Introduction
N-linked glycosylation is a well-studied protein
post-translational modification (PTM) that occurs at the Asn
residue in the consensus motif Asn-X-Ser/Thr, where X is any amino
acid except Pro (1).
N-glycosylation modulates the folding, stability, trafficking and
turnover of proteins, especially those of secreted or membrane
attached proteins, which are involved in various cell processes
such as cell-cell interaction or intracellular signaling (2–4).
As N-glycosylation is involved in important cell functions,
numerous N-glycosylation sites are evolutionarily conserved
(5).
We hypothesized that the losses of certain
ancestrally conserved N-glycosylation sites during evolution may
have been involved in the acquisition of novel human phenotypes.
The loss of N-glycosylation often disrupts the normal function of
proteins due to improper folding, trafficking, or activity of the
proteins (6,7). A proteome-wide analysis of
non-synonymous single-nucleotide variations in the N-glycosylation
motifs of human proteins revealed that 259 sites were lost because
of missense substitutions, some of which are involved in various
diseases (8). Although loss of a
glycosylation modification usually results in disadvantageous
phenotypes, some losses may be beneficial and fixed in humans
during evolution. For example, loss of the glycan moiety
N-glycolylneuraminic acid from cell surface proteins by the
inactivation of the CMAH gene, encoding
CMP-N-acetylneuraminic acid hydroxylase, was associated with the
evolution of resistance to a certain type of malaria in early
humans, although this loss subsequently led to susceptibility to
other pathogens (9,10).
A large number of N-glycosylation sites identified
from non-human animals and a suitable bioinformatics procedure are
necessary to identify cases where ancestrally conserved
N-glycosylation sites were lost during human evolution. An ideal
dataset for this analysis is the N-glycoproteome data obtained from
mouse tissues and plasma using high-throughput mass spectrometry
(11). Previously, a
bioinformatics method was used to identify novel gains of
N-glycosylation sites during human evolution (12). In the present study, the procedure
involved a simple modification to identify losses of ancestral
N-glycosylated Asn residues during human evolution following the
divergence of the Euarchonta lineage from the Glires lineage.
Additionally, a comprehensive literature survey was performed to
infer the possible functional outcomes of these changes, especially
for human-specific losses.
Materials and methods
Mouse N-glycosylation site data
For the N-linked glycosylation dataset from a
non-human proteome, we initially tested mouse data in the UniProt
database. However, there were only 419 experimentally verified
mouse N-glycosylation sites (as of December 20, 2013). Therefore,
mouse N-glycoproteome dataset from Zielinska et al was
utilized (11). This dataset
consisted of 6,367 N-linked glycosylation sites in 2,352 proteins.
Approximately 74% of the sites in the UniProt database were
re-identified in this data set.
Mammalian orthologous proteins
Mammalian orthologs of the mouse glycosylated
proteins were obtained from the University of California Santa Cruz
(UCSC) Genome Browser Database (http://genome.ucsc.edu). The 'CDS FASTA alignment from
multiple alignments' data, derived from the 'multiz100way'
alignment data prepared from 100 vertebrate genomes (13), were downloaded using the Table
Browser tool of the UCSC Genome Browser (14). Orthologous protein sequences from
62 mammalian species were extracted from these alignment datasets.
The selected mammalian species included humans, chimpanzees,
gorillas, orangutans, gibbons, rhesus macaques, crab-eating
macaques, baboons, green monkeys, marmosets, squirrel monkeys,
bushbabies, treeshrews, lesser Egyptian jerboas, prairie voles,
Chinese hamsters, golden hamsters, mice, rats, naked mole rats,
guinea pigs, chinchillas, brush-tailed rats, rabbits, pikas, pigs,
alpacas, Bactrian camels, dolphins, killer whales, Tibetan
antelopes, cattle such as cows, sheep, and goats, horses, white
rhinoceroses, cats, dogs, ferrets, pandas, Pacific walruses,
Weddell seals, black flying foxes, megabats, David's myotis bats,
microbats, big brown bats, hedgehogs, shrews, star-nosed moles,
elephants, cape elephant shrews, manatees, cape golden moles,
tenrecs, aardvarks, armadillos, opossums, Tasmanian devils,
wallabies and platypuses. Detailed information on species and
genome assemblies is available at the UCSC Genome Browser web site
(http://hgdownload.cse.ucsc.edu/goldenPath/hg19/multiz100way).
