Introduction
Diabetic vasculopathy, a major complication of
diabetes, is characteristic of impaired angiogenic response
(1,2). Nevertheless, the exact mechanisms
have not been thoroughly studied. Previous studies have shown that
vascular endothelial growth factor receptor-2 (VEGFR-2), which is
expressed mostly in vascular endothelial cells, plays an important
role in regulating endothelial cell proliferation, migration and
capillary-like tube formation, which are considered to be important
components of the angiogenic response (3–5).
When stimulated by vascular endothelial growth factor (VEGF),
autophosphorylation of VEGFR-2 occurs, leading to angiogenesis
(5,6). It has been recently reported that
the expression of VEGFR-2 in endothelial cells is decreased in
diabetic mice when reactive oxygen species (ROS) is induced by
hyperglycemia (7).
The ubiquitin-proteasome system (UPS), one method of
protein degradation, is responsible for 80–90% of proteolysis.
There are three enzymes in the UPS, namely ubiquitin-activating
enzyme (E1), ubiquitin-conjugating enzyme (E2), and ubiquitin
ligase (E3). E1 activates ubiquitin, which is accepted by E2, and
E3 targets the protein to which the ubiquitin is attached. The
protein is then degraded by the 26S proteasome. Thus, E3 is
responsible for specificity of protein turnover (8). β-transduction repeat-containing
protein (β-TrCP), an F-box component of the Skp1-Cul1-F-box protein
ubiquitin ligases which functions as a substrate recognition
subunit, has been previously reported to suppress angiogenesis by
promoting ubiquitination and degradation of VEGFR-2 in thyroid
cancer cells (9–11). However, the role of β-TrCP in the
regulation of VEGFR-2 and angiogenesis in endothelial cells where
hyperglycemia is induced is far from fully understood.
Additionally, β-TrCP recognizes a consensus sequence of
DSG(X)nS (X, any amino acid; n=2–4) in its substrates
and requires them to be phosphorylated before regulating
degradation (12). Interestingly,
the DSG(X)nS motif is present in VEGFR-2 (9). Moreover, as previously demomstrated,
glycogen synthase kinase-3β (GSK-3β) phosphorylates many β-TrCP
substrates and mediates their degradation (9,13,14). Accordingly, in the present study
we hypothesized that redundant ROS downregulated VEGFR-2 through
β-TrCP-induced UPS-dependent VEGFR-2 degradation, which was likely
mediated by GSK-3β.
Materials and methods
Cell culture and glucose or reagent
exposure
Human umbilical vein endothelial cells (HUVECs) were
obtained from American Type Culture Collection (Manassas, VA, USA).
The cells were routinely cultured in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% fetal bovine serum (both from
Gibco, Shanghai, China) and maintained at 37°C in a humidified
chamber with 5% CO2. The culture medium was changed for
fresh medium every other day. When they reached 80–90% confluence
after 12 h of incubation, cells were grouped into various groups:
normal glucose (NG, 6.6 mM), medium glucose (MG, 19.8 mM), high
glucose (HG, 33 mM), mannitol (Man, 33 mM), SU5416 (1.0 and 10
µM, respectively, as have been frequently adopted in
previous studies) and glucose oxidase (GO, 1 U/ml) (15–17). Cells exposed to various
concentrations of glucose or Man were incubated for 48 h. Treatment
with GO lasted for 15 min, as previously described (7).
Reagents and antibodies
Lipofectamine 2000 and TRIzol reagent were obtained
from Invitrogen Life Technologies, Inc. (Grand Island, NY, USA).
All the primary antibodies [phospho-VEGFR-2 (Tyr1175) (19A10)
rabbit monoclonal antibody (mAb) (#2478); VEGFR-2 (D5B1) rabbit mAb
(#9698); β-TrCP (D13F10) rabbit mAb (#4394); ubiquitin antibody
(#3933); phospho-GSK-3β (Ser9) (D85E12) rabbit mAb (#5558); GSK-3β
(D5C5Z) rabbit mAb (#12456)], secondary antibodies [anti-rabbit
IgG, HRP-linked antibody (#7074)], human vascular endothelial
growth factor 165 (hVEGF165) and MG132 (a potent
proteasome inhibitor) were purchased from Cell Signaling Technology
(Danvers, MA, USA). SU5416 (a specific VEGFR-2 inhibitor) and
SB216763 (a specific inhibitor of GSK-3β) was obtained from Tocris
Bioscience (Bristol, UK). GO was purchased from Sigma-Aldrich
Chemical (St. Louis, USA). β-TrCP siRNA (sc-37178) and lithium
chloride (LiCl) were obtained from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). LiCl was used to block GSK-3β activity. A
protease inhibitor phenylmethylsulfonyl fluoride (PMSF),
electro-chemiluminescence (ECL) kit and radio immunoprecipitation
assay (RIPA) buffer were purchased from the KeyGen Institute of
Biotechnology (Nanjing, China). A PrimeScript First Strand cDNA
Synthesis kit and SYBR-Green PCR Master Mix were purchased from
Takara Bio (Shiga, Japan).
Cell proliferation assay
Firstly, using a cell counting plate,
5×103 HUVECs were seeded into each well of a 96-well
plate and counted. The cells were incubated with NG, HG, Man,
SU5416 (1.0 and 10 µM, respectively) for 48 h (15–17). Subsequently, 10 µl
3-(4,5-dimethylthiazol-2yl)-2,5-diphenyltetrazolium bromide (MTT, 5
mg/ml) was added to each well and incubated for 4 h; 100 µl
formanzan solution was added to wells for an additional 4 h.
Absorbance was measured at 570 nm using a microplate reader (Gene
Company Limited, Chai Wan, Hong Kong). The cell proliferation rate
(%) was calculated using the following formula: cell proliferation
rate (%) = (OD570, sample − OD570, blank)/(OD570, control − OD570,
blank) ×100.
'Control' refers to the NG group. 'Sample' means
other groups in addition to NG group, and 'blank' means a group to
which only medium, MTT solution and DMSO were added.
Cell scratch wound-healing assay
A wound healing assay was performed as described
previously (18). Once HUVECs at
second passage reached approximately 80% confluence in 6-well
plates, a wound was made with a 10-µl pipette tip. The cells
were washed with PBS twice and incubated in DMEM with NG, MG, HG,
Man and SU5416 (1.0 and 10 µM, respectively), as previously
described (15). After 24 h of
incubation, cultured cells were photographed and te cell migration
activity was quantified by ImageJ software (version 1.37, National
Institutes of Health) by measuring the number of migrated cells in
each group.
