Introduction
Mitochondrial DNA (mtDNA) is comprised of a circular
molecule of approximately 16.6 kb, encoding RNAs and polypeptides
of the mitochondrial respiratory chain. Intact mtDNA is necessary
for the production of these polypeptides, which consist of the key
catalytic subunits of the mitochondrial respiratory chain complexes
and are essential for oxidative ATP production. The mtDNA is
frequently exposed to oxidative stress due to the process of
oxidative phosphorylation. During oxidative phosphorylation,
molecular oxygen is transformed into highly reactive oxygen species
(ROS) in the electron transport chain, resulting in the damage of
functional macromolecules including DNA (1).
The occurrence of mtDNA variation during evolution,
particularly with regard to the normal variation in its amount, is
closely associated with cell physiology and human health. Over the
past 20 years, researchers have found that a number of disorders in
humans are associated with severe mtDNA depletion and that
long-term exposure to low concentrations of ethidium bromide (EB)
establishes mtDNA-deficient (Rho° or ϱ°) cells (2). Thus, ϱ° cells have been used to
examine the roles of mtDNA and particularly the importance of mtDNA
in apoptosis (3). A previous
study showed that respiration-deficient cells resisted tumor
necrosis factor (TNF)/serum deprivation-induced apoptosis, whereas
apoptosis was initiated in parental cells and rescued cells with
normal mtDNA (4). These findings
suggest that mtDNA depletion is involved in a mechanism responsible
for resistance to apoptosis. Furthermore, studies by Amuthan et
al and Biswas et al demonstrated that mtDNA depletion
contributed to tumor progression and metastasis (5,6).
Thus, it is likely that mtDNA depletion prevents apoptosis and
generates cancer-related proteins.
Organisms may be exposed to numerous noxious agents
under various conditions. These agents not only passively
disintegrate cells, but also induce productive responses. In
particular, it has been shown in mammalian cells that several genes
are activated by ultraviolet (UV) irradiation (7).
Taking all of the above into consideration, this
study was designed to examine the hypothesis that mtDNA-depleted
mammalian cells resist UV-induced apoptosis and to explore the
possible mechanism responsible for this effect.
Materials and methods
Cell culture, reagents and
antibodies
The human parental osteosarcoma cell line 143B and
Rho°206 cells (mtDNA-depleted) were a gift from Professor Minxin
Guan (Zhejiang University, Zhejiang, China). The cells were
cultured in Dulbecco's modified Eagle's medium (Invitrogen,
Carlsbad, CA, USA) supplemented with 10% newborn calf serum (Gibco,
Carlsbad, CA, USA), 100 U/ml penicillin, 100 mg/ml streptomycin,
100 µg/ml bromodeoxyuridine and 50 µg/ml uridine
(Sigma-Aldrich, St. Louis, MO, USA). Rhodamine 123 (Rh123) and
dichlorodihydrofluorescein diacetate (DCFH-DA) were purchased from
Sigma Chemical Co. (St. Louis, MO, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenylte-trazolium bromide (MTT)
was purchased from Promega (Madison, WI, USA). We purchased
cytochrome c (Cyt c) antibodies (12963S) from Cell
Signaling Technology (CST; Beverly, MA, USA). Antibodies against
β-actin (AA128) and the secondary horseradish peroxidase
(HRP)-labeled antibodies (A0216), were purchased from Beyotime
Biotechnology (Jiangsu, China).
Irradiation procedure
The irradiation procedure was performed using a 1000
Watt Solar Oriel UV Simulator (Oriel, Stratford, CT, USA) with a
UVX digital radiometer (Ultra-Violet Products, Upland, CA, USA)
which was equipped with a UVX-310 sensor to measure UV radiation
intensity. The intensities of UVA and UVB were 2.95 and 56.6
mW/cm2, respectively, with doses ranging from 177
mJ/cm2 and 3.39 J/cm2 to 708
mJ/cm2 and 13.56 J/cm2, respectively
(Table I). The cells were washed
twice with phosphate-buffered saline (PBS) prior to UV irradiation
and then covered with a thin film of PBS during UV irradiation.
Following irradiation, PBS was removed and immediately replaced
with the maintenance medium. The sham-irradiated cells (control
group) were similarly treated; however, they were exposed to normal
room lighting. Finally, all the cells were incubated at 37°C in an
incubator with 5% CO2 (Thermo Fisher Scientific,
Waltham, MA, USA).
 | Table IUVA + UVB intensity, exposure time
and dosage. |
Table I
UVA + UVB intensity, exposure time
and dosage.
| Experimental
grouping | Intensity
(UVA + UVB mW/cm2) | Exposure
time
(min) | Dosage
(mJ/cm2+J/cm2) |
|---|
| Sham-irradiation
group (SIG) | 0+0 | 0 | 0+0 |
| Low-dose groups
(LDG) | | | |
| IG1 | 2.95+56.5 | 1 | 177+3.39 |
| IG2 | 2.95+56.5 | 2 | 354+6.78 |
| High-dose groups
(HDG) | | | |
| IG3 | 2.95+56.5 | 3 | 531+10.17 |
| IG4 | 2.95+56.5 | 4 | 708+13.56 |
Cell viability assay
As previously described, the MTT method (8) was performed to measure cell
viability. Prior to adding MTT tetrazolium salt to each well at a
working concentration of 5 mg/ml, the cells were seeded in 96-well
plates at a density of 5×105 cells/cm2
overnight and treated with UV as indicated in Table I with a 4 h incubation time in a
CO2 incubator. Subsequently, the medium in each well was
replaced with 150 µl DMSO (Sigma-Aldrich). The absorbance of
the dissolved formazan crystals in each well was measured using a
plate reader (Dynatech MR5000; Dynex Technologies, Chantilly, VA,
USA) at a test wavelength of 570 nm. Cell viability was calculated
using the following equation: (absorbance of the experiment
samples/absorbance of the control) ×100%.
