Introduction
Stroke ranks among the most lethal cerebrovascular
diseases worldwide and is associated with high morbidity and
long-term disability, which may cause irreversible brain damage and
loss of neuronal function (1).
Brain ischemia, characterized by the occlusion of blood vessels and
deprivation of energy and nutrition, leads to serious neuronal
injury and neurodegeneration, and further to learning and memory
impairment (2). Following brain
ischemia/reperfusion (I/R) injury, proinflammatory cytokines, such
as interleukin (IL)-1β, IL-6 and tumor necrosis factor (TNF)-α, are
immediately produced and secreted after the onset of cerebral
ischemia, eventually contributing to the pathogenesis and
exacerbation of brain tissue damage (3).
Astrocytes are one type of glial cell in the
mammalian central nervous system (CNS), and the most numerous cell
type of the brain (4). Under
physiologic conditions, astrocytes facilitate neuronal homeostasis
in the CNS whereas under pathologic conditions astrocytes promote
neuronal demise (5). In the
context of cerebral ischemia, all of the star-type cells, microglia
as well as endothelial cells, may produce cellular factors
(6,7).
Lysosomes consist of more than 50 acid hydrolases
and 120 membrane proteins, which participate in lysosomal digestion
or the maintenance of lysosomal integrity and the regulation of
lysosomal trafficking, fusion and intralysosomal pH (8). Lysosomal enzymes, including
cathepsins (Cats) (Cat B, D, K, L and so on) and some lipid
hydrolases, when released from the ruptured lysosomal membrane, may
cause the destruction of cellular components and even lead to cell
death (9). It has been
demonstrated that Cat B and D markedly increase in primary
astrocytes after oxygen-glucose deprivation (OGD) and this is
accompanied by the apoptosis of astrocytes (10). Moreover, Cat B and L activation
after brain ischemia triggered the tBid-mediated death of
astrocytes following brain I/R in vivo (11). Accumulating evidence has
demonstrated that the lysosome and its associated protein Cat D
play critical roles in the pathological process of secondary damage
following I/R damage (12).
However, the roles of Cat D during the exposure of astrocytes to
I/R remain unclear. In the present study, we examined the effects
of inflammatory molecules on the expression and activation of Cat D
as well as the role of Cat D in the OGD-induced death of astrocytes
and the underlying mechanism, in order to identify a novel strategy
for the treatment of brain damage following I/R injury.
Materials and methods
Astrocyte culture and OGD
Astrocytes were isolated from the cerebral cortex of
BALB/c mice (n=5) on postnatal days 1–3 according to previously
described protocols (13). All
the mice were obtained from the animal center of Xi'an Jiaotong
University (Xi'an, China). Briefly, all mice received general
anesthesia with isoflurane inhalation prior to sacrifice in order
to minimize their suffering. Ethics approval was obtained from the
Ethics Committee of Xi'an Jiaotong University. Cortical hemispheres
were dissociated with 0.25% trypsin in PBS. Following filtration,
the dissociated cells were placed on poly-L-lysine-coated plastic
flasks and maintained in Dulbecco's modified Eagle's medium
(DMEM)/nutrient mixture F-12 supplemented with 10% fetal bovine
serum (Gibco BRL, Gaithersburg, MD, USA), and cultivated at 37°C in
a humidified incubator in an atmosphere of 95% air and 5%
CO2 (Thermo Forma, Marietta, OH, USA). The cells were
maintained in complete culture medium for 7–8 days and the culture
medium was changed every three days. Astrocyte cultures were
passaged twice and the purity of astrocyte cultures consisted of at
least 95% astrocytes as determined by immunostaining against
astrocyte marker glial fibrillary acidic protein (GFAP, 1:200; Cell
Signaling Technology, Danvers, MA, USA). These cells were used in
subsequent experiments.
With regard to the establishment of an in
vitro model of OGD using astrocytes, the standard culture
medium was refreshed using a glucose-free DMEM buffer (Gibco,
Rockville, MD, USA), and the cells were then placed in a hypoxic
humidified incubator flushed with a gas mixture of 93%
N2/5% CO2/2% O2 (Thermo Forma) as
previously described (14). Four
hours later, the cells were cultured in standard medium containing
glucose under conditions of normoxia for reoxygenation for an
additional 48 h. The control group was comprised of naïve
astrocytes cultured under normal conditions and the OGD group was
defined as cells exposed to OGD without reperfusion. TNF-α/FasL (20
ng/ml) were added to naïve astrocytes and incubated for 24 h for
further experiments, and 1 µg/ml anti-Fas (anti-CD95-ZB4;
Millipore, Billerica, MA, USA) and anti-tumor necrosis factor
receptor 1 (TNFR1; R&D Systems, Minneapolis, MN, USA) were
added to astrocyte cultures 3 h prior to the OGD/reperfusion
(OGD/R) procedure.
Preparation and transfection of small
interfering RNA (siRNA)
Cat D knockdown was accomplished by transfecting
astrocytes with siRNA. Cat D and control siRNA were synthesized by
Wolsen Biological Co., Ltd. (Xi'an, China). Cells
(3×105) were transfected with 1 µg siRNA using
2.5 µg Lipofectamine 2000 reagent (Invitrogen, Carlsbad, CA,
USA). Cat D knockdown was analyzed 48 h after transfection by
western blot analysis.
Transient transfection of Cat D
plasmids
A pCMV-SPORT6 plasmid containing the Cat D encoding
region was purchased from Thermo Fisher Scientific (Ottawa, ON,
Canada). Cultured astrocytes were transfected with the Cat
D-overexpressing plasmid or a control plasmid using Lipofectamine
2000 according to the manufacturer's instructions. The
overexpression of Cat D was analyzed 48 h after transfection by
western blot analysis. The cells harboring Cat D or control vectors
were then subjected to OGD/R exposure.
