTACAA | 75 |
Statistical analysis
The results of the present study are expressed as
the means ± SEM values, and data were analyzed using the Student’s
t-test or one-way analysis of variance (ANOVA) followed by the
Bonferroni post-hoc test. A value of p<0.05 was considered to
indicate a statistically significant difference.
Results
Activation of the endogenous GLP-1 system
in injured isolated islets
Following the incubation of the isolated mouse
islets for 24, 48 or 72 h, treatment with 0.5 mmol/l palmitate
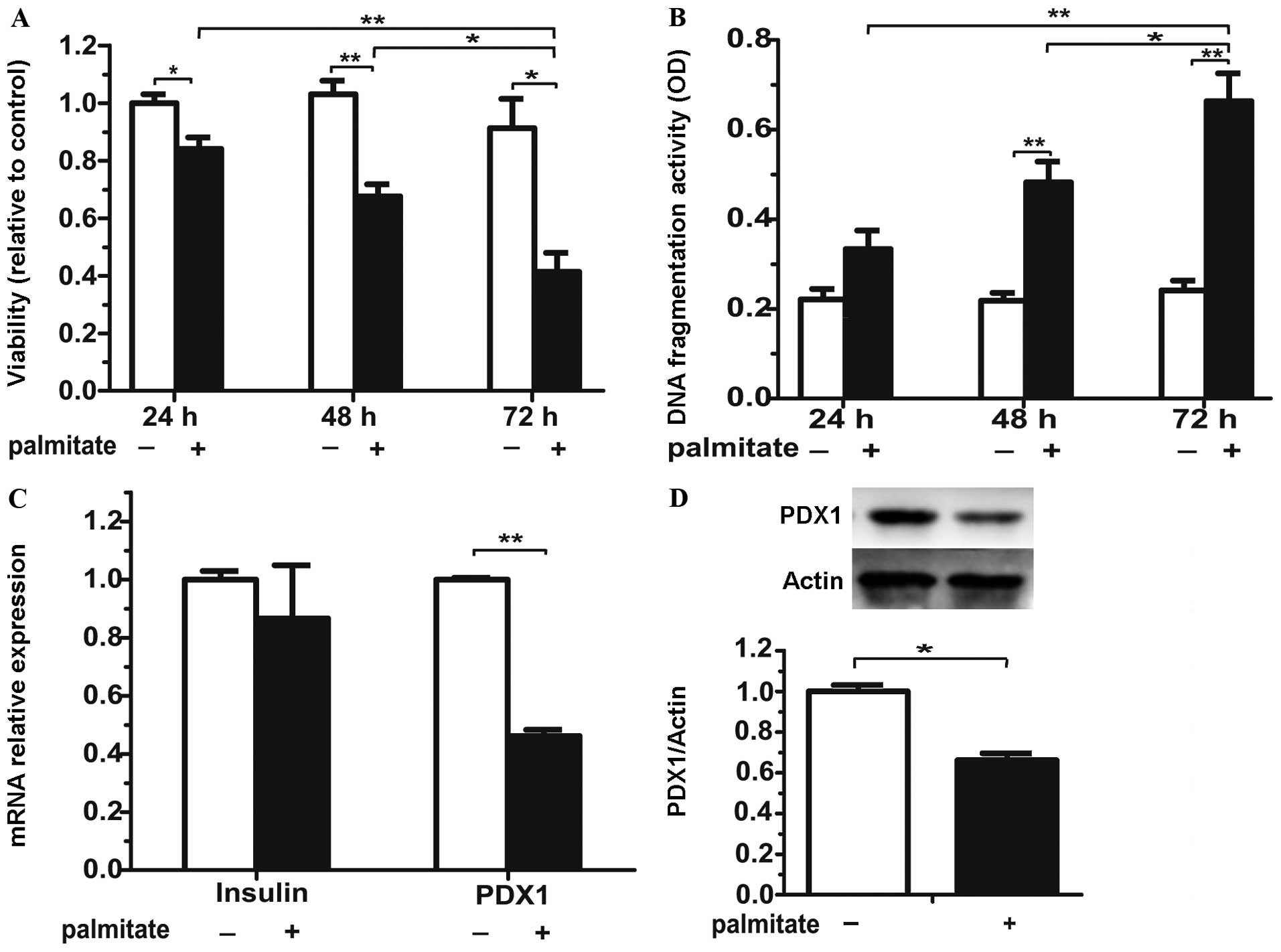
for24, 48 and 72 h led to a significant decrease in islet viability
and an increase in cell death in a time-dependent manner (Fig. 1A and B). Since the exposure of the
cultured islets for 72 h was severely damaging, we focused on the
effects of palmitate on β cell-specific transcription factors after
48 h of treatment. Incubation with palmitate for 48 h did not
significantly modify the mRNA expression of insulin (Fig. 1C). Nonetheless, both PDX1 mRNA and
protein (Fig. 1D) levels
decreased after 48 h of exposure to palmitate. Thus, prolonged
exposure to palmitate induces injury to isolated islets.
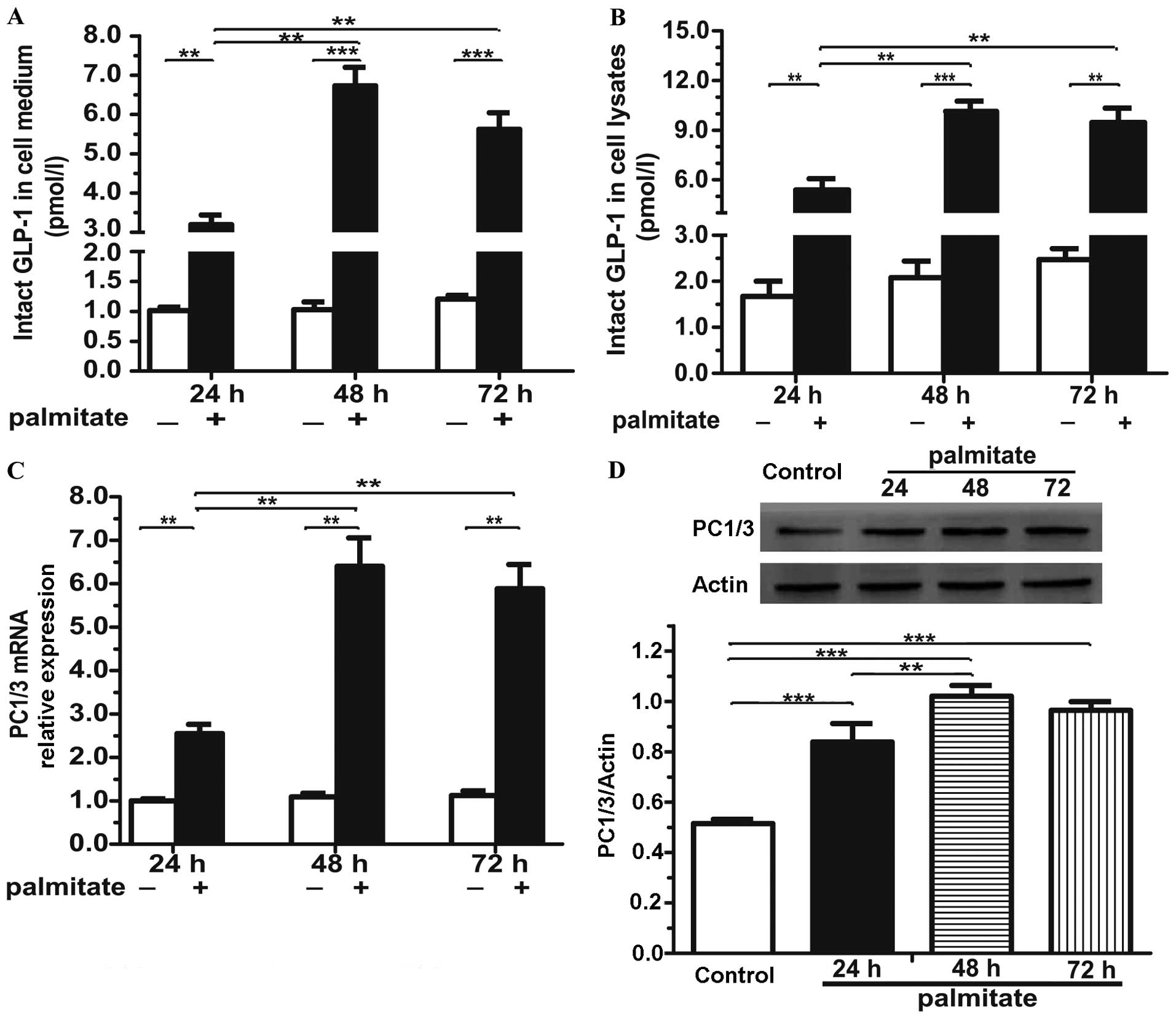
Importantly, incubation of the cells with 0.5 mmol/l
palmitate for 24, 48 or 72 h markedly induced the release of GLP-1
into the culture medium by 3.15-, 6.55- and 5.62-fold, respectively
(Fig. 2A). Moreover, in line with
the observations of the culture medium, the GLP-1 concentration in
the cell lysates was elevated and showed an even greater increase
(Fig. 2B). Nonetheless, the GLP-1
levels at 72 h in both the cell medium and the cell lysates were
almost equivalent to those at 48 h and even showed a decreasing
trend (Fig. 2A and B), which may
be due to the severe serious injury induced by 72 h of exposure to
palmitate (Fig. 1A and B;
increased apoptosis and decreased cell viability). Following
prolonged exposure to palmitate, the mRNA (Fig. 2C) and protein levels (Fig. 2D) of PC1/3, the key enzyme of
GLP-1 generation, also increased (48 h of exposure resulted in
higher mRNA and protein levels than 72 h of exposure).