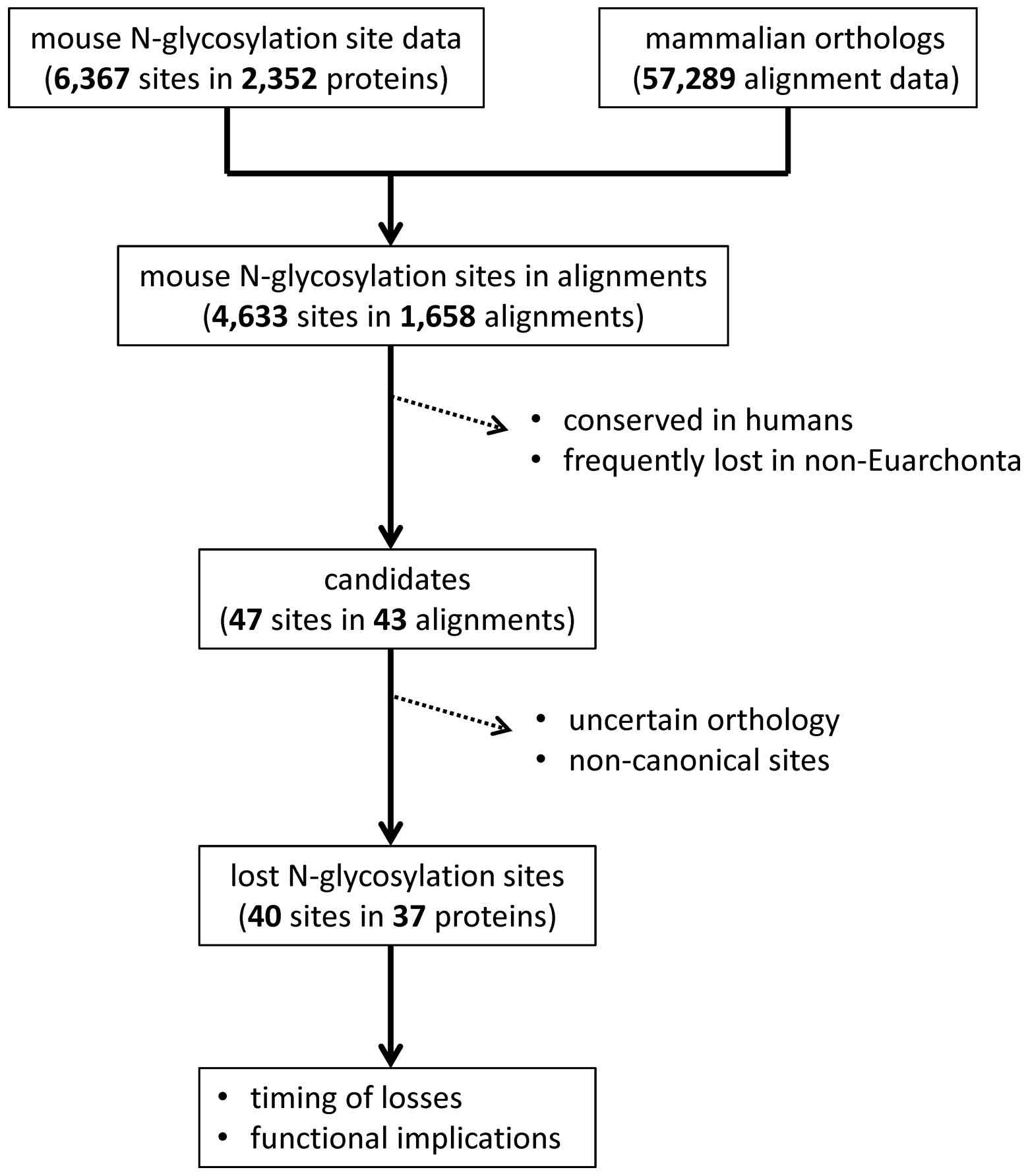
Computational screening for candidate
lost N-glycosylation sites
The total number of mouse N-glycosylation sites in
the data set from Zielinska et al was 6,367 (11). The 'multiz100way' alignment data,
containing 57,289 alignment sets, were analyzed to identify human
and other mammalian orthologs of each of the mouse N-glycosylated
proteins (Fig. 1). Ad hoc Perl
scripts were used to analyze the data. There were 1,658 orthologous
protein datasets containing human and mouse protein sequences. This
dataset covered 4,633 mouse N-glycosylation sites. From each
dataset, the mammalian sequences were extracted and realigned using
MUSCLE (http://www.drive5.com/muscle)
(15).
Each of the positions that aligned with a mouse
N-glycosylation site was examined using ad hoc Perl scripts. Sites
that were conserved in humans, where the human protein had a
consensus N-glycosylation motif, were discarded. Sites where ≥30%
non-Euarchonta mammals did not have an Asn residue, indicating a
frequent loss in these species, were also discarded. A total of 47
sites in 43 protein alignments were obtained after this
computational screening step.
Manual inspection to select lost
N-glycosylated Asn residues in the human lineage
As a final step, we manually scrutinized the 47
candidates to identify highly probable instances of N-glycosylation
site loss during evolution of the human lineage. In each dataset,
the species that had many gaps compared to other mammals were
removed. When the mouse sequence utilized from Zielinska et
al (11) differed from that
of the UCSC database by at least three residues, the case was
discarded as the orthology of the aligned proteins could not be
guaranteed. We also discarded cases in which the mouse
N-glycosylation site did not conform to the canonical sequence, or
cases showing low sequence conservation among mammals.
Finally, 40 ancestral N-glycosylation sites in 37
proteins were identified to be lost during human evolution. The
human and mouse protein sequences in the UCSC alignment were mapped
to UniProt database sequences to utilize the UniProt annotation
record. We examined the multiple sequence alignment and the
mammalian phylogenetic tree to infer the timing of the loss of the
N-glycosylated Asn residue.
Results and Discussion
Identification of N-glycosylation sites
lost during human evolution and timing of loss
We applied a bioinformatics procedure previously
developed to identify novel N-glycosylation sites during human
evolution, with modifications (12). Initially, there were 6,367
experimentally identified mouse N-glycosylation sites from 2,352
proteins in the dataset from Zielinska et al (11) and 57,289 orthologous protein
sequence alignments from 62 mammalian species extracted from the
UCSC 'multiz100way' data (13,14). These data were analyzed to collect
N-glycosylation sites lost during human evolution after the
Euarchonta (primates and treeshrews) diverged from the Glires
(rodents and rabbits).