MTT assay
The viability of the primary astrocytes subjected to
OGD/R or not were determined by MTT assay. In brief, the cells were
seeded in 96-well plates at a density of 1×104 cells in
300 µl medium. The control astrocytes were incubated in
standard condition and the OGD/R-subjected cells were exposed to 4
h of OGD followed by reperfusion. At each time point, 50 µl
of MTT solution (Sigma-Aldrich, St Louis, MO, USA) dissolved in
culture medium at a final concentration of 0.5 mmol/l were added to
each well and the plates were incubated at 37°C for a further 4 h.
Subsequently, the culture medium was discarded and 300 µl
DMSO (Sigma-Aldrich) was then added to each well, followed by
shaking for 20 min to solubilize the MTT tetrazolium crystal.
Finally, the absorbance was measured at 570 nm using a Benchmark
Plus microplate reader (Model 550; Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Cell apoptosis
Cells were incubated with Annexin V-FITC and
propidium iodide (BD Biosciences, Franklin Lakes, NJ, USA) for 10
min on ice. Apoptosis was analyzed using a FACSCalibur®
flow cytometer (BD Biosciences). Data were processed using FlowJo
software.
Western blot analysis
The astrocytes were harvested and lysed in lysis
buffer. The extracted proteins (30–50 µg) were separated on
SDS-PAGE gels and transferred to PVDF membranes. After blocking
with 5% skimmed milk, the blots were incubated overnight at 4°C
with antibodies against cleaved caspase-3 (Asp175; #9661; Cell
Signaling Technology), Bax (sc-7480) and β-actin (sc-47778) (Santa
Cruz Biotechnology, Santa Cruz, CA, USA). Total cytosolic and
mitochondrial protein extraction were performed as previously
described (7). The extracted
cytosolic Cat B (ab58802), D (ab6313) and L (ab58991) (Abcam,
Cambridge, UK) were detected. Finally, the immunoreactive bands
were visualized using an ECL detection system. The immunoreactive
bands were visualized using an ECL detection system (Amersham
Pharmacia Biotech, Uppsala, Sweden). Signals were quantified by a
grayscale scanner (GeneGnome XRQ; Syngene Corp., Cambridge,
UK).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the treated astrocytes
using TRIzol reagent (Invitrogen) and was reverse transcribed into
cDNA using the M-MLV Reverse Transcriptase kit (Invitrogen)
according to the manufacturer's instructions. Quantitative PCR
(qPCR) was run on a Bio-Rad CFX manager using SYBR Premix Ex Taq™
(Takara). The PCR primer sequences used were as follows: IL-6
sense, 5′-CTGCAAGAGACTTCCATCCAGTT-3′ and antisense,
5′-AGGGAAGGCCGTGGTTGT-3′; TNF-α sense, 5′-ACGTGCAGCTACTGCATGTGA-3′
and antisense, 5′-AGAAGGAACACGTTGTCAGCG-3′; FasL sense,
5′-AGCCCGTGAATTACCCATGTC-3′ and antisense,
5′-TGCTGGGGTTGGCTATTTGCT-3′; GAPDH sense,
5′-AGCAGTCCCGTACACTGGCAAAC-3′ and antisense,
5′-TCTCCTGTAAATGTAGTGGTGTCT-3′. PCR was performed on an iCycler iQ
(Bio-Rad Laboratories, Inc.) beginning at 95°C for 20 sec, followed
by 40 cycles: at 95°C for 1 sec, and at 60°C for 20 sec. Gene
expression was determined by the 2−ΔΔCt method.
Determination of Cat D activity
A Cat D activity assay kit (Abcam) was used to
determine the activity of Cat D following OGD/R exposure. The
substrate cleaved by Cat D in cell lysates releases fluorescence
which is captured by a fluorescence plate reader at Ex/Em = 328/460
nm.
LysoTracker assay
Following incubation with 1 µM LysoTracker
Red dye (Invitrogen) for 15 min, the astrocytes were trypsinized
and resuspended in PBS for FACS analysis (FACSCanto II; BD
Biosciences, San Jose, CA, USA). Cyflogic software (CyFlo Ltd.,
Turku, Finland) was used to analyze 20,000 events/run.
Evaluation of mitochondrial membrane
potential (ΔΨm) and reactive oxygen species (ROS)
production
Following exposure to OGD/R for 24 h, the astrocytes
were washed with PBS and evaluated for time-dependent changes in
ΔΨm by resuspension in freshly prepared JC-1-containing
(Invitrogen) medium, which was followed by 30 min of cultivation in
the dark at room temperature. Fluorescence intensity was measured
with excitation at 490 nm and emission at 530 and 590 nm using a
Bio-Rad microplate reader (Model 680; Bio-Rad Laboratories, Inc.).
The ratio between green and red fluorescence provides an estimate
of ΔΨm that is independent of mitochondrial mass. For
the ROS assay, treated cells were exposed to DCFH-DA for 15 min at
37°C. Fluorescence excitation and emission wavelengths were set at
480 and 530 nm, respectively, using a Bio-Rad microplate
reader.
Statistical analysis
Values are expressed as the means ± SEM. Data were
analyzed by one-way ANOVA followed by Fisher's LSD test and the
Bonferroni test using SPSS 13.0 software. P<0.05 was considered
to indicate a statistically significant difference. All experiments
were performed at least in triplicate.