Elevated expression of GLP-1 in local
pancreatic islets in vivo
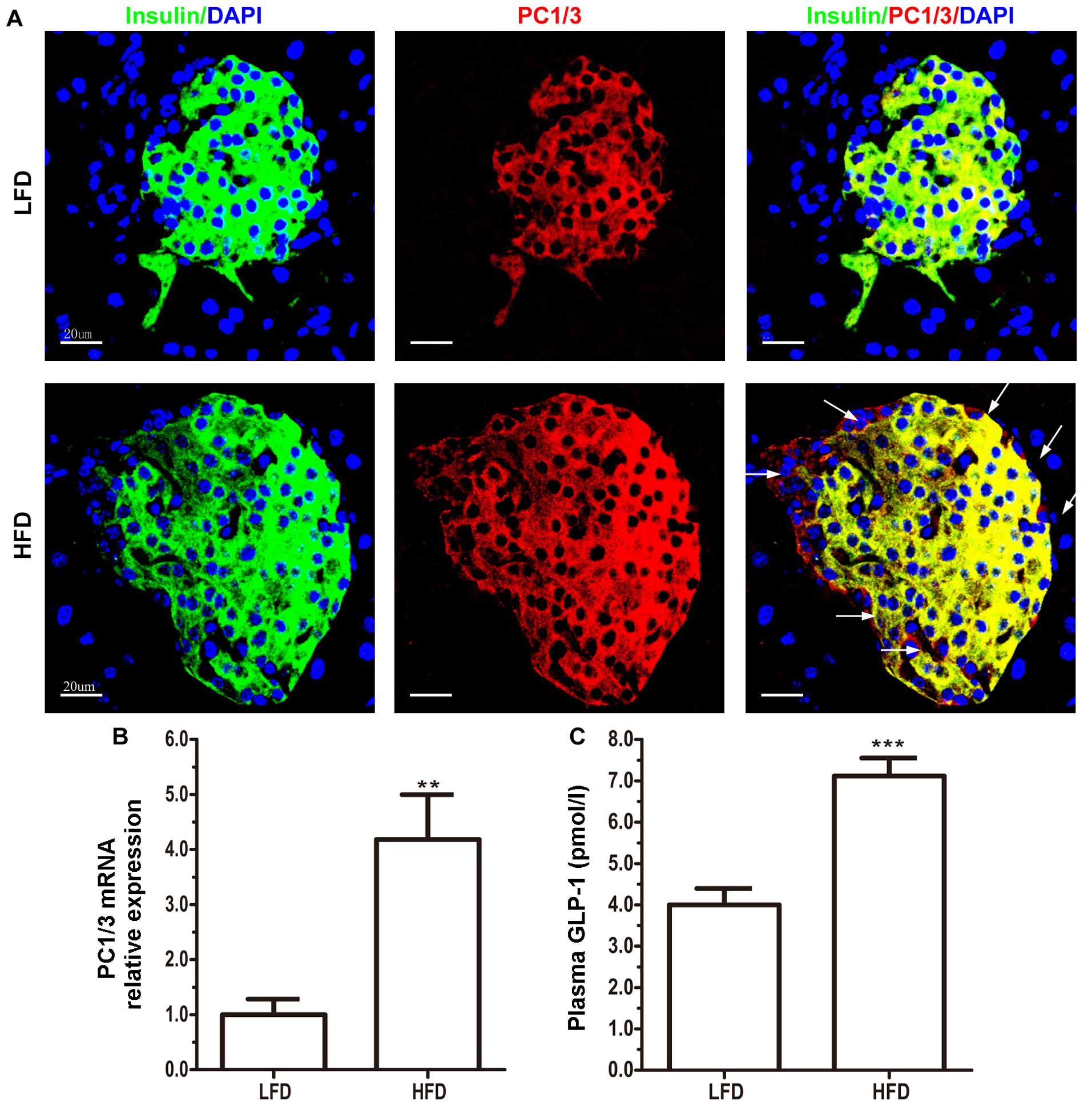
After 8 weeks of being fed a HFD, PC1/3 protein
expression in the HFD group markedly increased (Fig. 3A). Normally, insulin co-localizes
with PC1/3 (Fig. 3A), which is
known to cleave pro-insulin at the B-chain/C-peptide (26). However, we identified a few
PC1/3-positive cells in the extracellular β cell compartments
(Fig. 3A). When the
PC1/3-positive and insulin-negative α cells were quantified, an
increased number of these cells (163 cells) was found in the HFD
group (n=7) compared with the LFD group (n=6; 48 cells) in every 20
slices (data not shown). In addition to intestinal L cells, PC1/3,
the key enzyme governing GLP-1 formation, is expressed in islet β
cells and pro-α cells (27),
suggesting that the elevated number of PC1/3-positive and
insulin-negative cells may be pro-α cells. Notably, in the HFD
group, pancreatic PC1/3 mRNA expression was upregulated (Fig. 3B), and the plasma GLP-1
concentrations were increased compared with the LFD group (Fig. 3C), which was in accordance with
the increase in PC1/3 protein expression (Fig. 3A) and may be the result of the
activation of the intra-islet GLP-1 system. These results
demonstrated that a HFD also induced the formation of pro-α cells
and the release of GLP-1 through the upregulation of the expression
of PC1/3.
