Introduction
A hallmark of metabolic syndrome is the expansion of
the visceral adipose tissue, which is under a state of chronic
inflammation (1). Non-alcoholic
fatty liver disease (NAFLD) occurs in patients with components of
metabolic syndrome such as type 2 diabetes mellitus (T2DM),
obesity, hypertension and hyperlipidemia. Histological changes of
NAFLD range over a wide spectrum, extending from simple steatosis
to non-alcoholic steatohepatitis (NASH), liver cirrhosis and liver
failure, and sometimes, even hepatocellular carcinoma (2). In Western countries, the prevalence
of NAFLD in the general population ranges from 15–39% (3,4).
It is considered that the prevalence of NAFLD will continue to
increase. However, effective drug therapy for NAFLD has not yet
been established. Thus, studies aiming at exploring strategies for
the treatment of NAFLD are of utmost importance.
At present, the central pathophysiological problem
in patients afflicted with NAFLD is insulin resistance. The
incretin hormones glucagon-like peptide-1 (GLP-1) and
glucose-dependent insulinotropic polypeptide (GIP) are released
from the gastrointestinal tract in response to a meal (5,6).
GLP-1 regulates plasma glucose levels by inhibiting glucagon
secretion in a glucose-dependent manner, which results in reduced
food intake and delaying gastric emptying. The actions of GLP-1
in vivo are short-lived due to their rapid degradation and
inactivation by the enzyme dipeptidyl peptidase-4 (DPP-4) (7,8).
DPP-4 inhibitors have been demonstrated to improve glycemic
control, in particular postprandial hyperglycemic control, in
patients with T2DM. Several DPP-4 inhibitors have entered the
market and have been reported to improve postprandial hyperglycemia
(9–11). It has previously been reported
that sitagliptin, a DPP-4 inhibitor, prevents the development of
hepatic steatosis in mice (12).
Teneligliptin is an orally available and novel
chemotype prolylthiazolidine-based DPP-4 inhibitor. It was
synthesized in an effort to search for more potent and long-lasting
DPP-4 inhibitors (13). We aimed
to determine the effects of teneligliptin on the development of
NAFLD in ob/ob mice with diet-induced obesity and T2DM.
Materials and methods
Chemicals and diets
The DPP-4 inhibitor, teneligliptin hydro-genbromide
hydrate
(3-[(2S,4S)-4-[4-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl]pyrrolidin-2-yl-carbonyl]
thiazolidine hemipentahydrogenbromide hydrate) (teneligliptin),
used in this study was obtained from Tanabe Mitsubishi Pharma Corp.
(Osaka, Japan). Teneligliptin was administrated orally by premixing
with the high carbohydrate diet (HCD) to a concentration of 0.018%
as this concentration of teneligliptin has been used to treat
patients with T2DM. The HCD (Oriental Bio Service, Kyoto, Japan)
contains 5% of calories from fat, 21% from protein, and 69% 60%
fructose) from complex carbohydrates.
Animals
Obese male (ob/ob) 5-week-old mice were obtained
from Oriental Bio Service. These mice have been extensively used as
a naturally occurring model of hepatic steatosis. These mice are
leptin-deficient as a mutation in the ob gene encoding leptin
transcription prevents its biosynthesis (14).
Experimental design
After weaning, the mice were divided into 4
experimental groups for two purposes: one purpose was to determine
whether teneligliptin can be used as a preventive drug for the
development of NAFLD (experiment for prevention of NAFLD), and the
other was to determine whether teneligliptin can be used as a
treatment drug for NAFLD (experiment for treatment of NAFLD). The
experimental design was as follows: 5-week-old male ob/ob mice,
which develop T2DM and NAFLD by being fed a HCD, were divided into
a group in which they were fed HCD for 8 weeks (n=8) as controls
(control group 1), and another in which they were fed HCD
supplemented with 0.018% teneligliptin for 8 weeks (n=8)
(teneligliptin group 1) (experiment for prevention of NAFLD). In
addition, another 5-week-old male ob/ob mice were divided into a
group in which they were fed HCD for 12 weeks (n=8) as controls
(control group 2), and another group in which they were fed only
HCD for 4 weeks, and the HCD was then supplemented with 0.018%
teneligliptin for 8 weeks (n=8) (teligliptin group 2) (experiment
for treatment of NAFLD). Mice were allowed free access to food,
with a 12-h light/12-h dark cycle under conditions of controlled
temperature (22±1°C) and humidity (50±10%). Food intake was
measured daily, while individual body weight was recorded once a
week. Within each group, 8 mice were fasted overnight prior to
euthanasia. All mice were sacrificed after completing their
respective dietary regimens, and the livers of the individual
animals were weighed. The livers were removed, and part of the
samples were placed in formalin, while the remainder was
snap-frozen and stored at −80°C. All surgical and experimental
procedures were performed according to the guidelines for the care
and use of animals approved by Osaka Medical College, Takatsuki,
Japan.