Results
OGD/R injury triggers the production of
inflammatory mediators and the apoptosis of astrocytes
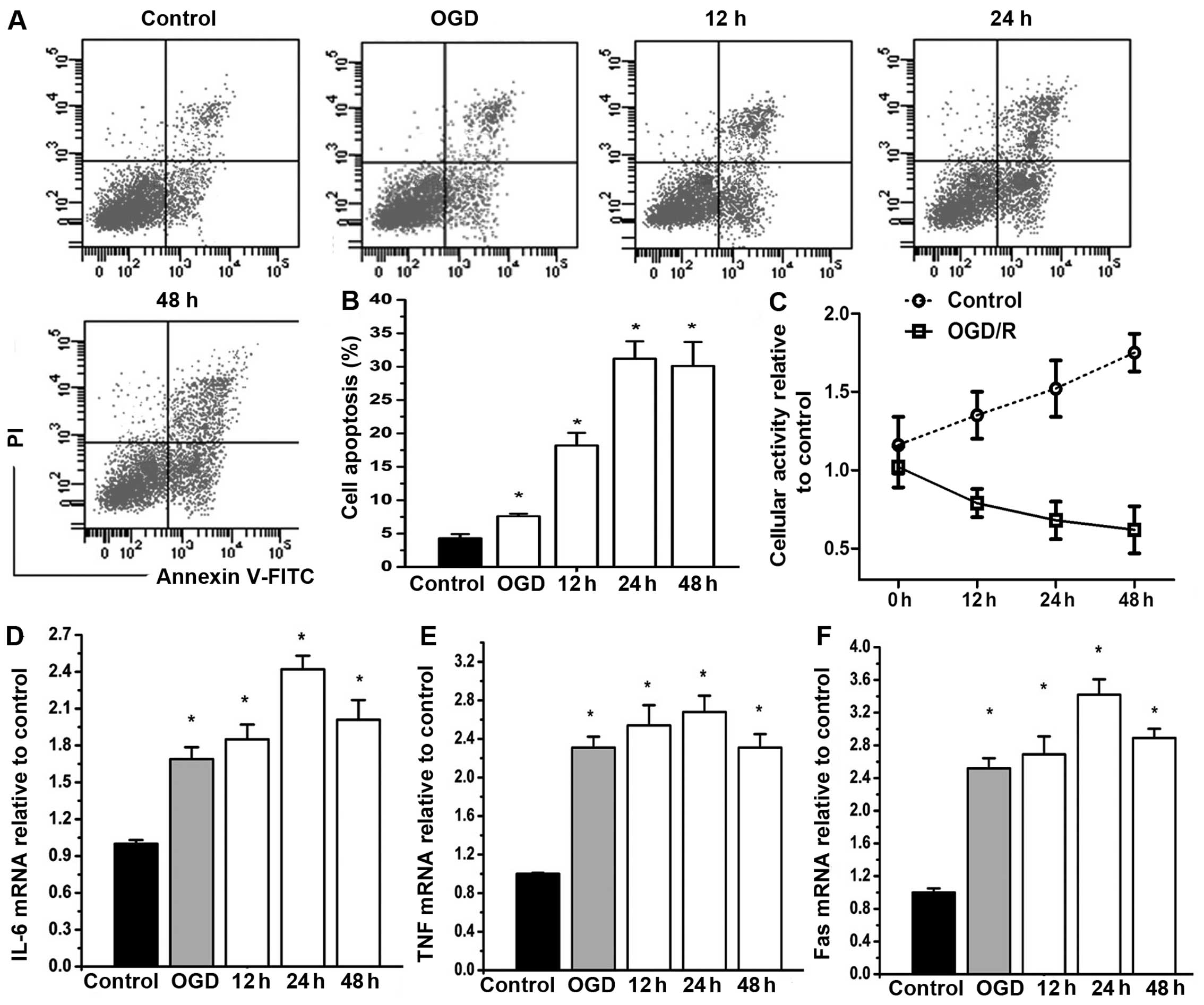
In order to examine the destructive effects of OGD/R
injury on astrocytes, cell apoptosis and the expression of
inflammatory factors, namely IL-6, TNF-α and FasL, were monitored
within 48 h of reperfusion following 4 h of OGD. Flow cytometric
analysis showed that compared with the OGD-exposed cells (4 h OGD
without reperfusion), the apoptosis of astrocytes was significantly
increased after 12 h of reperfusion. However, there was no clear
difference among the cells reoxygenated for 24 and 48 h (Fig. 1A and B). Moreover, the results of
MTT assay revealed that the viability of the control astrocytes was
significantly elevated after 24 h of reperfusion while the
OGD/R-exposed cells exhibited a gradual decrease in viability
against the reperfusion time (Fig.
1C). In addition, OGD/R resulted in an increase of IL-6, TNF-α
and FasL in a time-dependent manner with a peak at 24 h of
reperfusion (P<0.05) (Fig.
1D–F).