Inhibition of GLP-1R signaling
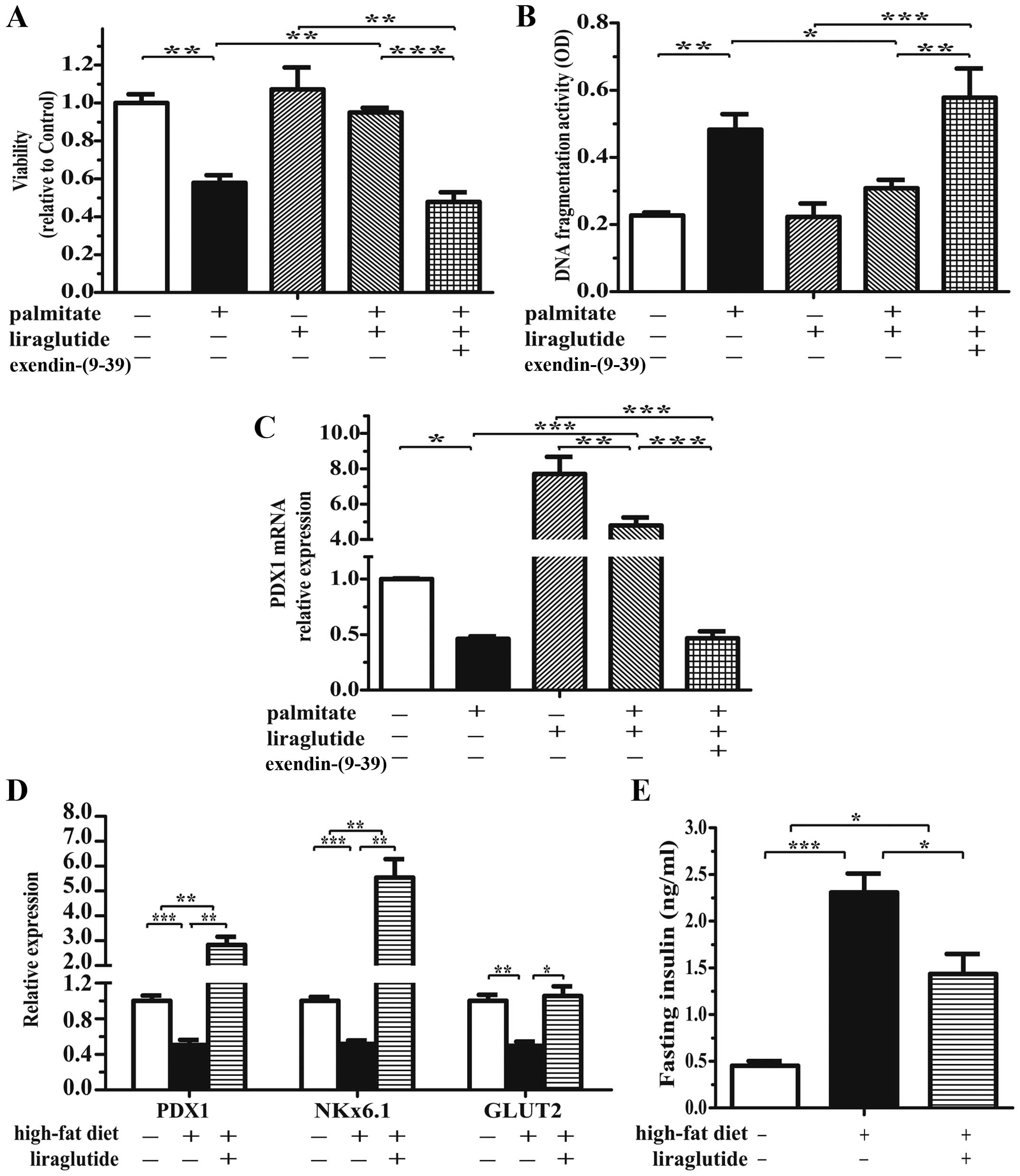
exacerbates the detrimental effects of palmitate
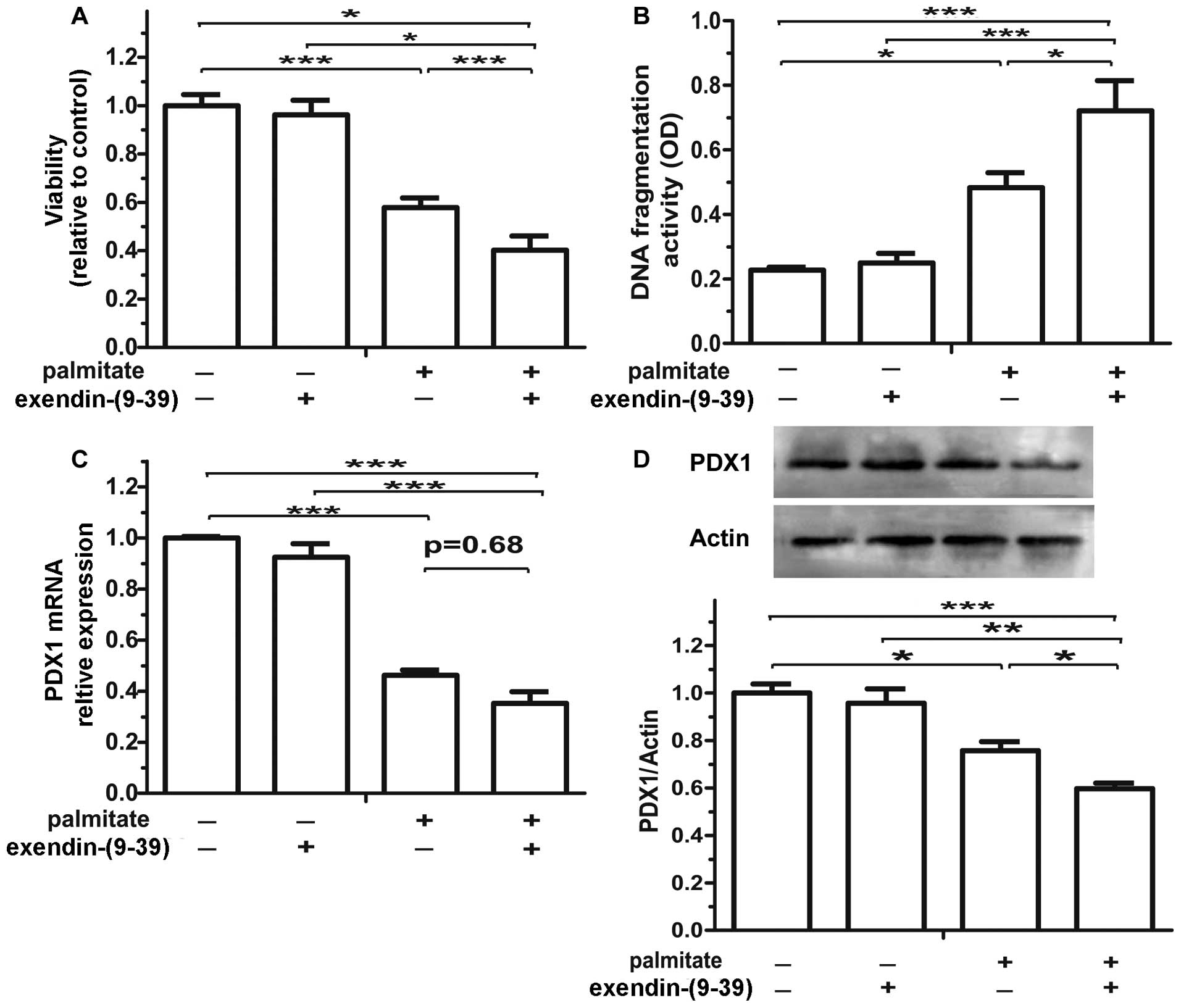
Treatment with exendin-(9-39)
alone, another GLP-1-derived peptide and a GLP-1R antagonist, did
not exert any effects on the isolated islets. When combined with
exposure to palmitate for 48 h, treatment with exendin-(9-39)
resulted in the progressive loss of islet cells that exhibited
decreased viability (Fig. 4A) and
higher apoptotic levels (Fig.