Measurement of mitochondrial membrane
potential (MMP)
The cationic lipophilic green fluorochrome Rh123 is
an effective reagent used to determine MMP (9). Prior to incubating with 10 µM
Rh123 at 37°C for 30 min, the cells were harvested and washed three
times with PBS. Following incubation, the cells were washed twice
with PBS and fluorescence was determined using a flow cytometer
(Beckman Coulter, Inc., Brea, CA, USA) with an excitation
wavelength of 488 nm through the FL-1 filter.
Determination of intracellular ROS
levels
DCFH-DA is a reliable reagent which was used to
determine intracellular ROS levels as previously described
(10). The cells were incubated
with DCFH-DA (10 µM) at 37°C for 30 min. Following
incubation, the cells were washed with PBS twice and the
fluorescence of dichlorofluorescein (DCF) generated by ROS was
analyzed immediately using a flow cytometer through the FL-1 filter
with an excitation wavelength of 488 nm.
Annexin V-PE/7-AAD staining
Apoptosis was determined by flow cytometric analysis
(BD Biosciences, Franklin Lakes, NJ, USA) with Annexin V-PE and
7-AAD double staining. After the cells were harvested and washed
twice with ice-cold PBS, the cells were stained and fluorescence
was measured at an excitation wavelength of 480 nm through the FL-1
filter (530 nm) and FL-2 filter (585 nm).
Assessment of cell morphology and PCR
identification
Cell morphology was assessed using a fluorescence
microscope and an electron microscope (Jeol, Peabody, MA, USA). For
microscope observations, the cells were firstly observed under
bright light after being cultured overnight in 24-well dishes and
exposed to UV radiation as described in Table I, and subsequently observed under
purple light after fixing and staining with DAPI (Beyotime
Biotechnology) for 15 min.
For electron microscopy, the cells were fixed with
2% paraformaldehyde/2% glutaraldehyde in 0.1 M PBS, followed by 1%
OsO4 (Sigma-Aldrich). Finally, the cells were stained
with uranyl acetate (Sigma-Aldrich) and lead citrate
(Sigma-Aldrich) for observation after dehydration, as previously
described (11).
A volume of 20 µl containing 10 µl
PrimeStar Max Premix 2X (Takara, Dalian, China) and 10 pmol of
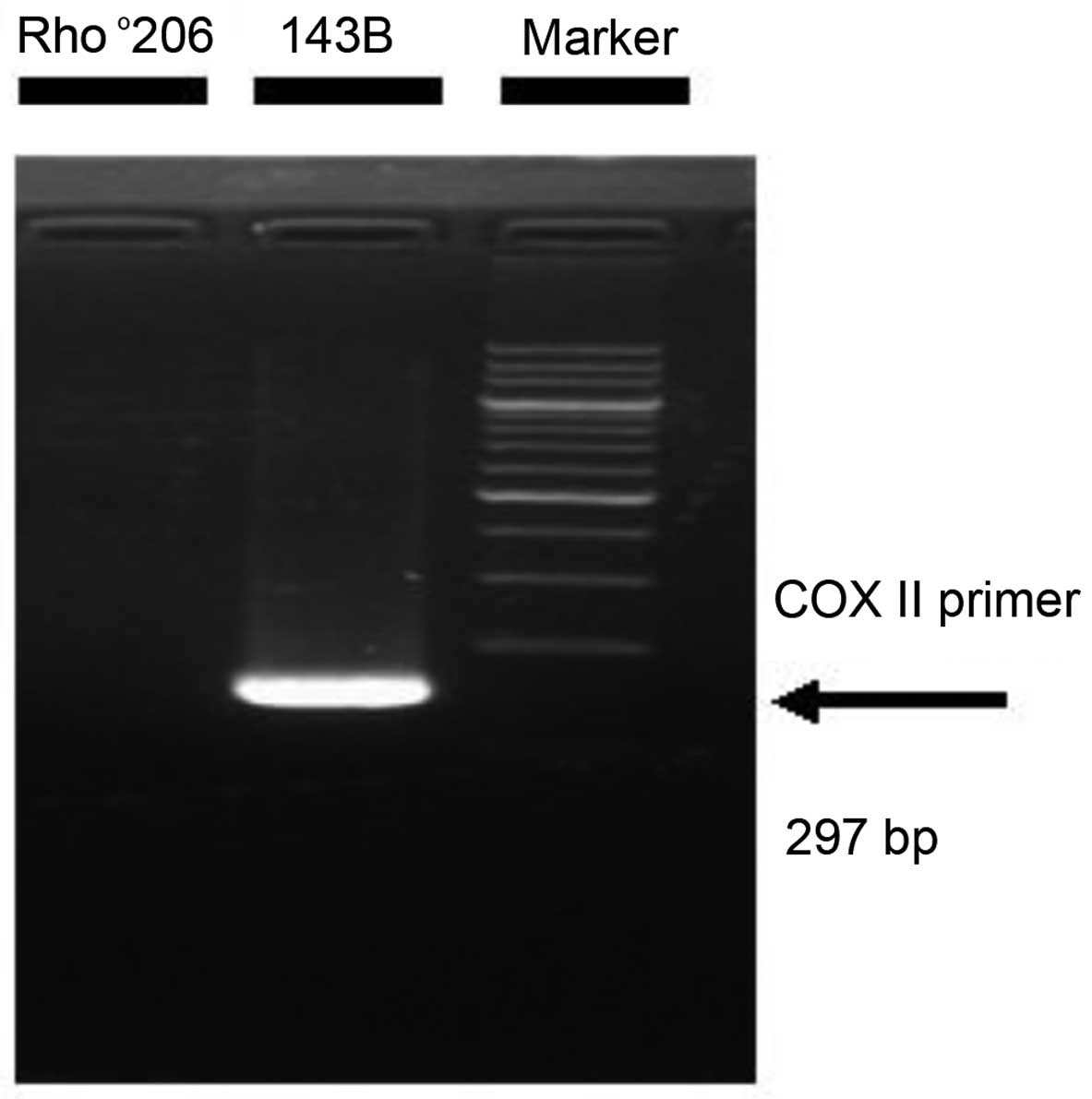
primer was added to amplify the COX II gene. The human
mitochondrial COX II primers used in our study have been reported
previously (12). The following
sequences of COX II primers were used: forward, 5′-ATC AAA TCA ATT
GGC CAC CAA TGG TA-3′ and reverse, 5′-TTG ACC GTA GTA TAC CCC CGG
TC-3′ (297 bp). PCR was performed according to the manufacturer's
instructions. The 200bp DNA Ladder (Dye Plus; Takara Bio, Otsu,
Japan) was used to verify the bands.