Assay for plasma hepatic and metabolic
parameters
Blood samples were obtained by cardiac puncture and
separated by centrifugation (12,000 rpm for 15 min) as plasma. The
levels of blood biochemical parameters, including aspartate
aminotransferase (AST), alanine aminotransferase (ALT), glico
albumin (GA), total-cholesterol (T-CHO), glucose and insulin were
measured by a local laboratory that performs clinical analyses
(Oriental Yeast Co. Ltd., Kyoto, Japan).
Assay for hepatic lipid content
Hepatic tissues were homogenized with a Janke and
Kunkel Polytron homogenizer (ULTRA-TURRAX TP18/1051;
IKA-Labortechnik, Staufen, Germany) in buffer (pH 7.4) containing
20 mM Tris HCl, 1 mM EGTA and 2 mM EDTA, and treated with protease
inhibitor (2 µg/ml, leupeptin cocktail). Hepatic tissue
triglyceride (TG) levels and free fatty acid (FFA) levels were
measured by a local laboratory that performs clinical analyses (SRL
Co. Ltd., Tokyo, Japan).
Assay for plasma GLP-1 concentration
Total plasma GLP-1 concentrations were measured
using a GLP-1, active from assay kit-IBL according to the
manufacture's instructions (Immuno-Biological Laboratories Co.,
Ltd., Gunma, Japan).
Assay for DPP-4 activity
To examine the potential of direct inhibitory
activity against DPP-4, 10 μl of plasma was mixed with 90
μl of assay buffer. Assays were reacted for 30 min at room
temperature, and the released AMC was determined fluoromerically
using Fuluorskan Ascent FL (375-nm excitation and 460-nm emissions;
Thermo Fisher Scientific, Waktham, MA, USA). DPP-4 activity was
determined with an AMC standard curve. The 50% inhibitory
concentration against DPP-4 was calculated from the enzyme reaction
curves the SAS system version 8.2 (SAS Institute, Cary, NC,
USA).
Histological analysis of hepatic
tissue
Liver sections were examined blindly from different
lobes of each mouse. Liver tissues were fixed in 10% buffered
formaldehyde, and then embedded in paraffin. A 4-mm-thick section
cut from a paraffin-embedded block was stained with hematoxylin and
eosin (H&E) or Oil Red O (both from Applied Medical Research,
Osaka, Japan). These sections were evaluated for fat content by the
absence of staining. For hepatic steatosis: grade 0, no fat; grade
1, steatosis occupying <33% of hepatic parenchyma; grade 2,
33–66% of the hepatic parenchyma; grade 3, >66% of the hepatic
parenchyma. For inflammatory cell infiltration: grade 0, none;
grade 1, 1–2 foci/field; grade 2, 3–4 foci/field; grade 3, >4
foci/field. For ballooning degeneration of the hepatocytes: grade
0, absent; grade 1, very mild inflammation; grade 2,
mild-to-moderate portal inflammation; grade 3, intraacinar
inflammation and moderate portal inflammation. For hepatic
fibrosis: stage 0, none; stage 1, mild, perisinusoidal; stage 2,
moderate, perisinusoidal fibrosis; stage 3, periportal fibrosis;
stage 4, bridging fibrosis (15).
Glucose tolerance test (GTT)
Mice used in the experiment for prevention of NAFLD
at 9 weeks of age and those in the experiment for the treatment of
NAFLD at 13 weeks of age were fasted overnight. After measuring the
body weight, 20% glucose was injected into each mouse i.p. at 100
μl/10 g body weight. The blood glucose level was measured
with a glucose meter 15-min intervals during a 2-h course. In order
to analyze insulin secretion during GTT, blood was collected 0, 15
and 120 min after glucose injection. The level of insulin was
measured by a local laboratory that does clinical analyses
(Oriental Yeast Co. Ltd.).
Real-time PCR
Tissue specimens were preserved in RNAlater reagent
(Qiagen, Valencia, CA, USA) until the isolation of total RNA. Total
RNA was isolated from the liver tissue using a QIA shredder and an
RNeasy kit (Qiagen). cDNA was prepared using the TaqMan reverse
transcriptase kit (Qiagen). Real-time PCR was performed on total
RNA using the StrataScript First Strand cDNA Synthesis kit and
FullVelocity SYBR-Green qPCR Master Mix (Stratagene, La Jolla, CA,
USA) according to the manufacturer's protocol. Primers for
real-time PCR were designed using Beacon Designer software version
2.12, according to the parameters outlined in the Bio-Rad iCycler
Manual, using reference mRNA sequences accessed through Gene Bank
and as shown in Table I. All
probes used in the TaqMan Gene Expression assays were purchased
from Applied Biosystems (Foster City, CA, USA). PCR reactions were
carried outin the iCycler Thermal Cycler (Bio-Rad Laboratories,
Hercules, CA, USA). PCR products were detected using the iCycler iQ
Real-Time PCR detection system (Bio-Rad Laboratories). The relative
amount of mRNA was calculated by comparative cycle time
determination with ribosomal protein RPL32 as the invariant
control. Gene expression values were calculated based on the ∆∆Ct
method. The results were expressed as a fold increase in expression
relative to the control group.