Capillary-like tube formation assay using
Matrigel
According to the manufacturer's instructions, growth
factor-reduced basement membrane matrix (BD Matrigel™; BD
Bioscience, Bedford, MA, USA) (300 µl) was added to each
well of the 24-well plate and gelation at 37°C for 0.5 h was
undertaken. HUVECs (1.2×105 cells) in 300 µl DMEM
were added to wells coated with Matrigel. The cells were divided
into NG, MG, HG, Man and SU5416 (1.0 and 10 µM,
respectively) groups and were incubated at 37°C in a humidified
chamber of 5% CO2 for 6 h to allow for tube formation.
Once tube formation images were captured using an inverted
microscope (Olympus, Tokyo, Japan), tube structures were quantified
by counting branch points in 3 random fields from each well, as
described in previous research (19). The average number of branch points
was calculated, and each experiment was repeated in triplicate.
MitoSOX™ Red and DAPI
Firstly, at an initial density of 5×103
cells/well, the cells seeded into each well of a 96-well plate were
incubated with medium containing 6.6 or 33 mM glucose for 48 h, or
1 U/ml GO for 15 min. The ROS levels of cells were monitored by
MitoSOX Red mitochondrial superoxide indicator (Life Technologies)
according to the manufacturer's instructions. The nucleus was
stained by DAPI (KeyGen Institute of Biotechnology). Images of
MitoSOX Red and DAPI, excited by green light and ultraviolet, were
obtained using a fluorescence microscope (Olympus, Tokyo, Japan)
and analyzed with ImageJ software.
siRNA transfection
For siRNA transfection, after cells reached 30–50%
confluence they were transfected with scrambled (used as negative
control) or β-TrCP siRNA (both from Santa Cruz Biotechnology, Inc.)
using Lipofectamine 2000, according to the manufacturer's
instructions. The cells were incubated for 48 h, subsequently
exposed to medium containing 1 U/ml GO for another 15 min and then
the collection of proteins was undertaken.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was first extracted from treated HUVECs
using TRIzol reagent according to the manufacturer's instructions.
In the present study, cDNA was synthesized through reverse
transcription and amplification using the following primers:
VEGFR-2 forward, 5′-TACCTCCACCATGCCAAGTG-3′ and reverse,
5′-GATGATTCTGCCCTCCTCCTT-3′; β-TrCP forward,
5′-AATTCCTCAGAGAGAGAAGACT-3′ and reverse,
5′-TCTGGCAAAACACTAAATATAT-3′; β-actin forward
5′-TGACGTGGACATCCGCAAAG-3′ and reverse, 5′-CTGGAAGGTGGACAGCGAGG-3′.
Amplification was performed according to the following protocol:
95°C for 30 sec, followed by 40 cycles at 95°C for 5 sec, 60°C for
34 sec. qPCR was conducted using the ABI 7500 RT-PCR system
(Applied Biosystems, Foster City, CA, USA) and SYBR-Green PCR
Master Mix, according to the manufacturer's instructions. Relative
mRNA expression was calculated using the 2−ΔΔCt method
and presented as fold-induction of the target gene normalized to
β-actin RNA.
Western blot analysis
Total cell lysates were obtained by harvesting the
cells in RIPA buffer with a protease inhibitor,
phenylmethylsulfonyl fluoride (PMSF), and a phosphatase inhibitor,
PhosStop (both purchased from KeyGen Institute of Biotechnology).
After incubation at 0°C for 20 min, the lysates were scraped and
centrifuged at 12,000 × g at 4°C for 20 min. The concentrations of
soluble proteins in the supernatants of total cell lysates were
determined using a BCA protein assay with bovine serum albumin as
the standard. The protein samples containing 50 µg total
protein were separated on 10% sodium dodecyl sulfate-polyacrylamide
gel electrophoresis (SDS-PAGE) and transferred to PVDF membranes.
The membranes were incubated for 2 h with 5% skim milk in TBST
buffer (20 mol/l Tris-HCl, pH 7.6, 150 mmol/l NaCl, and 0.05%
Tween-20) to block non-specific binding. The membranes were
subsequently incubated with specific primary antibodies and gently
shaken at 4°C overnight, which was followed by incubation with the
secondary antibody for 1 h. Immunoreactive protein bands were
visualized by chemiluminescence using a Syngene Bio Imaging Device
(Syngene, Cambridge, UK).
Confocal microscopy
The co-localization of β-TrCP and VEGFR-2 was
detected by confocal microscopy. HUVECs were incubated with primary
antibody for anti-β-TrCP (Cell Signaling Technology) and
anti-VEGFR-2 (Abcam, Cambridge, UK), then with Cy3 goat anti-rabbit
antibody and FITC goat anti-mouse. Nuclei were stained with DAPI
for 10 min at room temperature. Cells were imaged using a LSM710
laser scanning confocal microscope (Leica Microsystems, Wetzlar,
Germany).
Co-immunoprecipitation assay
In the present study, HUVECs were incubated with 200
µl RIPA buffer and 1 µl protease inhibitor at 0°C for
1 h, homogenized, and centrifuged at 500 rpm at 4°C for 10 min. The
supernatant was incubated with VEGFR-2 antibody at a final
concentration of 4 µg/ml for each at 4°C overnight incubated
with 20 µl Protein G Plus-Agarose (Beyotime, Shanghai,
China) and then washed twice with RIPA and protease inhibitor
buffer. The immunoprecipitates were collected and eluted from
agarose with 30 µl SDS-PAGE loading buffer per tube.
Statistical analysis
The data are expressed as the means ± SD. The
student's t-test was employed to compare data between two groups.
One-way ANOVA analysis, followed by Dunnett's test, was used to
compare multiple groups. In the present study, a P-value <0.05
was considered to indicate a statistically significant difference.
Moreover, statistical analysis was performed using SPSS 16.0
statistical software (SPSS Inc., Chicago, IL, USA).
