Introduction
Angiogenesis is the process whereby new blood
vessels form from pre-existing vessels via microvascular sprouting
(1,2). It is a continuous process, regulated
by various angiogenic factors. Abnormal angiogenesis causes a
number of diseases, such as diabetes, rheumatoid arthritis,
atherosclerosis and malignant tumors (3–5).
Tumor angiogenesis has been shown to play an important role in the
occurrence, development and metastasis of tumors (6,7).
Folkman first attempted to treat tumors by inhibiting angiogenesis
and achieved significant clinical outcomes (8). Since then, the development of
angiogenesis inhibitors has become a key area of study for cancer
treatment.
p43, also known as proEMAPII protein (9,10),
is a cofactor of mammalian aminoacyl-tRNA synthetase (ARS). It is a
34-kDa single-chain protein, containing 312 amino acids (11–14). It harbours an N-terminal domain,
of which the function remains unclear, and a C-terminal domain,
which encodes EMAPII protein (15–19) and is comprised of a
super-secondary structure of an OB-fold and a β-barrel structure
(10). Previous reports have
revealed two primary functions of p43. Firstly, p43 effectively
inhibits angiogenesis. p43 was found to suppress development of new
blood vessels in chick embryo chorioallantoic membrane at a low
dose and inhibit in vitro tubule formation and cell
migration in human microvascular endothelial cells-1 (HMEC-1) cells
(20). Secondly, p43 is secreted
into the extracellular space as a cytokine and plays a role in a
number of cellular processes. For example, p43 was found to prevent
tumor growth by activating tumor necrosis factor (TNF), interleukin
(IL)-8 and other TNFs (20,21). Therefore, p43 may potentially
serve as a clinical drug target for the treatment of solid tumors
such as lung, gastric, prostate and breast cancers as well as
nasopharyngeal carcinoma. However, the underlying mechanisms
responsible for the inhibition of angiogenesis by p43 remain
unclear. Previous findings have suggested that p43 inhibits
angiogenesis by blocking the proliferation of endothelial cells and
inducing the apoptosis of endothelial cells (22). The C-terminus of p43 is reported
to contain a heparin-binding region. By binding to heparin, p43
interacts with the α-subunit of ATP synthase in endothelial cells;
thus, repressing blood vessel growth (22).
Although the structure and function of p43 has been
identified, the roles of p43 in diverse biological processes,
particularly in angiogenesis, remain poorly understood. In the
present study, we examined the effects of p43 in HMEC-1 cells using
Affymetrix Human Genome chips. Our microarray data showed that p43
upregulated multiple cytokines, the majority of which are
associated with the Janus kinase (JAK)-signal transducer and
activator of transcription (STAT) pathway. Interferon-inducible
protein 10 (IP-10), also known as C-X-C motif chemokine 10
(CXCL10), is a member of the chemokine CXC family (23), and it was previously shown to
inhibit angiogenesis and repress tumor growth through the JAK-STAT
signaling pathway in vitro and in vivo (24–27). In the present study, we aimed to
determine the mechanism through which p43 regulates both
angiogenesis in HMEC-1 cells and IP-10 expression as well as the
role of the JAK-STAT signaling pathway.
Materials and methods
Cell lines and reagents
The HMEC-1 cell line was purchased from Shanghai
Sine Pharmaceutical Laboratories (Shanghai, China) and cultured in
MCDB 131 medium (HyClone, Logan, UT, USA) with 10% fetal bovine
serum (FBS) at 37°C and at 5% CO2 in a humidified
incubator. AG490 was purchased from Sigma (St. Louis, MO, USA).
Screening for differentially expressed
genes using microarray analysis
p43 protein was added to HMEC-1 cells at 80%
confluence, and the final p43 concentration was 50 µg/ml
(28). PBS-treated cells served
as controls. Following 8 h of treatment, the cells were washed once
with PBS, followed by the addition of 3 ml TRIzol reagent in order
to the digest cells over a 15-min period. Cell lysates (1 ml) were
transferred to a 1.5 ml RNase-free centrifuge tube. The collected
samples were analyzed by CapitalBio Corporation (Beijing, China)
using the Affymetrix Human Genome U133 Plus 2.0 Array. The
differentially expressed genes in response to p43 treatment of
HMEC-1 cells were analyzed.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
When HMEC-1 cells were at approximately 80%
confluence, p43 protein was added. PBS was used as a control.
Following 8 h of treatment, cells were collected. Total RNAs were
isolated using an RNeasy Plus Mini kit (Qiagen, Hilden, Germany).