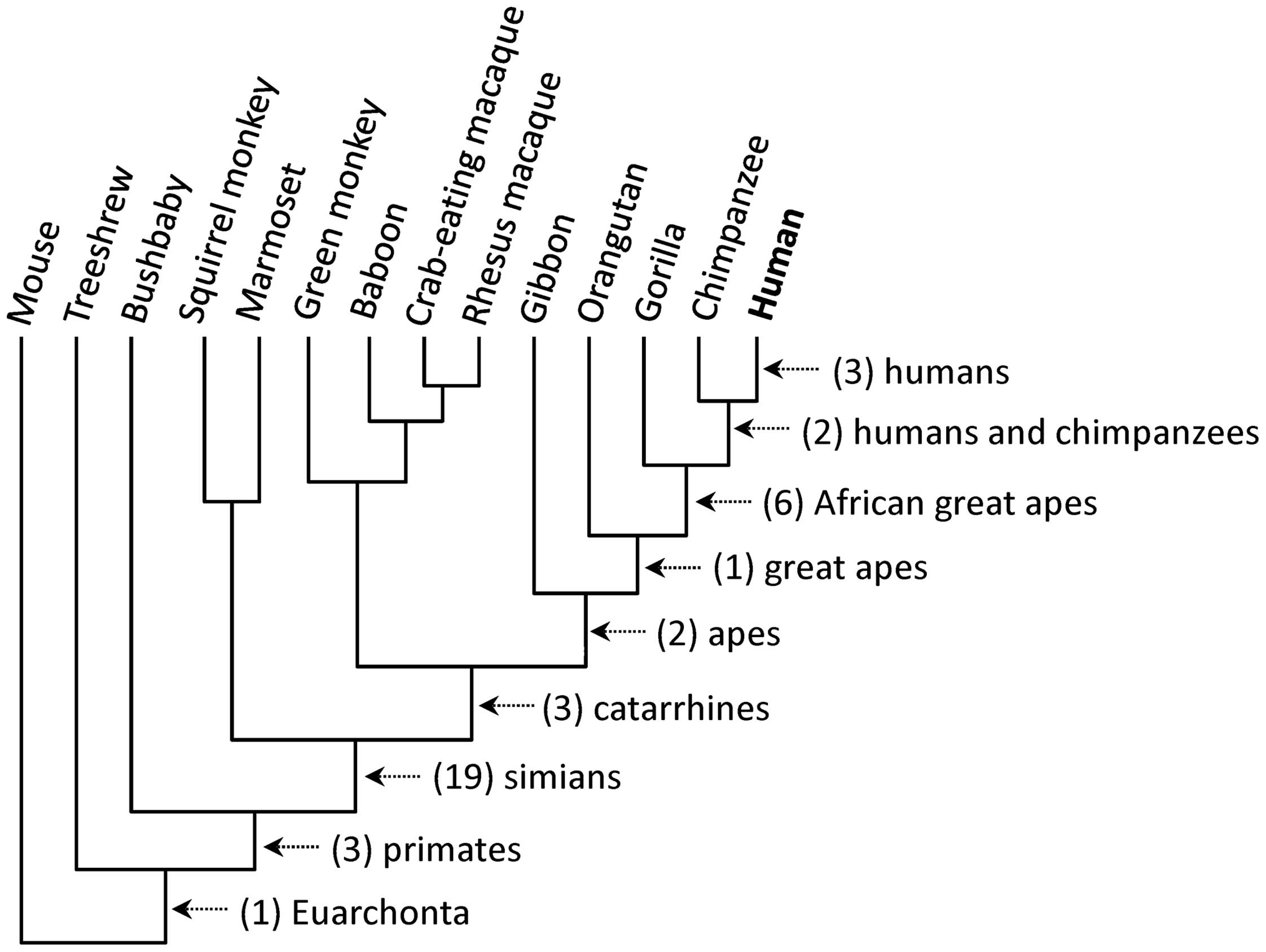
As a result, 40 N-glycosylation sites in 37 proteins
were identified to have been lost during human evolution (Table I). Of the 37 proteins, three
proteins encoded by the ICAM1, LRP2 and MASP2
genes had each lost two N-glycosylation sites (nos. 13 and 14 for
ICAM1, 23 and 24 for LRP2, and 27 and 28 for
MASP2), and the remaining 34 proteins had lost one site
each. Fig. 2 shows the number of
N-glycosylation sites that have been lost in each common ancestor
along the human lineage: humans, three; humans and chimpanzees,
two; African great apes, six; great apes, one; apes, two;
catarrhines, three; simians, 19; primates, three; and Euarchonta,
one.
 | Table IList of ancestral N-glycosylation
sites that were lost during human evolution. |
Table I
List of ancestral N-glycosylation
sites that were lost during human evolution.