Results
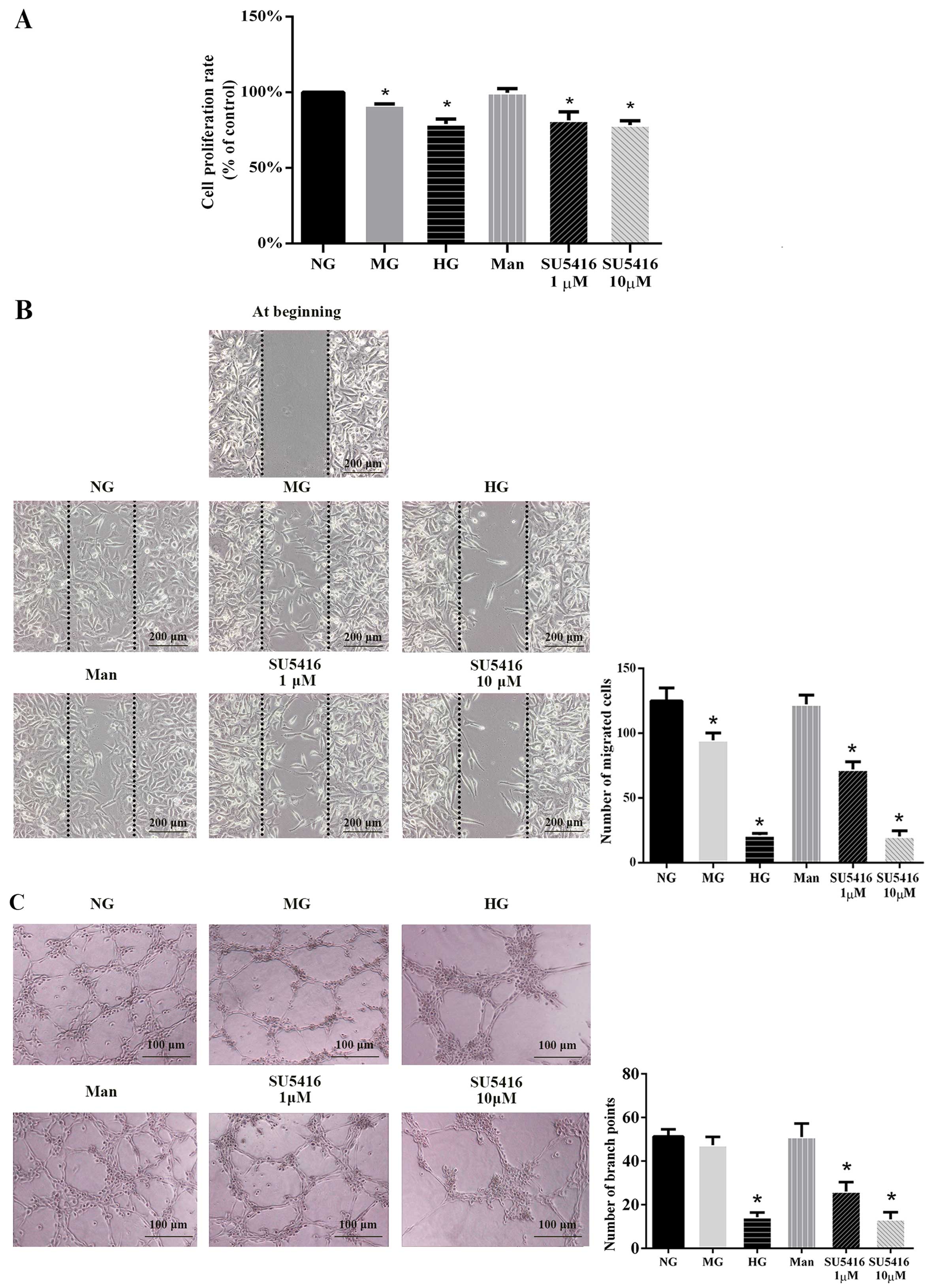
Hyperglycemia and VEGFR-2 inhibitor
impair angiogenesis
After exposure for 48 h to MG, HG and SU5416 (a
specific VEGFR-2 inhibitor, at 1 and 10 µM), but not Man,
the HUVEC proliferation rate was significantly decreased compared
with the NG group (Fig. 1A). To
further investigate the role of hyperglycemia in inhibiting the
angiogenic response, we preformed a cell migration assay. As shown
in Fig. 1B, HUVECs exposed to MG,
HG and SU5416 exhibited a lower migration rate compared with cells
exposed to NG. In the capillary-like tube formation assay, fewer
branch points of formed tubes were observed in MG, HG, SU5416 (both
in 1 and 10 µM)-stumulated groups, compared with the NG
group (Fig. 1C). Taken together,
the data in Fig. 1 indicate the
role of the impaired VEGF-VEGFR-2 signaling transduction pathway in
angiogenesis upon hyperglycemia induction. To investigate the
mechanism of the phenomenon above, we subsequently examined the
expression level of VEGFR-2 and phosphorylated VEGFR-2 under
hyperglycemic conditions.