 | Table IPrimer sequences used for the
real-time polymerase chain reaction. |
Table I
Primer sequences used for the
real-time polymerase chain reaction.
| Gene | Primer sequences
(sense) | Primer sequences
(antisense) |
|---|
| TNF-α |
5′-ACCTTGTTGCCTCCTCTT-3′ |
5′-GTTCAGTGATGTAGCGACAG-3′ |
| IL-1β |
5′-TCCAGGATGAGGACATGAGCAC-3′ |
5′-GAACGTCACACACCAGCAGGTTA-3′ |
| IL-6 |
5′-TTCCTCACTGTGGTCAGA-3′ |
5′-CATTCATATTGTCAGTTCTTCGTA-3′ |
| IFN-γ |
5′-CGGCACAGTCATTGAAAGCCTA-3′ |
5′-GTTGCTGATGGCCTGATTGTC-3′ |
| SREBP-1c |
5′-GGTACCTGCGGGACAGCTTA-3′ |
5′-CCGTGAGCTACCTGGACTGAA-3′ |
| FAS |
5′-TACAGATGGCAGCAAGGA-3′ |
5′-TGATACAGAGAGCAGATGAGT-3′ |
| ChREBP |
5′-TCGTGTAGACAACAAC-3′ |
5′-ATATTGAACCGCCTCT-3′ |
| PPAR-α |
5′-ATGGCAGCAATATCAGAG-3′ |
5′-AGCAGTAAAGTATCATATCAAAG-3′ |
| PPAR-γ |
5′-GAAGACAGAGACAGACAT-3′ |
5′-GCAATCAATAGAAGGAACA-3′ |
| SCD-1 |
5′-CTGGCTGGAGAGTCATCA-3′ |
5′-TAACGAGGACGACAATACAATC-3′ |
| CPT1A |
5′-GACTACTTGCTAACCTCTGT-3′ |
5′-GACACTGGAGACCTGAGA-3′ |
| ACC1 |
5′-ATCATAACTCGTATAACATCCA-3′ |
5′-CAGGTTAAGGCTCAGACT-3′ |
| ACL |
5′-ACCCAGACATGCGAGTGCAG-3′ |
5′-CCGTCCACATTCAGGATAAGATTTG-3′ |
| MCAD |
5′-CGGAGGAACCTGTCTTCA-3′ |
5′-GGCTAAGGACCAATCATTGT-3′ |
| LCAD |
5′-GACATCTGCCTACATCCT-3′ |
5′-TCTCTCCCTGTGTTAATCTT-3′ |
| ECAH |
5′-AGTCTATTCAAGTCACAAGT-3′ |
5′-ATGGCATTCCTCTTCTCT-3′ |
| HACDH |
5′-ATCTTAACCATCACTGTC-3′ |
5′-TAGTAGAGTCAATTCATAGG-3′ |
| bKACTB |
5′-AGGTTGTCACGCTACTCA-3′ |
5′-ATCCCAGTCCCGATACAC-3′ |
| PI3K |
5′-GCAGTTAAGAAGCACA-3′ |
5′-GTATGAAGCAGGAGAG-3′ |
| IRS-1 |
5′-GCCTTCCATATAGTTA-3′ |
5′-GAACCATCATCATCTC-3′ |
| IRS-2 |
5′-CCATCCTTTGCCCACA-3′ |
5′-GTACTGCTGCCTTCAC-3′ |
| AMPK |
5′-AAGCCGACCCAATGACATCA-3′ |
5′-CTTCCTTCGTACACGCAAAT-3′ |
| G6Pase |
5′-GTGGCAGTGGTCGGAGACT-3′ |
5′-ACGGGCGTTGTCCAAAC-3′ |
| PEPCK |
5′-TGCCCGGGTGGAAGGTCGAA-3′ |
5′-TGGGCACATGGTTCCGCGTC-3′ |
Statistical analysis
Data are presented as the means ± standard error of
the mean. Statistical analyses were performed using the Student's
t-test. Values of p<0.05 were considered to indicate
statistically significant differences.