| No. | UniProt ID | Position | Sequencea | Clade | Gene | Protein |
|---|
| 1 | ABCA1_HUMAN | 1499 |
LPPPQRKQNTADILQDLTGRNISDYLVKTYV | Simians | ABCA1 | ATP-binding cassette
sub-family A member 1 |
| ABCA1_MOUSE | 1499 |
LPPPQRKQKTADILQNLTGRNISDYLVKTYV | | | |
| 2 | ADAM9_HUMAN | 636 |
TKCGAGKICRNFQCVDASVLNYDCDVQKKCH | Primates | ADAM9 | Disintegrin and
metalloproteinase domain-containing protein 9 |
| ADAM9_MOUSE | 636 |
TKCDAGKICRNFQCVNASVLNYDCDIQGKCH | | | |
| 3 | ASM3A_HUMAN | 367 |
QYYLNLTEANLKGESIWKLEYILTQTYDIED | Simians | SMPDL3A | Acid
sphingomyelinase-like phosphodiesterase 3a |
| ASM3A_MOUSE | 364 |
QYYLNLTEANLKGESNWTLEYVLTQAYSVAD | | | |
| 4 | C4BPA_HUMAN | 67 |
PPTLSFAAPM-DITLTETRFKTGTTLKYTCL | Simians | C4BPA | C4b-binding protein α
chain |
| C4BPA_MOUSE | 74 |
PPAIPNALPA-D–VNRTDFESHTTLKYECL | | | |
| 5 | CAD13_HUMAN | 489 |
GPVFYPDPMMVTRQEDLSVGSVLLTVNATDP | African great
apes | CDH13 | Cadherin-13 |
| CAD13_MOUSE | 489 |
GPVFYPDPMMVTKQENISVGSVLLTVNATDP | | | |
| 6 | CBG_HUMAN | 224 |
QPFDLASTREENFYVDETTVVKVPMMLQSST | Simians | SERPINA6 |
Corticosteroid-binding globulin |
| CBG_MOUSE | 217 |
LPFSPENTREEDFYVNETSTVKVPMMVQSGN | | | |
| 7 | CELR1_HUMAN | 2140 |
QVDGARALQLVRALRSATQHTGTLFGNDVRT | Humans | CELSR1 | Cadherin EGF LAG
seven-pass G-type receptor 1 |
| CELR1_MOUSE | 2155 |
RMDGNRSLRLAKALRNATQGNSTLFGNDVRT | | | |
| 8 | CPN2_HUMAN | 311 |
LTHNQLETVAEGTFAHLSNLRSLMLSYNAIT | Primates | CPN2 | Carboxypeptidase N
subunit 2 |
| CPN2_MOUSE | 311 |
LSYNQLETIPEGAFTNLSRLVSLTLSHNAIT | | | |
| 9 | CSF1R_HUMAN | 493 |
EHNQTYECRAHNSVGSGSWAFIPISAGAHTH | Simians | CSF1R | Macrophage
colony-stimulating factor 1 receptor |
| CSF1R_MOUSE | 491 |
KHNMTYFCKTHNSVGNSSQYFRAVSLGQSKQ | | | |
| 10 | CTL4_HUMAN | 198 |
TNV–TPPALPGITNDTTIQQGISGLIDSLN | Euarchonta | SLC44A4 | Choline
transporter-like protein 4 |
| CTL4_MOUSE | 196 |
PNI–TLPEDLRI-NNTTVSNGISGLLDSIN | | | |
| 11 | DCC_HUMAN | 60 |
EPSDAVTMRGGNVLLDCSAESDRGVPVIKWK | Primates | DCC | Netrin receptor
DCC |
| DCC_MOUSE | 60 |
EPSDAVTMRGGNVLLNCSAESDRGVPVIKWK | | | |
| 12 | FETUA_HUMAN | 99 |
TLETTCHVLDPTPVARCSVRQLKEHAVEGDC | Catarrhines | AHSG |
α-2-HS-glycoprotein |
| FETUA_MOUSE | 99 |
TLETTCHALDPTPLANCSVRQLTEHAVEGDC | | | |
| 13 | ICAM1_HUMAN | 359 |
GVPAQPLGPRAQLLLKATPEDNGRSFSCSAT | Simians | ICAM1 | Intercellular
adhesion molecule 1 |
| ICAM1_MOUSE | 362 |
GVEPRPPTPQVQFTLNASSEDHKRSFFCSAA | | | |
| 14 | ICAM1_HUMAN | 47 |
SPSKVILPRGGSVLVTCSTSCDQPKLLGIET | African great
apes | ICAM1 | Intercellular
adhesion molecule 1 |
| ICAM1_MOUSE | 47 |
HPREAFLPQGGSVQVNCSSSCKEDLSLGLET | | | |
| 15 | IGSF5_HUMAN | 160 |
FIPSVNLVVAENEPCEVTCLPSHWTRLPDIS | Simians | IGSF5 | Immunoglobulin
superfamily member 5 |
| IGSF5_MOUSE | 146 |
NIPSNNLIVTEGEPCNVTCYAVGWTSLPDIS | | | |
| 16 | ITB5_HUMAN | 479 |
GCSVGLEPNSARCNGSGTYVCGLCECSPGYL | Simians | ITGB5 | Integrin β-5 |
| ITB5_MOUSE | 479 |
GCSTGL-PNSARCSGNGTYTCGLCECDPGYL | | | |
| 17 | LAMA1_HUMAN | 1337 |
IKASYGQGLQQSRISDISMEVGRKAEKLHPE | Simians | LAMA1 | Laminin subunit
α-1 |
| LAMA1_MOUSE | 1344 |
IKASYGQGLQQSRIANISMEVGRKAVELPAE | | | |
| 18 | LAMA2_HUMAN | 923 |
DAVDAKNCQPCRCNAGGSFSEVCHSQTGQCE | Great apes | LAMA2 | Laminin subunit
α-2 |
| LAMA2_MOUSE | 919 |
DAVNAKNCQPCRCNINGSFSEICHTRTGQCE | | | |
| 19 | LAMP5_HUMAN | 102 |
IALTRGAEVKGRCGHSQSELQVFWVDRAYAL | Simians | LAMP5 | Lysosome-associated
membrane glycoprotein 5 |
| LAMP5_MOUSE | 102 |
ISLTRGAEVKGHCGHNESELEVFWVDHAYTL | | | |
| 20 | LAT3_HUMAN | 54 |
ILKNEGFYSSTCPAESSTNTTQDEQRRWPGC | African great
apes | SLC43A1 | Large neutral amino
acids transporter small subunit 3 |
| LAT3_MOUSE | 54 |
MLKKEGFYSSLCPAENRTNTTQDEQHQWTSC | | | |
| 21 | LCAP_HUMAN | 447 |
NWGLLTFREETLLYDSNTSSMADRKLVTKII | Humans and
chimpanzees | LNPEP | Leucyl-cystinyl
aminopeptidase |
| LCAP_MOUSE | 447 |
NWGLLTFREETLLYDNATSSVADRKLVTKII | | | |
| 22 | LPP2_HUMAN | 156 |
SVYVQLEKVCRGNPADVTEARLSFYSGHSSF | Simians | PPAP2C | Lipid phosphate