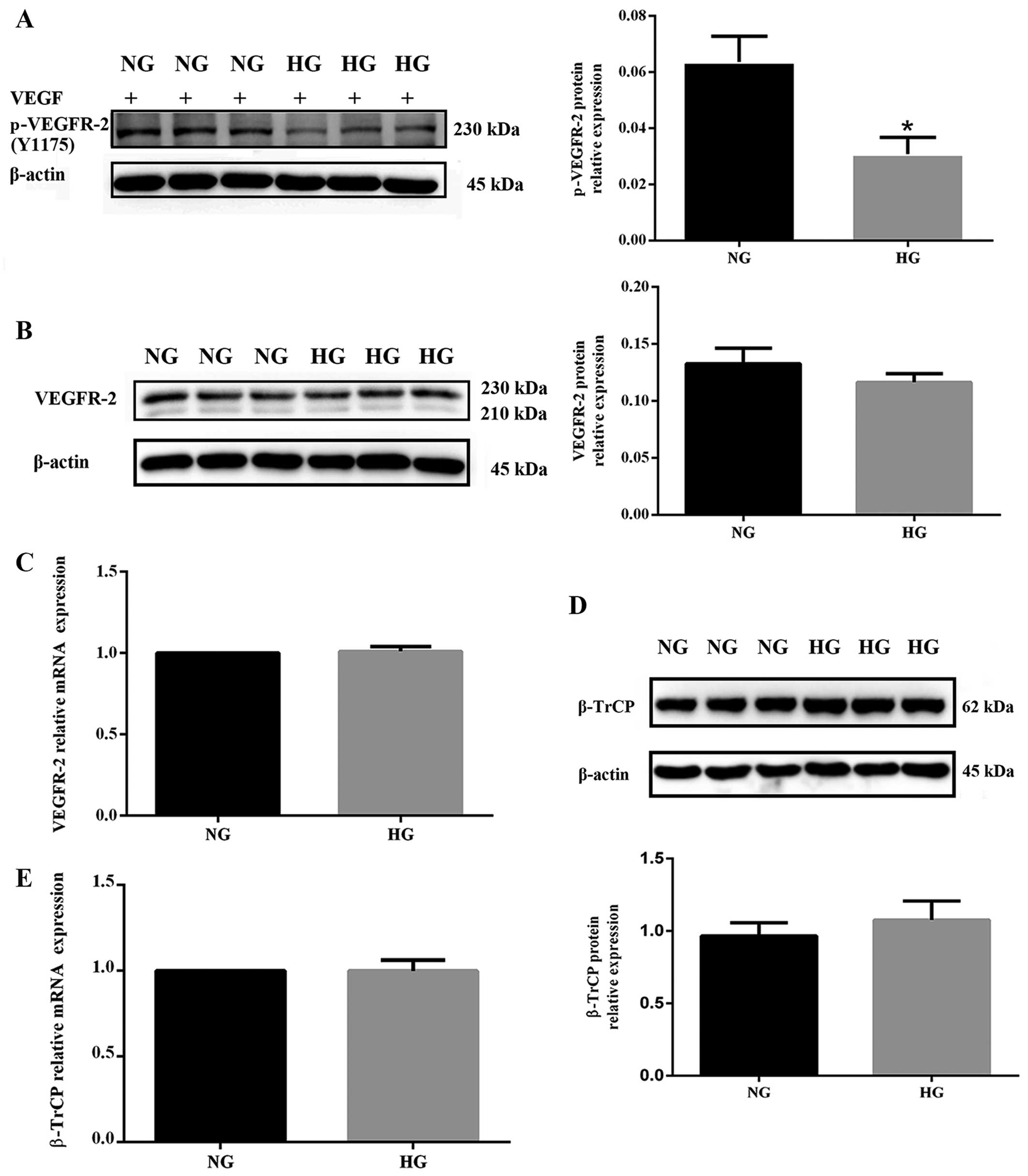
Hyperglycemia impairs VEGFR-2 activation
without markedly affecting its expression
The level of phosphorylated VEGFR-2 was decreased in
HUVECs cultured under hyperglycemic conditions for 48 h (P<0.05
vs. NG) when activated by VEGF (100 ng/ml for 5 min), a specific
ligand for VEGFR-2 (Fig. 2A).
Although in HUVECs exposed to high glucose the response to VEGF was
impaired, total VEGFR-2 protein was not significantly decreased
(P>0.05 vs. NG) (Fig. 2B).
Accordingly, the mRNA expression level of VEGFR-2 remained almost
unchanged in the HG group (P>0.05 vs. NG) (Fig. 2C). These findings indicate that
high glucose decreases VEGF-induced phosphorylation of VEGFR-2 but
has no marked effect on its total expression, both at protein and
mRNA levels. In addition, the protein and mRNA levels of β-TrCP in
NG and HG groups were measured. Although β-TrCP was slightly
increased in the HG group, there was no significant difference
between the two groups (P>0.05 vs. NG) (Fig. 2D and E).
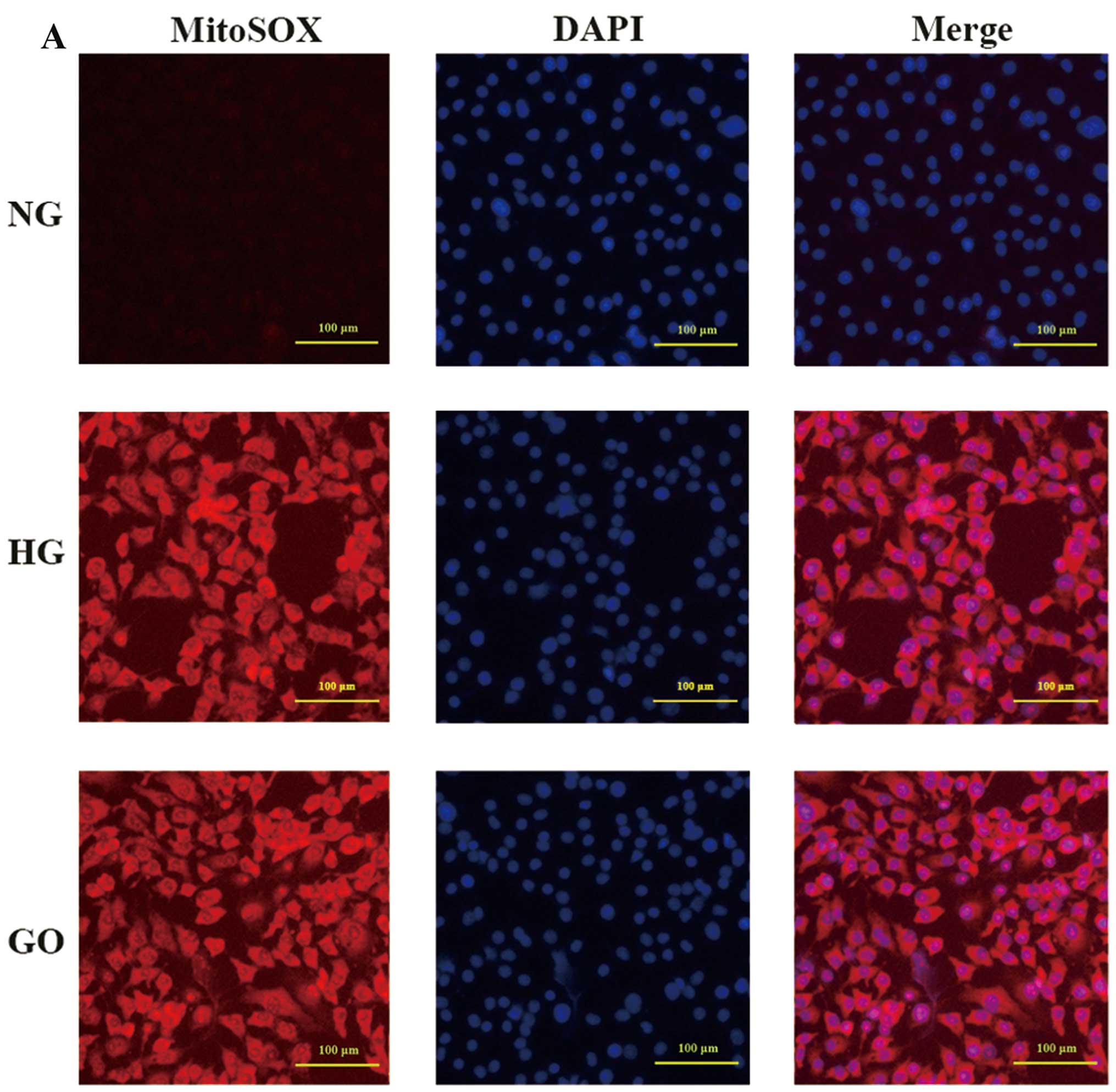
VEGFR-2 degradation is induced by ROS
overproduction
A previous study has indicated that increased
oxidative stress is one of the mechanisms underlying
hyperglycemia-induced endothelial dysfunction (20). In the present study, to
investigate the role of ROS in regulating VEGFR-2, we employed GO,
which catalyzes the oxidation of glucose into
H2O2, in order to create an excess ROS model
while excluding the effects of other aspects of diabetic
vasculopathy mechanisms. As shown in Fig. 3A, endothelial cells exposed to