Results
Effect of treatment and diets on body
weight and liver/body weight ratio of mice in each experimental
group
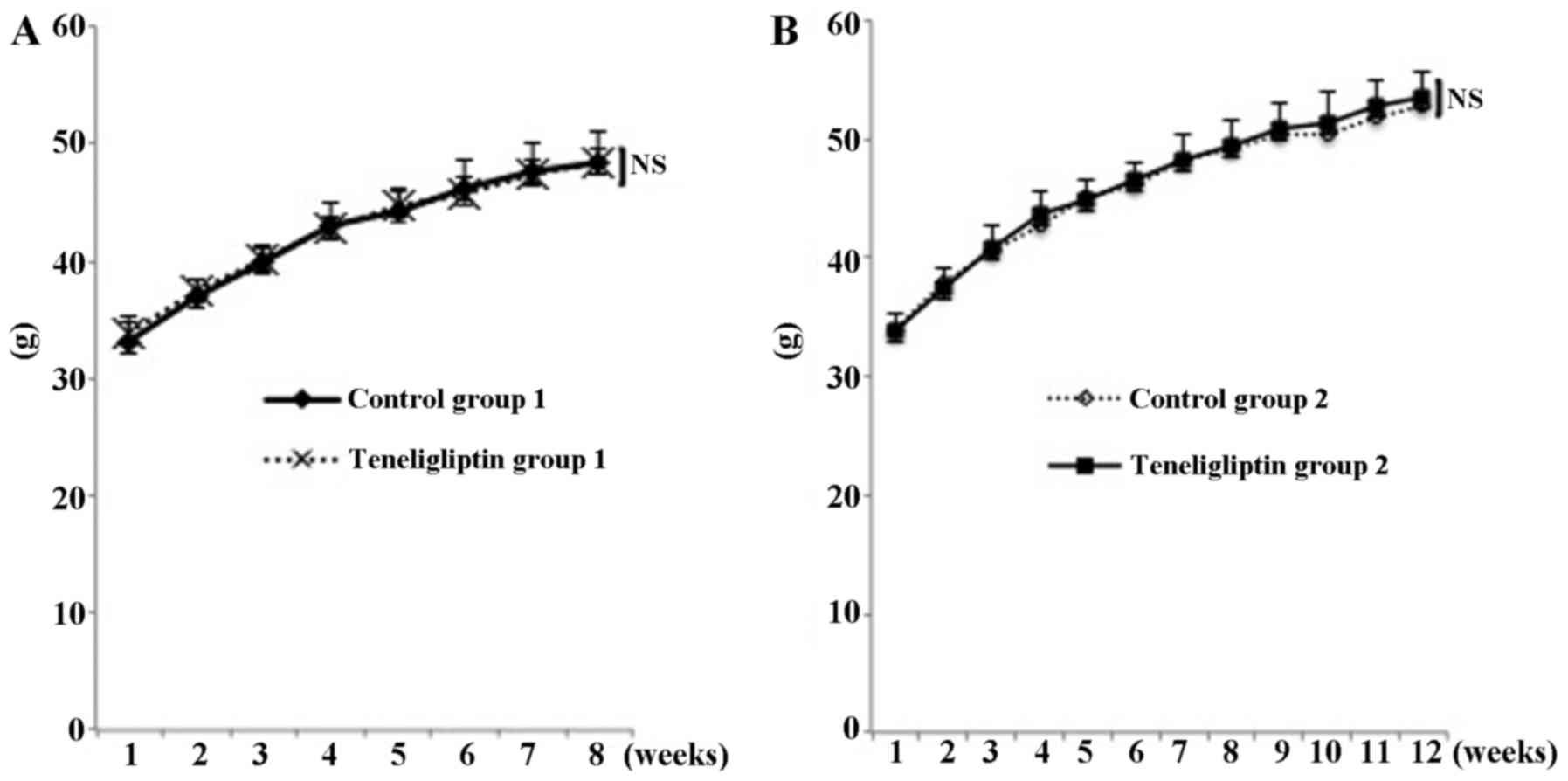
As shown Fig. 1A,
at the end of the experimental period, there was no significant
difference in the weight of the mice in both groups (48.40±2.74 and
48.35±1.15 g in both groups of mice, respectively; p<0.05)
(experiment for prevention of NAFLD). In addition, as shown
Fig. 1B, at the end of
experimental period, there was also no significant difference in
the weight of mice in the both groups (52.75±2.33 and 53.58±2.07 g
in both groups of mice, respectively; p<0.05) (experiment for
treatment of NAFLD). As shown Table
II-A, the ratio of liver weight to body weight was not
significantly different in both groups of mice (5.82±1.10 and
5.80±0.98% in both group of mice, respectively; p<0.05)
(experiment for prevention of NAFLD). In addition, as shown
Table II-B, the ratio of liver
weight to body weight was not significantly difference in both
groups of mice (6.81±1.08 and 6.80±1.08% in both group of mice,
respectively; p<0.05) (experiment for treatment of NAFLD).
| Table IIOutcome of liver/body weight ratio
and biochemical parameters in all mice in both types of
experiments. |
Table II
Outcome of liver/body weight ratio
and biochemical parameters in all mice in both types of
experiments.