cDNA was obtained by reverse transcription using a Transcriptor
First Strand cDNA Synthesis kit (Qiagen). The reaction was
performed using the LightCycler 2.0 Real-Time PCR system (Roche,
Mannheim, Germany). Roche FastStart Universal SYBR-Green Master
(Rox) kits were used. The following PCR program was used:
95°C for 2 min; 45 cycles of 95°C for 2 sec,
57–60°C for 3 sec, 72°C for 11 sec, 80°C for 3
sec and 65°C for 30 sec. The reaction for each sample was
performed in triplicate. GAPDH was used as a control gene. Gene
expression was calculated using the following formula:
2−(ΔCtsample − ΔCtcontrol). The sequences for the PCR
primers are presented in Table
I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Primer
sequences |
|---|
| STAT1 |
5′-CCCTTCTGGCTTTGGATTGA-3′ |
|
5′-GAAGTCAGGTTCGCCTCCGT-3′ |
| GAPDH |
5′-TGAAGGTCGGAGTCAACGGAT-3′ |
|
5′-CTGGAAGATGGTGATGGGATT-3′ |
| IP-10 |
5′-AGGAACCTCCAGTCTCAGCA-3′ |
|
5′-CAAAATTGGCTTGCAGGAAT-3′ |
| SOCS1 |
5′-TTTTCGCCCTTAGCGTGAAGA-3′ |
|
5′-GAGGCAGTCGAAGCTCTCG-3′ |
| SOCS3 |
5′-CAGCATCTCTGTCGGAAGACC-3′ |
|
5′-GCTGGGTGACTTTCTCATAGGAG-3′ |
| TYK2 |
5′-TCGCTGTCATCCTCATTGCT-3′ |
|
5′-ACACTTGGCGGTTCTTTCG-3′ |
| IRF9 |
5′-CATCTTCGACTTCAGAGTCTTCTTC-3′ |
|
5′-TCGGCACAGCCAGGGTT-3′ |
| SMAD2 |
5′-AAGGGGTTCGCCGTTCTC-3′ |
|
5′-TGGCAGCGCAGTTCAGTG-3′ |
| SMAD3 |
5′-CTGCGCTGCCAGTGCTT-3′ |
|
5′-GGTGCTCCCCTTGTTCAGTATCT-3′ |
| IL-6 |
5′-ATGAGGAGACTTGCCTGGTGA-3′ |
|
5′-GGCATTTGTGGTTGGGTCA-3′ |
| ISG15 |
5′-ATGGGCTGGGACCTGACG-3′ |
|
5′-ACCGCTCGGGTGGACAG-3′ |
| STAT2 |
5′-CCCCAAACTCCCCATCG-3′ |
|
5′-CAAAGTCCCAAATCAAACCCT-3′ |
Western blot analysis
When HMEC-1 cells were at approximately 80%
confluence, p43 (Shanghai Sine Pharmaceutical Laboratories) and/or
the JAK inhibitor (AG490) at different concentrations were used to
treat the cells for 8 h. The cells were then lysed in
radioimmunoprecipation buffer (RIPA) supplemented with protease and
phosphatase inhibitors. The cell lysates was centrifuged, and the
resulting supernatant was loaded onto SDS-PAGE gels. Proteins were
resolved on PAGE gels and transferred to PVDF membranes. Membranes
were blocked at room temperature for 1 h. Primary anti-JAK1
(ab125051), anti-phospho-JAK1 (ab138005), anti-STAT1 (ab31369),
anti-phospho-JAK2 (ab32101) and anti-IP-10 (ab9807) antibodies
(AG490) were all 1:1,000 diluted and used to probe membranes for 2
h. The membranes were then washed three times with TBS/Tween-20
(TBST). Goat anti-mouse (ab6789) and goat anti-rabbit (ab6721) IgG
H&L [horseradish peroxidase (HRP)]-conjugated secondary
antibodies (1:4,000) were then used to probe the membranes (at room
temperature for 1 h). β-actin antibody (ab8227) was used as the
loading control. All antibodies were from Abcam, Cambridge, UK.
Subsequently, the membranes were visualized using chemiluminescent
ECL detection reagents (GE Healthcare, Pittsburgh, PA, USA).
Cell migration assays
HMEC-1 cell migration assays were conducted as
previously described (29).
Briefly, Transwell cell culture chambers were pre-coated with 20
µl fibronectin (500 µg/ml). HMEC-1 cells in the
logarithmic growth phase were digested using trypsin, which was
followed by re-suspension with serum-free MCDB 131 medium to a
final density of 5.5×105 cells/ml. The cell suspension
(180 µl) was added to the inner chambers and MCDB 131 medium
supplemented with 10% FBS was added to the outer chambers (540
µl/well). Then, 20 µl p43 protein plus JAK inhibitors
at different concentrations was added to the inner chambers, and 60
µl of the mixture was added to the outer chambers. PBS was
used as a negative control. The system was incubated at 37°C
in 5% CO2 in an incubator for 16-18 h. The xCELLigence
Real Time Cell Analysis (RTCA) DP instrument (ACEA Biosciences, San
Diego, CA, USA) was used to record real-time cell migration. When
the reaction was complete, the media were removed. The cells were
fixed in 90% ethanol and stained with 0.1% crystal violet
(Sigma-Aldrich, Shanghai, China). Migration was observed and images
were captured using a DP instrument (ACEA Biosciences). Surplus dye
was washed off with PBS. Then, 10% acetic acid was applied for cell
extraction for 10 min. The absorbance (OD value) was measured at
595 nm using a microplate reader (Thermo Scientific, Pittsburgh,
PA, USA). The migration inhibition rate was calculated using the
following formula: migration inhibition rate (%) = (OD
valuecontrol group − ODvaluedrug group)/OD
valuecontrol group ×100.
Endothelial cell tubule formation
assays
ECM Matrigel (Sigma) was placed on ice to melt and
then 1:2 diluted with serum-free media. The Matrigel was then added
to pre-cooled 96-well plates (60 µl/well) and allowed to
solidify at 37°C for 30 min. Trypsin digestion was applied
to the cells, which were then resuspended in MCDB 131 medium
supplemented with 10% FBS to attain a final concentration of
3×105 cells/ml. Then, 90 µl of cell suspension
was added to each well. Moreover, the mixtures of p43 plus AG490 at
different concentrations were added to the wells, and PBS was used
as a negative control. The plates were cultured at 37°C in
an incubator with 5% CO2 for 8 h. The fluorescent dye
calcein AM (Thermo Scientific) was employed to stain the cells.
Tubule formation was observed under a fluorescence microscope
(Olympus, Snanghai, China) at ×40 magnification. Five fields were
randomly selected and images were captured.
Enzyme linked immunosorbent assay
(ELISA)
Cells in the logarithmic growth phase were treated
with p43 protein at different concentrations (0, 10, 30, 50, 70 and
100 µg/ml) at 37°C in an incubator with 5%
CO2 for 8 h. PBS was used as a negative control. ELISA
assays were performed using ELISA kits (Mercodia AB, Uppsala,
Sweden) to measure IL-6 and IP-10 protein expression.
Statistical analysis
Data were collected from three or more independent
experiments. Statistical significance was assessed using the
Student's unpaired t-test. For all analyses, SAS software version
8.0 was used. P<0.05 was considered to indicate a statistically
significant difference.
