Introduction
The pathogenesis of inflammatory skin diseases
involves interactions between immune cells and keratinocytes.
Keratinocytes perform important functions in the regulation of
inflammation and respond to environmental and pro-inflammatory
stimuli, including the cytokines interleukin (IL)-17 and IL-22
which are produced by T helper 17 (Th17) cells. IL-17 binds to a
heterodimeric receptor complex consisting of IL-17RA and IL-17RC.
The adaptor protein Act1 binds to IL-17RA and mediates
IL-17-induced signaling pathways. Following stimulation with IL-17,
Act1 is recruited to the IL-17 receptor, followed by the activation
of the kinase transforming growth factor β-activated kinase 1
(TAK1) and the IκB kinase (IKK) complex, which subsequently
activates nuclear factor-κB (NF-κB). Previously, we have observed
that IL-17 enhanced skin inflammation by stimulating the secretion
of IL-1β by keratinocytes through the NLR family, pyrin domain
containing 3 (NLRP3)-caspase-1 pathway (1). It has been reported that Act1 is a
client protein of heat shock protein 90 (HSP90), and that HSP90
activity is required for IL-17 signaling (2).
HSP90 functions as a chaperone that facilitates the
folding and assembly of its client proteins. Loss of HSP90
chaperone function results in the degradation of its client
proteins. HSP90 is constitutively expressed in human keratinocytes
and fibroblasts in vitro (3) and is focally expressed in epidermal
layers in vivo. The epidermal expression of HSP90 is
upregulated by external stimuli, such as heat (4), chemical stress and tape stripping
(5). In addition, increased HSP90
expression in keratinocytes and mast cells from the skin of
patients with psoriasis has been reported (6).
Balneotherapy involves immersing the patient in
mineral water baths or pools. Inflammatory skin diseases, including
psoriasis and atopic dermatitis, are commonly and successfully
treated using balneotherapy (7,8).
Although the mechanisms through which spa therapy exerts beneficial
effects on inflammatory skin diseases have not been fully
elucidated, they are likely to involve chemical, thermal,
mechanical and immunomodulatory processes (9).
Based on the involvement of HSP90 in IL-17-mediated
inflammatory skin diseases, we hypothesized that following
stimulation with IL-17, thermal stimulation may affect keratinocyte
function through HSP90. Thus, we stimulated the human keratinocyte
cell line HaCaT with IL-17 in the presence or absence of the HSP90
inhibitor, 17-N-allylamino-17-demethoxygeldanamycin (17-AAG).
Following thermal stimulation, we compared the expression of
antimicrobial peptides and chemokines.
Materials and methods
Cell culture
The human immortal keratinocyte cell line, HaCaT
(kindly provided by Dr Tae-Yoon Kim, College of Medicine, The
Catholic University of Korea, Bucheon, Korea), was cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS) and 100 U/ml penicillin/streptomycin (all
from Welgene Inc., Dalseogu, Korea). The cells were maintained at
37°C in a 5% CO2 incubator. The cells were grown until
they were approximately 70–80% confluent and treated for 24 h with
100 ng/ml IL-17 (R&D Systems, Inc., Minneapolis, MN, USA),
followed by the addition of 1 µM 17-AAG (Sigma, St. Louis,
MO, USA) for 1 h. For thermal stimulation, the cells were incubated
at 42°C for an additional hour after the addition of 17-AAG.
Immunoprecipitation assays
The cells were lysed in lysis buffer [0.5% Triton
X-100, 20 mM HEPES (pH 7.4), 150 mM NaCl, 12.5 mM
β-glycerophosphate, 1.5 mM MgCl2, 10 mM NaF, 2 mM
dithiothreitol, 1 mM sodium orthovanadate
(Na3VO4)] supplemented with 10 µg
protease inhibitor cocktail (P8340; Sigma) and 2 mM
Na3VO4. The cleared cell lysates were
incubated with antibodies (2 µg) and protein G sepharose
beads (10 µl of a 50% slurry in lysis buffer). Following
incubation on a rotating shaker at 4°C for 2 h, the protein G
sepharose beads were washed twice with lysis buffer. Finally,
reducing sample buffer was added prior to performing sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and western
blot analysis (also known as immunoblot analysis).
Western blot analysis
The cells were lysed in lysis buffer [1% Triton
X-100, 150 mM NaCl, 20 mM Tris (pH 7.5)] supplemented with protease
inhibitors, and the proteins (10 µg) were separated via
SDS-PAGE on 10% gels. Resolved proteins were transferred to
polyvinylidene fluoride (PVDF) membranes (Millipore Corp., Bedford,
MA, USA). The membranes were blocked with 5% skim milk in
Tris-buffered saline and incubated with the following primary
antibodies: anti-HSP90-α (D7a, mouse monoclonal IgG1; Abcam,
Cambridge, UK), anti-Act1 (C-16, goat monoclonal IgG; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), and anti-β-actin (AC-40,
mouse monoclonal; Sigma). After washing, the membranes were
incubated with horseradish peroxidase-conjugated anti-mouse,
anti-rabbit, or anti-goat IgG secondary antibodies, and the signals
were visualized using an enhanced chemiluminescence (ECL) kit
(Thermo Fisher Scientific, Waltham, MA, USA) according to the
manufacturer's instructions.