A, Outcome of
liver/body weight ratio and biochemical parameters in mice in the
experiment for NAFLD prevention
|
|---|
| Parameter | Control group
1 | Teneligliptin group
1 |
|---|
| Liver/body weight
ratio (%) | 5.82±1.10 | 5.80±0.98 |
| AST (IU/l) | 293.88±93.28 |
183.20±86.71a |
| ALT (IU/l) | 239.24±80.91 |
150.76±81.99a |
| T-CHO (mg/dl) | 216.0±17.0126 |
198.29±12.93a |
| Glucose
(mg/dl) | 314.0±34.98 |
264.33±31.63a |
| Insulin
(ng/ml) | 3.91±3.59 | 1.03±0.47a |
| GA (%) | 2.22±0.16 | 1.72±0.62a |
| Hepatic TG
(mg/dl) | 105.69±4.89 | 86.85±6.61a |
| Hepatic FFA
(μEq/l) | 111.66±5.71 | 82.81±3.93a |
B, Outcome of
liver/body weight ratio and biochemical parameters in mice in the
experiment of the treatment of NAFLD
|
|---|
| Parameter | Teneligliptin group
2 | Control group
2 |
|---|
| Liver/body weight
ratio (%) | 6.81±1.08 | 6.80±1.08 |
| AST (IU/l) | 140.09±61.53 | 125.87±73.80 |
| ALT (IU/l) | 121.03±52.21 |
233.80±70.09b |
| T-CHO (mg/dl) | 227.88±16.97 |
270.83±37.06b |
| Glucose
(mg/dl) | 278.50±31.52 | 313.38±44.14 |
| Insulin
(ng/ml) | 1.24±0.37 | 1.16±0.38 |
| GA (%) | 2.07±0.12 | 1.50±0.71 |
| Hepatic TG
(mg/dl) | 107.71±5.62 | 108.91±5.79 |
| Hepatic FFA
(μEq/l) | 132.66±3.17 | 134.18±3.92 |
Plasma and hepatic biochemical
parameters
To examine whether teneligliptin if used as a
preventive drug for NAFLD affects liver damage and steatosis, we
quantified the plasma levels of AST, ALT, T-CHO hepatic TG and FFA.
The plasma AST (293.88±93.28 vs. 183.2±86.71 IU/l, p<0.05) and
ALT levels (239.24±80.91 vs. 150.76±81.99 IU/l, p<0.05) were
significantly different in the teneligliptin group 1 from the
control group 1 (Table II-A).
Mice fed the diet containing teneligliptin had lower plasma levels
of T-CHO (216.0±1 7.01 vs. 198.29±12.93 mg/dl, p<0.05), a lower
hepatic TG content (105.69±4.89 vs. 86.85±6.61 mg/dl, p<0.05)
and a lower hepatic FFA content (111.66±5.71 vs. 82.81±3.93
μEq/l, p<0.05) (Table
II-A).
Next, to determine whether teneligliptin if used as
a treatment drug for NAFLD affects liver damage and steatosis, we
quantified the plasma levels of AST, ALT, T-CHO hepatic TG and FFA.
The plasma AST (140.09±61.53 vs. 125.87±73.80 IU/l, p<0.05)
levels were not significantly different in both groups. The ALT
levels (121.03±52.21 vs. 233.80±70.09 IU/l, p<0.05) were
significantly different in the teneligliptin group 2 compared to
the control group 2 (Table
II-B). Mice fed the diet containing teneligliptin had higher
plasma levels of T-CHO compared to the HCD-fed mice (227.88±16.97
vs. 270.83±37.06 mg/dl, p<0.05). The hepatic TG content
(107.71±5.62 vs. 108.91±5.79 mg/dl, p<0.05) and hepatic FFA
content (132.66±3.17 vs. 134.18±3.92 μEq/l, p<0.05) were
not significantly different in the both groups (Table II-B).
Plasma glucose, insulin and GA
Mice fed the diet containing teneligliptin as a
preventive drug for NAFLD had lower fasting blood glucose levels
(314.0±34.98 vs. 264. 33±31.63 mg/dl, p<0.05), lower fasting
plasma insulin levels (3.91±3.59 vs. 1.03±0.47 ng/dl, p<0.05)
and a lower level of GA (2.22±0.16 vs. 1.72±0.62%, p<0.05)
(Table II-A). Mice fed the diet
containing teneligliptin as a treatment drug for NAFLD exhibited no
significant difference in fasting blood glucose levels
(278.50±31.52 vs. 313.38±44.14 mg/dl, p<0.05), fasting plasma
insulin levels (1.24±0.37 vs. 1.16±0.38 ng/dl, p<0.05) and the
level of GA (2.07±0.12 vs. 1.50±0.71%, p<0.05), compared with
the mice fed the HCD alone (Table
II-B).