Results
p43 regulates gene expression in HMEC-1
cells
HMEC-1 cells were treated with p43 protein (50
µg/ml) for 8 h. Gene expression was analyzed using the
Affymetrix Human Genome U133 Plus 2.0 microarray. A total of 132
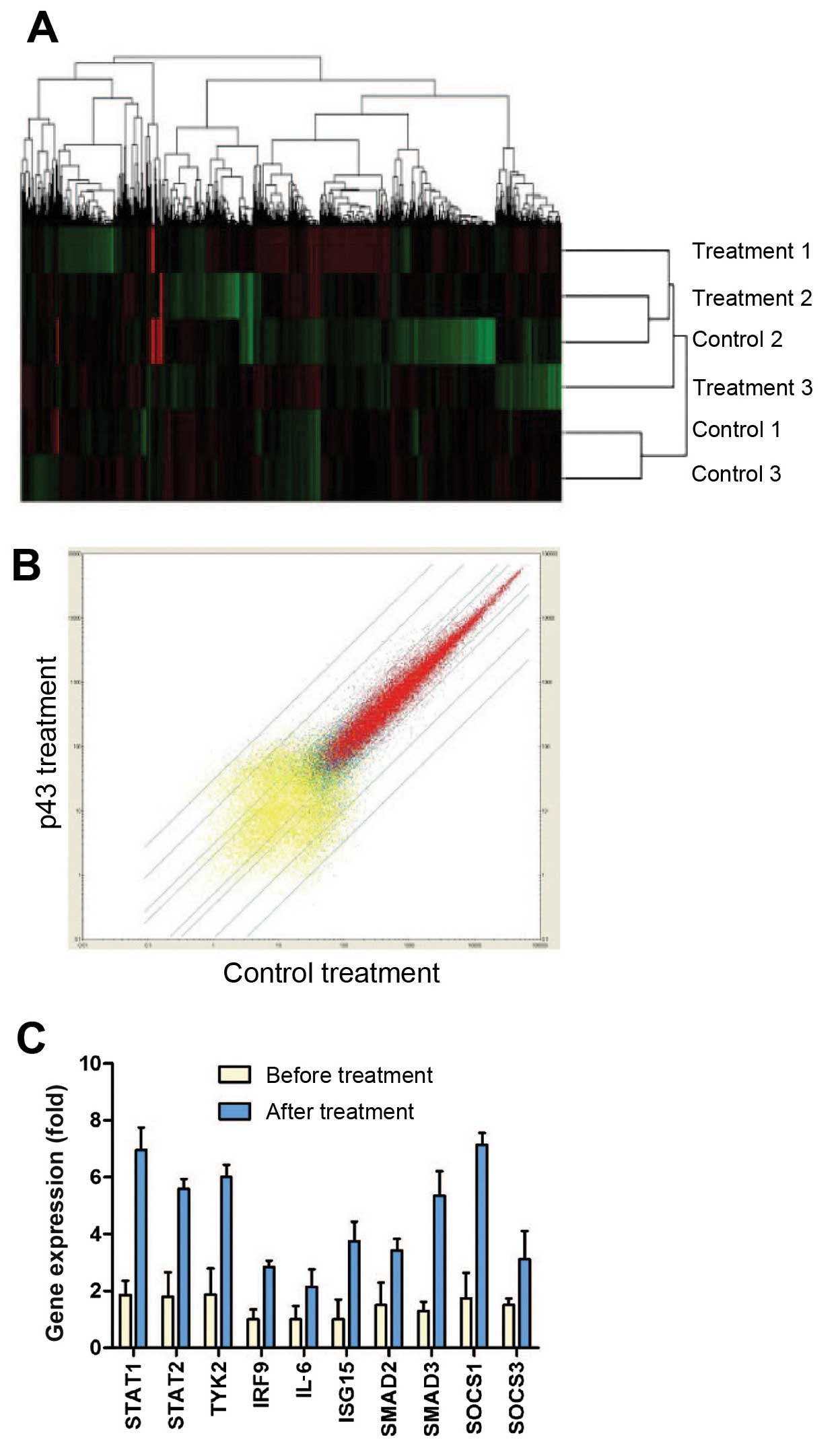
differentially expressed genes were identified (Fig. 1A and B). Among them, 123 genes
were upregulated and 9 genes were downregulated. These genes were
classified according to their functions listed in the Gene Ontology
(GO) classification (Table II).
Among the GO terms, the JAK-STAT signaling pathway was identified
and provided a functional link between p43 and angiogenesis.
Furthermore, we validated the identified genes in the JAK-STAT
pathway by performing RT-qPCR. GAPDH was used as a reference
control. The cells were treated with p43 (50 µg/ml) for 8 h,
and total RNA was extracted for qPCR. The PCR results were
consistent with the microarray data, showing that the identified
genes in the JAK-STAT pathway (STAT1, STAT2, TYK2, IRF9, IL-6,
ISG15, SMAD2, SMAD3, SOCS1 and SOCS3) were induced following p43
treatment (Fig. 1C).
| Table IIUpregulated genes participating in
the JAK-STAT pathway. |
Table II
Upregulated genes participating in
the JAK-STAT pathway.
| Gene | Gene ID | Change ratio |
|---|
| ISG15 | NM_005101 | 3.75 |
| STAT2 | NM_005419 | 3.109 |
| TYK2 | NM_003331 | 3.194 |
| SMAD2 | NM_005901 | 2.28 |
| SOCS1 | NM_003745 | 4.086 |
| IL-6 | NM_000600 | 2.15 |
| STAT1 | NM_007315 | 3.73 |
| IRF9 | NM_006084 | 2.84 |
| SMAD3 | NM_005902 | 4.126 |
| SOCS3 | NM_003955 | 2.061 |
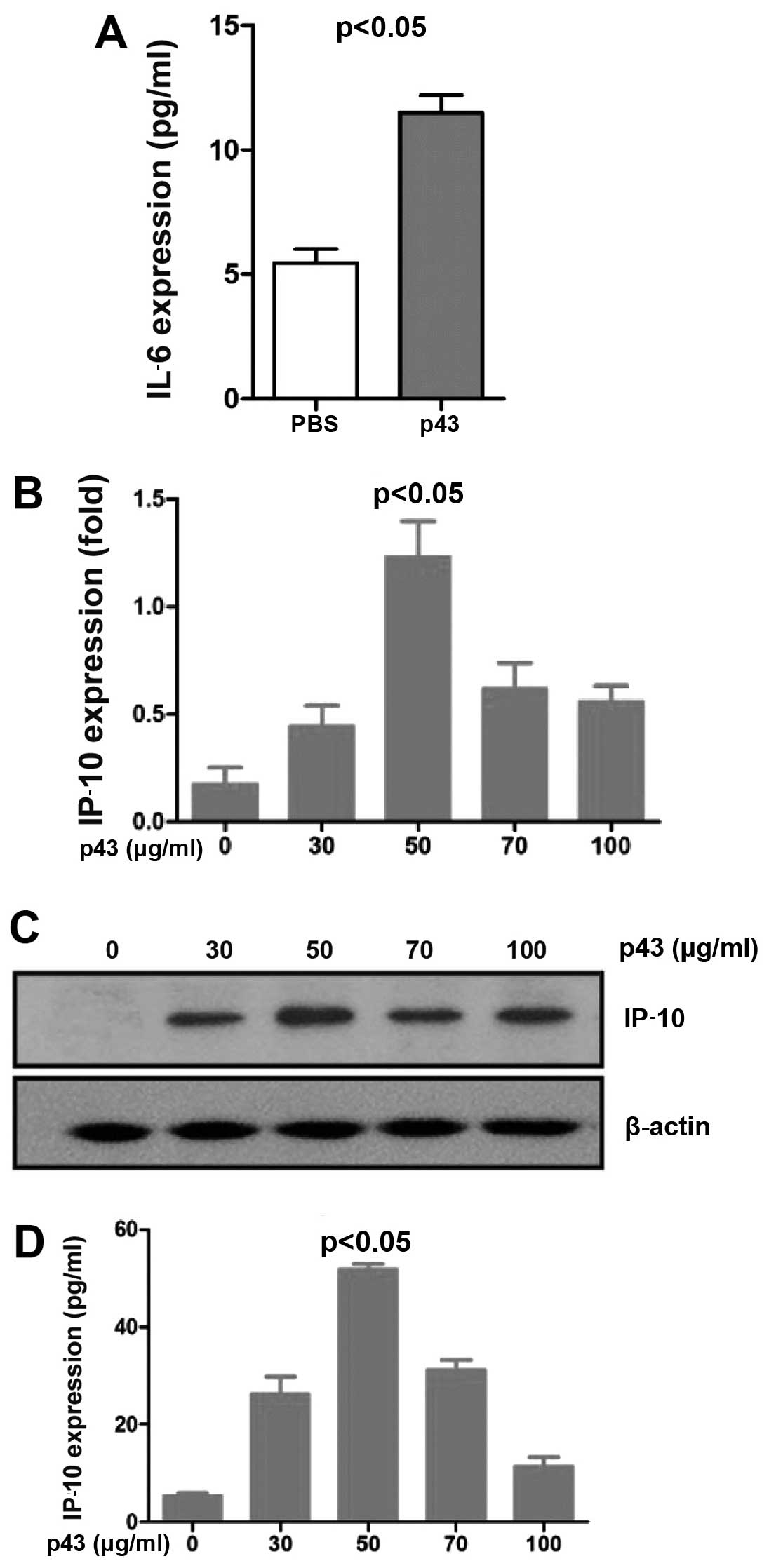
p43 regulation of IP-10 expression
To validate the cellular supernatant from the
treatment, we measured the protein level of IL-6, a secreted
cytokine, by ELISA. The results showed that the IL-6 protein level
was 1-fold higher in the drug-treatment group than in the control
group (PBS) (Fig. 2A). IP-10 was
previously shown to inhibit angiogenesis and repress tumor growth