phosphohydrolase 2 |
| LPP2_MOUSE | 155 |
SGYVQLE-VCRGSPANVTEARLSFYSGHSSF | | | |
| 23 | LRP2_HUMAN | 1450 |
SLLLLVASQNKIIADSVTSQVHNIYSLVENG | Catarrhines | LRP2 | Low-density
lipoprotein receptor-related protein 2 |
| LRP2_MOUSE | 1451 |
NLLLVVASRDKIIMDNITAHTHNIYSLVQDV | | | |
| 24 | LRP2_HUMAN | 3838 |
CLDASDEADCPTRFPDGAYCQATMFECKNHV | Simians | LRP2 | Low-density
lipoprotein receptor-related protein 2 |
| LRP2_MOUSE | 3840 |
CLDASDESACPTRFPNGTYCPAAMFECKNHV | | | |
| 25 | LYAM1_HUMAN | 226 |
THPLGNFSFSSQCAFSCSEGTNLTGIEETTC | African great
apes | SELL | L-selectin |
| LYAM1_MOUSE | 226 |
IHPLGNFSFQSKCAFNCSEGRELLGTAETQC | | | |
| 26 | MA2B1_HUMAN | 345 |
KNLDKLIRLVNAQQAKGSSVHVLYSTPACYL | Simians | MAN2B1 | Lysosomal
α-mannosidase |
| MA2B1_MOUSE | 345 |
KNMDKLIRLVNAQQVNGSLVHVLYSTPTCYL | | | |
| 27 | MASP2_HUMAN | 103 |
TLCGQESTDTERAPGKDTFYSLGSSLDITFR | African great
apes | MASP2 | Mannan-binding
lectin serine protease 2 |
| MASP2_MOUSE | 103 |
TLCGQESTDTEQAPGNDTFYSLGPSLKVTFH | | | |
| 28 | MASP2_HUMAN | 642 |
DSCRGDSGGALVFLDSETERWFVGGIVSWGS | Apes | MASP2 | Mannan-binding
lectin serine protease 2 |
| MASP2_MOUSE | 641 |
DSCRGDSGGALVFLDNETQRWFVGGIVSWGS | | | |
| 29 | MERTK_HUMAN | 97 |
QVTSVESKPLPPLAFKHTVGHIILSEHKGVK | Simians | MERTK | Tyrosine-protein
kinase Mer |
| MERTK_MOUSE | 91 |
QVTSTASKLLPPVAFNHTIGHIVLSEHKNVK | | | |
| 30 | MET_HUMAN | 358 |
FGVFAQSKPDSAEPMDRSAMCAFPIKYVNDF | Simians | MET | Hepatocyte growth
factor receptor |
| MET_MOUSE | 357 |
FGVFAQSKPDSAEPVNRSAVCAFPIKYVNDF | | | |
| 31 | PTPRB_HUMAN | 709 |
VRECSFSSLTPGRLYTVTITTRSGKYENHSF | Simians | PTPRB | Receptor-type
tyrosine-protein phosphatase β |
| PTPRB_MOUSE | 710 |
VSECSFSSLTPGRLYNVTVTTKSGNYASHSF | | | |
| 32 | PTPRF_HUMAN | 950 |
AWDPPVLAERNGRIISYTVVFRDINSQQELQ | Simians | PTPRF | Receptor-type
tyrosine-protein phosphatase F |
| PTPRF_MOUSE | 941 |
TWDPPVLAERNGHITNYTVVYRDINSQLELQ | | | |
| 33 | SIAT9_HUMAN | 280 |
LFKSVDFNWLQAMVKKETLPFWVRLFFWKQV | Humans | ST3GAL5 | Lactosylceramide
α-2,3-sialyltransferase |
| SIAT9_MOUSE | 279 |
LFKSVDFKWLQAMVKNESLPFWVRLFFWKQV | | | |
| 34 | ST14_HUMAN | 489 |
WADCTDHSDELNCSCDAGHQFTCKNKFCKPL | Catarrhines | ST14 | Suppressor of
tumorigenicity 14 protein |
| ST14_MOUSE | 489 |
WADCPDYSDERYCRCNATHQFTCKNQFCKPL | | | |
| 35 | STAB2_HUMAN | 63 |
LNLGVKCPDGYTMITSGSVGVRDCRYTFEVR | Apes | STAB2 | Stabilin-2 |
| STAB2_MOUSE | 71 |
VNIAVKCPDGYIKITNGTVGVRDCRYSLKIQ | | | |
| 36 | SUSD2_HUMAN | 703 |
FCNFDVAATGSLSTGTATRVAHQLHQRRMQS | African great
apes | SUSD2 | Sushi
domain-containing protein 2 |
| SUSD2_MOUSE | 700 |
FCILDVMSTGSSSVGNATRIAHQLHQHRLKS | | | |
| 37 | TMM62_HUMAN | 384 |
SGPIFVLKWNPRNYSSGTHNIEVIVQDSAGR | Simians | TMEM62 | Transmembrane
protein 62 |
| TMM62_MOUSE | 384 |
SGPIFILKWNPRNYSNGTHTIEVFVQDSAGR | | | |
| 38 | VGFR3_HUMAN | 582 |
ELLEGQPVLLSCQADSYKYEHLRWYRLNLST | Simians | FLT4 | Vascular
endothelial growth factor receptor 3 |
| VGFR3_MOUSE | 582 |
DPLEGQSVRLSCRADNYTYEHLRWYRLNLST | | | |
| 39 | VNN1_HUMAN | 146 |
NSIYVVANIGDKKPCDTSDPQCPPDGRYQYN | Humans and
chimpanzees | VNN1 | Pantetheinase |
| VNN1_MOUSE | 148 |
NSIYVVANMGDKKPCNTSDSHCPPDGRFQYN | | | |
| 40 | VSI10_HUMAN | 100 |
ATSLHIESLSLGDEGIYTCQEILNVTQWFQV | Humans | VSIG10 | V-set and
immunoglobulin domain-containing protein 10 |
| VSI10_MOUSE | 121 |
AGALRIEALRLEDDGNYTCQEVLNETHWFPV | | | |
Of the 37 N-glycosylation sites that were lost in
the human lineage since the divergence of the Euarchonta and the
Glires, three events occurred in human proteins after the
divergence of humans and chimpanzees (Table I, nos. 7, 33 and 40 and Fig. 3). The residue positions for these
human-specific losses are Ser-2140 in cadherin EGF LAG seven-pass
G-type receptor 1 encoded by the CELSR1 gene, Lys-280 in
lactosylceramide α-2,3-sialyltransferase encoded by the
ST3GAL5 gene, and Ile-100 in the V-set and immunoglobulin
domain-containing protein 10 encoded by the VSIG10 gene.