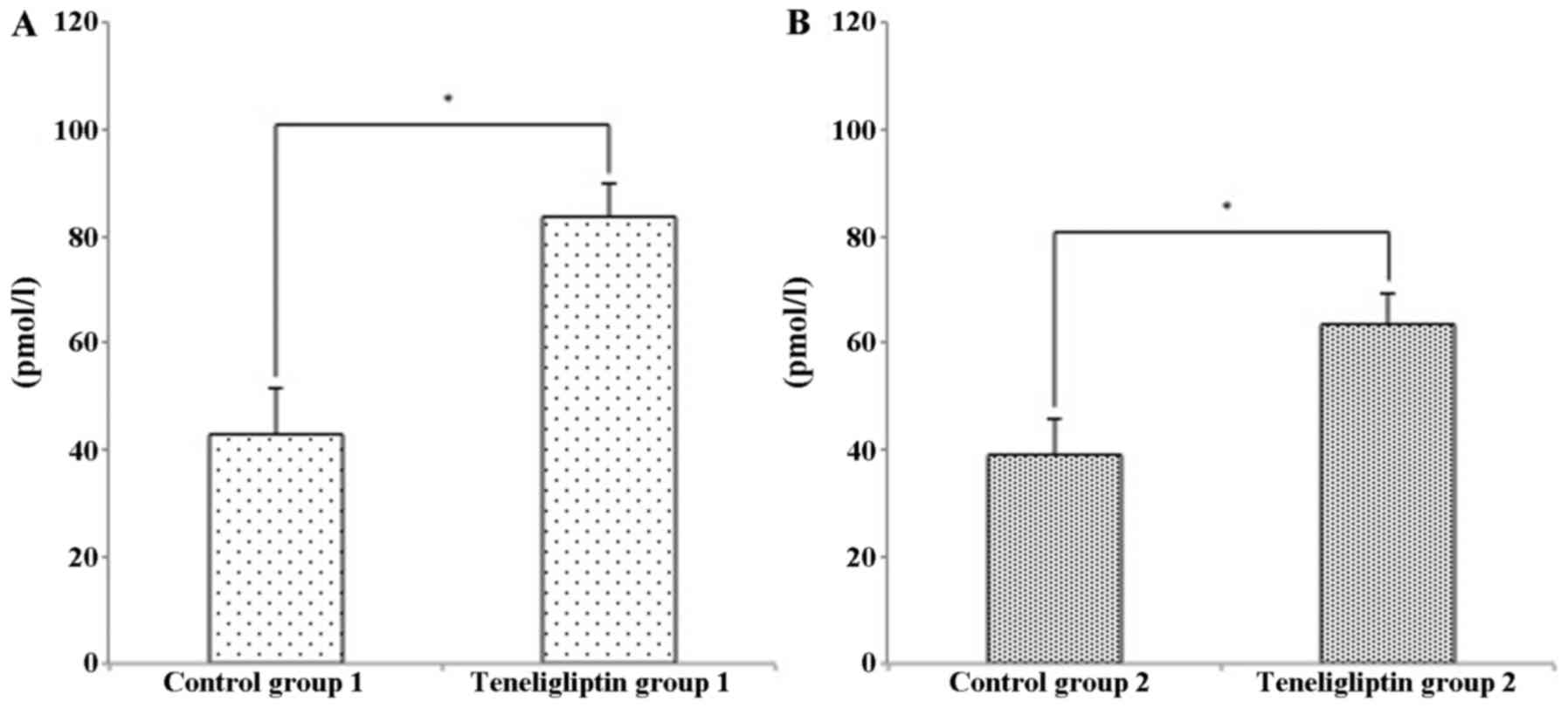
Concentration of plasma GLP-1
Since a previous study demonstrated that GLP-1
prevents the development of NAFLD (16), we thus examined the concentration