via the JAK-STAT signaling pathway in vitro and in
vivo (20). We examined
whether p43 affected IP-10 expression. The HMEC-1 cells were
treated with p43 protein at different concentrations (0, 30, 50, 70
and 100 µg/ml) for 8 h. RT-qPCR was performed to evaluate
the mRNA expression of IP-10, and ELISA and western blot analysis
were applied to evaluate the protein expression of IP-10. Total RNA
was extracted from the treated cells and then cDNA was generated by
reverse transcription and used for RT-qPCR. The results suggest
that IP-10 gene expression was apparently enhanced in a
dose-dependent manner (Fig. 2B).
RIPA lysis buffer containing protease and phosphatase inhibitors
was added to the cells. Following centrifugation, the cell
supernatant was collected and used for western blot analysis to
determine IP-10 protein expression. Anti-IP-10 antibodies were used
to detect endogenous IP-10. The results showed that IP-10 bands
appeared at the appropriate molecular weight. IP-10 expression was
augmented by p43 (Fig. 2C). IP-10
ELISA kits were used to further detect IP-10 protein expression.
IP-10 was upregulated, and the highest expression was detected at a
p43 concentration of 50 µg/ml (Fig. 2D).
Expression of key JAK-STAT pathway
factors in p43-treated HMEC-1 cells
It was determined that p43 increased IP-10
expression. To determine whether p43 upregulates IP-10 expression
through the JAK-STAT pathway, we measured the expression of key
JAK-STAT pathway factors in p43-treated HMEC-1 cells.
STAT1 plays an important role in the JAK-STAT
pathway. The HMEC-1 cells were treated with p43 at different
concentrations (0, 30, 50, 70 and 100 µg/ml) for 8 h and
then total RNA was extracted. cDNA was obtained by reverse
transcription and used for qPCR in order to detect the mRNA
expression of STAT1. The RT-qPCR results indicated that p43
augmented the mRNA expression of STAT1 in a dose-dependent manner
(Fig. 3A).