4B). Furthermore, treatment with exendin-(9-39)
exacerbated the detrimental effects of palmitate on β cell survival
by decreasing PDX1 mRNA (Fig. 4C)
(p=0.68) and protein expression (Fig.
4D). These results suggest that the activation of the
intra-islet GLP-1 system ameliorates the detrimental effects of
palmitate.
GLP-1R agonist attenuates
lipotoxicity-induced islet dysfunction in vitro and in vivo
Considering the short biological half-life of
exendin-(9-39) [Kieffer et al (28)], we used the stable, long-lived
GLP-1 analog, liraglutide, which is an agonist of GLP-1R.
Intriguingly, compared to treatment with palmitate alone, treatment
with liraglutide significantly increased islet viability (Fig. 5A) and decreased islet cell
apoptosis (Fig. 5B) to levels
close to those of the controls. Liraglutide also markedly
upregulated PDX1 mRNA expression by 7.70-fold, and this increase
was attenuated by treatment with palmitate (Fig. 5C). Moreover, the protective
effects of liraglutide were completely abrogated by
exendin-(9-39) (Fig.
5A–C). We also investigated the effects of liraglutide on islet
function in mice fed a HFD. In the HFD group, liraglutide also
increased the mRNA expression of the β cell markers, PDX1, Nkx6.1
and glucose transporter 2 (GLUT2) (Fig. 5D). Compared to the mice fed a HFD
alone, treatment with liraglutide reduced the plasma insulin
concentration (Fig. 5E).
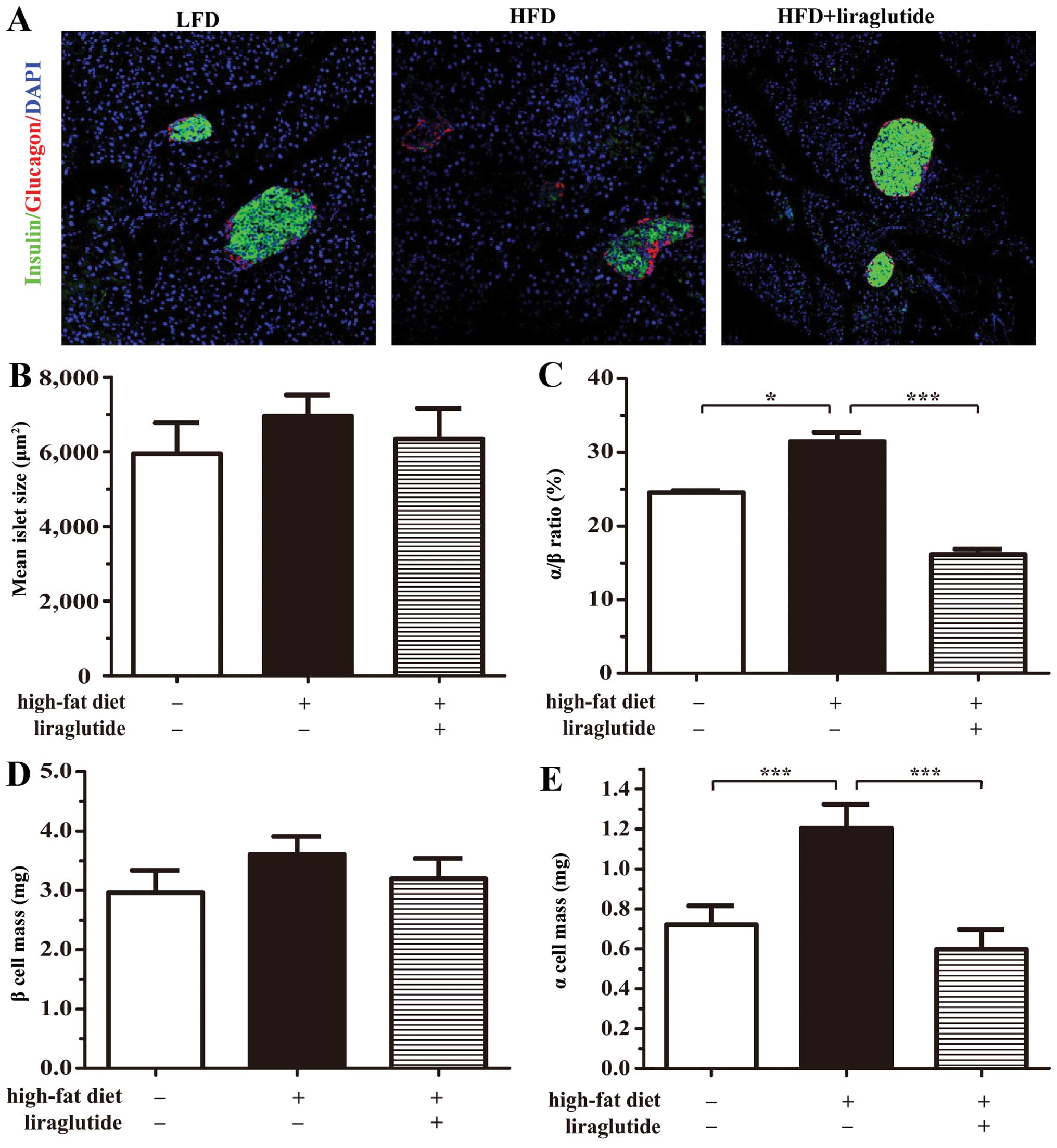
Liraglutide normalizes the islet
architecture of mice fed a HFD
Unlike the defined α cell mantle and β cell core
characteristics of the islets from the mice fed a LFD, the islets
from the mice fed a HFD maintained a more scattered organization
and a higher percentage of α cells. Furthermore, in the HFD group,
there was a greater difference in the expression of insulin
(Fig. 6A). As expected, treatment
with liraglutide reverted the cell structure to a more normal islet
structure (Fig. 6A). Despite the
differences in the mean islet areas of the 3 groups (Fig. 6B), the elevated proportions of
medium islets (5,000–10,000 µm2) and large islets
(>5,000 µm2), as well as the mean area of
small islets (<5,000 µm2) that were induced by
a HFD were altered by treatment with liraglutide (Table II). In accordance with an
elevated α/β cell ratio, the HFD group exhibited a significant
increase in α cell mass (Fig. 6C and
E). Treatment with liraglutide decreased the β cell mass
(Fig. 6D). However, these results
do not completely agree with previously published findings
(29), possibly due to the
different experimental conditions used, the increased β cell
proportion and the inhibitory effects of a HFD on insulin
expression.
| Table IIThe percentage and mean area
(µm2) of small, medium and large-sized islets per
group. |
Table II
The percentage and mean area
(µm2) of small, medium and large-sized islets per
group.