of GLP-1 in this study. The plasma concentration of GLP-1 was
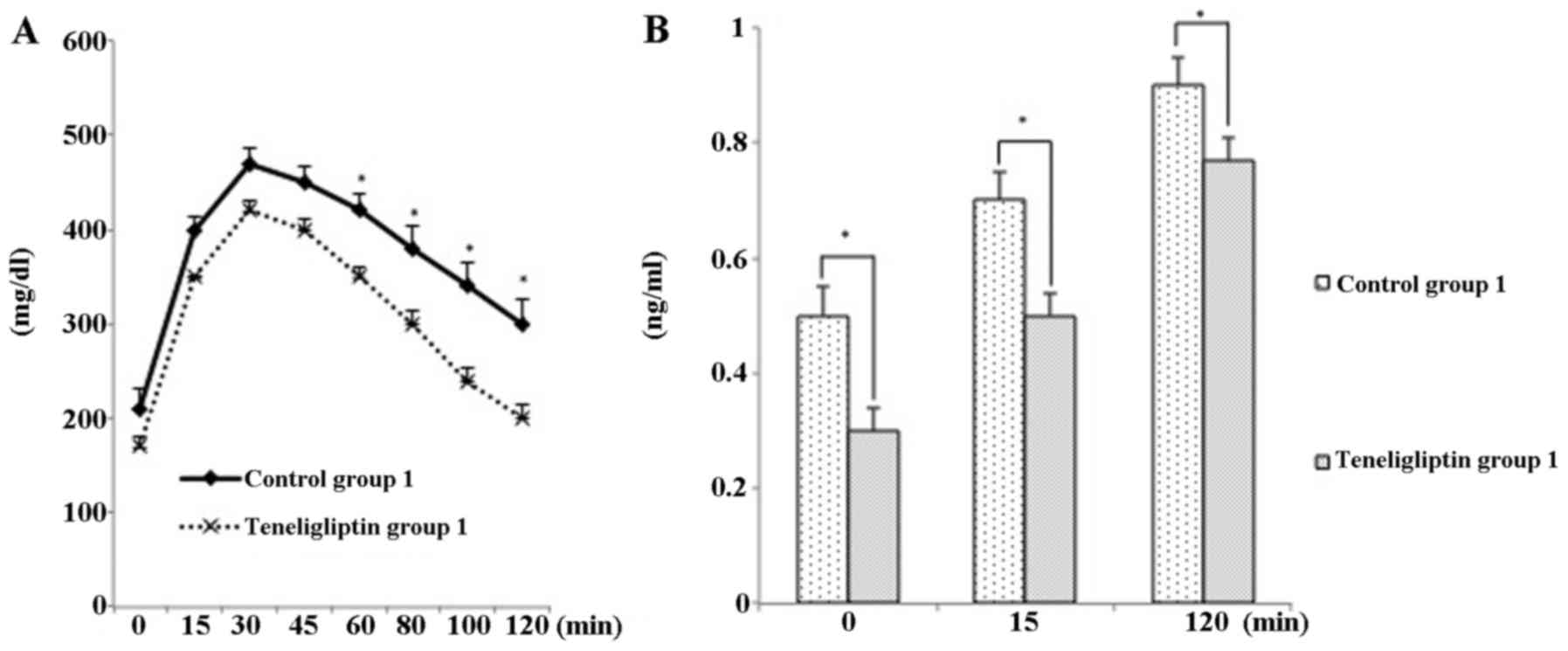
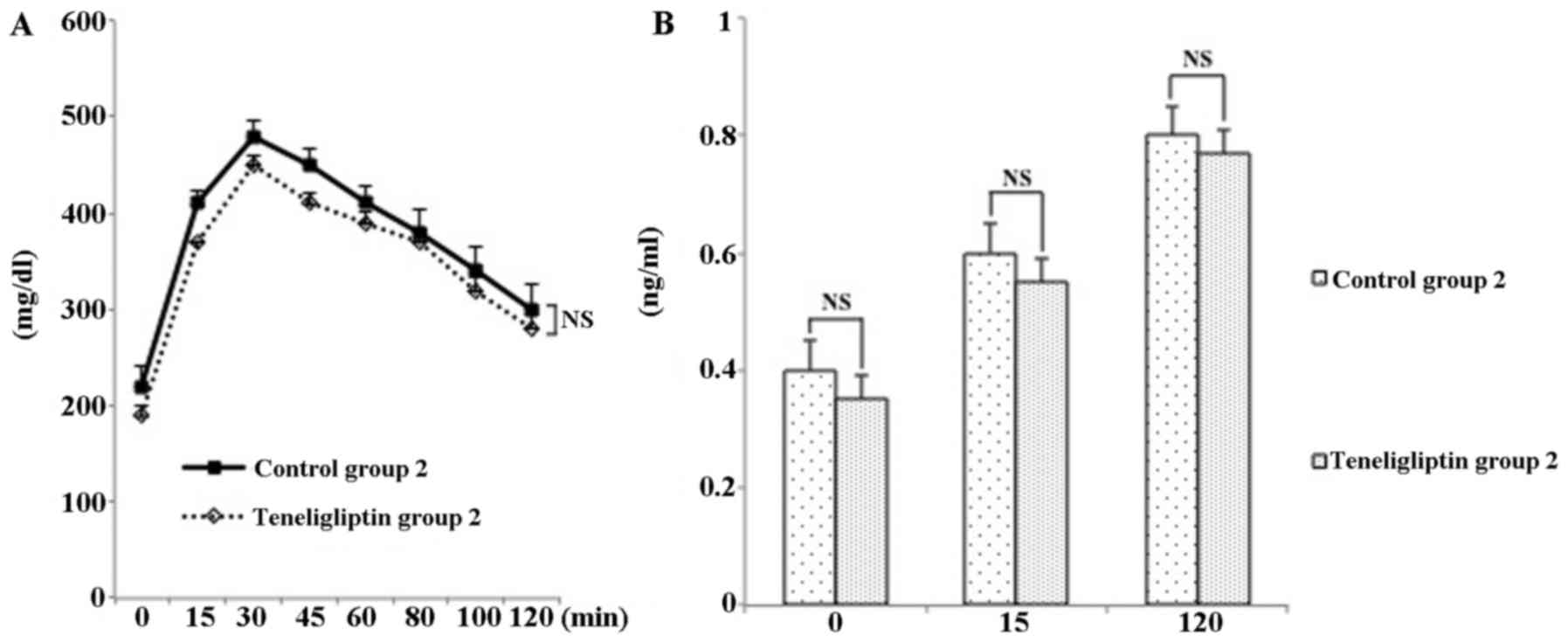
higher in both teneligliptin groups (Fig. 2).