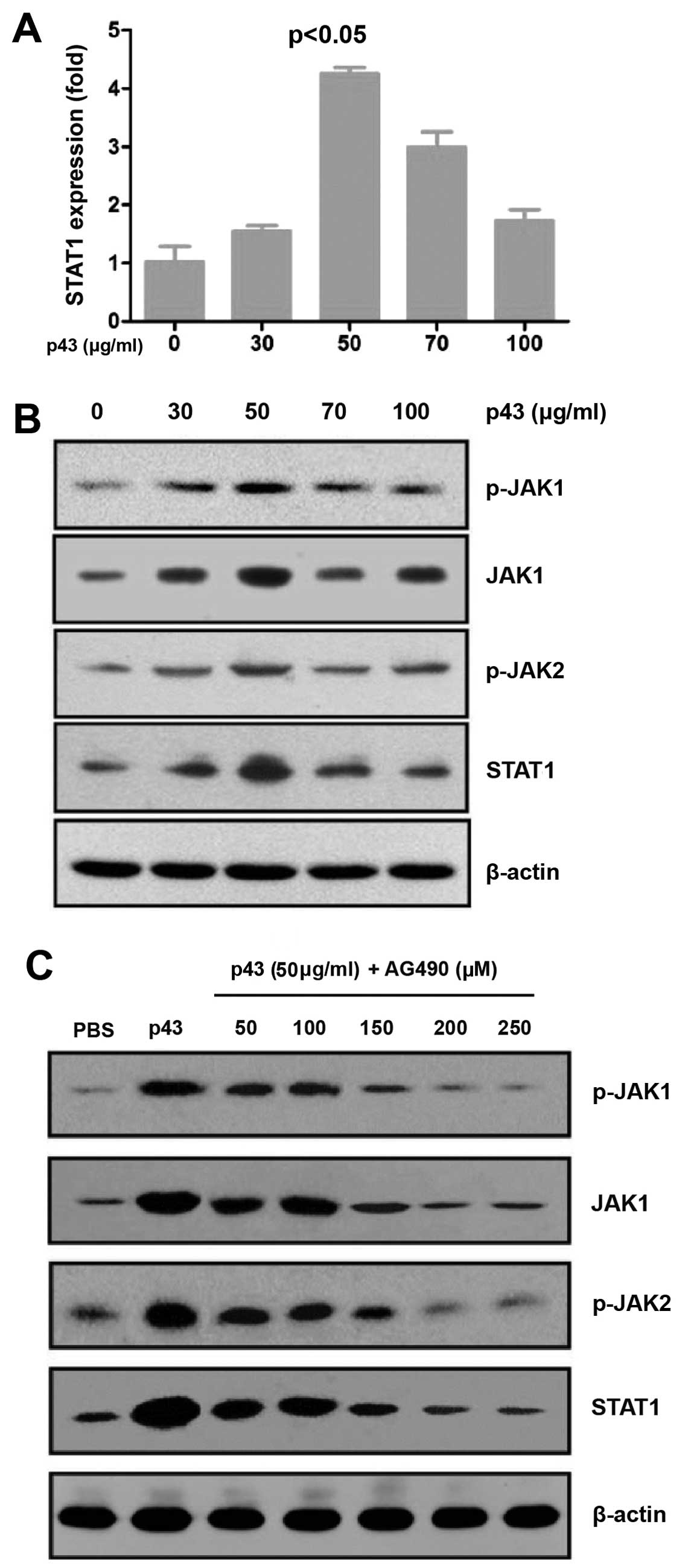 | Figure 3p43 mediates the JAK-STAT signaling
pathway in human microvascular endothelial cells-1 (HMEC-1) cells.
(A) p43 regulates the mRNA expression of STAT1. HMEC-1 cells were
treated with p43 protein at different concentrations (0, 30, 50, 70
and 100 µg/ml) for 8 h. The mRNA expression of STAT1 was
determined by quantitative PCR. (B) p43 regulates key regulators in
the JAK-STAT signaling pathway. The protein expression of JAK1,
STAT1, phosphorylated (p-)JAK1 and p-JAK2 were evaluated by western
blot analysis. (C) AG490 inhibits the effects of p43 in HMEC-1
cells. HMEC-1 cells were treated with PBS (negative control), 50
µg/ml p43 (positive control), or p43 (50 µg/ml) +
AG490 at 50, 100, 150, 200, 250 and 300 µM, for 8 h. The
protein levels of JAK1, STAT1, p-JAK1 and p-JAK2 were measured by
western blot analysis. |
The HMEC-1 cells were treated with p43 at different
concentrations (0, 30, 50, 70 and 100 µg/ml) for 8 h. RIPA
lysis buffer supplemented with protease and phosphatase inhibitors
was added to the cells. Following centrifugation, the cell
supernatant was collected and used for western blot analysis to
determine the protein levels of JAK1, STAT1, phosphorylated
(p-)JAK1 and p-JAK2. All proteins were detected at the appropriate
molecular weights (Fig. 3B).
Therefore, p43 may function through the JAK-STAT pathway, which
supports our hypothesis.
AG490 inhibits the effects of p43 protein
on the key factors in the JAK-STAT pathway
AG490 is a JAK inhibitor, which blocks the JAK-STAT
pathway. We showed that p43 may function through the JAK-STAT
pathway; therefore we hypothesized that AG490 inhibits the effects
of p43. We added p43 protein (50 µg/ml) to 80% confluent
HMEC-1 cells. The cells were also treated with AG490 at different
concentrations (0, 50, 100, 150, 200 and 250 µM). PBS was
used as a negative control. The cells were cultured at 37°C
in 5% CO2 for 8 h. RIPA lysis buffer supplemented with
protease and phosphatase inhibitors was added to the cells.
Following centrifugation, the cell supernatant was collected and
used for western blot analysis to determine the protein levels of
JAK1, STAT1, p-JAK1 and p-JAK2. The results showed that the
expression of the above-mentioned proteins was reduced in the
presence of AG490; thus, suggesting AG490 inhibits the effect of
p43 on these proteins (Fig.
3C).
AG490 suppresses the inhibitory effect of
p43 on tubule formation and the migration of HMEC-1 cells
AG490 is an inhibitor of the JAK-STAT pathway,
specifically suppressing JAK. To further confirm that p43 increases
IP-10 through the JAK-STAT pathway, we performed a tubule formation
assay and cell migration experiments.
HMEC-1 cells at an appropriate stage are capable of
forming a complete lumen on Matrigel. The HMEC-1 cells were treated
with PBS (negative control), p43 or p43+ AG490 for 8 h. The
fluorescent dye calcein AM was used to stain the cells. Tubule
formation was observed under a fluorescent microscope and images
were captured (Fig. 4A). The
results showed that p43 significantly inhibited tubule formation in
the HMEC-1 cells. AG490 minimally affected tubule formation at
concentrations of 25, 50, 75 and 100 µM; however, AG490
markedly induced cell death and decreased tubule formation at a
concentration of 150 µM (data not shown). The cells treated
with both p43 and AG490 formed more tubules than the cells treated
with p43 alone. In addition, the number of tubules formed was
increased at higher concentrations of AG490. A concentration of
AG490 of 100 µM produced optimal effects, forming a similar
amount of tubules compared to the negative control. The findings
indicate that p43 functions through the JAK-STAT pathway at least
in the range of the tested AG490 concentrations. The higher the
AG490 concentration, the weaker the inhibitory effect of p43 on
HMEC-1 cell tubule formation. This finding suggests that AG490
suppresses the inhibitory effect of p43 on tubule formation in
HMEC-1 cells, and for this effect to occur, the optimal
concentration of AG490 is 100 µM.