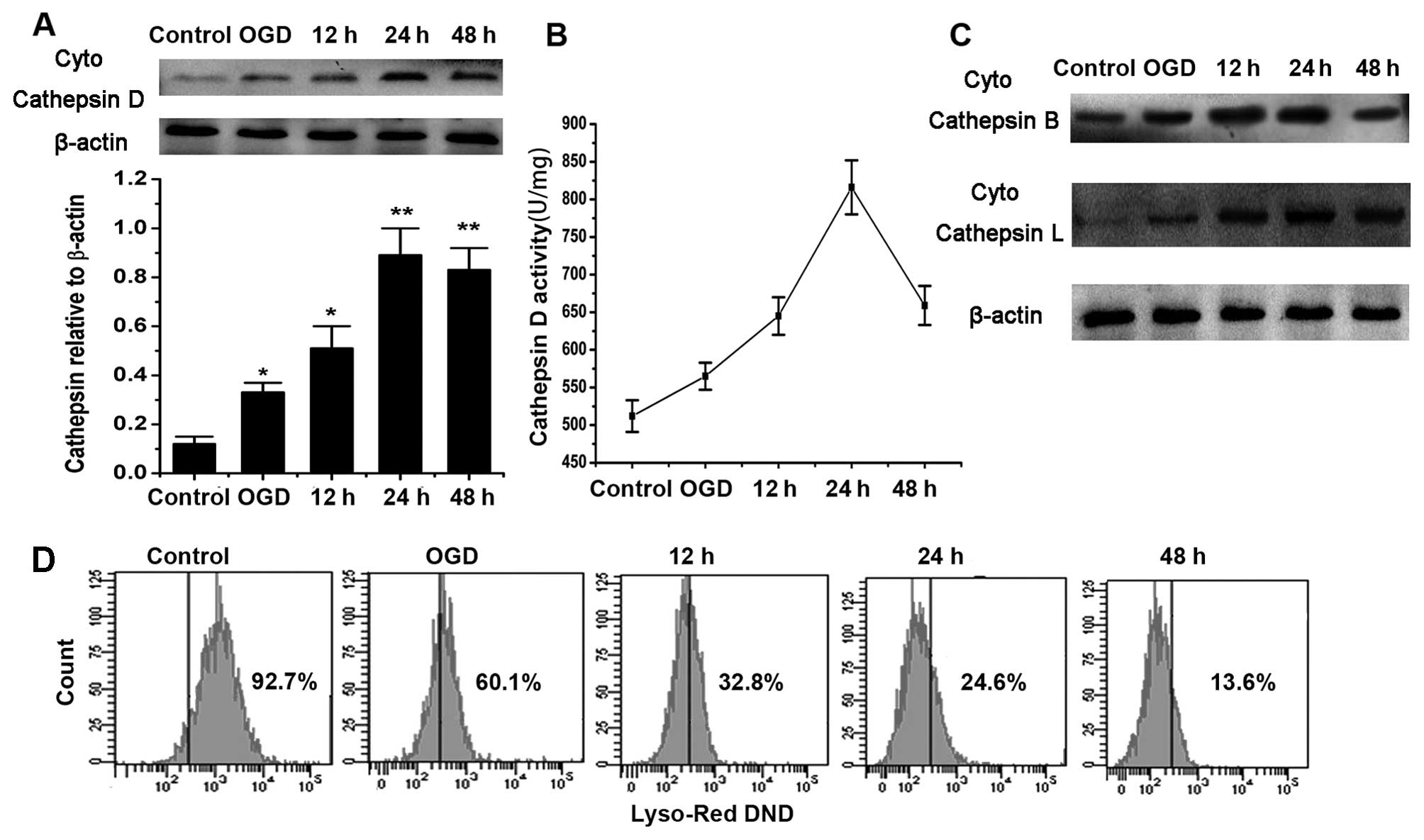
OGD/R induces Cat D upregulation and
lysosomal dysfunction
As shown in Fig. 2A
and B, the active cytosolic levels and the activity of Cat D
significantly increased after 12 h of reperfusion and peaked at 24
h following the reoxygenation compared with the control cells.
Furthermore, we examined other Cat proteases and the change in
acidic conditions in lysosomes at various time points after OGD/R.
The results showed that Cat B and L were also released from
lysosomes in a similar manner to Cat D (Fig. 2C). Moreover, OGD/R disturbed the
innate acidic conditions of lysosomes in a time-dependent manner
and this was illustrated by LysoTracker labeling (Fig. 2D).
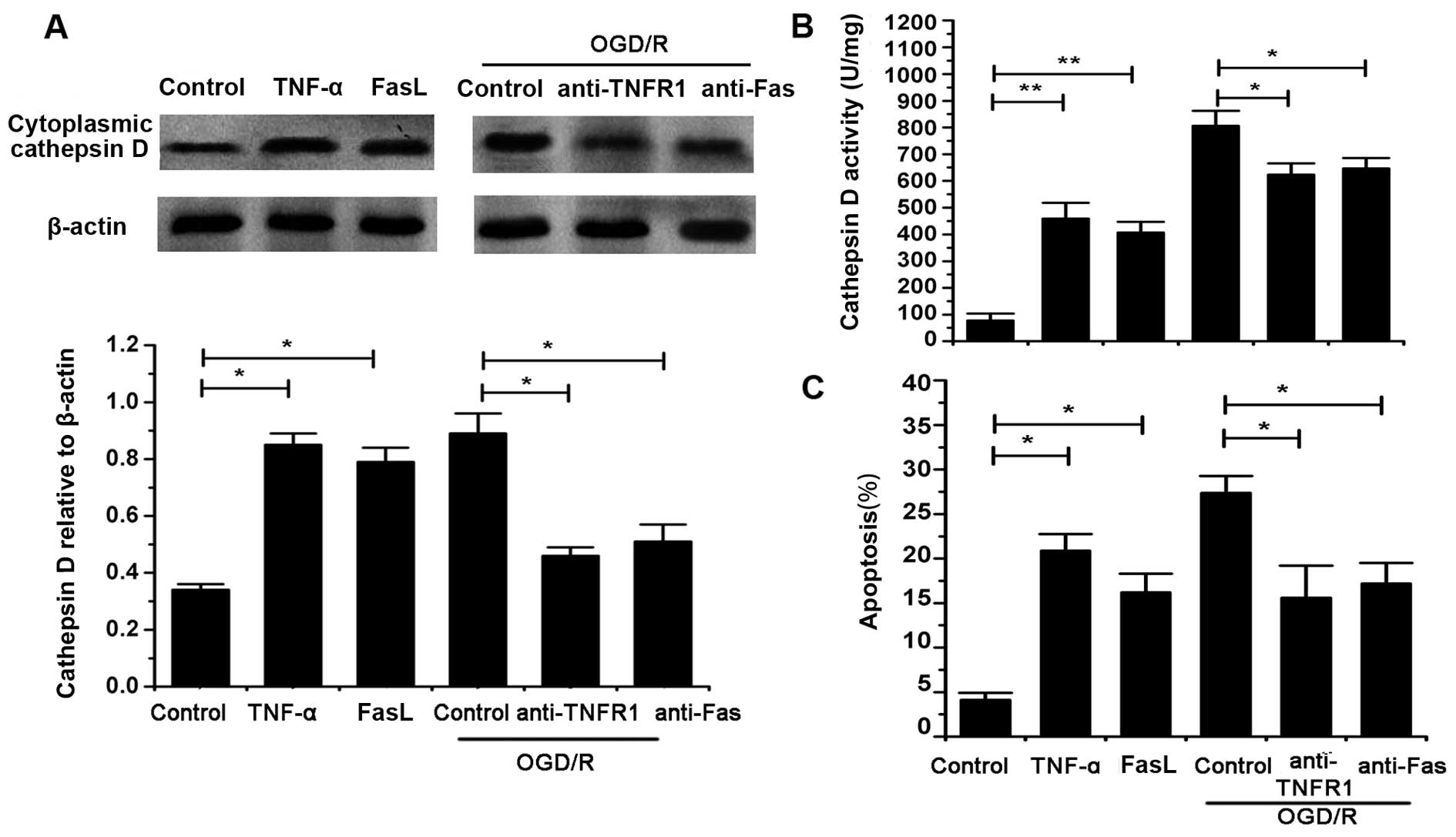
TNF-α and FasL mediate Cat D upregulation
and cell apoptosis
It was shown that TNF-α and FasL significantly
increased the cytosolic level and activity of Cat D in astrocytes
without OGD/R exposure (P<0.05). Moreover, we identified that
the inhibition of TNFR1 and Fas using specific antibodies, markedly
reversed the OGD/R-induced changes in Cat D levels and activity
(P<0.05) (Fig. 3A and B).