| LFD
| HFD
| HFD + liraglutide
|
|---|
| Percentage | Mean area | Percentage | Mean area | Percentage | Mean area |
|---|
| Small islets
(<5,000 µm2) | 70.37 |
1470.86±115.034 | 54.40 |
2122.65±154.12a | 66.67 |
2027.76±148.49a |
| Medium islets
(5,000–10,000 µm2) | 15.43 | 7696.40±256.16 | 25.82 | 7562.77±209.21 | 20.60 | 8101.04±256.92 |
| Large islets
(>10,000 µm2) | 14.20 |
26220.00±3448.25 | 19.78 |
19460.94±1306.30 | 12.73 |
26757.57±3244.72 |
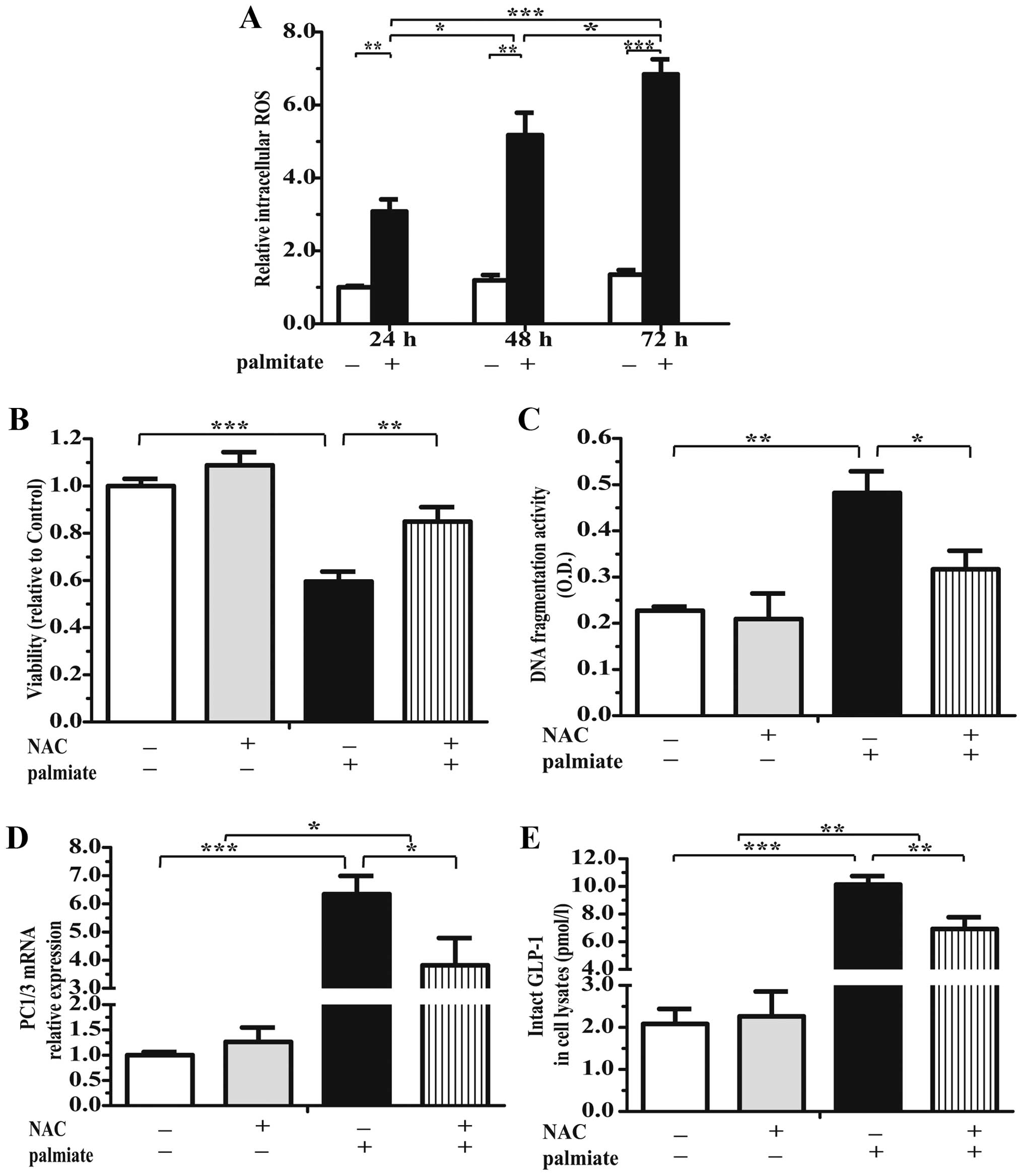
The excess production of ROS induced by
palmitate activates the GLP-1 system
Incubation of the cells with 0.5 mmol/l palmitate
for 24, 48 or 72 h markedly increased the intra-islet ROS levels by
approximately 3.01-, 5.12- and 6.45-fold, respectively (Fig. 7A). In addition, pre-incubation
with 5 mmol/l NAC, neutralized the harmful effects of palmitate,
increased islet viability (by 1.43-fold; Fig. 7B) and decreased apoptosis by
approximately 64.84% (Fig. 7C) in
the presence of 0.5 mmol/l palmitate. Importantly, NAC
significantly decreased the PC1/3 mRNA levels (Fig. 7D) and the GLP-1 concentration in
the cell lysates (Fig. 7E), which
may be due to the attenuation of β cell injury. Nonetheless, the
elevated levels of PC1/3 mRNA (Fig.
7D) and GLP-1 protein, which were induced by palmitate, did not
return to normal after NAC treatment (Fig. 7E), suggesting that the inhibition
did not completely reverse the detrimental effects of palmitate
overload in the pancreatic islets.
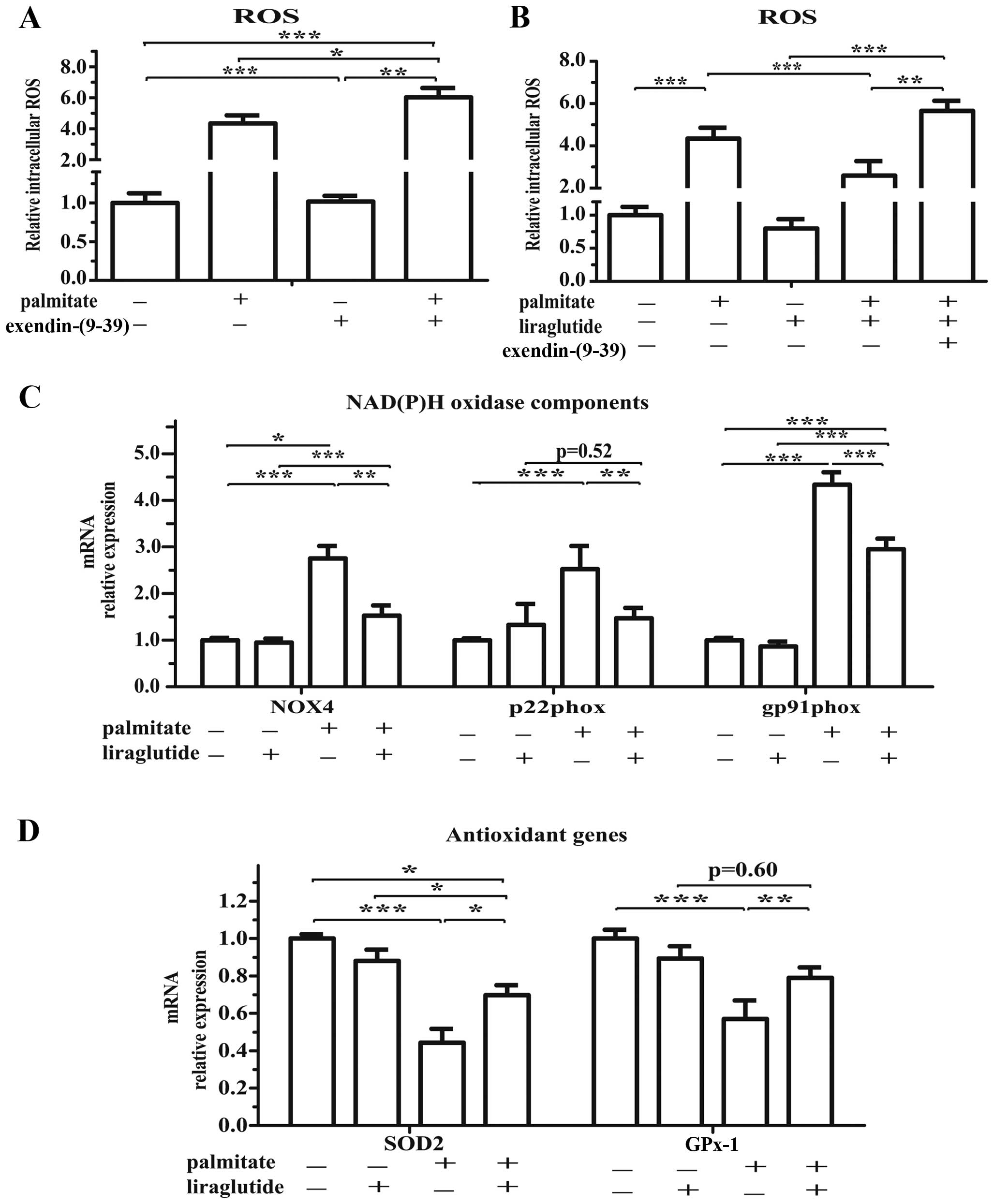
GLP-1R signaling helps to maintain the
oxidative balance
Given the intermediary role of oxidative stress in
palmitate-induced injury, we hypothesized that GLP-1R signaling may
re-shape the oxidative balance by suppressing the generation of ROS
and enhancing antioxidant defenses. The inhibition of GLP-1R
signaling by exendin-(9-39) (in combination with palmitate) also
slightly increased the production of ROS (p=0.045; Fig. 8A). Furthermore, the activation of
GLP-1R by liraglutide decreased the ROS levels in the isolated
islets (p=0.038; Fig. 8B).