Reverse transcription-quantitative
polymerase chain reaction (RT-PCR)
Total RNA was extracted from the cells using TRIzol
(Invitrogen, Carlsbad, CA, USA) in accordance with the
manufacturer's instructions. A total of 1 µg RNA was
transcribed to cDNA using a reverse transcription system (Promega,
Madison, WI, USA). qPCR with SYBR green detection was conducted
using a Maxime PCR PreMix kit (Toyobo, Osaka, Japan) and an ABI
PRISM 7000 sequence detection system (Applied Biosystems Life
Technologies, Foster City, CA, USA). cDNA (50 ng) was amplified
using the following primers: 5′-TGA GAT GGC CTC AGG TGG TA-3′ and
5′-CGG GCA GGC AGA ATA GAG AC-3′ for human β-defensin 1 (BD1; 106
bp); 5′-GGG GCT CCT TTG ACA TCA GT-3′ and 5′-TGG GTA CAA GAT TCC
GCA AA-3′ for human LL-37 (153 bp); 5′-GCC GTC TAC AGG GAT GAC
CT-3′ and 5′-TTT GTG GCT TTC ATG GC-3′ for human S100A8 (204 bp);
5′-CAA AGA GCT GGT GCG AAA AG-3′ and 5′-CGA AGC TCA GCT TGT CT-3′
for human S100A9 (125 bp); 5′-GCT GCT CCT GCT CCT GGT A-3′ and
5′-CTT TCC GCC CAT TCT TGA GT-3′ for human chemokine (C-X-C motif)
ligand (CXCL)1 (200 bp); 5′-TGC G-TTG CGT TTG TTT ACA G-3′ and
5′-GAA AAG GGG CTT CTG GAT CA-3′ for human CXCL5 (162 bp); and
5′-GCA GCT CTG TGT GAA GGT GC-3′ and 5′-TCT GCA CCC AGT TTT CCT
T-G-3′ for human CXCL8 (220 bp).
Small interfering RNA (siRNA) silencing
of Act1 expression
The HaCaT cells were transfected with siRNA
oligonucleotides (80 pmol; sc-29634; Santa Cruz Biotechnology,
Inc.) to reduce endogenous Act1 expression. The cells were
transfected with either an siRNA oligonucleotide against Act1 or a
non-targeted control siRNA oligonucleotide, and maintained at 37°C
under standard tissue culture conditions. Twenty hours after
transfection, the cells were treated with IL-17 or IL-22 (100