Moreover, cell apoptosis was evaluated with or without OGD/R injury
in the presence and absence of TNF-α and FasL as well as the
antibodies against TNFR1 and Fas. We found that both TNF-α and FasL
significantly increased the apoptosis of naïve cells whereas TNFR1
and Fas blocking antibodies inhibited the cell death induced by
OGD/R (P<0.05) (Fig. 3C).
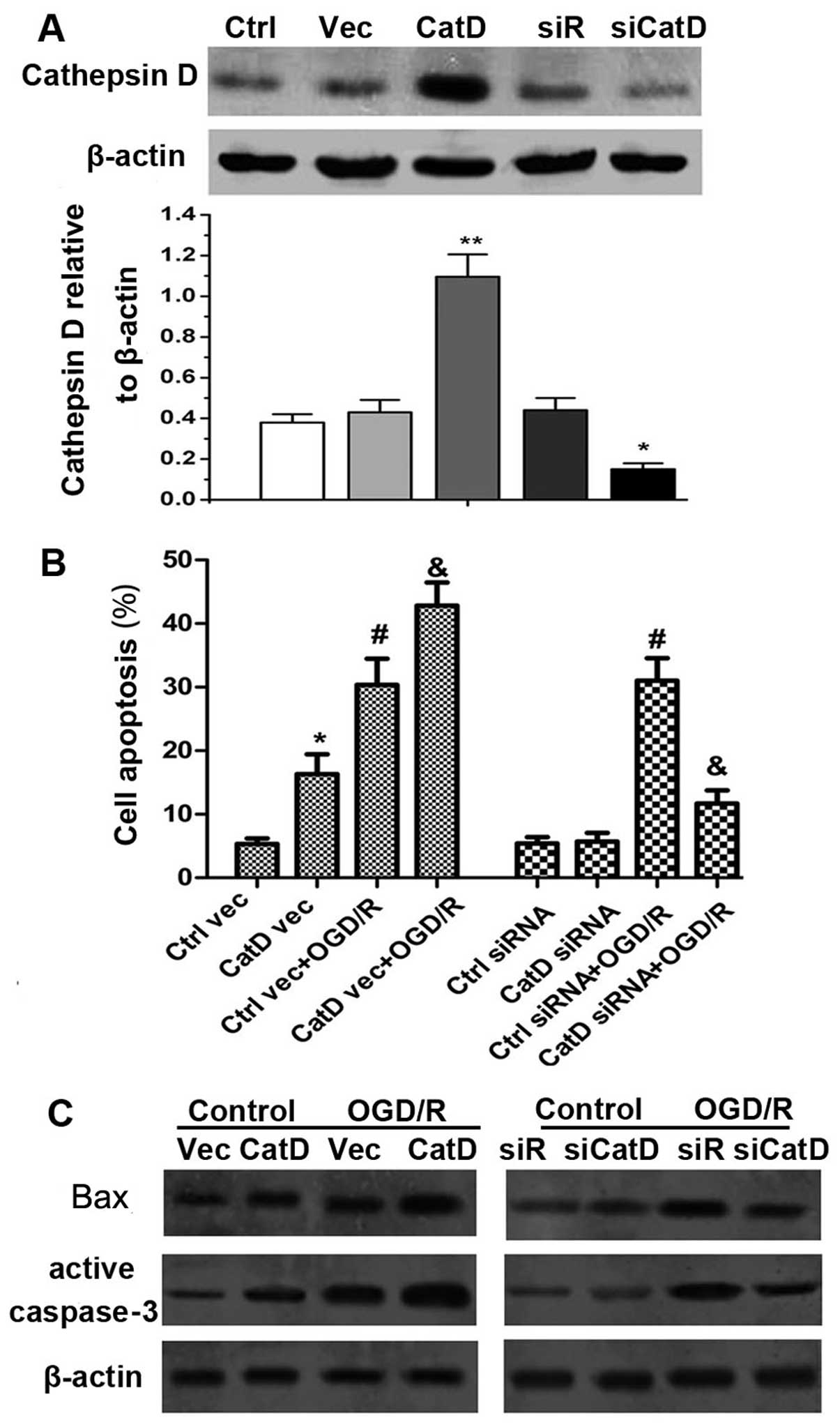
OGD/R-induced cell death associated with
Cat D is caspase-dependent
Cat D-overexpressing vector and siRNA were created
and introduced into astrocytes and the overall expression of mature
Cat D was examined as shown in Fig.