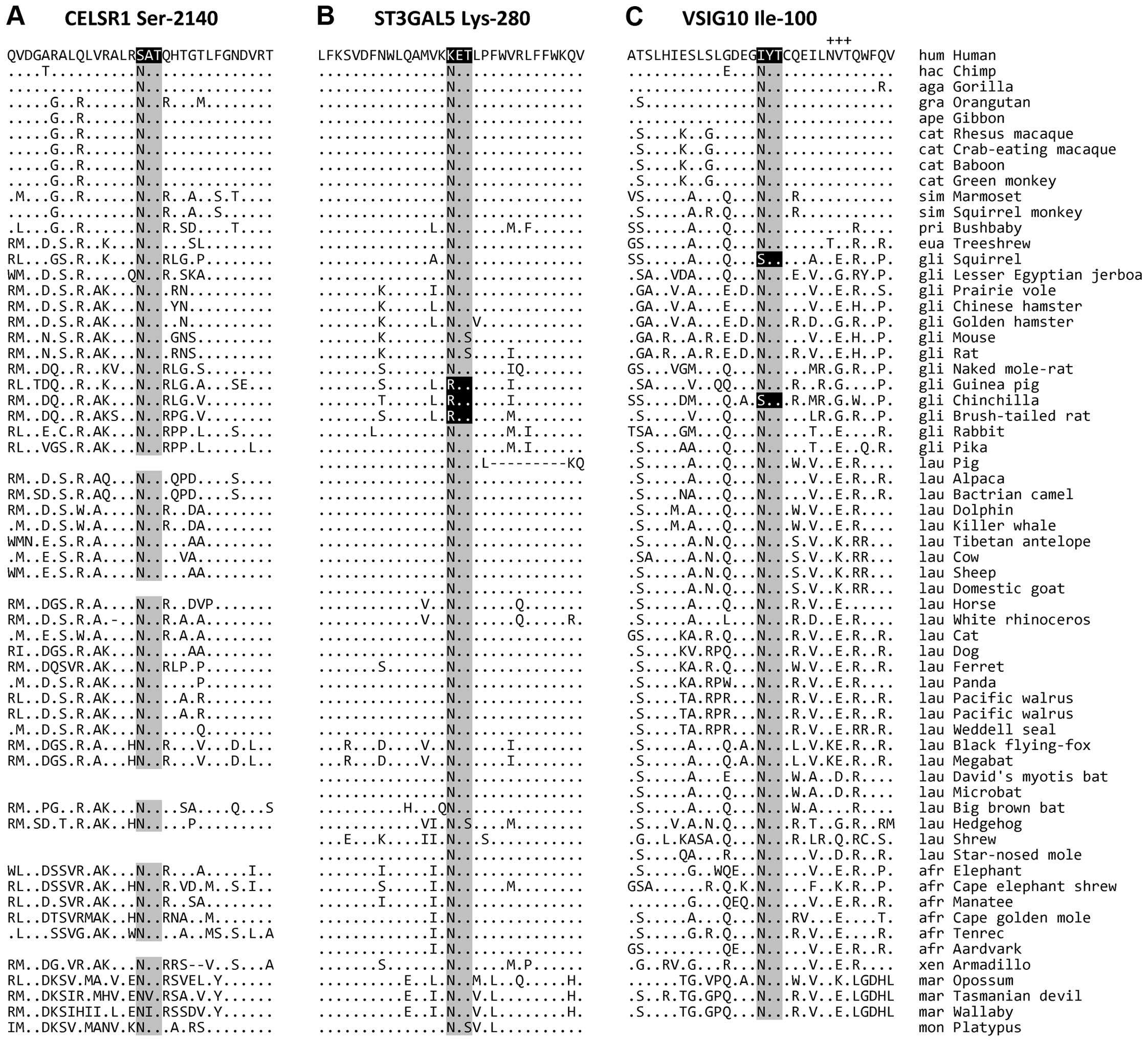 | Figure 3Human-specific losses of ancestral
N-glycosylation sites. The ancestral N-glycosylation sites and the
surrounding regions of (A) CELSR1 Ser-2140, (B) ST3GAL5 Lys-280 and
(C) VSIG10 Ile-100 are presented. The ancestral N-glycosylation
consensus sequences are highlighted in grey, and corresponding
sequences that lost the consensus, in black. The adjacent conserved
N-glycosylation site Asn-108 in VSIG10 is indicated by plus signs
(+++). The residues that are identical to those in the human
sequence are indicated by dots (.). Dashes (−) denote alignment
gaps. In some species, the sequences were not determined. hum,
humans; hac, humans and chimpanzees; aga, African great apes; gra,
great apes; ape, apes; cat, catarrhines; sim, simians; pri,
primates; eua, Euarchonta; gli, Glires; lau, Laurasiatheria; afr,
Afrotheria; xen, Xenarthra; mar, Marsupialia; and mon,
Monotremata. |
Human-specific loss of N-glycosylation at
the amino acid position 2140 of CELSR1
The human cadherin EGF LAG seven-pass G-type
receptor 1 or CELSR1, encoded by the CELSR1 gene, is a
heavily glycosylated protein with 20 glycosylation sites
(http://www.uniprot.org/uniprot/Q9NYQ6). Sequence
comparison revealed that an ancestrally conserved glycosylation
site at position 2140 was altered from Asn to Ser in humans
following the human-chimpanzee divergence (Fig. 3A). The other mammals examined have
a conserved Asn residue, conforming to the N-glycosylation motif
consensus.