ng/ml) and maintained for 24 h. Thereafter, the cell lysates and
the supernates were harvested for RT-qPCR and western blot
analysis. For the detection of Act1 (85 bp) by RT-qPCR in siRNA
gene knockdown experiments, we used the following specific primers:
5′-CAC AGA GAG ACT GCT CCT TTC C-3′ (forward) and 5′-CCA GGA GCA
CCA GCT CTA TTA-3′ (reverse). As a control for input,
glyceraldehyde 3-phosphate dehydrogenase (GAPDH; 395 bp) was
amplified using the following primers: 5′-GTC TTC TCC ACC ATG GAG
AAG GCT-3′ (forward) and 5′-CAT GCC AGT GAG CTT CCC GTT CA-3′
(reverse).
Statistical analysis
Values are expressed as the means ± standard error
of the mean (SEM). Non-parametric Mann-Whitney tests or two-way
analysis of variance (ANOVA) were conducted to determine
significance using GraphPad Prism software (GraphPad Software Inc.,
San Diego, CA, USA). A P-value <0.05 was considered to indicate
a statistically significant difference.
Results
Act1 binding to HSP90α is increased in
HaCaT cells after thermal stimulation
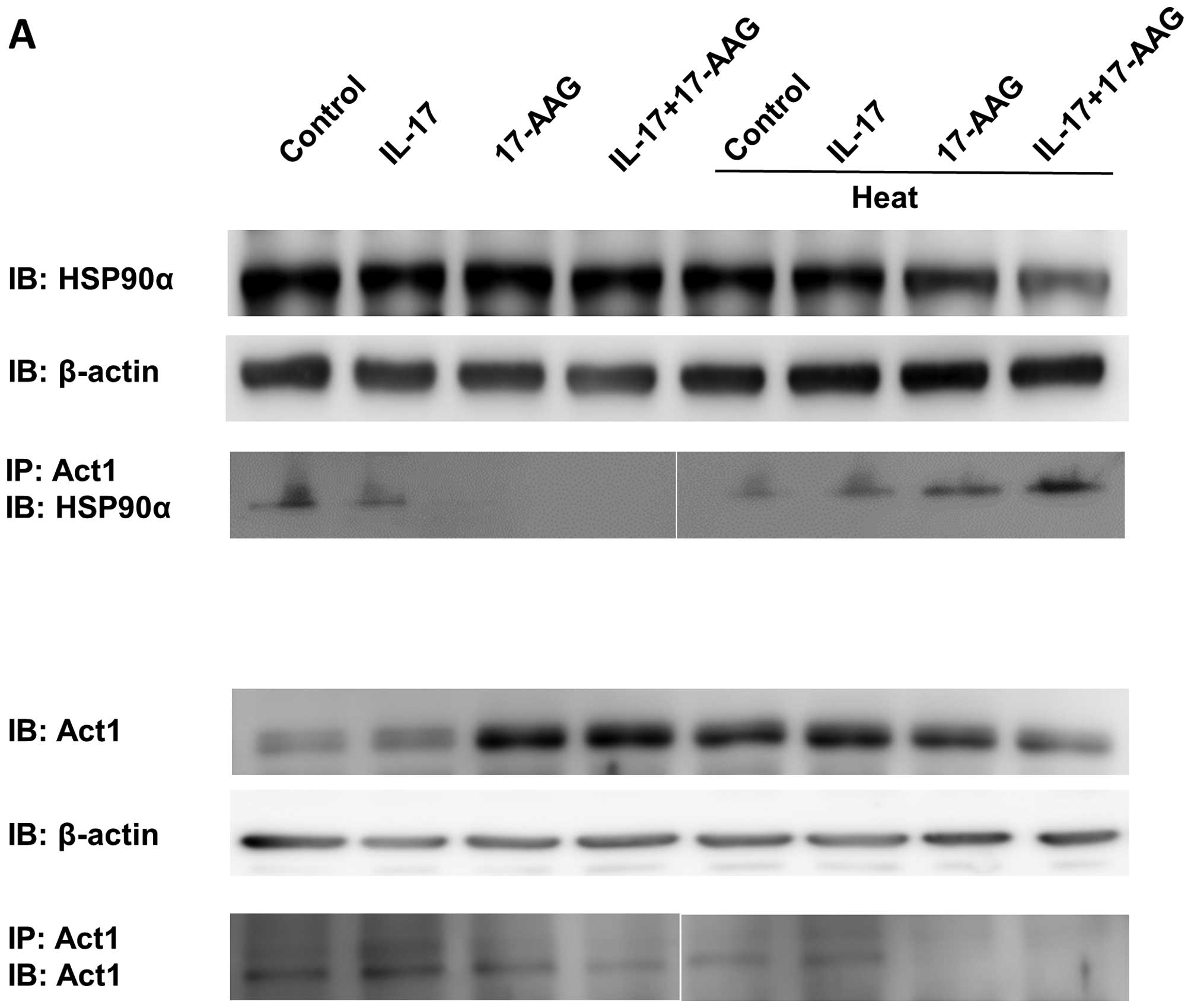
Firstly, we examined whether the heat treatment of
IL-17-stimulated HaCaT cells affects the outcome of IL-17
receptor-mediated signaling. To analyze signaling in
IL-17-stimulated HaCaT cells after heat treatment, we first
compared the amount of Act1 binding to HSP90 in the presence or
absence of heat. We immunoprecipitated HaCaT cell lysates with the
anti-Act1 antibody following IL-17 stimulation, with or without
heat stimulation (Fig. 1A).
HSP90α expression was decreased after heat treatment when the
HSP90α inhibitor, 17-AAG, was added with IL-17 (Fig. 1B). We also found that Act1-bound
HSP90α levels were increased after thermal stimulation; however,