high glucose for 48 h exhibited excess ROS production, as detected
by MitoSOX Red mitochondrial superoxide indicator. A similar result
was also obtained when endothelial cells were treated with 1 U/ml
GO for 15 min (Fig. 3A), which
concurs with previous research (7). The fluorescence intensity was
comparable between the HG and GO group, and it was observed that
both were significantly higher than the NG group (P<0.05 vs.
NG). By contrast to Fig. 2B,
VEGFR-2 expression was significantly reduced by GO (P<0.05 vs.
GO) (Fig. 3B). Consistent with
Fig. 2C, we noted that VEGFR-2
mRNA expression was not markedly altered in the presence of GO,
though it was very slightly increased (P>0.05 vs. GO) (Fig. 3C). Furthermore, the β-TrCP protein
and mRNA expression levels were both assessed. As was clearly
demonstrated in Fig. 3D and E, no
significant difference between the two groups (NG and GO) was
observed (P>0.05 vs. GO).
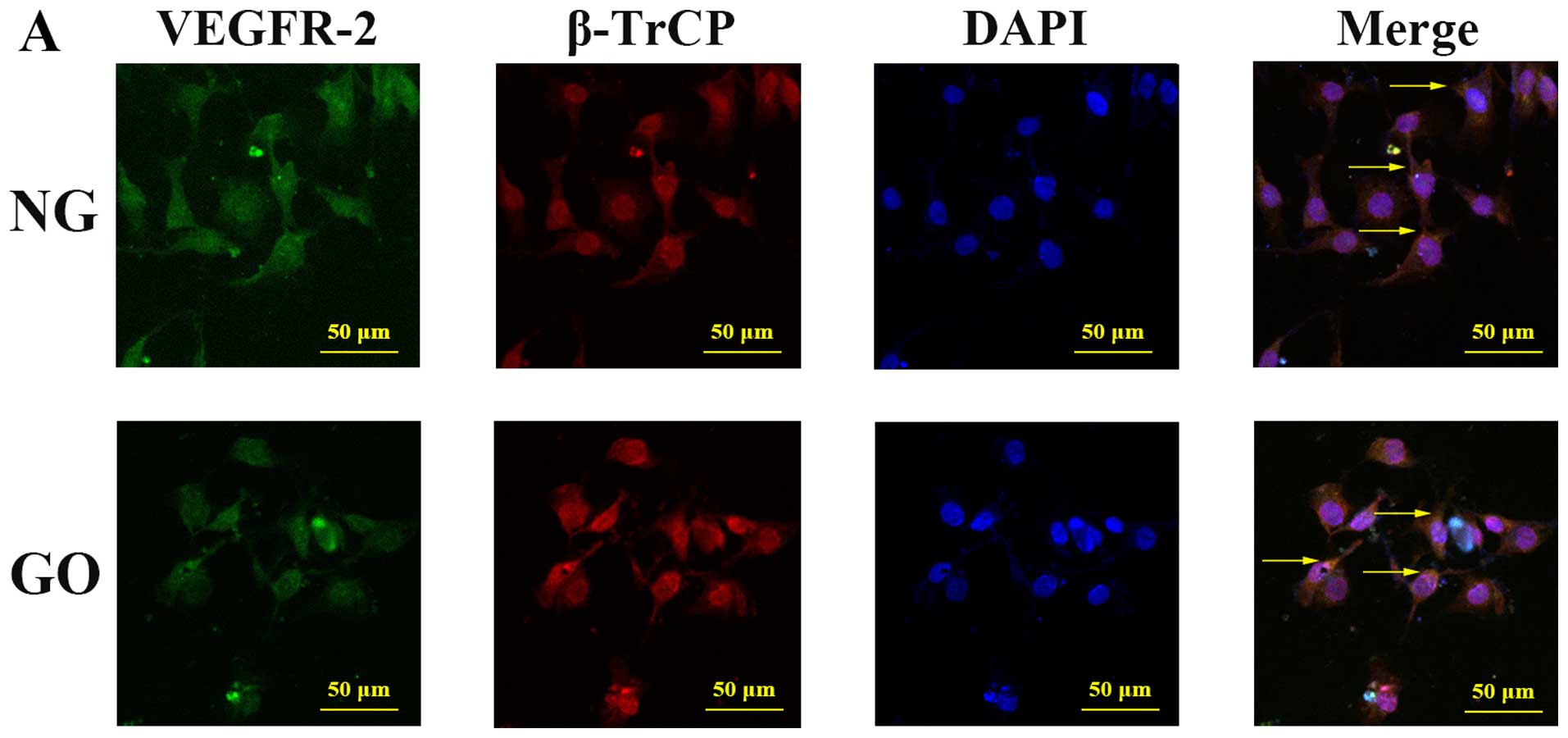
Participation of β-TrCP in the
downregulation of VEGFR-2 by ROS
Confocal fluorescence microscopy was used to explore
whether there was co-location of VEGFR-2 and β-TrCP. In HUVECs in
both the NG and GO groups, cytomembrane expression of VEGFR-2 and
cytoplasmic and nuclear expression of β-TrCP were noted. Moreover,
co-localization of VEGFR-2 and β-TrCP existed both under normal and
high ROS conditions. To determine and compare the level of
ubiquitinated VEGFR-2 protein in the NG and GO groups, we performed
a co-immunoprecipitation assay using anti-VEGFR-2 antibodies,
followed by western blot analysis using anti-ubiquitin antibody.