4A. After 24 h of reperfusion, cell death was evaluated. It was
shown that Cat D overexpression enhanced cell apoptosis whereas Cat
D inhibition inhibited this effect with OGD/R exposure (P<0.05)
(Fig. 4B). In order to identify
the underlying mechanism responsible for Cat D-mediated cell death,
we examined the levels of apoptogenic proteins, namely Bax and
caspase-3. The results showed that after 24 h of reperfusion, Cat D
overexpression significantly increased the expression of Bax and
caspase-3 in naïve cells and following OGD/R injury, whereas Cat D
silencing reduced the expression of Bax and caspase-3 (Fig. 4C).
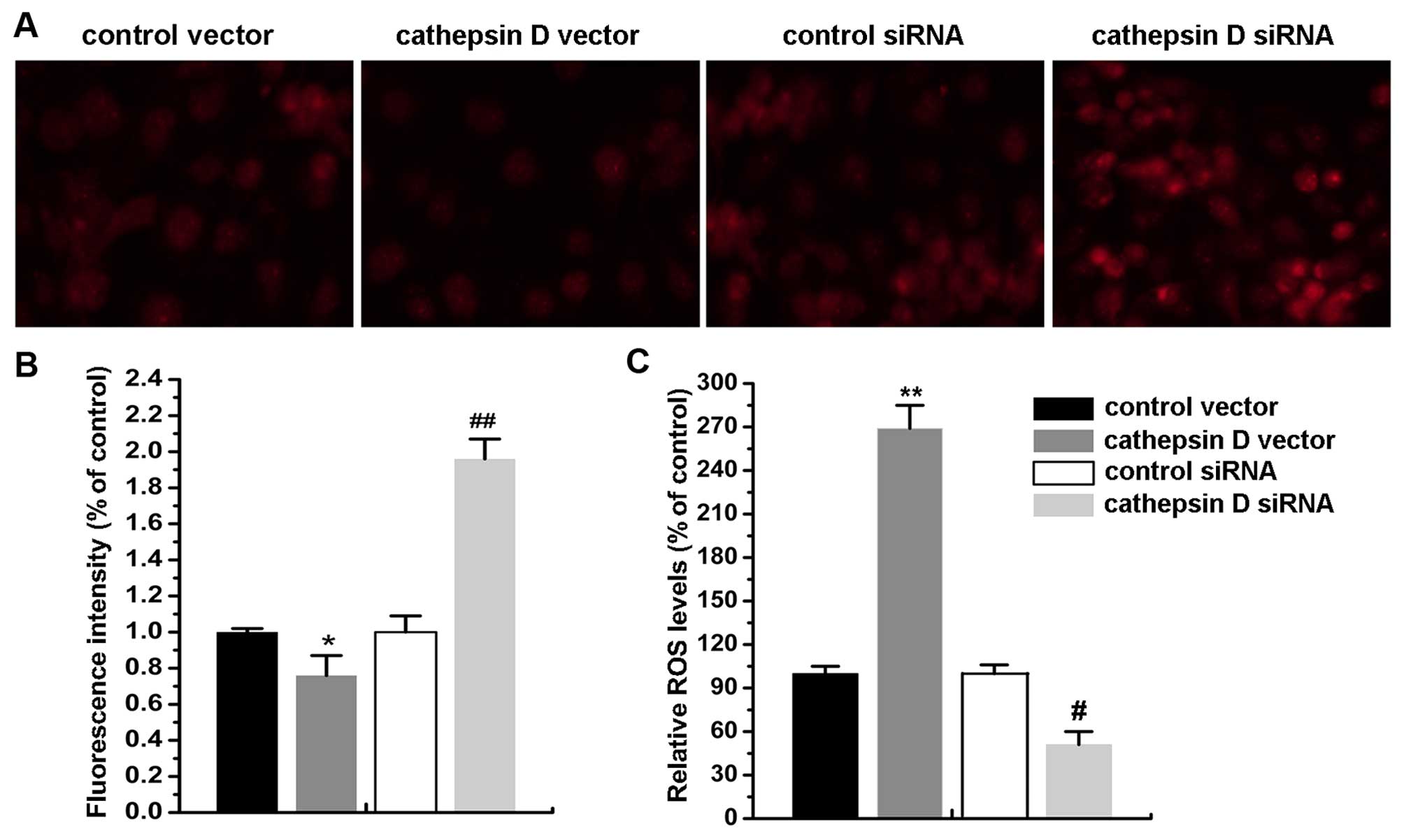
Cat D upregulation disrupts
ΔΨm and induces ROS production
ΔΨm disturbance is an early feature of
apoptosis which is indicative of mitochondrial dysfunction and the
loss of membrane integrity. Cat D overexpression resulted in a
profound decrease in ΔΨm following reperfusion at 24 h
compared with the control. This was accompanied by a significant
increase in the production of ROS, and this decrease lasted for 24
h. Simultaneously, Cat D silencing markedly reversed these effects
(P<0.05) (Fig. 5A and B).
Discussion
Preventing astrocytes from I/R-induced damage is
critical for maintaining the survival and function of neurons. In
the current study, we demonstrated that Cat D expression after
OGD/R contributes to the apoptosis of astrocytes and that elevated
levels of TNF-α and FasL reinforced the expression and activity of
Cat D. Moreover, Cat D disrupted ΔΨm and induced ROS
production, which was accompanied by the upregulation of
apoptogenic proteins, namely Bax and caspase-3.