there were no differences between the control and the IL-17-treated
groups (Fig. 1B). Although the
expression of Act1 did not significantly differ, the
immunoprecipitated amount of Act1 following IL-17 stimulation and
17-AAG treatment decreased (Fig.
1C). Thus, under 37°C culture conditions, IL-17 or the HSP90α
inhibitor 17-AAG did not affect Act1 or HSP90α expression in HaCaT
cells; however, following heat treatment, Act1-bound HSP90α
significantly increased in the presence of IL-17 and 17-AAG.
Effect of Act1 knockdown on HSP90
expression in HaCaT cells following heat treatment
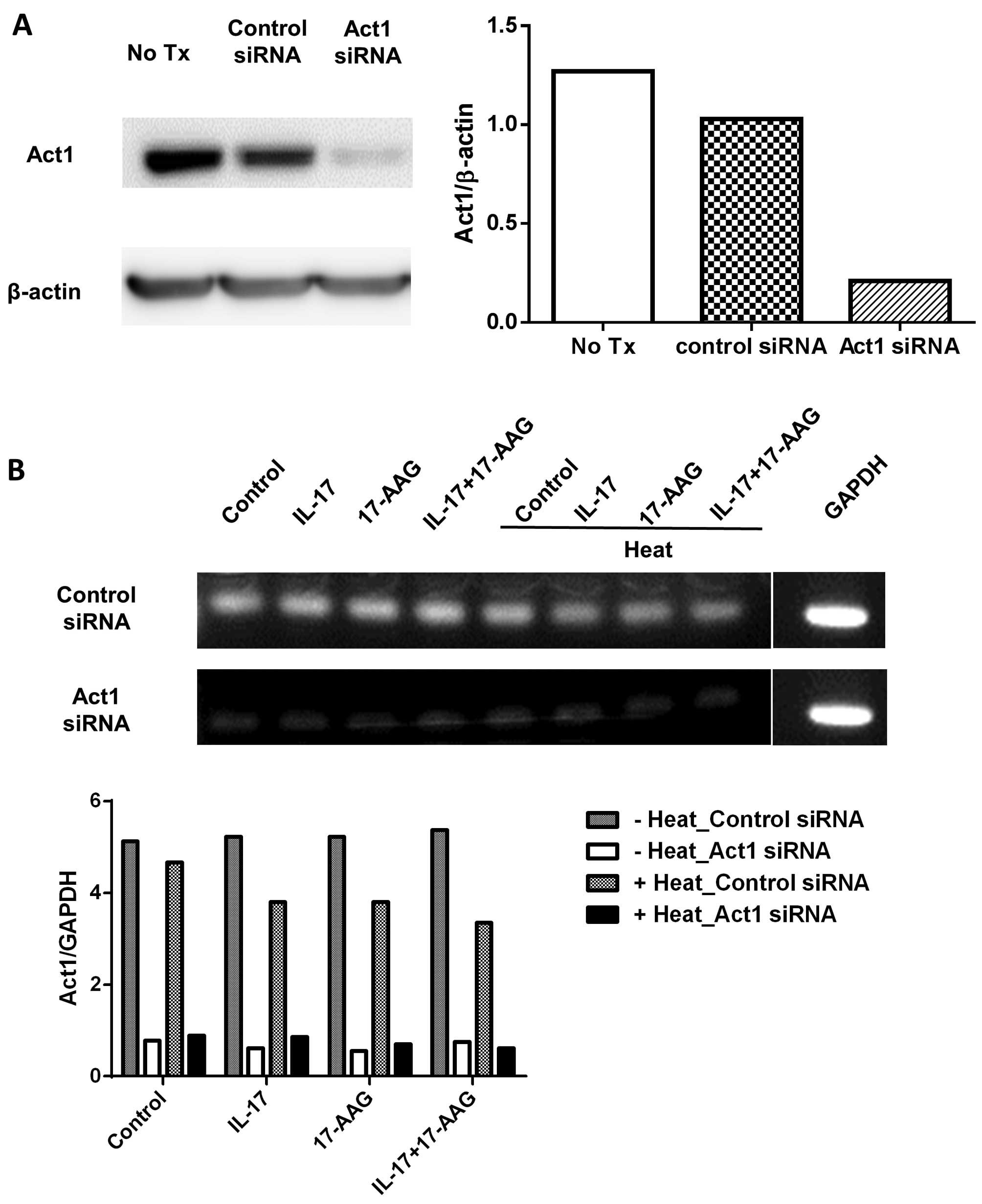
In order to compare the levels of HSP90 following
the knockdown of Act1 in the HaCaT cells, we transfected HaCaT
cells with Act1 siRNA, heated the cells, and confirmed the
decreased expression of Act1 using western blot analysis and
RT-qPCR (Fig. 2). We then
transfected HaCaT cells with Act1 siRNA which was followed by heat
treatment and the addition of 17-AAG. We compared the levels of
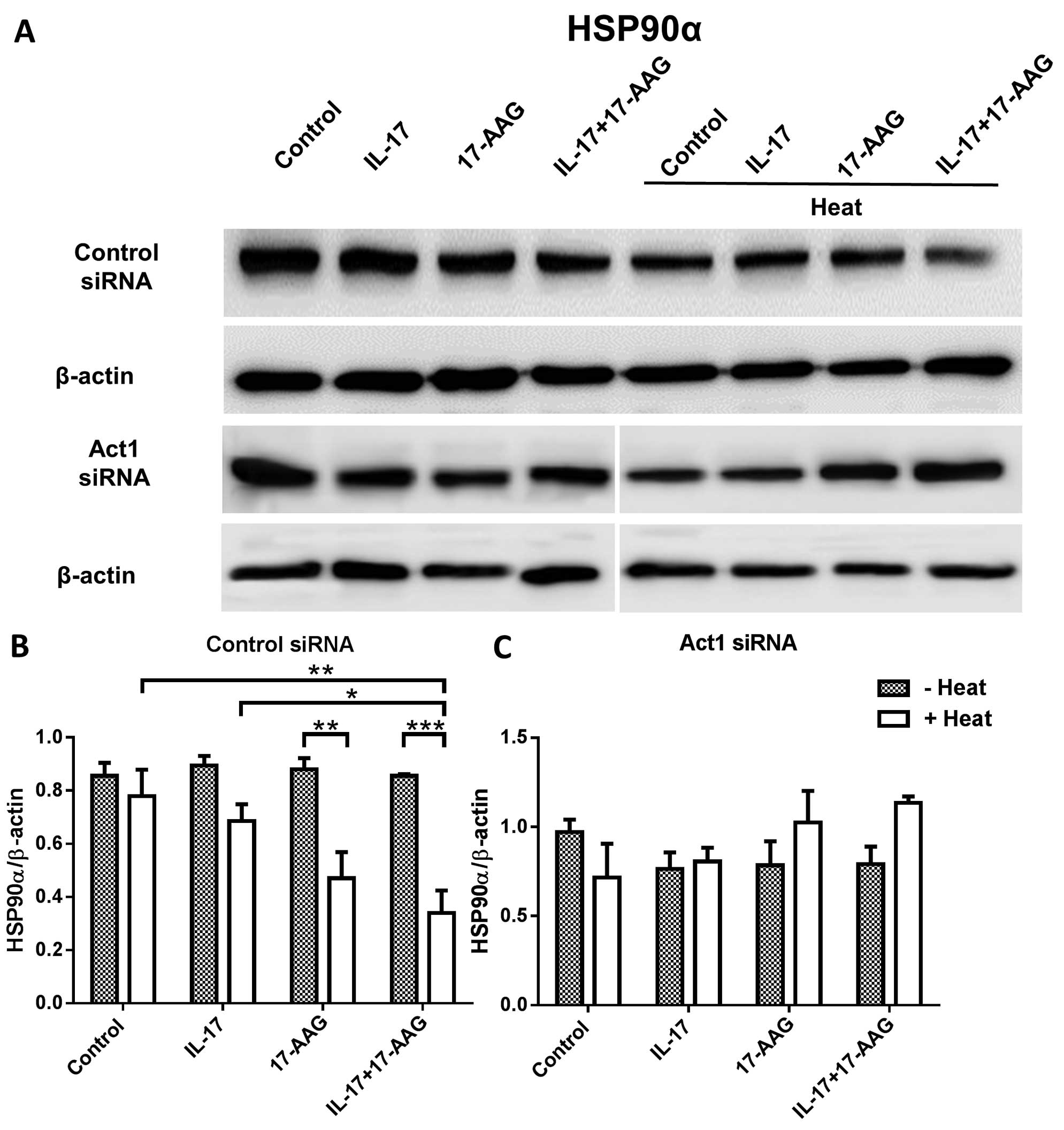
HSP90 and Act1 in these cells. We found that after thermal
stimulation, HSP90α expression was decreased in the control
siRNA-transfected HaCaT cells in the presence of IL-17 and 17-AAG,
which is nearly identical to the result shown in Fig. 1B. However, in the Act1 knockdown
HaCaT cells following heat treatment, HSP90 expression did not
significantly differ among the groups (Fig. 3A–C). Finally, we compared Act1
expression in the HaCaT cells following heat treatment, in the
presence or absence of the HSP90 inhibitor, following Act1 siRNA
transfection. Following thermal stimulation, we observed increased
Act1 levels in the control cells and those treated with IL-17
(Fig. 3D–F).