Western blot analysis
The cells, at a density of 1×107
cells/ml, were exposed to UV radiation as described in Table I. Following the lysis procedure,
the lysates were centrifuged to obtain the supernatants. The
protein concentrations of the supernatants were then determined
using BCA Protein assay reagent (Beyotime Biotechnology). Western
blot analysis was performed to analyze certain proteins in the
supernatants (50 µg) of each sample.
Statistical analysis
Data are expressed as the means ± SD and analyzed
using the Student's t-test (two-tailed). A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
Rho°206 cells exhibit greater resistance
to apoptosis compared with 143B cells following exposure to UV
radiation
Previous studies have shown that transient mtDNA
depletion enhances the invasive ability of several types of cancer
cells as well as resistance to apoptosis (5,13).
Thus, it is evident that mtDNA-depleted cells play roles in tumor
progression and metastasis. To the best of our knowledge, there are
no studies regarding the effect of mtDNA depletion in human
osteosarcoma 143B cells. To determine the anti-apoptotic effects of
mtDNA depletion in 143B cells, we used Rho°206 cells, an
mtDNA-depleted 143B cell type, as shown in Fig. 1.
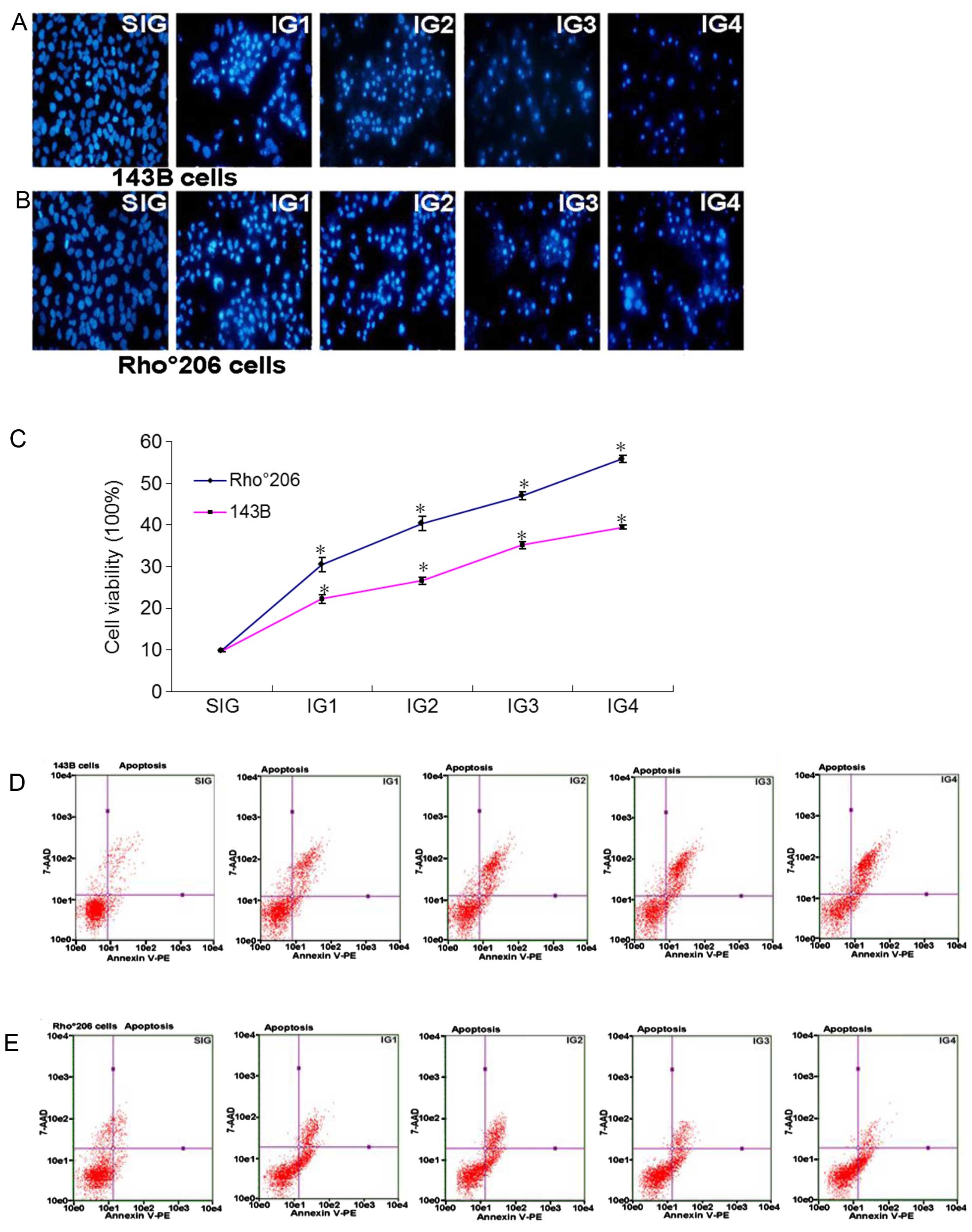
Firstly, we observed the decrease in cell numbers in
the two cell lines following exposure to UV radiation as described
in Table I under a light
microscope and a fluorescence microscope. Fig. 2A and B show that the numbers of
143B cells clearly decreased in comparison with the number of
Rho°206 cells. Furthermore, MTT analysis showed the differences in
the cell viability of Rho°206 and 143B cells following exposure to
UV radiation. The cell viability of 143B cells was lower than that
of Rho°206 cells (Fig. 2C).