There was an increase in VEGFR-2 ubiquitination in endothelial
cells exposed to GO (P<0.05 vs. NG) (Fig. 4B). Forty-eight hours after β-TrCP
small interfering RNA (siRNA) (50 nM) was transfected into HUVECs,
the β-TrCP protein level was suppressed (P<0.05) compared with
scrambled siRNA, whereas VEGFR-2 expression was increased
(P<0.05) (Fig. 4C). GO-induced
VEGFR-2 degradation was abolished by a 4-h treatment with 10
µM MG132, a proteasome inhibitor (P<0.05) (Fig. 4D).
ROS-activated GSK-3β induces VEGFR-2
degradation in HUVECs
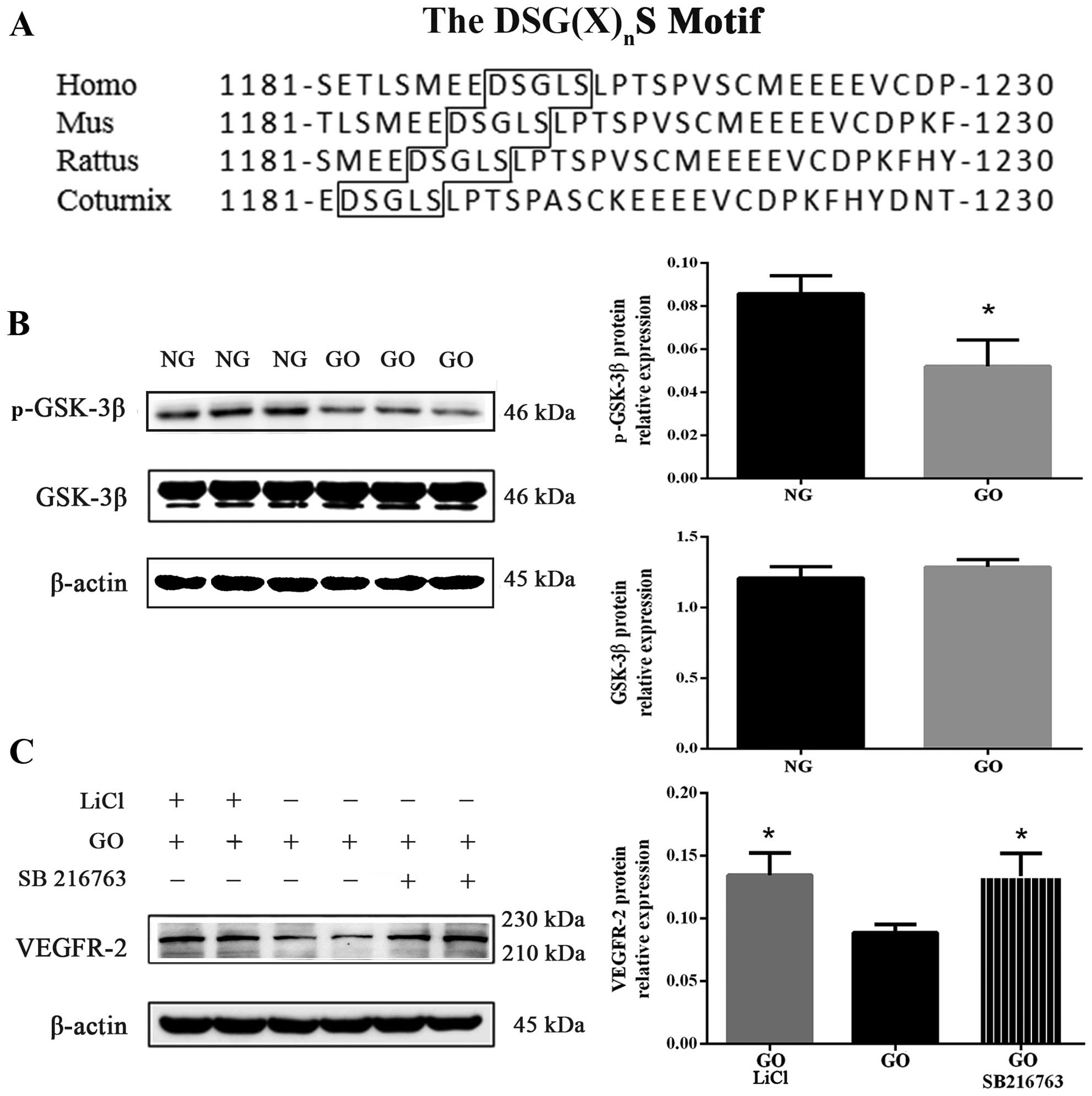
Sequence alignment of the DSG(X)nS
binding motif was obtained using the Database of the National
Center for Biotechnology Information. The DSG(X)nS motif
is well conserved in VEGFR-2 across many species, including humans,
mice, rats and coturnix (Fig.
5A). After 15 min incubation with 1 U/ml GO, GSK-3β
phosphorylation (Serine 9) was attenuated (P<0.05 vs. NG) but
total GSK-3β protein expression was almost unchanged (Fig. 5B). As is well known,
phosphorylated GSK-3β acts as an inactivated form, and decreased
phosphorylated GSK-3β upon exposure to GO, as shown in Fig. 5B, was noted; this signified that
GSK-3β was enhanced by excess ROS.
In this study, to verify whether activated GSK-3β
was involved in the degradation process of VEGFR-2, we exposed
HUVECs for 30 min to GSK-3β inhibitor, LiCl (20 mM) and SB216763
(20 mM), before exposure to GO, as also previously described
(21). We noted that VEGFR-2
protein expression of cells co-cultured with GO and GSK-3β
inhibitors (LiCl and SB216763) increased compared with the GO group
(P<0.05) (Fig. 5C). These
findings demonstrate that GSK-3β plays a key role in regulating
VEGFR-2 degradation.