Effect of 17-AAG on antimicrobial peptide
and cytokine expression in HaCaT cells following Act1 knockdown and
thermal stimulation
To examine the effect of IL-17 receptor-mediated
expression of antimicrobial peptides or inflammatory chemokines in
HaCaT cells, Act1 siRNA-transfected HaCaT cells were stimulated
with heat and 17-AAG was added. RNA was extracted to compare the
levels of BD1, LL-37, S100A8, S100A9, CXCL1, CXCL5 and CXCL8
(Fig. 4). At 37°C, Act1 knockdown
with IL-17 stimulation tended to decrease LL-37, S100A8, S100A9,
CXCL1 and CXCL5 expression; however, the expression of these
antimicrobial peptides and chemokine increased after incubation at
42°C. This thermal-mediated effect was suppressed upon treatment
with 17-AAG.
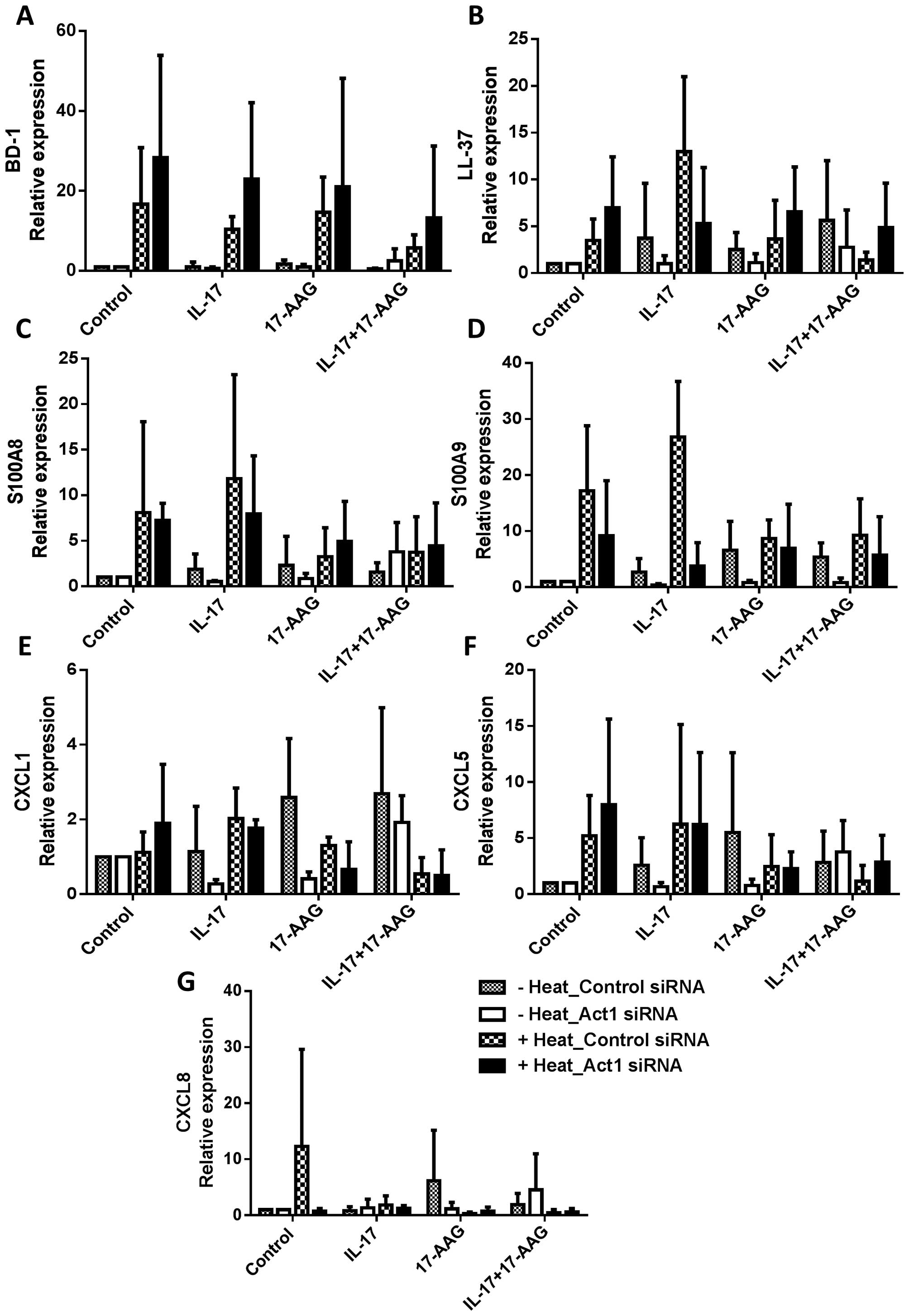 | Figure 4Antimicrobial peptide and chemokine
expression in the HaCaT cells following treatment with interleukin
(IL)-17 and 17-N-allylamino-17-demethoxygeldanamycin (17-AAG), with
or without thermal stimulation. Control siRNA- or Act1
siRNA-transfected HaCaT cells were treated with IL-17, 17-AAG, and
heat, and RNA was extracted for RT-qPCR. The results of (A)
β-defensin 1 (BD1), (B) cathelicidin LL-37, (C) S100A8, (D) S100A9,
(E) chemo-kine (C-X-C motif) ligand (CXCL)1, (F) CXCL5 and (G)
CXCL8 are shown. Data are expressed as the means ± SEM of the three
independent experiments and compared with the results from the 'no
heat' and 'heat treatment' groups (*P<0.05, and
**P<0.001). |
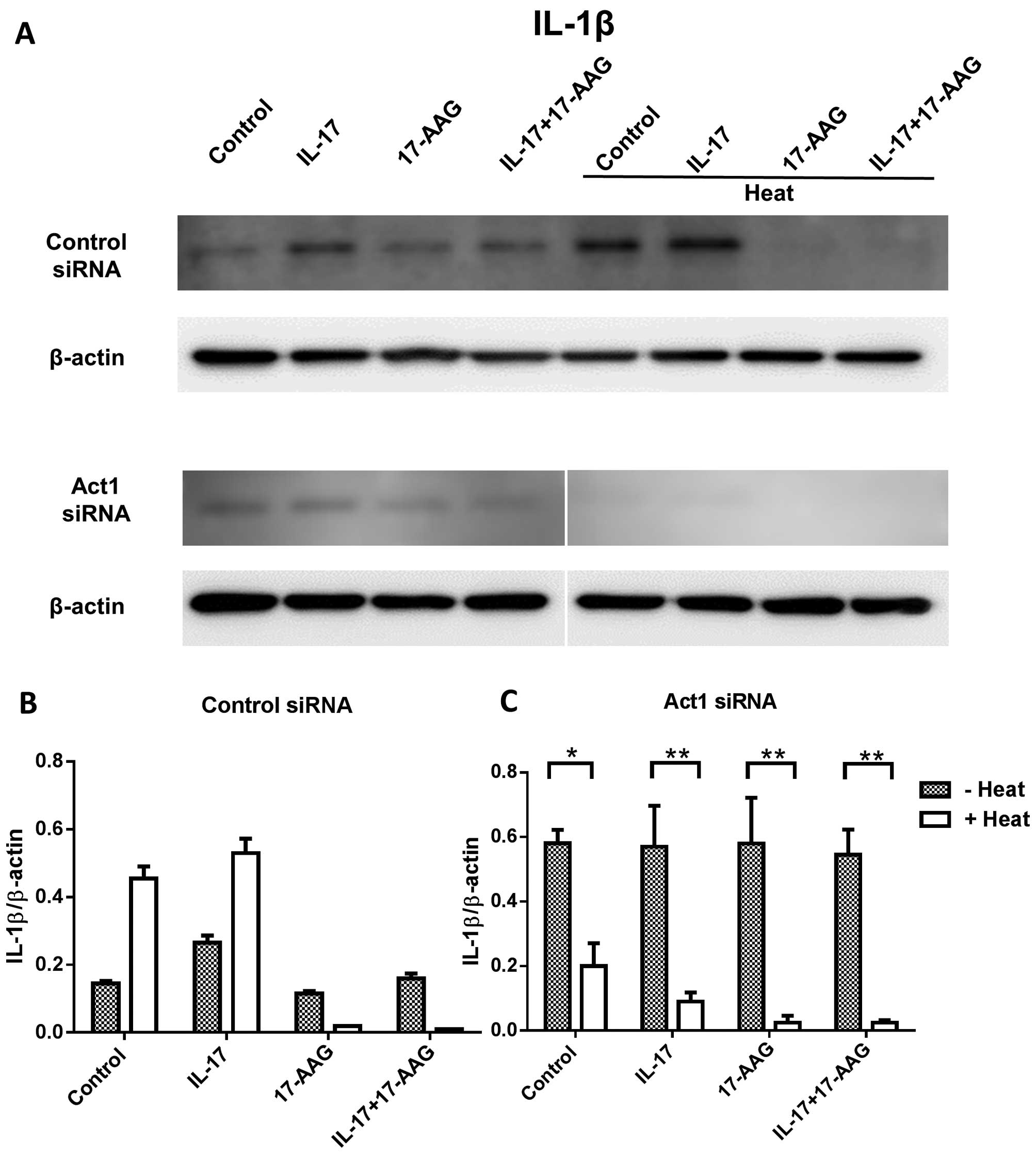