Additionally, Annexin V-PE and 7-AAD double staining by flow
cytometry shows that the 143B cells endured more severe apoptosis
compared with the Rho°206 cells following exposure to UV radiation
(Fig. 2D and E). This may
indicate that Rho°206 cells are more resistant to apoptosis.
UV-induced disruption of MMP is reduced
in Rho°206 cells compared with that in 143B cells
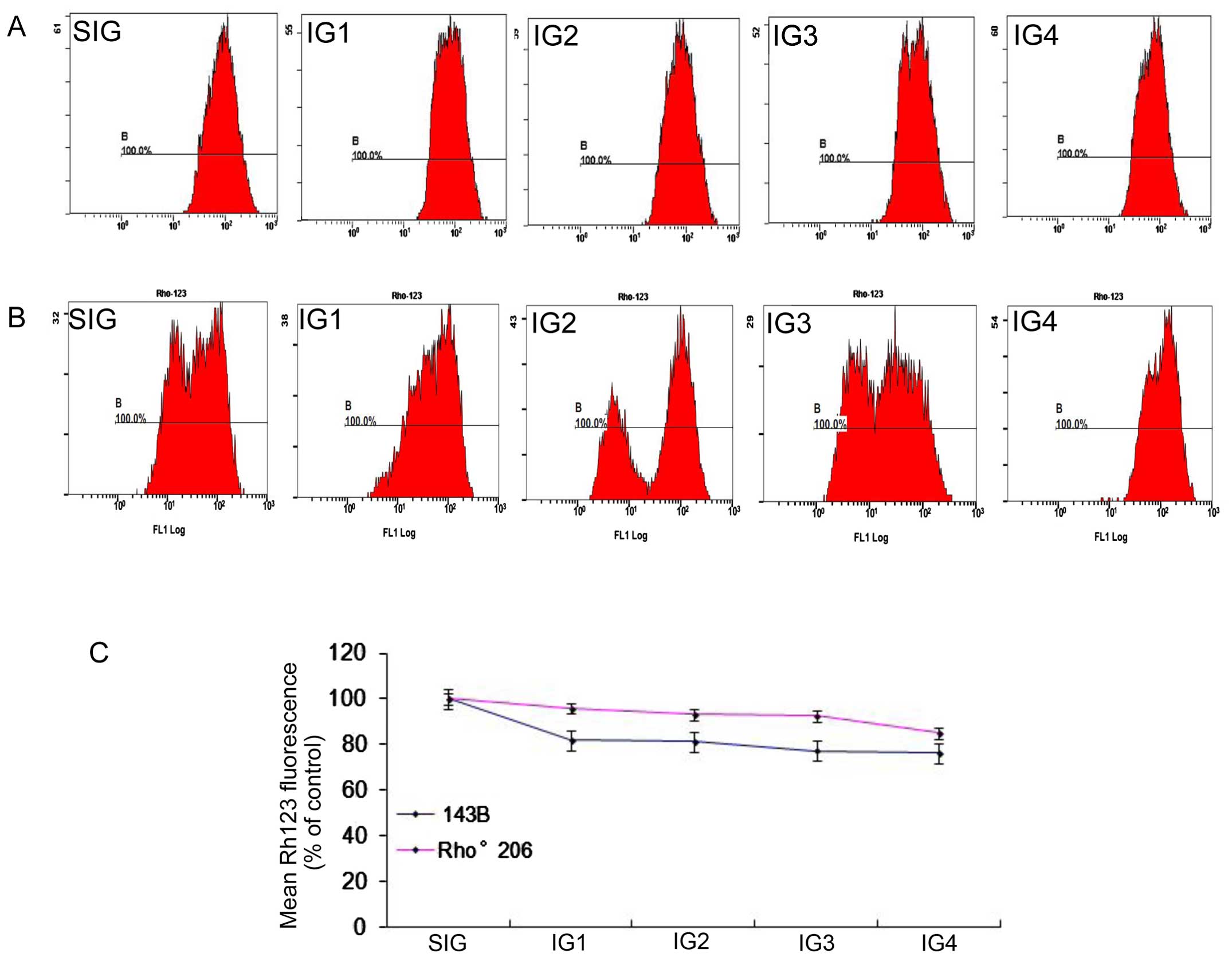
To detect changes in MMP in the cells following UV
irradiation in the present study, we measured MMP by assessing the
uptake of Rh123 using a flow cytometer.