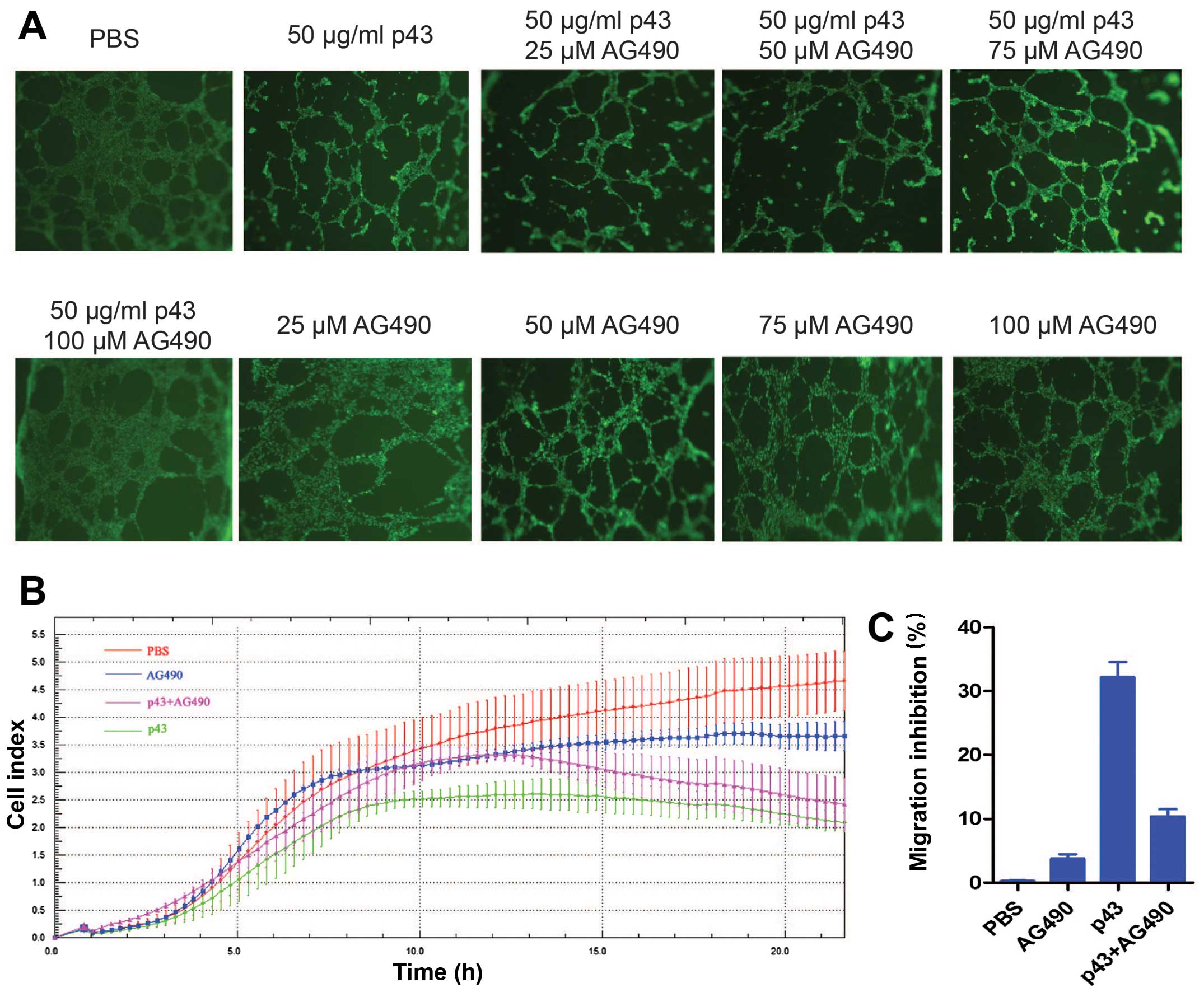 | Figure 4AG490 suppresses the inhibitory
effect of p43 on tubule formation and the migration of HMEC-1
cells. (A) p43 inhibition affects tubule formation in human
microvascular endothelial cells-1 (HMEC-1) cells. HMEC-1 cells were
treated with PBS (negative control), 50 µg/ml p43, 50
µg/ml p43+25 µM AG490, 50 µg/ml p43+50
µM AG490, 50 µg/ml p43+75 µM AG490, 50
µg/ml p43+100 µM AG490, 25 µM AG490, 50
µM AG490, 75 µM AG490 and 100 µM AG490. Tubule
formation was measured after 8 h of treatment. Five fields were
randomly selected. Each treatment was performed in triplicate.
Calcein AM was used to stain cells. The morphology was observed
under a fluorescent microscope (×40 magnification). (B) p43
inhibits the migration of HMEC-1 cells. HMEC-1 cells were treated
with PBS (negative control), 100 µM AG490, 50 µg/ml
p43 (positive control), or p43 (50 µg/ml) + AG490 (100
µM) for 16-18 h. The migration of HMEC-1 cells was measured
by real-time monitoring. The data are representative of the results
from three independent experiments. Error bars indicate standard
deviations; *P<0.05. (C) Inhibitory effects of p43 on
the migration of HMEC-1 cells. |
Cell migration was measured by performing Transwell
migration assays (30). As the
results of the tubule formation experiment suggested that the
optimal concentration of AG490 was 100 µM, we used the same
concentration for cell migration experiments. The HMEC-1 cells in
the logarithmic growth phase were treated with PBS, AG490 (100
µM), p43 (50 µg/ml), or p43 (50 µg/ml) + AG490
(100 µM) at 37°C in 5% CO2 for 16–18 h.
The xCELLigence RTCA DP instrument was used to monitor cell
migration (Fig. 4B). Crystal
violet was used for staining and acetic acid was used for
extraction. The absorbance was measured at 595 nm using a
microplate reader and the migration inhibition rate was calculated
as described in the Materials & methods (Fig. 4C). We determined that AG490 (100
µM) alone had no impact on cell migration; however, p43 (50
µg/ml) significantly inhibited the migration of HMEC-1
cells. The inhibitory effect of p43 on the migration of HMEC-1
cells was reduced in the presence of AG490, suggesting that AG490
decreased the inhibitory effect of p43 on migration. Statistical
analysis showed that the differences were significant
(P<0.05).