We also examined IL-1β expression after Act1
knockdown in thermal-stimulated HaCaT cells treated with IL-17 and
17-AAG. We found that IL-1β expression decreased, and 17-AAG
synergistically affected the control siRNA cells after heat
stimulation. Act1 siRNA-treated cells showed a significant decrease
in IL-1β expression after heat treatment (Fig. 5).
Discussion
In the present study, we evaluated the effect of
heat on keratinocytes following IL-17 stimulation. We found that
under heat-treated conditions, Act1-bound HSP90α significantly
increased in the presence of IL-17 (100 ng/ml) and 17-AAG (1
µM). Antimicrobial peptide and chemokine expression
generally increased after heat treatment; however, Act1 knockdown
and 17-AAG treatment inhibited the production of antimicrobial
peptides and chemokines.
A previous study of hyperthermia (41±0.5°C for 1 h),
applied to normal human skin has indicated that the HSPs 27, 60,
72i and 90 were upregulated, with maximal increases noted 24 h
following hyperthermia (4).
However, increased HSP expression poorly correlated with
thermotolerance, and did not improve cell survival after heat
shock. In addition, it has been shown that the upregulation of
HSP90, HSP70, IL-33, TNF-α and CXCL8 (also known as IL-8) mRNA
after tape stripping induced the production of alarmins from
keratinocytes (5). Although the
possible roles of several HSPs under various stresses in skin
diseases remain unclear, their importance in cutaneous aging, skin
cancer and wound healing is increasingly recognized (10).