Following exposure to UV radiation as described in
Table I, mitochondrial activity
was decreased in both cell lines. As shown in Fig. 3, following exposure to different
intensities of radiation, the 143B cells displayed various
disruptions of MMP. Compared with the control, mitochondrial
activity decreased to 76.35% after 708 mJ/cm2 UVA and
13.56 J/cm2 UVB treatment (IG4 group) in the 143B cells,
and to 85.1% in the Rho°206 cells. The impact of the combination of
708 mJ/cm2 UVA and 13.56J/cm2 UVB on
mitochondrial activity in the 143B cells (Fig. 3B and C) suggested that UV caused
the decline in mitochondrial activity. At the same time, no
significant disruption of MMP was detected after 708
mJ/cm2 UVA and 13.56 J/cm2 UVB treatment in
both cell lines (data not shown), indicating that UV may exert a
minimal effect on the MMP and mitochondrial dysfunction.
Furthermore, Fig.
3 shows clearly that the MMP in the 143B cells dropped more
quickly than in the Rho°206 cells following UV irradiation from
which we concluded that mtDNA depletion may protect cells against
UV-induced disruption of MMP.
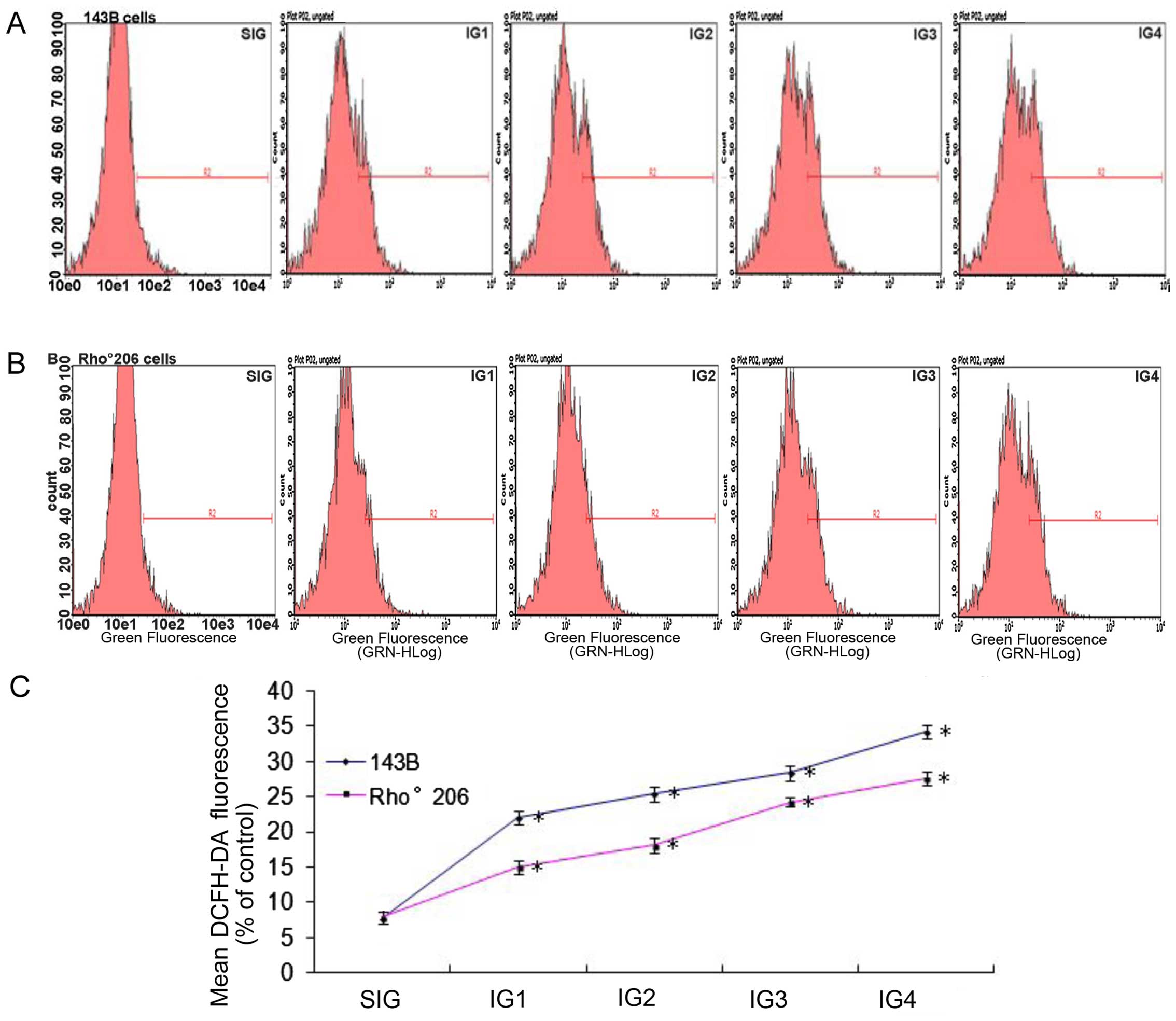
UV irradiation induces less ROS
production in Rho°206 cells compared with that in 143B cells
Disruptions in MMP and cell apoptosis are associated
with the production of ROS (14,15). Thus, in the present study, we
measured ROS production in the 143B cells and the Rho°206 cells
following exposure to UV radiation as described in Table I. The oxidation sensitive
fluorescent dye DCFH-DA was applied in order to evaluate ROS
production. ROS production was significantly increased in both cell
lines following exposure to increasing doses of UV radiation. In
the 143B cells, the generation of ROS was faster compared with that
in the control group after UV exposure, (an approximate 3–5-fold
increase), as shown in Fig. 4A and
C. In the Rho°206 cells, there was an approximate 2–4-fold
increase in the generation of ROS compared with the control group
(Fig. 4B and C). These results
show that ROS production increased more rapidly in 143B cells
compared with that in Rho°206 cells following UV irradiation.