Discussion
Angiogenesis is a complex and continuous process. It
is the result of the proliferation, migration and remodeling of
vascular endothelial cells. The inhibition of tumor angiogenesis
has been a novel approach for the treatment of cancer. It is known
that proEMAPII/p43 is a precursor of EMAPII, which is the active
polypeptide in endothelial monocytes. p43 is a cofactor of
mammalian ARS and it was first identified by Quevillon et al
in 1997 (9). The p43 protein has
multiple biological functions. Previous studies have shown that p43
is also an effective angiogenesis inhibitor (30) exhibiting strong antitumor activity
in vitro and in vivo (31). Therefore, the p43 protein is a
potential drug target for the treatment of solid tumors, such as
lung, gastric, prostate and breast cancers as well as
nasopharyngeal carcinoma.
Some studies have revealed the structure and
function of the p43 protein; however, the detailed roles of p43 and
the related mechanisms remain undefined. Furthermore, the p43
receptors remain unknown. The mechanism whereby p43 protein
inhibits angiogenesis also remains unknown. Thus, we treated HMEC-1
cells with 50 µg/ml p43 protein for 8 h. The Affymetrix
Human Genome microarray was applied to analyze differential gene
expression. The data showed that p43 regulated many cytokines. The
majority of the factors upregulated by p43 are associated with the
JAK-STAT pathway. IP-10, also known as CXCL10, is a member of the
chemokine CXC family (24). It is
activated by the JAK-STAT pathway.
IP-10 significantly inhibits angiogenesis in
vitro, which is consistent with the role of p43. Therefore,
IP-10 was selected as a candidate gene. We performed experiments to
reveal the associations among p43, IP-10 and the JAK-STAT pathway.
Firstly, quantitative PCR was used to detect the mRNA expression of
IP-10 in p43-treated HMEC-1 cells, and ELISA as well as western
blot analysis were applied to determine the protein expression of
IP-10. The results indicated that p43 increased IP-10 expression in
a dose-dependent manner. At a p43 protein concentration of 50
µg/ml, the expression of IP-10 was the highest.
IP-10 functions through the JAK-STAT pathway;
therefore, we examined this pathway to find possible mechanisms
responsible for the augmentation of IP-10 levels. The JAK-STAT
pathway (32) consists of two
main families: the JAK family of tyrosine kinases and the STAT
family. The STAT family is comprised of transcription factors,
which play critical roles in the JAK-STAT pathway. The binding of
cytokines to their specific receptors on the cell surface activates
JAK proteins, which are phosphorylated and form dimers. Activated
JAKs subsequently activate STATs through particular domains.
Functional STATs are transferred into the nucleus to regulate
target gene expression. Recent studies have shown that STAT
proteins are important regulators of the signaling pathway
controlling tumor development (33–36). Various products of cancer genes
may continuously activate specific STAT proteins, which play
crucial roles in the occurrence of tumors. Therefore, we examined
STAT1 expression using quantitative PCR. Western blot analysis was
adopted to determine the protein levels of JAK1, STAT1, p-JAK1 and
p-JAK2. The results showed that p43 protein upregulated the
expression of the above-mentioned genes. They suggest that p43 may
increase IP-10 expression through the JAK-STAT pathway; this
finding requires confirmation by further studies.
To confirm our hypothesis, we treated cells with p43
as well as an inhibitor of the JAK-STAT pathway and the effect of
the inhibitor on the JAK-STAT pathway was detected. AG490 and
WP1022 are two common inhibitors of the JAK-STAT pathway,
specifically repressing JAK. HMEC-1 cells were treated with p43 (50
µg/ml) + AG490 for 8 h. The inhibitory effect of AG490 on
the p43-treated cells was examined using several approaches.
Western blot analysis showed that AG490 prevented the
phosphorylation of JAK1 and JAK2 by p43. We then performed in
vitro tubule formation and cell migration experiments in HMEC-1
cells. The tubule formation mediated by p43 was inhibited by AG490.
The cells treated with both AG490 and p43 formed more tubules than
cells treated with p43 alone. Moreover, the migration inhibition
rate was reduced in the presence of the inhibitor. These findings
indicate that AG490 inhibits the phosphorylation of JAK1 and JAK2
by p43 and blocks the JAK-STAT pathway. The results suggest that
p43 inhibits angiogenesis by increasing IP-10 expression through
the JAK-STAT pathway.
Our findings revealed that the p43 protein inhibits
angiogenesis mainly through the JAK-STAT pathway. However, whether
the JAK-STAT pathway is the only pathway participating in the
p43-mediated inhibition of angiogenesis remains unknown. In
addition, the p43 receptors remain unclear. These issues warrant
further investigation.
References
|
1
|
Bdolah Y, Sukhatme VP and Karumanchi SA:
Angiogenic imbalance in the pathophysiology of preeclampsia: newer
insights. Semin Nephrol. 24:548–556. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bussolino F, Mantovani A and Persico G:
Molecular mechanisms of blood vessel formation. Trends Biochem Sci.
22:251–256. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bouck N, Stellmach V and Hsu SC: How
tumors become angiogenic. Adv Cancer Res. 69:135–174. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Carmeliet P: Mechanisms of angiogenesis
and arteriogenesis. Nat Med. 6:389–395. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Goldmann E: The growth of malignant
disease in man and the lower animals, with special reference to the
vascular system. Proc R Soc Med. 1(Surg Sect): 1–13.
1908.PubMed/NCBI
|
|
7
|
Algire GH and Legallais FY: Vascular
reactions of normal and malignant tissues in vivo. IV. The effect
of peripheral hypotension on transplanted tumors. J Natl Cancer
Inst. 12:399–421. 1951.PubMed/NCBI
|
|
8
|
Folkman J: Tumor angiogenesis: therapeutic
implications. N Engl J Med. 285:1182–1186. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Quevillon S, Agou F, Robinson JC and
Mirande M: The p43 component of the mammalian multi-synthetase
complex is likely to be the precursor of the endothelial
monocyte-activating polypeptide II cytokine. J Biol Chem.