Psoriasis is a chronic inflammatory skin disease
characterized by keratinocyte hyperplasia, dermal leukocyte
infiltration and vascular enhancement. The cytokine milieu, through
activation of Th1 and Th17 cells, contributes to the establishment
of skin inflammation. Keratinocytes exposed to IL-17 or IL-22 are
further activated, which has been demonstrated to result in the
abnormal expression of antimicrobial peptides and defensins
(11). It has been previously
reported that Act1 is a client protein of the molecular chaperone
HSP90, and that HSP90 activity is required for IL-17 signaling
(2). In addition, it has been
demonstrated that there is increased expression of HSP90 in
keratinocytes isolated from patients with psoriasis. The HSP90
levels were decreased following therapy with ustekinumab (blocking
antibody to p40 of the IL-12 and IL-23 receptor) (6). The HSP90 inhibitor, 17-AAG, has been
found to repress the differentiation of HaCaT keratinocytes
(12). It has been suggested that
HSP90 inhibitors may offer a novel therapeutic approach for
treating inflammatory skin diseases.
HSP90 is a molecular chaperone that is critical for
the function and stability of proteins necessary for cell survival.
Many molecules involved in signal transduction pathways are client
proteins of HSP90, including tyrosine-kinase receptors, adapter
proteins, transcription factors, cell cycle proteins and
anti-apoptotic proteins (13).
HSP90 inhibitors have been used to target the function of HSP90 in
cell proliferation and survival signaling pathways (14–16). Currently, HSP90 inhibitors include
derivatives of geldanamycin and resorcinol as well as purine
analogues and other synthetic inhibitors. The first class of HSP90
inhibitors, the benzoquinone ansamycin antibiotics, includes
17-AAG. The HSP90 inhibitor binds to HSP90 and leads to proteasomal
degradation of the HSP90 client protein, thereby disrupting the
signaling pathway. Thus, HSP90 inhibitors are promising novel
treatments targeting cancer (13)
and immune diseases. Clinically, they have been used to target the
leukemic blast phase of cells to induce apoptosis and
differentiation (17). Elevated
HSP90 expression has been reported in systemic lupus erythematosus
(SLE) patients, and HSP90 inhibition has been shown to lessen
disease in the MRL/lpr mouse model of SLE (18). HSP90 inhibitors have been
demonstrated to be effective in murine models of sepsis (19), arthritis (20), uveitis (21), multiple sclerosis (22), and inflammatory bowel disease
(23).
A previous study has found that cathelicidin LL-37
posseses antimicrobial and immunomodulatory activity, and the
expression of this type of antimicrobial peptide may ameliorate
skin inflammation (24). S100A8
and S100A9 are known mediators of human keratinocyte growth
stimulation, and these peptides have been implicated in the onset
and progression of hyperproliferative skin diseases, such as
psoriasis (25,26). Although the precise role of
antimicrobial peptides in inflammatory skin diseases is not fully
known and often controversial, we found that antimicrobial peptides
(BD1, LL-37, S100A8 and S100A9), chemokines (CXCL1, CXCL5 and
CXCL8), and IL-1β were elevated upon IL-17 addition or thermal
stimulation, and inhibited by 17-AAG treatment or Act1 knockdown
(Figs. 4 and 5).
In conclusion, our data suggests that HSP90 is
involved in IL-17-mediated keratinocyte activation through Act1
following thermal stimulation. Although HSP90 enhanced the
expression of antimicrobial peptides or chemokines in keratinocytes
after heat treatment, HSP90 inhibitors reversed these effects.
Thus, the administration of HSP90 inhibitors following heat
stimulation (thermal baths) may be a beneficial therapeutic
approach for IL-17-mediated inflammatory skin diseases.
Abbreviations:
|
17-AAG
|
17-N-allylamino-17-demethoxygeldanamycin
|
|
HSP90
|
heat shock protein 90
|
|
BD1
|
β-defensin 1
|
Acknowledgments
The present study was supported by the Basic Science
Research Program through the National Research Foundation of Korea
(NRF) funded by the Ministry of Science, ICT and Future Planning
(2013R1A1A3A04006995).
References
|
1
|
Cho KA, Suh JW, Lee KH, Kang JL and Woo
SY: IL-17 and IL-22 enhance skin inflammation by stimulating the
secretion of IL-1β by keratinocytes via the ROS-NLRP3-caspase-1
pathway. Int Immunol. 24:147–158. 2012. View Article : Google Scholar
|
|
2
|
Wang C, Wu L, Bulek K, Martin BN, Zepp JA,
Kang Z, Liu C, Herjan T, Misra S, Carman JA, et al: The
psoriasis-associated D10N variant of the adaptor Act1 with impaired
regulation by the molecular chaperone hsp90. Nat Immunol. 14:72–81.
2013. View
Article : Google Scholar
|
|
3
|
Edwards MJ, Marks R, Dykes PJ, Merrett VR,
Morgan HE and O'Donovan MR: Heat shock proteins in cultured human
keratinocytes and fibroblasts. J Invest Dermatol. 96:392–396. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wilson N, McArdle A, Guerin D, Tasker H,
Wareing P, Foster CS, Jackson MJ and Rhodes LE: Hyperthermia to
normal human skin in vivo upregulates heat shock proteins 27, 60,
72i and 90. J Cutan Pathol. 27:176–182. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dickel H, Gambichler T, Kamphowe J,
Altmeyer P and Skrygan M: Standardized tape stripping prior to
patch testing induces upregulation of Hsp90, Hsp70, IL-33, TNF-α
and IL-8/CXCL8 mRNA: new insights into the involvement of
'alarmins'. Contact Dermat. 63:215–222. 2010. View Article : Google Scholar
|
|
6
|
Kakeda M, Arock M, Schlapbach C and
Yawalkar N: Increased expression of heat shock protein 90 in
keratinocytes and mast cells in patients with psoriasis. J Am Acad
Dermatol. 70:683–690.e1. 2014. View Article : Google Scholar
|
|
7
|
Halevy S and Sukenik S: Different
modalities of spa therapy for skin diseases at the Dead Sea area.