Lysosomal enzymes, including Cats and some lipid
hydrolases, play key roles in the digestion of substrates, antigen
processing and extracellular matrix degradation. Research has
focused on the roles of these enzymes in the initiation of
apoptosis (15). When secreted
following the rupture of the lysosomal membrane, Cats and lipid
hydrolases may be harmful to the cellular environment, and result
in pathological destruction of cellular structures (16). It has been demonstrated that the
permeabilization of lysosomal membranes, and the resultant leakage
of proteases play important roles in ischemic brain damage, and the
leakage of lysosomal proteins was noted as an early event in the
progression of brain damage (17). Cats are representative lysosomal
proteases involved in ischemic astrocyte injury and their roles in
cell apoptosis and necrosis have been well documented (18). A convincing study in rat models of
permanent middle cerebral artery occlusion (pMCAO) showed that Cat
B and L activation after brain ischemia triggered mitochondrial
tBid-mediated astrocyte death (11). Moreover, Zhang and Li reported
that Cat B activated the Rho kinase-JNK pathway to initiate cell
apoptosis (19). Cat D is a
lysosomal aspartic proteinase which plays roles in the degradation
of proteins and in apoptotic processes induced by oxidative stress,
cytokines, and aging (20). Conus
et al reported that Cat D is involved in initiating the
apoptosis of neutrophils during the resolution of inflammation
(21). In addition, it has been
demonstrated that Cat D overexpression in cancer cells enhances
apoptosis-dependent chemosensitivity (22). Therefore, lysosomal membrane
permeabilization, the leakage of Cats and the subsequent
apoptogenic signals contribute to I/R-induced cell death. In our
study, following OGD/R injury, both cytoplasmic Cat D levels and
the enzymatic activity of Cat D markedly increased with the
duration of reperfusion, and this was accompanied by the increased
production of inflammatory factors, namely TNF-α, FasL and IL-6. A
convincing study has demonstrated that Cat D inhibition by specific
siRNA protected HeLa cells from interferon-γ (IFN-γ) and
Fas/APO-1-induced death (23).
Moreover, Heinrich et al reported that Cat D mediated the
TNF-induced apoptosis of fibroblasts (24). We also found that blocking TNFR1
and Fas suppressed the levels of cytosolic Cat D and the apoptosis
of astrocytes following OGD/R injury, indicating that the
inflammatory factors induced by OGD/R are responsible for the
lysosome leakage and the subsequent cell apoptosis.
It has been proved that excessive ROS production and
oxidative stress overloading cause lysosomal destabilization and
membrane rupture, which in turn lead to propagation of apoptosis
when the lysosomal contents are released into the cytosol (25). The crosstalk between lysosomes and
mitochondria in cell apoptosis has attracted increasing attention
over the last ten years. The release of lysosomal hydrolases may
cause mitochondrial damage through the activation of phospholipases
or pro-apoptotic proteins directly or indirectly. Simultaneously,
lysosomal rupture appears to be a consequence of a transient
oxidative stress of mitochondrial origin that follows the attack by
lysosomal hydrolases and/or phospholipases, creating an amplifying
loop system (26). It has been
found that Cat D activates Bax in T cells and is involved in the
release of cytochrome c from mitochondria in fibroblasts
(27). In this study, we found
that Cat D upregulation induced mitochondrial membrane damage and
the excessive production of ROS. Moreover, the expression of
apoptogenic proteins, namely Bax and caspase-3, increased following
Cat D overexpression. Liu et al reported that lysosome Cat D
exocytosis from glioma cells facilitated their migration and
invasion (28). However, whether
lysosome Cat D is transported outside of the astrocytes following
OGD/R as well as the roles of Cat D exocytosis in the crosstalk
among glia and neurons following I/R injury remain poorly
understood. Therefore, elucidating the roles of Cat D in mediating
I/R damage warrants further investigation.
In conclusion, our study showed that Cat D
participated in the OGD/R-induced damage to astrocytes. OGD/R
induced elevated cell apoptosis and the excessive production of
inflammatory cytokines, namely TNF-α, IL-6 and FasL, which
contributed to the leakage of Cat D from the lysosome. Moreover,
Cat D overexpression exacerbated OGD/R-triggered disturbances in
ΔΨm, ROS production and the expression of apoptogenic
proteins, including Bax and caspase-3.
References
|
1
|
Tu Q, Cao H, Zhong W, Ding B and Tang X:
Atorvastatin protects against cerebral ischemia/reperfusion injury
through anti-inflammatory and antioxidant effects. Neural Regen
Res. 9:268–275. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Luo Y, He Q, Kuang G, Jiang Q and Yang J:
PPAR-alpha and PPAR-beta expression changes in the hippocampus of
rats undergoing global cerebral ischemia/reperfusion due to
PPAR-gamma status. Behav Brain Funct. 10:212014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sun BZ, Chen L, Wu Q, Wang HL, Wei XB,
Xiang YX and Zhang XM: Suppression of inflammatory response by
flurbiprofen following focal cerebral ischemia involves the NF-κB
signaling pathway. Int J Clin Exp Med. 7:3087–3095. 2014.
|
|
4
|
Almeida AS, Queiroga CS, Sousa MF, Alves
PM and Vieira HL: Carbon monoxide modulates apoptosis by
reinforcing oxidative metabolism in astrocytes: role of Bcl-2. J
Biol Chem. 287:10761–10770. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Hong S, Son MR, Yun K, Lee WT, Park KA and
Lee JE: Retroviral expression of human arginine decarboxylase
reduces oxidative stress injury in mouse cortical astrocytes. BMC
Neurosci. 15:992014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun J, Zhu Y, Zhang L and Ma Y: Effects of
xuelian injection on cerebral TNF-α, IL-1β and MMP-9 in rats
experienced focal cerebral ischemia/reperfusion. Int J Clin Exp
Med. 7:2632–2638. 2014.