Notably, these changes were directly confirmed by qPCR, which
revealed a clear and widespread reshaping of the oxidative balance.
Treatment with liraglutide attenuated the palmitate-induced
activation of NAD(P)H oxidase components, including NADPH oxidase 4
(NOX4), p22phox and gp91phox (Fig.
8C). Simultaneously, treatment with liraglutide upregulated the
expression of mitochondrial-specific superoxide dismutase 2 (SOD2)
and glutathione peroxidase 1 (GPx-1) (Fig. 8D).
Liraglutide helps to attenuate islet
inflammation
Based on the fact that the nuclear factor-κB (NF-κB)
activation by ROS (30) and
inflammation are key to the development of β cell failure (31), we examined the effects of
liraglutide on inflammatory factors and the NF-κB pathway in
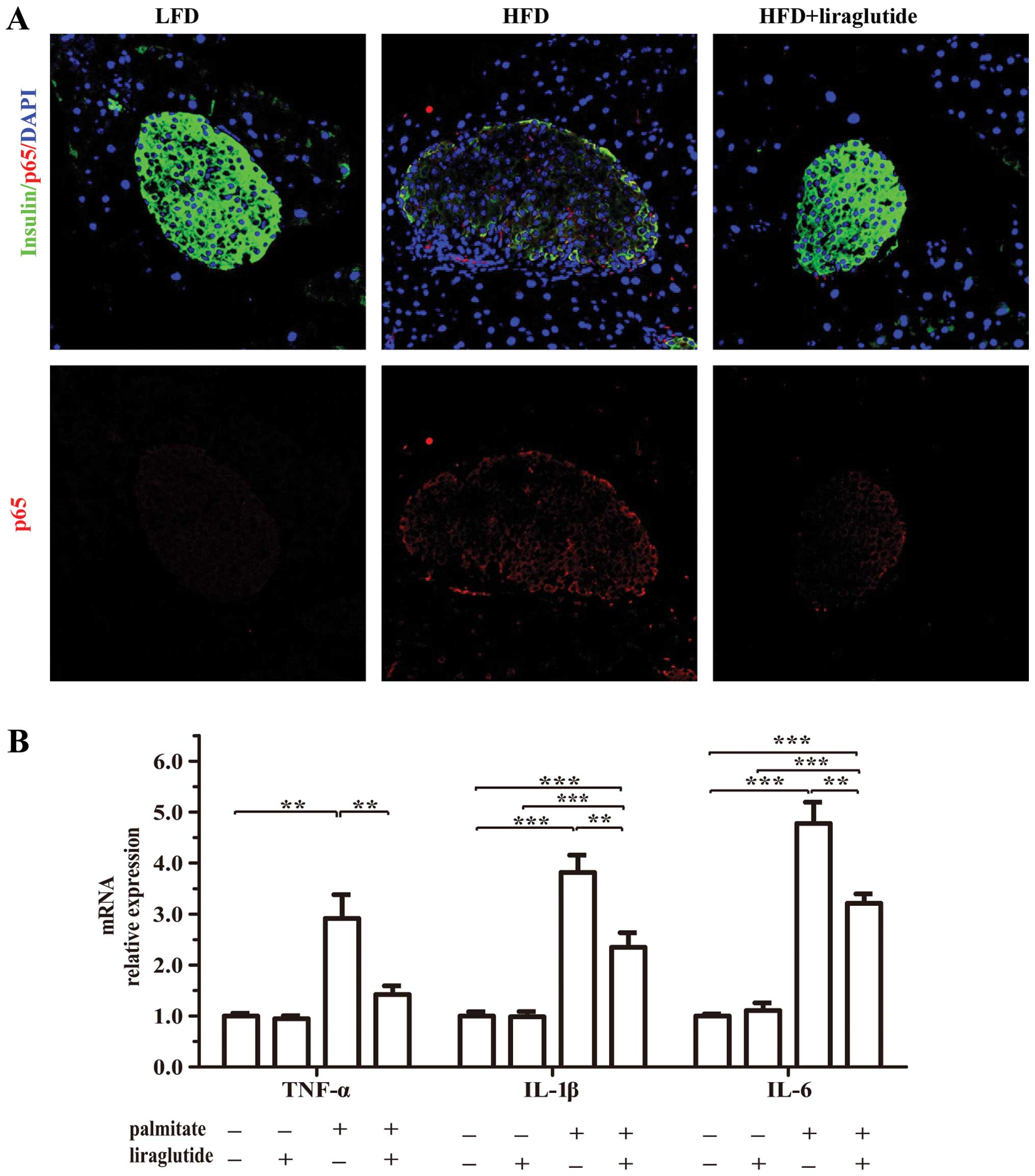
pancreatic islets. Double immunostaining revealed that the p65
protein expression levels were markedly increased in the pancreatic
islets of the mice fed a HFD compared to those of the islets of the
mice fed a LFD (Fig. 9A).
Furthermore, in the isolated islets, treatment with liraglutide
suppressed the palmitate-induced expression of inflammatory
factors, including tumor necrosis factor-α (TNF-α), interleukin
(IL)-1β and IL-6 (Fig. 9B).
Discussion
To the best of our knowledge, the present study for
the first time examined the hypothesis that lipotoxicity directly
stimulates the generation of immature pro-α cells, resulting in
pro-α cells producing endogenous GLP-1 to facilitate β cell
survival by maintaining the oxidative balance and by inhibiting
islet inflammation. Prolonged exposure to palmitate induced
lipotoxicity, and a HFD induced PC1/3 expression and, in turn,
increased the synthesis and release of GLP-1, which were partly
mediated by lipotoxicity-induced oxidative stress. The activation
of GLP-1R signaling was attributed to the normalization of islet
function and structure. Furthermore, GLP-1 exerted protective
effects against lipid overload, partially by increasing antioxidant
gene expression and decreasing the levels of ROS, NF-κB and
inflammatory factors.