272:32573–32579. 1997. View Article : Google Scholar
|
|
10
|
Renault L, Kerjan P, Pasqualato S,
Ménétrey J, Robinson JC, Kawaguchi S, Vassylyev DG, Yokoyama S,
Mirande M and Cherfils J: Structure of the EMAPII domain of human
aminoacyl-tRNA synthetase complex reveals evolutionary dimer
mimicry. EMBO J. 20:570–578. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Otani A, Slike BM, Dorrell MI, Hood J,
Kinder K, Ewalt KL, Cheresh D, Schimmel P and Friedlander M: A
fragment of human TrpRS as a potent antagonist of ocular
angiogenesis. Proc Natl Acad Sci USA. 99:178–183. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kao J, Fan YG, Haehnel I, Brett J,
Greenberg S, Clauss M, Kayton M, Houck K, Kisiel W, Seljelid R, et
al: A peptide derived from the amino terminus of
endothelial-monocyte-activating polypeptide II modulates
mononuclear and polymorphonuclear leukocyte functions, defines an
apparently novel cellular interaction site, and induces an acute
inflammatory response. J Biol Chem. 269:9774–9782. 1994.PubMed/NCBI
|
|
13
|
Park H, Park SG, Lee JW, Kim T, Kim G, Ko
YG and Kim S: Monocyte cell adhesion induced by a human
aminoacyl-tRNA synthetase-associated factor, p43: identification of
the related adhesion molecules and signal pathways. J Leukoc Biol.
71:223–230. 2002.PubMed/NCBI
|
|
14
|
Norcum MT and Warrington JA: The cytokine
portion of p43 occupies a central position within the eukaryotic
multisynthetase complex. J Biol Chem. 275:17921–17924. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Behrensdorf HA, van de Craen M, Knies UE,
Vandenabeele P and Clauss M: The endothelial monocyte-activating
polypeptide II (EMAP II) is a substrate for caspase-7. FEBS Lett.
466:143–147. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shalak V, Kaminska M, Mitnacht-Kraus R,
Vandenabeele P, Clauss M and Mirande M: The EMAPII cytokine is
released from the mammalian multisynthetase complex after cleavage
of its p43/proEMAPII component. J Biol Chem. 276:23769–23776. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wakasugi K and Schimmel P: Highly
differentiated motifs responsible for two cytokine activities of a
split human tRNA synthetase. J Biol Chem. 274:23155–23159. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rosen HR, Moroz C, Reiner A, Stierer M,
Svec J, Reinerova M, Schemper M and Jakesz R: Expression of p43 in
breast cancer tissue, correlation with prognostic parameters.
Cancer Lett. 67:35–45. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Park H, Park SG, Kim J, Ko YG and Kim S:
Signaling pathways for TNF production induced by human
aminoacyl-tRNA synthetase-associating factor, p43. Cytokine.
20:148–153. 2002. View Article : Google Scholar
|
|
20
|
Park SG, Kang YS, Ahn YH, Lee SH, Kim KR,
Kim KW, Koh GY, Ko YG and Kim S: Dose-dependent biphasic activity
of tRNA synthetase-associating factor, p43, in angiogenesis. J Biol
Chem. 277:45243–45248. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ko YG, Park H, Kim T, Lee JW, Park SG,
Seol W, Kim JE, Lee WH, Kim SH, Park JE and Kim S: A cofactor of
tRNA synthetase, p43, is secreted to up-regulate proinflammatory
genes. J Biol Chem. 276:23028–23033. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chang SY, Park SG, Kim S and Kang CY:
Interaction of the C-terminal domain of p43 and the alpha subunit
of ATP synthase. Its functional implication in endothelial cell
proliferation. J Biol Chem. 277:8388–8394. 2002. View Article : Google Scholar
|
|
23
|
Luster AD, Unkeless JC and Ravetch JV:
Gamma-interferon transcriptionally regulates an early-response gene
containing homology to platelet proteins. Nature. 315:672–676.
1985. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Neville LF, Mathiak G and Bagasra O: The
immunobiology of interferon-gamma inducible protein 10 kD (IP-10):
a novel, pleiotropic member of the C-X-C chemokine superfamily.
Cytokine Growth Factor Rev. 8:207–219. 1997. View Article : Google Scholar
|
|
25
|
Belperio JA, Keane MP, Arenberg DA,
Addison CL, Ehlert JE, Burdick MD and Strieter RM: CXC chemokines
in angiogenesis. J Leukoc Biol. 68:1–8. 2000.PubMed/NCBI
|
|
26
|
Strieter RM, Burdick MD, Gomperts BN,
Belperio JA and Keane MP: CXC chemokines in angiogenesis. Cytokine
Growth Factor Rev. 16:593–609. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rosenkilde MM and Schwartz TW: The
chemokine system - a major regulator of angiogenesis in health and
disease. APMIS. 112:481–495. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xing YH, Liu DT, Tan JJ, Hu LD, Liu G, Fu
XQ and Chen HP: Construction and screening of truncated mutants of
recombinant human anti-angiogenic protein proEMAP/p43. Prog Biochem
Biophys. 41:567–574. 2014.
|
|
29
|
Staton CA, Stribbling SM, Tazzyman S,
Hughes R, Brown NJ and Lewis CE: Current methods for assaying
angiogenesis in vitro and in vivo. Int J Exp Pathol. 85:233–248.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Park SG, Jung KH, Lee JS, Jo YJ, Motegi H,
Kim S and Shiba K: Precursor of pro-apoptotic cytokine modulates
aminoacylation activity of tRNA synthetase. J Biol Chem.
274:16673–16676. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee YS, Han JM, Kang T, Park YI, Kim HM
and Kim S: Antitumor activity of the novel human cytokine AIMP1 in
an in vivo tumor model. Mol Cells. 21:213–217. 2006.PubMed/NCBI
|
|
32
|
Wilks AF and Oates AC: The JAK/STAT
pathway. Cancer Surv. 27:139–163. 1996.PubMed/NCBI
|
|
33
|
Yoshikawa H, Matsubara K, Qian GS, Jackson
P, Groopman JD, Manning JE, Harris CC and Herman JG: SOCS-1, a
negative regulator of the JAK/STAT pathway, is silenced by
methylation in human hepatocellular carcinoma and shows
growth-suppression activity. Nat Genet. 28:29–35. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Lin TS, Mahajan S and Frank DA: STAT
signaling in the pathogenesis and treatment of leukemias. Oncogene.
19:2496–2504. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu CL and Burakoff SJ: Involvement of
proteasomes in regulating Jak-STAT pathways upon interleukin-2
stimulation. J Biol Chem. 272:14017–14020. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guschin D, Rogers N, Briscoe J, Witthuhn
B, Watling D, Horn F, Pellegrini S, Yasukawa K, Heinrich P, Stark
GR, et al: A major role for the protein tyrosine kinase JAK1 in the
JAK/STAT signal transduction pathway in response to interleukin-6.
EMBO J. 14:1421–1429. 1995.PubMed/NCBI
|