|
|
7
|
Zhang X, Yan H, Yuan Y, Gao J, Shen Z,
Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, et al: Cerebral
ischemia-reperfusion-induced autophagy protects against neuronal
injury by mitochondrial clearance. Autophagy. 9:1321–1333. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou J, Tan SH, Nicolas V, Bauvy C, Yang
ND, Zhang J, Xue Y, Codogno P and Shen HM: Activation of lysosomal
function in the course of autophagy via mTORC1 suppression and
autophagosome-lysosome fusion. Cell Res. 23:508–523. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qin AP, Zhang HL and Qin ZH: Mechanisms of
lysosomal proteases participating in cerebral ischemia-induced
neuronal death. Neurosci Bull. 24:117–123. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Qin AP, Liu CF, Qin YY, Hong LZ, Xu M,
Yang L, Liu J, Qin ZH and Zhang HL: Autophagy was activated in
injured astrocytes and mildly decreased cell survival following
glucose and oxygen deprivation and focal cerebral ischemia.
Autophagy. 6:738–753. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu M, Yang L, Rong JG, Ni Y, Gu WW, Luo Y,
Ishidoh K, Katunuma N, Li ZS and Zhang HL: Inhibition of cysteine
cathepsin B and L activation in astrocytes contributes to
neuroprotection against cerebral ischemia via blocking the
tBid-mitochondrial apoptotic signaling pathway. Glia. 62:855–880.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Windelborn JA and Lipton P: Lysosomal
release of cathepsins causes ischemic damage in the rat hippocampal
slice and depends on NMDA-mediated calcium influx, arachidonic acid
metabolism, and free radical production. J Neurochem. 106:56–69.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
García Samartino C, Delpino MV, Pott Godoy
C, Di Genaro MS, Pasquevich KA, Zwerdling A, Barrionuevo P, Mathieu
P, Cassataro J, Pitossi F, et al: Brucella abortus induces the
secretion of proinflammatory mediators from glial cells leading to
astrocyte apoptosis. Am J Pathol. 176:1323–1338. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li CY, Li X, Liu SF, Qu WS, Wang W and
Tian DS: Inhibition of mTOR pathway restrains astrocyte
proliferation, migration and production of inflammatory mediators
after oxygen-glucose deprivation and reoxygenation. Neurochem Int.
83–84:9–18. 2015. View Article : Google Scholar
|
|
15
|
Česen MH, Pegan K, Špes A and Turk B:
Lysosomal pathways to cell death and their therapeutic
applications. Exp Cell Res. 318:1245–1251. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lee SJ, Park MH, Kim HJ and Koh JY:
Metallothionein-3 regulates lysosomal function in cultured
astrocytes under both normal and oxidative conditions. Glia.
58:1186–1196. 2010.PubMed/NCBI
|
|
17
|
Lipton P: Lysosomal membrane
permeabilization as a key player in brain ischemic cell death: a
'lysosomocentric' hypothesis for ischemic brain damage. Transl
Stroke Res. 4:672–684. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yamashima T and Oikawa S: The role of
lysosomal rupture in neuronal death. Prog Neurobiol. 89:343–358.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang ZB and Li ZG: Cathepsin B and
phospo-JNK in relation to ongoing apoptosis after transient focal
cerebral ischemia in the rat. Neurochem Res. 37:948–957. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Haendeler J, Popp R, Goy C, Tischler V,
Zeiher AM and Dimmeler S: Cathepsin D and
H2O2 stimulate degradation of thioredoxin-1:
implication for endothelial cell apoptosis. J Biol Chem.
280:42945–42951. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Conus S, Perozzo R, Reinheckel T, Peters
C, Scapozza L, Yousefi S and Simon HU: Caspase-8 is activated by
cathepsin D initiating neutrophil apoptosis during the resolution
of inflammation. J Exp Med. 205:685–698. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Beaujouin M and Liaudet-Coopman E:
Cathepsin D overexpressed by cancer cells can enhance
apoptosis-dependent chemo-sensitivity independently of its
catalytic activity. Hormonal Carcinogenesis V. Li JJ, Li S, Mohla
S, Rochefort H and Maudelonde T: 617. Springer; New York, NY: pp.
453–461. 2008, View Article : Google Scholar
|
|
23
|
Deiss LP, Galinka H, Berissi H, Cohen O
and Kimchi A: Cathepsin D protease mediates programmed cell death
induced by interferon-gamma, Fas/APO-1 and TNF-alpha. EMBO J.
15:3861–3870. 1996.PubMed/NCBI
|
|
24
|
Heinrich M, Neumeyer J, Jakob M, Hallas C,
Tchikov V, Winoto-Morbach S, Wickel M, Schneider-Brachert W,
Trauzold A, Hethke A, et al: Cathepsin D links TNF-induced acid
sphingomyelinase to Bid-mediated caspase-9 and -3 activation. Cell
Death Differ. 11:550–563. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Repnik U and Turk B:
Lysosomal-mitochondrial cross-talk during cell death.
Mitochondrion. 10:662–669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Terman A, Gustafsson B and Brunk UT: The
lysosomal-mitochondrial axis theory of postmitotic aging and cell
death. Chem Biol Interact. 163:29–37. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bidère N, Lorenzo HK, Carmona S, Laforge
M, Harper F, Dumont C and Senik A: Cathepsin D triggers Bax
activation, resulting in selective apoptosis-inducing factor (AIF)
relocation in T lymphocytes entering the early commitment phase to
apoptosis. J Biol Chem. 278:31401–31411. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Liu Y, Zhou Y and Zhu K: Inhibition of
glioma cell lysosome exocytosis inhibits glioma invasion. PLoS One.
7:e459102012. View Article : Google Scholar : PubMed/NCBI
|