Arch Dermatol. 134:1416–1420. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Choi YJ, Lee HJ, Lee H, Woo SY, Lee KH,
Yun ST, Kim JM, Kim HJ and Kim JW: Therapeutic effects and
immunomodulation of Suanbo mineral water therapy in a murine model
of atopic dermatitis. Ann Dermatol. 25:462–470. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Matz H, Orion E and Wolf R: Balneotherapy
in dermatology. Dermatol Ther (Heidelb). 16:132–140. 2003.
View Article : Google Scholar
|
|
10
|
Vidal Magalhães W, Gouveia Nogueira MF and
Kaneko TM: Heat shock proteins (HSP): dermatological implications
and perspectives. Eur J Dermatol. 22:8–13. 2012.PubMed/NCBI
|
|
11
|
Zaba LC, Cardinale I, Gilleaudeau P,
Sullivan-Whalen M, Suárez-Fariñas M, Fuentes-Duculan J, Novitskaya
I, Khatcherian A, Bluth MJ, Lowes MA and Krueger JG: Amelioration
of epidermal hyperplasia by TNF inhibition is associated with
reduced Th17 responses. J Exp Med. 204:3183–3194. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Miyoshi S, Yamazaki S, Uchiumi A and
Katagata Y: The Hsp90 inhibitor 17-AAG represses calcium-induced
cytokeratin 1 and 10 expression in HaCaT keratinocytes. FEBS Open
Bio. 2:47–50. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Garcia-Carbonero R, Carnero A and Paz-Ares
L: Inhibition of HSP90 molecular chaperones: moving into the
clinic. Lancet Oncol. 14:e358–e369. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
An WG, Schulte TW and Neckers LM: The heat
shock protein 90 antagonist geldanamycin alters chaperone
association with p210bcr-abl and v-src proteins before their
degradation by the proteasome. Cell Growth Differ. 11:355–360.
2000.PubMed/NCBI
|
|
15
|
Mimnaugh EG, Chavany C and Neckers L:
Polyubiquitination and proteasomal degradation of the p185c-erbB-2
receptor protein-tyrosine kinase induced by geldanamycin. J Biol
Chem. 271:22796–22801. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sauvageot CM, Weatherbee JL, Kesari S,
Winters SE, Barnes J, Dellagatta J, Ramakrishna NR, Stiles CD, Kung
AL, Kieran MW and Wen PY: Efficacy of the HSP90 inhibitor 17-AAG in
human glioma cell lines and tumorigenic glioma stem cells. Neuro
Oncol. 11:109–121. 2009. View Article : Google Scholar :
|
|
17
|
Nimmanapalli R, O'Bryan E and Bhalla K:
Geldanamycin and its analogue
17-allylamino-17-demethoxygeldanamycin lowers Bcr-Abl levels and
induces apoptosis and differentiation of Bcr-Abl-positive human
leukemic blasts. Cancer Res. 61:1799–1804. 2001.PubMed/NCBI
|
|
18
|
Shimp SK III, Chafin CB, Regna NL, Hammond
SE, Read MA, Caudell DL, Rylander M and Reilly CM: Heat shock
protein 90 inhibition by 17-DMAG lessens disease in the MRL/lpr
mouse model of systemic lupus erythematosus. Cell Mol Immunol.
9:255–266. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chatterjee A, Dimitropoulou C,
Drakopanayiotakis F, Antonova G, Snead C, Cannon J, Venema RC and
Catravas JD: Heat shock protein 90 inhibitors prolong survival,
attenuate inflammation, and reduce lung injury in murine sepsis. Am
J Respir Crit Care Med. 176:667–675. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Prakken BJ, Wendling U, van der Zee R,
Rutten VP, Kuis W and van Eden W: Induction of IL-10 and inhibition
of experimental arthritis are specific features of microbial heat
shock proteins that are absent for other evolutionarily conserved
immunodominant proteins. J Immunol. 167:4147–4153. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Poulaki V, Iliaki E, Mitsiades N,
Mitsiades CS, Paulus YN, Bula DV, Gragoudas ES and Miller JW:
Inhibition of Hsp90 attenuates inflammation in endotoxin-induced
uveitis. FASEB J. 21:2113–2123. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dello Russo C, Polak PE, Mercado PR,
Spagnolo A, Sharp A, Murphy P, Kamal A, Burrows FJ, Fritz LC and
Feinstein DL: The heat-shock protein 90 inhibitor
17-allylamino-17-deme-thoxygeldanamycin suppresses glial
inflammatory responses and ameliorates experimental autoimmune
encephalomyelitis. J Neurochem. 99:1351–1362. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Collins CB, Aherne CM, Yeckes A, Pound K,
Eltzschig HK, Jedlicka P and de Zoeten EF: Inhibition of N-terminal
ATPase on HSP90 attenuates colitis through enhanced Treg function.
Mucosal Immunol. 6:960–971. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Reinholz M, Ruzicka T and Schauber J:
Cathelicidin LL-37: an antimicrobial peptide with a role in
inflammatory skin disease. Ann Dermatol. 24:126–135. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Di Cesare A, Di Meglio P and Nestle FO:
The IL-23/Th17 axis in the immunopathogenesis of psoriasis. J
Invest Dermatol. 129:1339–1350. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Park CC, Kim KJ, Woo SY, Chun JH and Lee
KH: Comparison of the expression profile of JunB, c-Jun, and S100A8
(calgranulin A) in psoriasis vulgaris and guttate psoriasis. Ann
Dermatol. 21:35–38. 2009. View Article : Google Scholar : PubMed/NCBI
|