Evidence examining the striking innate plasticity of
islets has recently received significant attention as the
dedifferentiation of hyperplastic α cells maintains an immature
pro-α phenotype in response to β cell stress or injury (16). As α cells constitutively express
proglucagon, their plasticity is manifested in the expression of
glucagon or GLP-1 and depends on the relative levels of PC2 and
PC1/3 (16). In models of insulin
resistance or diabetes, there is a significant, progressive
increase in intra-islet GLP-1 expression based on the upregulation
of PC1/3 expression (32,33). The present study similarly
demonstrated that lipotoxicity upregulated GLP-1 expression in
response to β cell injury. Generally, the increase in GLP-1
expression was attributed to pro-α cells, which are derived from
the dedifferentiation of hyperplastic α cells. β cells may serve as
an alternative source of pro-α cells since the dedifferentiation of
β cells to progenitor-like cells caused by metabolic stress may
also represent a distinct ‘pro-α’ cell differentiation stage
(34,35). Therefore, the identification of a
set of pro-α cells that contribute to the endogenous GLP-1 system
supports the view that it is advantageous for islet cells to
dedifferentiate to facilitate their survival.
Unequivocally, in our study, intra-islet GLP-1
enhanced β cell survival against lipotoxicity through a pleiotropic
mechanism. Initially, intra-islet GLP-1/GLP-1R signaling partially
decreased the palmitate-induced damage to β cells. The GLP-1R
antagonist, exendin-(9-39), further suppressed islet viability,
increased apoptosis and inhibited PDX1 transcription in the
presence of palmitate after 48 h. Additionally, our data
demonstrated that GLP-1/GLP-1R signaling attenuated the
transcription of β cell markers, such as insulin, PDX1, Nkx6.1 and
GLUT2. GLP-1R signaling upregulates the insulin gene promoter by
inhibiting p38 mitogen-activated protein kinase (p38 MAPK)
(8). The shielding action of
GLP-1 also involves the activation of the phosphoinositide 3-kinase
(PI3K) and extracellular signal-regulated kinase (ERK) pathways and
the upregulation of PDX-1 transcription (36). Finally, GLP-1 normalizes islet
structure by promoting proliferation, inhibiting apoptosis and
normalizing the distribution of islet cells. GLP-1 protects
pancreatic β cells against lipotoxicity-induced apoptosis by
activating peroxisome proliferator-activated receptor (PPAR)-β/δ
(7) and promotes β cell
proliferation mediated through the EGF pathway (5). Therefore, the activation of the
endogenous GLP-1 system appears to be a ‘self-defense’ response in
pancreatic islets. However, whether there are other more specific
‘protectors’ in the prevention and treatment of lipotoxicity
remains to be determined.
Our data suggest that oxidative stress plays a role
as the bridge between lipid overload and the intra-islet GLP-1
system. Oxidative stress is a common biochemical trigger of
stress-sensitive signaling pathways, including NF-κB, p38 MAPK and
JNK (37,38). Moreover, β cells are particularly
susceptible to the damage inflicted by oxidative stress due to low
levels of free-radical scavenging enzymes (37,39). Exendin-4, another GLP-1R agonist,
protects endothelial cells from palmitate-induced apoptosis by
modulating stress-sensitive signaling pathways (40). In a previous study, in rats with
tacrolimus-induced diabetes, the dipeptidyl peptidase-4 (DPP4)
inhibitor, MK-0626, decreased the levels of 8-OHdG (a marker of
oxidative DNA damage) and increased the levels of manganese
superoxide dismutase and heme oxygenase-1 (41). In rats with streptozotocin-induced
diabetes, liraglutide was shown to directly protect the rats
against oxidative stress through the inhibition of NAD(P)H oxidases
(11). Consistent with our
results, Lotfy et al (9)
reported that another GLP-1R signaling agonist, exenatide, elevated
the expression of catalase and glutathione reductase in the
pancreata of diabetic rats. Small GLP-1-derived peptides also
modulate nutrient homeostasis by suppressing oxidative stress
(42,43). In addition, ROS activate NF-κB and
induce the generation of inflammatory factors, and our data, as
well as previous findings demonstrate that liraglutide decreases
p65 expression and the expression of inflammatory factors, such as
TNF-α and IL-1β (30).
In conclusion, the findings of the present study
suggest that endogenous GLP-1/GLP-1R signaling is a ‘self-defense’
pathway that facilitates islet survival against lipotoxicity. The
protective mechanism of intra-islet GLP-1 may be the achievement of
oxidative balance, as well as the inhibition of islet inflammation.
Further research on the intra-islet GLP-1 system is required to
obtain a better understanding of the mechanisms through which adult
β cells maintain or change their identity due to lipotoxicity.
Acknowledgments
The present study was supported by a grant provided
by the National Natural Science Foundation of China (no.
81370880).
References
|
1
|
Kusminski CM, Shetty S, Orci L, Unger RH
and Scherer PE: Diabetes and apoptosis: Lipotoxicity. Apoptosis.
14:1484–1495. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim JW and Yoon KH: Glucolipotoxicity in
pancreatic β-cells. Diabetes Metab J. 35:444–450. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rial E, Rodríguez-Sánchez L, Gallardo-Vara
E, Zaragoza P, Moyano E and González-Barroso MM: Lipotoxicity,
fatty acid uncoupling and mitochondrial carrier function. Biochim
Biophys Acta. 1797:800–806. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kim W and Egan JM: The role of incretins
in glucose homeo-stasis and diabetes treatment. Pharmacol Rev.
60:470–512. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Buteau J: GLP-1 receptor signaling:
Effects on pancreatic beta-cell proliferation and survival.
Diabetes Metab. 34(Suppl 2): S73–S77. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lamont BJ and Andrikopoulos S: Hope and
fear for new classes of type 2 diabetes drugs: Is there preclinical
evidence that incretin-based therapies alter pancreatic morphology?
J Endocrinol. 221:T43–T61. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang Y, Tong Y, Gong M, Lu Y, Wang C, Zhou
M, Yang Q, Mao T and Tong N: Activation of PPARβ/δ protects
pancreatic β cells from palmitate-induced apoptosis by upregulating
the expression of GLP-1 receptor. Cell Signal. 26:268–278. 2014.
View Article : Google Scholar
|
|
8
|
Kemp DM and Habener JF: Insulinotropic
hormone glucagon-like peptide-1 (GLP-1) activation of insulin gene
promoter inhibited by p38 mitogen-activated protein kinase.
Endocrinology. 142:1179–1187. 2001.PubMed/NCBI
|
|
9
|
Lotfy M, Singh J, Rashed H, Tariq S,
Zilahi E and Adeghate E: Mechanism of the beneficial and protective
effects of exenatide in diabetic rats. J Endocrinol. 220:291–304.
2014. View Article : Google Scholar
|
|
10
|
Chen LN, Lyu J, Yang XF, Ji WJ, Yuan BX,
Chen MX, Ma X and Wang B: Liraglutide ameliorates glycometabolism
and insulin resistance through the upregulation of GLUT4 in
diabetic KKAy mice. Int J Mol Med. 32:892–900. 2013.PubMed/NCBI
|
|
11
|
Hendarto H, Inoguchi T, Maeda Y, Ikeda N,
Zheng J, Takei R, Yokomizo H, Hirata E, Sonoda N and Takayanagi R:
GLP-1 analog liraglutide protects against oxidative stress and
albu-minuria in streptozotocin-induced diabetic rats via protein
kinase A-mediated inhibition of renal NAD(P)H oxidases. Metabolism.
61:1422–1434. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liu Z, Stanojevic V, Avadhani S, Yano T
and Habener JF: Stromal cell-derived factor-1 (SDF-1)/chemokine
(C-X-C motif) receptor 4 (CXCR4) axis activation induces
intra-islet glucagon-like peptide-1 (GLP-1) production and enhances
beta cell survival. Diabetologia. 54:2067–2076. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pugazhenthi U, Velmurugan K, Tran A,
Mahaffey G and Pugazhenthi S: Anti-inflammatory action of exendin-4
in human islets is enhanced by phosphodiesterase inhibitors:
Potential therapeutic benefits in diabetic patients. Diabetologia.