The CELSR1 protein is a member of the flamingo
cadherin protein family, which are proteins located at the plasma
membrane with seven transmembrane domains (16,17). It has nine cadherin domains, seven
epidermal growth factor-like repeats and two laminin A G-type
repeats. This gene is highly expressed during mouse embryonic
development, especially in the central nervous system (16,17). Mutations in this protein were
reported to cause neural tube defects and caudal agenesis in humans
(18,19). Therefore, CELSR1 may play an
important role in contact-mediated signaling during nervous system
formation in early embryogenesis. CELSR1 also plays an important
role in the development of other organs, such as lung branching
morphogenesis (20), intraluminal
valve formation in lymphatic vessels (21), and hair follicle polarization and
orientation (22).
Therefore, changes in the CELSR1 protein may be
involved in the evolution of the nervous system, lung, lymphatic
system, or hair patterns. However, a probable direct phenotypic
consequence of the loss of the N-glycosylation site at position
2140 in humans remains to be determined.
Human-specific loss of N-glycosylation at
the amino acid position 280 of ST3GAL5
The human lactosylceramide α-2,3-sialyltransferase,
encoded by the ST3GAL5 gene, which is also known as
ganglioside GM3 synthase or sialyltransferase 9 (SIAT9), has three
N-glycosylation sites (http://www.uniprot.org/uniprot/Q9UNP4). A sequence
comparison revealed that the human protein lost a conserved
N-glycosylation site at 280 (Asn to Lys) following the
human-chimpanzee divergence (Fig.
3B). All of the other mammals analyzed, except three, have the
N-glycosylation consensus sequence at this site. A loss of the
N-glycosylation consensus motif was also identified in guinea pigs,
chinchillas, and brush-tailed rats (also known as degus), which
have a Gly residue instead of Asn at the corresponding position.
The three species belong to the rodent clade Caviomorpha (23), suggesting that the Asn-to-Gly
change occurred in an ancestor of the three mammals.
The ST3GAL5 gene encodes a sialyltransferase,
a type II membrane protein that catalyzes the formation of GM3, a
glycosphingolipid enriched in neural tissue, by adding sialic acid
to lactosylceramide (24,25). GM3 is known to participate in the
induction of cell differentiation, modulation of cell
proliferation, and integrin-mediated cell adhesion.
Mutations in this gene are associated with several
neurological disorders, such as Amish infantile epilepsy syndrome
(26), Salt and Pepper syndrome
characterized by severe intellectual disability, epilepsy,
scoliosis, choreoathetosis, dysmorphic facial features and altered
dermal pigmentation (25), or
disruption of the structural integrity and function of cochlear
hair cells (27). Therefore, the
ST3GAL5 enzyme is crucial for normal neural development and
function. The loss of an ancestrally conserved N-glycosylation site
may be associated with a novel phenotype in the nervous system and
function in humans, which may be demonstrated by molecular
functional analysis.
Human-specific loss of N-glycosylation at
position 100 of VSIG10
The VSIG10 gene encodes for V-set and
immunoglobulin domain-containing protein 10. The human VSIG10
protein has nine N-glycosylation sites (http://www.uniprot.org/uniprot/Q8N0Z9). In the present
study, we found that this protein lost an ancestrally conserved
site at position 121, specifically, an Asn-to-Ile mutation
abolished the N-glycosylation consensus (Fig. 3C). Of note, the consensus motif
was also independently lost in squirrels and chinchillas. VSIG10 is
a single-pass type I membrane protein containing a V-set domain,
two immunoglobulin domains, and an I-set domain, which is present
in cell adhesion molecules. No known molecular or biological
function of VSIG10 has been reported.
In conclusion, we have identified 40 cases for loss
of ancestrally conserved N-glycosylation sites, three of which are
human-specific. Two human-specific losses occurred in the CELSR1
and ST3GAL5 proteins, which play indispensable roles in the normal
development and function of the nervous systems. This finding
suggests that the loss of N-glycosylation sites in these proteins
may be associated with the evolution of human cognitive function.
We suggest that a loss of ancestrally conserved N-glycosylation
sites may result in the evolution of novel phenotypes, and the
cases identified in the present study may serve as immediate
targets for functional analyses to elucidate the molecular basis
for an explanation of human phenotype evolution.
Acknowledgments
This study was supported by the National Research
Foundation of Korea (NRF) grant (NRF-2012R1A1B3001513) funded by
the Ministry of Education, Science and Technology, Republic of
Korea.
References
|
1
|
Schwarz F and Aebi M: Mechanisms and
principles of N-linked protein glycosylation. Curr Opin Struct
Biol. 21:576–582. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Helenius A and Aebi M: Intracellular
functions of N-linked glycans. Science. 291:2364–2369. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dennis JW, Nabi IR and Demetriou M:
Metabolism, cell surface organization, and disease. Cell.
139:1229–1241. 2009. View Article : Google Scholar
|
|
4
|
Scott H and Panin VM: The role of protein
N-glycosylation in neural transmission. Glycobiology. 24:407–417.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Park C and Zhang J: Genome-wide
evolutionary conservation of N-glycosylation sites. Mol Biol Evol.