Rho°206 cells resist UV-induced apoptosis
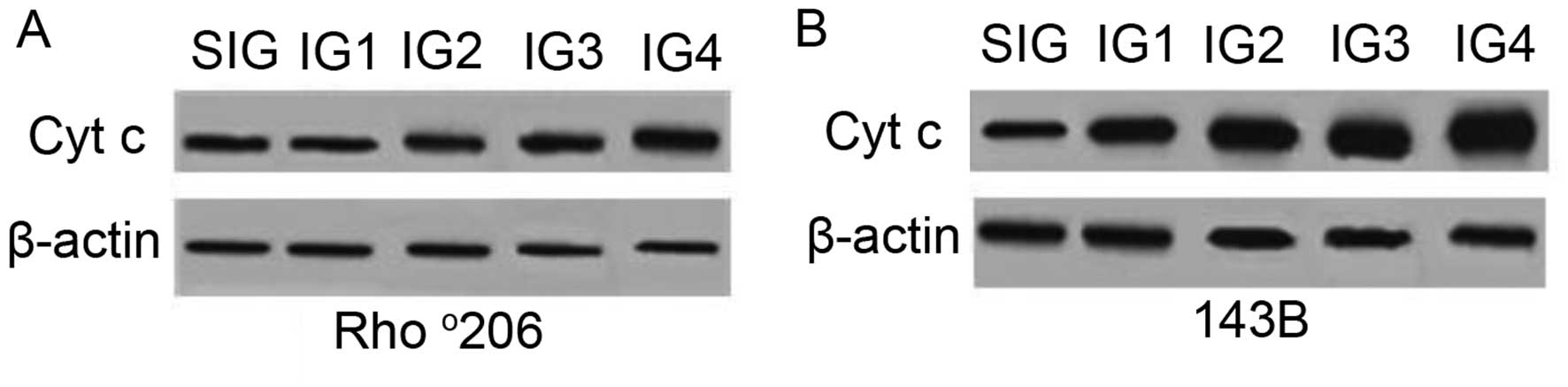
through decreasing Cyt c release
During apoptosis, Cyt c is released from
mitochondria into the cytoplasm (16). To examine whether Cyt c was
released during UVA combined with UVB-induced cell apoptosis, the
expression of cytosolic Cyt c was measured using western
blot analysis. Increased levels of Cyt c in the cytoplasm
were detected following exposure to a combination of UVA and UVB
(Fig. 5) compared with the
controls. This increase was more evident in the parental 143B cells
than in the mtDNA-depleted Rho°206 cells, indicating a potential
resistance to Cyt c release in the Rho°206 cells.
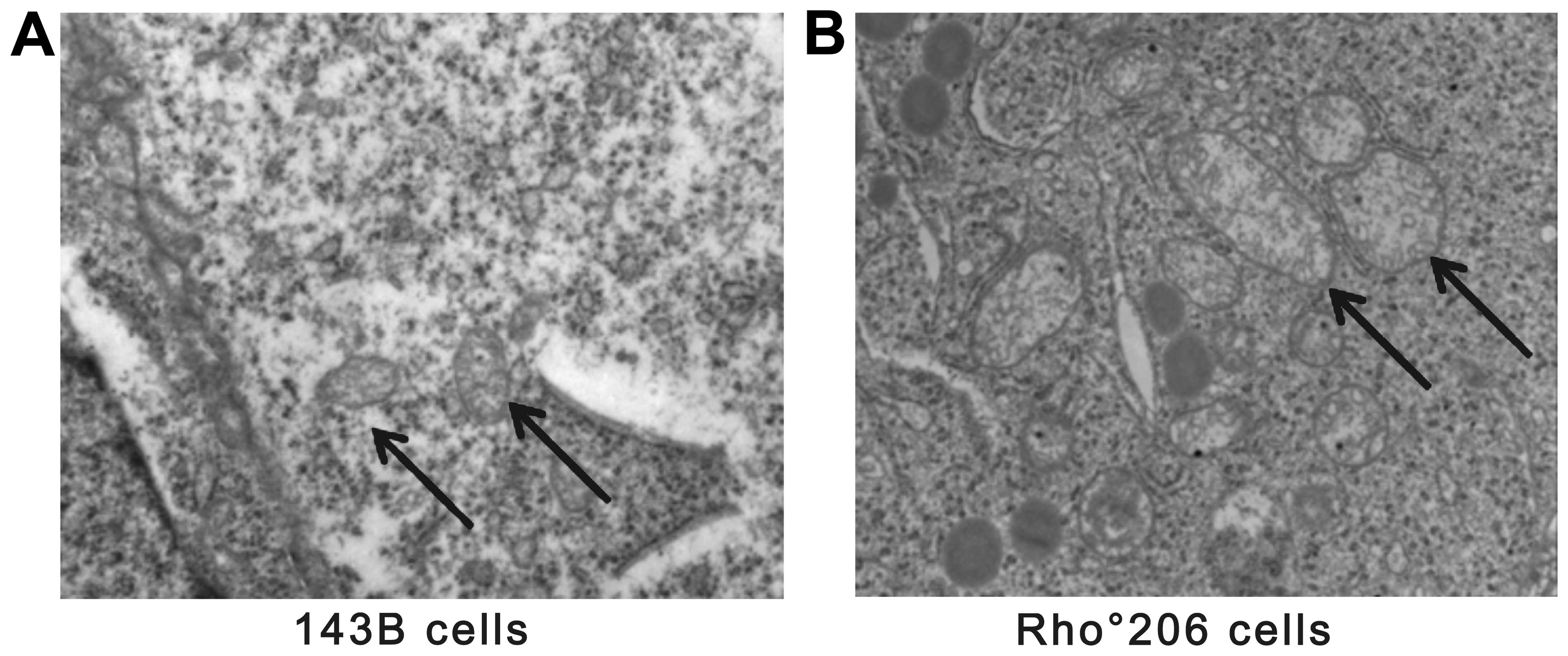
Furthermore, the morphological changes observed in the mitochondria
of the Rho°206 cells (Fig. 6) may
not be conducive to Cyt c release in these cells.