Discussion
VEGFR-2 plays an important role in the angiogenic
response (3–5). In the present study, we demonstrated
the impairment of angiogenic function in endothelial cells when
exposed to VEGFR-2 inhibitor (SU5416) as well as high glucose
stimulation. The protein expression of VEGFR-2 was not
significantly altered in the HG group, whereas it was significantly
decreased after GO exposure (Figs.
2B and 3B). Warren et
al have reported that stimulation with high glucose for 48 h
results only in decreased abundance of plasma membrane-localized
VEGFR-2. As noted in the present study, ROS production was induced
under hyperglycemic conditions, and we suggest that downregulation
of membrane VEGFR-2 expression occurred due to disruption of
cytoplasm trafficking from Golgi apparatus to the membrane. The
total protein level of VEGFR-2 is significantly altered only when
the Golgi-localized pool of VEGFR-2 is decreased under conditions
such as chronic exposure to hyperglycemia and ROS, rather than a
relatively transient exposure to hyperglycemia (7). Moreover, we suggest that the
impaired angiogenic response in the short term was partially due to
downregulation of the phosphorylated VEGFR-2 (Threonine 1175),
leading to inhibition of the VEGF-VEGFR-2 signaling pathways
(Fig. 2A).
GSK-3β acts as a key point for convergent insulin
signaling pathways in endothelial cells to angiogenic responses;
the enzyme is a downstream target of PI3K/Akt signaling pathway and
is inactivated by phosphorylation (22). Previous studies have noted that
GSK-3β activity is upregulated in the skeletal muscle of T2DM
patients and in diabetic mice, indicating that activated GSK-3β
causes dysfunction of insulin signaling and then diabetes (23,24). SB216763, a specific inhibitor of
GSK-3β, does not affect other relevant protein kinase, including
PDK-1 and PKB (25). Another
inhibitor, LiCl, has been reported to exert various effects on
other protein kinases, while in the present study, upon exposure to
GO LiCl clearly ameliorated VEGFR-2 degradation just as SB216763
also did (26,27). Moreover, LiCl is known to
stimulate glucose uptake, glycogen synthesis and normalize insulin
sensitivity in diabetic rats (28). Clinical results have also
demonstrated that LiCl improved impaired wound healing in diabetic
patients by inducing the Wnt signaling pathway (29,30). The data of the present study
indicated that GSK-3β expression was slightly enhanced and
subsequently induced the degradation of VEGFR-2. The insulin
receptor signaling is diminished by ROS (31). However, whether β-TrCP is directly
regulated by GSK-3β has not yet been explored.
β-TrCP, an F-box component of the Skp1-Cul1-F-box
protein ubiquitin ligases, functioning as a substrate recognition
subunit, has been reported to suppress angiogenesis by promoting
ubiquitination and degradation of VEGFR-2 in thyroid cancer cells
(9–11). Our results showed that VEGFR-2 and
β-TrCP were co-located in the cytomembrane under high GO
conditions. We noted that β-TrCP protein expression in HUVECs was
suppressed by β-TrCP siRNA, whereas VEGFR-2 expression increased.
GSK-3β acts as a mediator in the degradation of several proteins
induced by β-TrCP (13,14). Further experiments should be
undertaken to determine whether β-TrCP and GSK-3β play roles in the
degradation of VEGFR-2 in endothelial cells in which hyperglycemia
has been induced. Moreover, we noted that the degradation of
VEGFR-2 induced by GO was abolished by MG132, a proteasome
inhibitor, as well as by LiCl. GO attenuated GSK-3β phosphorylation
in HUVECs whereas the protein expression of GSK-3β and β-TrCP was
not significantly altered upon exposure to GO. GO catalyzes the
oxidation of D-glucose to produce gluconic and hydrogen peroxide
(H2O2) (7).
In this study, we noted that H2O2 induced by
high glucose stimulation impaired VEGF-2 function and decreased
p-GSK-3β, but not β-TrCP. The results suggest that the
downregulation of VEGFR-2 induced by β-TrCP depends on GSK-3β
activity.
Certain VEGFR-2 phosphorylation sites are
responsible for ubiquitination and degradation, such as Serine
1188/Serine 1191 (Ser 1188/Ser 1191) (32); according to this study by Meyer
et al, the phosphorylation of Ser 1188/Ser 1191 is
upregulated, which mediates the degradation of VEGFR-2. Taking this
into consideration, we predict that GSK-3β phosphorylated VEGFR-2
at sites within the DSG(X)nS motif.
Our results demonstrate that reduction of VEGFR-2
under hyperglycemic conditions induced by excess ROS was regulated
by GSK-3β coupling with β-TrCP. We suggest that GSK-3β inhibitors
act as a promising therapeutic target. In conclusion, these
findings provide novel insights into the mechanisms of vascular
pathophysiology and may potentially be used in the clinical
treatment of diabetes mellitus.
Acknowledgments
The present study was supported by grants from the
National Natural Scientific Foundation of China (nos. 81270191 and
81370304).
References
|
1
|
Waltenberger J: VEGF resistance as a
molecular basis to explain the angiogenesis paradox in diabetes
mellitus. Biochem Soc Trans. 37:1167–1170. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schiekofer S, Galasso G, Sato K, Kraus BJ
and Walsh K: Impaired revascularization in a mouse model of type 2
diabetes is associated with dysregulation of a complex
angiogenic-regulatory network. Arterioscler Thromb Vasc Biol.
25:1603–1609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Shibuya M: Vascular endothelial growth
factor and its receptor system: physiological functions in
angiogenesis and pathological roles in various diseases. J Biochem.