53:2357–2368. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Marchetti P, Lupi R, Bugliani M,
Kirkpatrick CL, Sebastiani G, Grieco FA, Del Guerra S, D’Aleo V,
Piro S, Marselli L, et al: A local glucagon-like peptide 1 (GLP-1)
system in human pancreatic islets. Diabetologia. 55:3262–3272.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Habener JF and Stanojevic V: Alpha cells
come of age. Trends Endocrinol Metab. 24:153–163. 2013. View Article : Google Scholar
|
|
16
|
Habener JF and Stanojevic V: α-cell role
in β-cell generation and regeneration. Islets. 4:188–198. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Nie Y, Nakashima M, Brubaker PL, Li QL,
Perfetti R, Jansen E, Zambre Y, Pipeleers D and Friedman TC:
Regulation of pancreatic PC1 and PC2 associated with increased
glucagon-like peptide 1 in diabetic rats. J Clin Invest.
105:955–965. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhang Y, Zhang Y, Bone RN, Cui W, Peng JB,
Siegal GP, Wang H and Wu H: Regeneration of pancreatic non-β
endocrine cells in adult mice following a single diabetes-inducing
dose of streptozotocin. PLoS One. 7:e366752012. View Article : Google Scholar
|
|
19
|
Yano T, Liu Z, Donovan J, Thomas MK and
Habener JF: Stromal cell derived factor-1 (SDF-1)/CXCL12 attenuates
diabetes in mice and promotes pancreatic beta-cell survival by
activation of the prosurvival kinase Akt. Diabetes. 56:2946–2957.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Thyssen S, Arany E and Hill DJ: Ontogeny
of regeneration of beta-cells in the neonatal rat after treatment
with streptozotocin. Endocrinology. 147:2346–2356. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lau T, Carlsson PO and Leung PS: Evidence
for a local angiotensin-generating system and dose-dependent
inhibition of glucose-stimulated insulin release by angiotensin II
in isolated pancreatic islets. Diabetologia. 47:240–248. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang HW, Mizuta M, Saitoh Y, Noma K, Ueno
H and Nakazato M: Glucagon-like peptide-1 and candesartan
additively improve glucolipotoxicity in pancreatic β-cells.
Metabolism. 60:1081–1089. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jaksch C and Thams P: A critical role for
CK2 in cytokine-induced activation of NFκB in pancreatic β cell
death. Endocrine. 47:117–128. 2014. View Article : Google Scholar :
|
|
24
|
Yuan L, Lu CL, Wang Y, Li Y and Li XY: Ang
(1-7) protects islet endothelial cells from palmitate-induced
apoptosis by AKT, eNOS, p38 MAPK, and JNK pathways. J Diabetes Res.
2014:3914762014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Fraulob JC, Ogg-Diamantino R,
Fernandes-Santos C, Aguila MB and Mandarim-de-Lacerda CA: A mouse
model of metabolic syndrome: Insulin resistance, fatty liver and
non-alcoholic fatty pancreas disease (NAFPD) in C57BL/6 mice fed a
high fat diet. J Clin Biochem Nutr. 46:212–223. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Davidson HW: (Pro)Insulin processing: A
historical perspective. Cell Biochem Biophys. 40(Suppl 3): 143–158.
2004.PubMed/NCBI
|
|
27
|
Portela-Gomes GM, Grimelius L and
Stridsberg M: Prohormone convertases 1/3, 2, furin and protein 7B2
(Secretogranin V) in endocrine cells of the human pancreas. Regul
Pept. 146:117–124. 2008. View Article : Google Scholar
|
|
28
|
Kieffer TJ, McIntosh CH and Pederson RA:
Degradation of glucose-dependent insulinotropic polypeptide and
truncated glucagon-like peptide 1 in vitro and in vivo by
dipeptidyl peptidase IV. Endocrinology. 136:3585–3596.
1995.PubMed/NCBI
|
|
29
|
Mondragon A, Davidsson D, Kyriakoudi S,
Bertling A, Gomes-Faria R, Cohen P, Rothery S, Chabosseau P, Rutter
GA and da Silva Xavier G: Divergent effects of liraglutide,
exendin-4, and sitagliptin on beta-cell mass and indicators of
pancreatitis in a mouse model of hyperglycaemia. PLoS One.
9:e1048732014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gloire G, Legrand-Poels S and Piette J:
NF-kappaB activation by reactive oxygen species: Fifteen years
later. Biochem Pharmacol. 72:1493–1505. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Quan W, Jo EK and Lee MS: Role of
pancreatic β-cell death and inflammation in diabetes. Diabetes Obes
Metab. 15(Suppl 3): 141–151. 2013. View Article : Google Scholar
|
|
32
|
O’Malley TJ, Fava GE, Zhang Y, Fonseca VA
and Wu H: Progressive change of intra-islet GLP-1 production during
diabetes development. Diabetes Metab Res Rev. 30:661–668. 2014.
View Article : Google Scholar
|
|
33
|
Hansen AM, Bödvarsdottir TB, Nordestgaard
DN, Heller RS, Gotfredsen CF, Maedler K, Fels JJ, Holst JJ and
Karlsen AE: Upregulation of alpha cell glucagon-like peptide 1
(GLP-1) in Psammomys obesus - an adaptive response to
hyperglycaemia? Diabetologia. 54:1379–1387. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Talchai C, Xuan S, Lin HV, Sussel L and
Accili D: Pancreatic β cell dedifferentiation as a mechanism of
diabetic β cell failure. Cell. 150:1223–1234. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wang Z, York NW, Nichols CG and Remedi MS:
Pancreatic β cell dedifferentiation in diabetes and
redifferentiation following insulin therapy. Cell Metab.
19:872–882. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Tortosa F and Dotta F: Incretin hormones
and beta-cell mass expansion: What we know and what is missing?
Arch Physiol Biochem. 119:161–169. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Evans JL, Goldfine ID, Maddux BA and
Grodsky GM: Oxidative stress and stress-activated signaling
pathways: A unifying hypothesis of type 2 diabetes. Endocr Rev.
23:599–622. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yuzefovych LV, LeDoux SP, Wilson GL and
Rachek LI: Mitochondrial DNA damage via augmented oxidative stress
regulates endoplasmic reticulum stress and autophagy: Crosstalk,
links and signaling. PLoS One. 8:e833492013. View Article : Google Scholar :
|
|
39
|
Gehrmann W, Elsner M and Lenzen S: Role of
metabolically generated reactive oxygen species for lipotoxicity in
pancreatic β-cells. Diabetes Obes Metab. 12(Suppl 2): 149–158.
2010. View Article : Google Scholar
|
|
40
|
Erdogdu O, Eriksson L, Xu H, Sjöholm A,
Zhang Q and Nyström T: Exendin-4 protects endothelial cells from
lipo-apoptosis by PKA, PI3K, eNOS, p38 MAPK, and JNK pathways. J
Mol Endocrinol. 50:229–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Jin L, Lim SW, Doh KC, Piao SG, Jin J, Heo
SB, Chung BH and Yang CW: Dipeptidyl peptidase IV inhibitor MK-0626
attenuates pancreatic islet injury in tacrolimus-induced diabetic
rats. PLoS One. 9:e1007982014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Tomas E, Stanojevic V and Habener JF:
GLP-1-derived nonapeptide GLP-1(28-36)amide targets to mitochondria
and suppresses glucose production and oxidative stress in isolated
mouse hepatocytes. Regul Pept. 167:177–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu Z, Stanojevic V, Brindamour LJ and
Habener JF: GLP1-derived nonapeptide GLP1(28-36)amide protects
pancreatic β-cells from glucolipotoxicity. J Endocrinol.
213:143–154. 2012. View Article : Google Scholar : PubMed/NCBI
|