Discussion
Previous findings have demonstrated that mtDNA
encodes pivotal catalytic subunits and its complete depletion in
cells leads to several morphological changes. Fig. 6 shows that the mitochondrial
density of mtDNA-depleted Rho°206 cells was reduced and that the
mitochondria were larger and more elongated compared with those in
the parental 143B cells. Hojo et al also observed similar
changes in cyclosporin-treated cells (17). Furthermore, a major block also
occurs in the normal electron flow in mtDNA-depleted cells.
Mitochondria are the principal source of
intracellular ROS under adverse conditions (18,19). This, in combination with the fact
that excision repair frequently occurs in mtDNA (20), suggests that mtDNA is liable to
attack under conditions of oxidative and chemical stress. A number
of studies regarding homoplasmic/heteroplasmic mtDNA
depletion/mutations have been reported in human tumors which
support this view (21–27). However, it remains unclear whether
cancer progression results in (28) or from (29) the depletion/mutations. The results
presented in this study suggest that mtDNA-depleted cells possess a
survival advantage following environmental exposure to UV
irradiation.
UV irradiation is a pivotal factor that increases
levels of ROS (30–35), and simultaneously decreases
antioxidant enzymes (36),
causing oxidative stress to initiate cellular signal transduction.
Redundant ROS are likely to lead to cell death by oxidizing and
then damaging functional macromolecules such as DNA and protein.
Herein, we found that UV exposure induced ROS production in human
mammalian cells and mtDNA-depleted cells resisted the release of
ROS thereby escaping greater death compared with the normal cells.
Notably, UV irradiation in 143B cells and Rho°206 cells had minor
effects on MMP, since MMP disruption is always followed by
increasing ROS levels (37). This
paradox warrants further investigation. We also observed Cyt
c release, as previously described (37). Notably, the 143B cells released
more Cyt c from the mitochondria into the cytoplasm than the
Rho°206 cells following UV irradiation. Therefore, it is possible
that DNA depletion in mitochondria induces resistance to UV-induced
apoptosis by decreasing ROS production, which plays a role in the
upstream apoptotic mechanism following Cyt c release and the
activation of caspases.
Acknowledgments
The present study was supported by the National
Science Foundation of China (NSFC, grant no. 30972652), and the
Science and Technology Department of Guangdong Province Program
(no. 2013B021800053).
References
|
1
|
Orrenius S, Gogvadze V and Zhivotovsky B:
Mitochondrial oxidative stress: implications for cell death. Annu
Rev Pharmacol Toxicol. 47:143–183. 2007. View Article : Google Scholar
|
|
2
|
King MP and Attardi G: Human cells lacking
mtDNA: repopulation with exogenous mitochondria by complementation.
Science. 246:500–503. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chandel NS and Schumacker PT: Cells
depleted of mitochondrial DNA (ϱ°) yield insight into physiological
mechanisms. FEBS Lett. 454:173–176. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Higuchi M, Aggarwal BB and Yeh ET:
Activation of CPP32-like protease in tumor necrosis factor-induced
apoptosis is dependent on mitochondrial function. J Clin Invest.
99:1751–1758. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Amuthan G, Biswas G, Zhang SY,
Klein-Szanto A, Vijayasarathy C and Avadhani NG:
Mitochondria-to-nucleus stress signaling induces phenotypic
changes, tumor progression and cell invasion. EMBO J. 20:1910–1920.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Biswas G, Anandatheerthavarada HK, Zaidi M
and Avadhani NG: Mitochondria to nucleus stress signaling: a
distinctive mechanism of NFkappaB/Rel activation through
calcineurin-mediated inactivation of IkappaBbeta. J Cell Biol.
161:507–519. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Friedberg E, Walker G and Siede W: DNA
repair and mutagenesis. ASM Press; Washington, DC: 1995
|
|
8
|
Shi Y, Wang CH and Gong XG:
Apoptosis-inducing effects of two anthraquinones from Hedyotis
diffusa WILLD. Biol Pharm Bull. 31:1075–1078. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu M, Gong X, Lu Y, Guo J, Wang C and Pan
Y: Molecular cloning and functional characterization of a
cell-permeable superoxide dismutase targeted to lung adenocarcinoma
cells. Inhibition cell proliferation through the Akt/p27kip1
pathway. J Biol Chem. 281:13620–13627. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Li J, Xu Z, Tan M, Su W and Gong XG:
3-(4-(Benzo[d) thiazol-2-yl)-1-phenyl-1H-pyrazol-3-yl) phenyl
acetate induced Hep G2 cell apoptosis through a ROS-mediated
pathway. Chem Biol Interact. 183:341–348. 2010. View Article : Google Scholar
|
|
11
|
Gong K, Chen C, Zhan Y, Chen Y, Huang Z
and Li W: Autophagy-related gene 7 (ATG7) and reactive oxygen
species/extracellular signal-regulated kinase regulate
tetrandrine-induced autophagy in human hepatocellular carcinoma. J
Biol Chem. 287:35576–35588. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lewis LD, Hamzeh FM and Lietman PS:
Ultrastructural changes associated with reduced mitochondrial DNA
and impaired mitochondrial function in the presence of
2′3′-dideoxycytidine. Antimicrob Agents Chemother. 36:2061–2065.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Amuthan G, Biswas G, Ananadatheerthavarada
HK, Vijayasarathy C, Shephard HM and Avadhani NG: Mitochondrial
stress-induced calcium signaling, phenotypic changes and invasive
behavior in human lung carcinoma A549 cells. Oncogene.