153:13–19. 2013. View Article : Google Scholar
|
|
4
|
Shibuya M: VEGFR and type-V RTK activation
and signaling. Cold Spring Harb Perspect Biol. 5:a0090922013.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koch S and Claesson-Welsh L: Signal
transduction by vascular endothelial growth factor receptors. Cold
Spring Harb Perspect Med. 2:a0065022012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shibuya M and Claesson-Welsh L: Signal
transduction by VEGF receptors in regulation of angiogenesis and
lymphangiogenesis. Exp Cell Res. 312:549–560. 2006. View Article : Google Scholar
|
|
7
|
Warren CM, Ziyad S, Briot A, Der A and
Iruela-Arispe ML: A ligand-independent VEGFR2 signaling pathway
limits angiogenic responses in diabetes. Sci Signal. 7:ra12014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen YS and Qiu XB: Ubiquitin at the
crossroad of cell death and survival. Chin J Cancer. 32:640–647.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shaik S, Nucera C, Inuzuka H, Gao D,
Garnaas M, Frechette G, Harris L, Wan L, Fukushima H, Husain A, et
al: SCF(β-TRCP) suppresses angiogenesis and thyroid cancer cell
migration by promoting ubiquitination and destruction of VEGF
receptor 2. J Exp Med. 209:1289–1307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wei S, Lin LF, Yang CC, Wang YC, Chang GD,
Chen H and Chen CS: Thiazolidinediones modulate the expression of
beta-catenin and other cell-cycle regulatory proteins by targeting
the F-box proteins of Skp1-Cul1-F-box protein E3 ubiquitin ligase
independently of peroxisome proliferator-activated receptor gamma.
Mol Pharmacol. 72:725–733. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wei S, Yang HC, Chuang HC, Yang J, Kulp
SK, Lu PJ, Lai MD and Chen CS: A novel mechanism by which
thiazolidinediones facilitate the proteasomal degradation of cyclin
D1 in cancer cells. J Biol Chem. 283:26759–26770. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Cardozo T and Pagano M: The SCF ubiquitin
ligase: insights into a molecular machine. Nat Rev Mol Cell Biol.
5:739–751. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li X, Liu J and Gao T: beta-TrCP-mediated
ubiquitination and degradation of PHLPP1 are negatively regulated
by Akt. Mol Cell Biol. 29:6192–6205. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu C, Li Y, Semenov M, Han C, Baeg GH,
Tan Y, Zhang Z, Lin X and He X: Control of beta-catenin
phosphorylation/degradation by a dual-kinase mechanism. Cell.
108:837–847. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bischof M, Abdollahi A, Gong P, Stoffregen
C, Lipson KE, Debus JU, Weber KJ and Huber PE: Triple combination
of irradiation, chemotherapy (pemetrexed), and VEGFR inhibition
(SU5416) in human endothelial and tumor cells. Int J Radiat Oncol
Biol Phys. 60:1220–1232. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Yang BR, Hong SJ, Lee SM, Cong WH, Wan JB,
Zhang ZR, Zhang QW, Zhang Y, Wang YT and Lin ZX: Pro-angiogenic
activity of notoginsenoside R1 in human umbilical vein endothelial
cells in vitro and in a chemical-induced blood vessel loss model of
zebrafish in vivo. Chin J Integr Med. Dec 22–2014.Epub ahead of
print. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cukiernik M, Hileeto D, Evans T, Mukherjee
S, Downey D and Chakrabarti S: Vascular endothelial growth factor
in diabetes induced early retinal abnormalities. Diabetes Res Clin
Pract. 65:197–208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ettenson DS and Gotlieb AI: Centrosomes,
microtubules, and microfilaments in the reendothelialization and
remodeling of double-sided in vitro wounds. Lab Invest. 66:722–733.
1992.PubMed/NCBI
|
|
19
|
Pollman MJ, Naumovski L and Gibbons GH:
Endothelial cell apoptosis in capillary network remodeling. J Cell
Physiol. 178:359–370. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Choi SE, Jang HJ, Kang Y, Jung JG, Han SJ,
Kim HJ, Kim DJ and Lee KW: Atherosclerosis induced by a high-fat
diet is alleviated by lithium chloride via reduction of VCAM
expression in ApoE-deficient mice. Vascul Pharmacol. 53:264–272.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kobayashi T, Matsumoto T and Kamata K: The
PI3-K/Akt pathway: roles related to alterations in vasomotor
responses in diabetic models. J Smooth Muscle Res. 41:283–302.
2005. View Article : Google Scholar
|
|
23
|
Nikoulina SE, Ciaraldi TP, Mudaliar S,
Mohideen P, Carter L and Henry RR: Potential role of glycogen
synthase kinase-3 in skeletal muscle insulin resistance of type 2
diabetes. Diabetes. 49:263–271. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Eldar-Finkelman H and Krebs EG:
Phosphorylation of insulin receptor substrate 1 by glycogen
synthase kinase 3 impairs insulin action. Proc Natl Acad Sci USA.
94:9660–9664. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Coghlan MP, Culbert AA, Cross DA, Corcoran
SL, Yates JW, Pearce NJ, Rausch OL, Murphy GJ, Carter PS, Roxbee
Cox L, et al: Selective small molecule inhibitors of glycogen
synthase kinase-3 modulate glycogen metabolism and gene
transcription. Chem Biol. 7:793–803. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Mora A, Sabio G, Risco AM, Cuenda A,
Alonso JC, Soler G and Centeno F: Lithium blocks the PKB and GSK3
dephosphorylation induced by ceramide through protein
phosphatase-2A. Cell Signal. 14:557–562. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Klein PS and Melton DA: A molecular
mechanism for the effect of lithium on development. Proc Natl Acad
Sci USA. 93:8455–8459. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rossetti L: Normalization of insulin
sensitivity with lithium in diabetic rats. Diabetes. 38:648–652.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dejana E: The role of wnt signaling in
physiological and pathological angiogenesis. Circ Res. 107:943–952.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qi W, Yang C, Dai Z, Che D, Feng J, Mao Y,
Cheng R, Wang Z, He X, Zhou T, et al: High levels of pigment
epithelium-derived factor in diabetes impair wound healing through
suppression of Wnt signaling. Diabetes. 64:1407–1419. 2015.
View Article : Google Scholar
|
|
31
|
Hou Q, Lei M, Hu K and Wang M: The effects
of high glucose levels on reactive oxygen species-induced apoptosis
and involved signaling in human vascular endothelial cells.
Cardiovasc Toxicol. 15:140–146. 2015. View Article : Google Scholar
|
|
32
|
Meyer RD, Srinivasan S, Singh AJ, Mahoney
JE, Gharahassanlou KR and Rahimi N: PEST motif serine and tyrosine
phosphorylation controls vascular endothelial growth factor
receptor 2 stability and downregulation. Mol Cell Biol.
31:2010–2025. 2011. View Article : Google Scholar : PubMed/NCBI
|