28:2351–2357. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Winterpacht A, Hilbert K, Stelzer C,
Schweikardt T, Decker H, Segerer H, Spranger J and Zabel B: A novel
mutation in FGFR-3 disrupts a putative N-glycosylation site and
results in hypochondroplasia. Physiol Genomics. 2:9–12.
2000.PubMed/NCBI
|
|
7
|
Wujek P, Kida E, Walus M, Wisniewski KE
and Golabek AA: N-glycosylation is crucial for folding,
trafficking, and stability of human tripeptidyl-peptidase I. J Biol
Chem. 279:12827–12839. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mazumder R, Morampudi KS, Motwani M,
Vasudevan S and Goldman R: Proteome-wide analysis of
single-nucleotide variations in the N-glycosylation sequon of human
genes. PLoS One. 7:e362122012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deng L, Song J, Gao X, Wang J, Yu H, Chen
X, Varki N, Naito-Matsui Y, Galán JE and Varki A: Host adaptation
of a bacterial toxin from the human pathogen Salmonella Typhi.
Cell. 159:1290–1299. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rich SM, Leendertz FH, Xu G, LeBreton M,
Djoko CF, Aminake MN, Takang EE, Diffo JL, Pike BL, Rosenthal BM,
et al: The origin of malignant malaria. Proc Natl Acad Sci USA.
106:14902–14907. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zielinska DF, Gnad F, Wiśniewski JR and
Mann M: Precision mapping of an in vivo N-glycoproteome reveals
rigid topological and sequence constraints. Cell. 141:897–907.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim DS and Hahn Y: The acquisition of
novel N-glycosylation sites in conserved proteins during human
evolution. BMC Bioinformatics. 16:292015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Blanchette M, Kent WJ, Riemer C, Elnitski
L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green
ED, et al: Aligning multiple genomic sequences with the threaded
blockset aligner. Genome Res. 14:708–715. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Karolchik D, Hinrichs AS, Furey TS, Roskin
KM, Sugnet CW, Haussler D and Kent WJ: The UCSC Table Browser data
retrieval tool. Nucleic Acids Res. 32:D493–D496. 2004. View Article : Google Scholar :
|
|
15
|
Edgar RC: MUSCLE: Multiple sequence
alignment with high accuracy and high throughput. Nucleic Acids
Res. 32:1792–1797. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hadjantonakis AK, Sheward WJ, Harmar AJ,
de Galan L, Hoovers JM and Little PF: Celsr1, a neural-specific
gene encoding an unusual seven-pass transmembrane receptor, maps to
mouse chromosome 15 and human chromosome 22qter. Genomics.
45:97–104. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hadjantonakis AK, Formstone CJ and Little
PF: mCelsr1 is an evolutionarily conserved seven-pass transmembrane
receptor and is expressed during mouse embryonic development. Mech
Dev. 78:91–95. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Allache R, De Marco P, Merello E, Capra V
and Kibar Z: Role of the planar cell polarity gene CELSR1 in neural
tube defects and caudal agenesis. Birth Defects Res A Clin Mol
Teratol. 94:176–181. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lei Y, Zhu H, Yang W, Ross ME, Shaw GM and
Finnell RH: Identification of novel CELSR1 mutations in spina
bifida. PLoS One. 9:e922072014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yates LL, Schnatwinkel C, Murdoch JN,
Bogani D, Formstone CJ, Townsend S, Greenfield A, Niswander LA and
Dean CH: The PCP genes Celsr1 and Vangl2 are required for normal
lung branching morphogenesis. Hum Mol Genet. 19:2251–2267. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tatin F, Taddei A, Weston A, Fuchs E,
Devenport D, Tissir F and Makinen T: Planar cell polarity protein
Celsr1 regulates endothelial adherens junctions and directed cell
rearrangements during valve morphogenesis. Dev Cell. 26:31–44.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Devenport D and Fuchs E: Planar
polarization in embryonic epidermis orchestrates global asymmetric
morphogenesis of hair follicles. Nat Cell Biol. 10:1257–1268. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Upham NS and Patterson BD: Diversification
and biogeography of the Neotropical caviomorph lineage
Octodontoidea (Rodentia: Hystricognathi). Mol Phylogenet Evol.
63:417–429. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ishii A, Ohta M, Watanabe Y, Matsuda K,
Ishiyama K, Sakoe K, Nakamura M, Inokuchi J, Sanai Y and Saito M:
Expression cloning and functional characterization of human cDNA
for ganglioside GM3 synthase. J Biol Chem. 273:31652–31655. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Boccuto L, Aoki K, Flanagan-Steet H, Chen
CF, Fan X, Bartel F, Petukh M, Pittman A, Saul R, Chaubey A, et al:
A mutation in a ganglioside biosynthetic enzyme, ST3GAL5, results
in salt and pepper syndrome, a neurocutaneous disorder with altered
glycolipid and glycoprotein glycosylation. Hum Mol Genet.
23:418–433. 2014. View Article : Google Scholar
|
|
26
|
Simpson MA, Cross H, Proukakis C,
Priestman DA, Neville DC, Reinkensmeier G, Wang H, Wiznitzer M,
Gurtz K, Verganelaki A, et al: Infantile-onset symptomatic epilepsy
syndrome caused by a homozygous loss-of-function mutation of GM3
synthase. Nat Genet. 36:1225–1229. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yoshikawa M, Go S, Suzuki S, Suzuki A,
Katori Y, Morlet T, Gottlieb SM, Fujiwara M, Iwasaki K, Strauss KA,
et al: Ganglioside GM3 is essential for the structural integrity
and function of cochlear hair cells. Hum Mol Genet. 24:2796–2807.
2015. View Article : Google Scholar : PubMed/NCBI
|