21:7839–7849. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Xiong Y, Liu X, Lee CP, Chua BH and Ho YS:
Attenuation of doxorubicin-induced contractile and mitochondrial
dysfunction in mouse heart by cellular glutathione peroxidase. Free
Radic Biol Med. 41:46–55. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
López E, Arce C, Oset-Gasque MJ, Cañadas S
and González MP: Cadmium induces reactive oxygen species generation
and lipid peroxidation in cortical neurons in culture. Free Radic
Biol Med. 40:940–951. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hojo M, Morimoto T, Maluccio M, Asano T,
Morimoto K, Lagman M, Shimbo T and Suthanthiran M: Cyclosporine
induces cancer progression by a cell-autonomous mechanism. Nature.
397:530–534. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kehrer JP: Free radicals as mediators of
tissue injury and disease. Crit Rev Toxicol. 23:21–48. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lenaz G: Role of mitochondria in oxidative
stress and ageing. Biochim Biophys Acta. 1366:53–67. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pinz KG and Bogenhagen DF: Efficient
repair of abasic sites in DNA by mitochondrial enzymes. Mol Cell
Biol. 18:1257–1265. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Allen JA and Coombs MM: Covalent binding
of polycyclic aromatic compounds to mitochondrial and nuclear DNA.
Nature. 287:244–245. 1980. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Backer JM and Weinstein IB: Mitochondrial
DNA is a major cellular target for a dihydrodiol-epoxide derivative
of benzo[a) pyrene. Science. 209:297–299. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Niranjan BG, Bhat NK and Avadhani NG:
Preferential attack of mitochondrial DNA by aflatoxin B1 during
hepatocarcinogenesis. Science. 215:73–75. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Horton TM, Petros JA, Heddi A, Shoffner J,
Kaufman AE, Graham SD Jr, Gramlich T and Wallace DC: Novel
mitochondrial DNA deletion found in a renal cell carcinoma. Genes
Chromosomes Cancer. 15:95–101. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Polyak K, Li Y, Zhu H, Lengauer C, Willson
JK, Markowitz SD, Trush MA, Kinzler KW and Vogelstein B: Somatic
mutations of the mitochondrial genome in human colorectal tumours.
Nat Genet. 20:291–293. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fliss MS, Usadel H, Caballero OL, Wu L,
Buta MR, Eleff SM, Jen J and Sidransky D: Facile detection of
mitochondrial DNA mutations in tumors and bodily fluids. Science.
287:2017–2019. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Yeh JJ, Lunetta KL, van Orsouw NJ, Moore
FD Jr, Mutter GL, Vijg J, Dahia PL and Eng C: Somatic mitochondrial
DNA (mtDNA) mutations in papillary thyroid carcinomas and
differential mtDNA sequence variants in cases with thyroid tumours.
Oncogene. 19:2060–2066. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Loeb LA: A mutator phenotype in cancer.
Cancer Res. 61:3230–3239. 2001.PubMed/NCBI
|
|
29
|
Rasmussen AK, Chatterjee A, Rasmussen LJ
and Singh KK: Mitochondria-mediated nuclear mutator phenotype in
Saccharomyces cerevisiae. Nucleic Acids Res. 31:3909–3917. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Masaki H, Atsumi T and Sakurai H:
Detection of hydrogen peroxide and hydroxyl radicals in murine skin
fibroblasts under UVB irradiation. Biochem Biophys Res Commun.
206:474–479. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jurkiewicz BA and Buettner GR: EPR
detection of free radicals in UV-irradiated skin: mouse versus
human. Photochem Photobiol. 64:918–922. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Barber LA, Spandau DF, Rathman SC, Murphy
RC, Johnson CA, Kelley SW, Hurwitz SA and Travers JB: Expression of
the platelet-activating factor receptor results in enhanced
ultraviolet B radiation-induced apoptosis in a human epidermal cell
line. J Biol Chem. 273:18891–18897. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brenneisen P, Wenk J, Klotz LO, Wlaschek
M, Briviba K, Krieg T and Scharffetter-Kochanek K: Central role of
ferrous/ferric iron in the ultraviolet B irradiation-mediated
signaling pathway leading to increased interstitial collagenase
(matrix-degrading metalloprotease (MMP)-1) and stromelysin-1
(MMP-3) mRNA levels in cultured human dermal fibroblasts. Biol
Chem. 273:5279–5287. 1998. View Article : Google Scholar
|
|
34
|
Yasui H and Sakurai H: Chemiluminescent
detection and imaging of reactive oxygen species in live mouse skin
exposed to UVA. Biochem Biophys Res Commun. 269:131–136. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kang S, Chung JH, Lee JH, Fisher GJ, Wan
YS, Duell EA and Voorhees JJ: Topical N-acetyl cysteine and
genistein prevent ultraviolet-light-induced signaling that leads to
photoaging in human skin in vivo. J Invest Dermatol. 120:835–841.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yamamoto Y: Role of active oxygen species
and antioxidants in photoaging. J Dermatol Sci. 27:1–4. 2001.
View Article : Google Scholar
|
|
37
|
Chandra D, Liu JW and Tang DG: Early
mitochondrial activation and cytochrome c up-regulation during
apoptosis. J Biol Chem. 277:50842–50854. 2002. View Article : Google Scholar : PubMed/NCBI
|