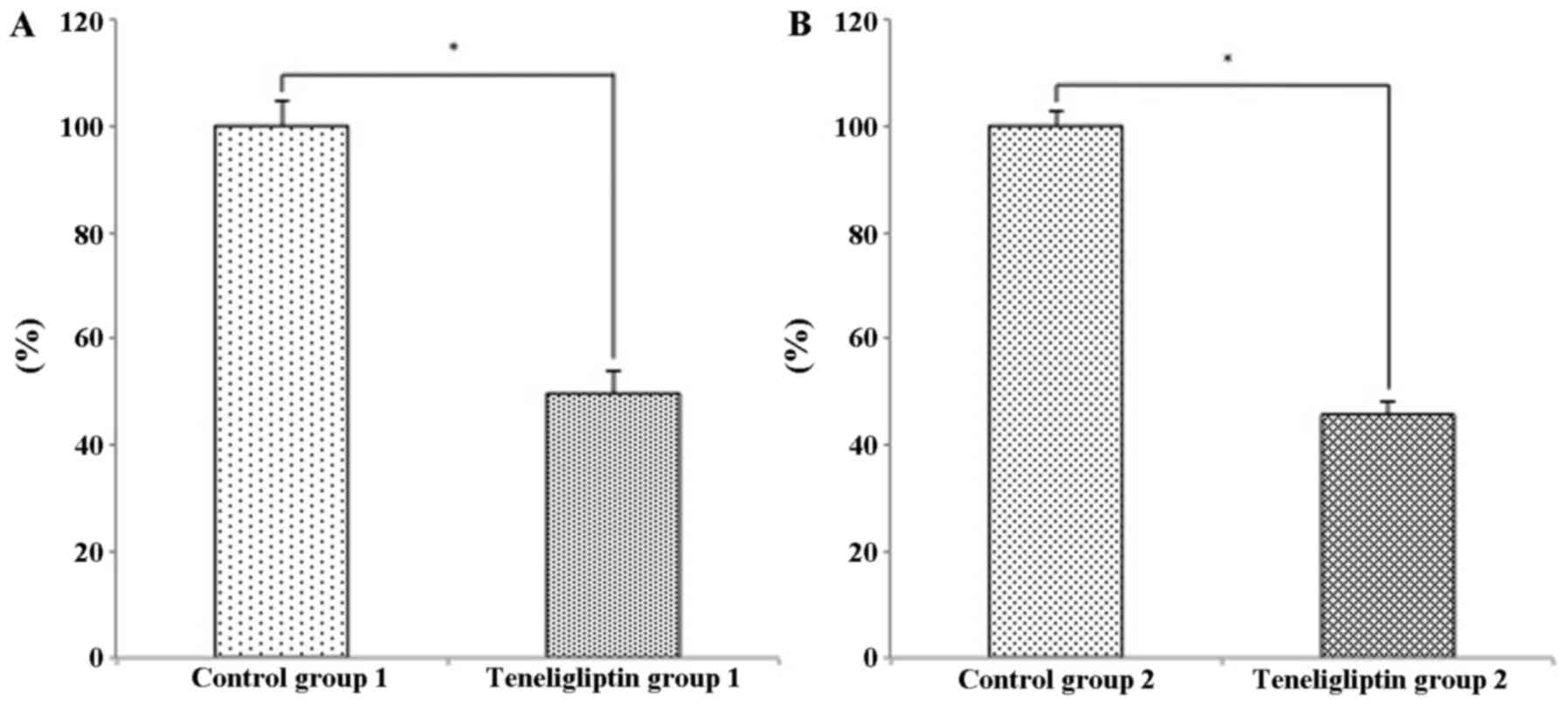
Effects of teneligliptin on DPP-4
activity
We wished to examine whether teneligliptin affects
plasma DPP-4 activity in the two experimental groups. Plasma DPP-4
activity in the mice in teneligliptin group 1 was significantly
decreased 0.49-fold, compared with that of mice in control group 1
(p<0.05; Fig. 3A). In
addition, plasma DPP-4 activity in the mice in teneligliptin group
2 was significantly decreased 0.45-fold, compared with that of mice
in control group 2 (p<0.05; Fig.
3B).
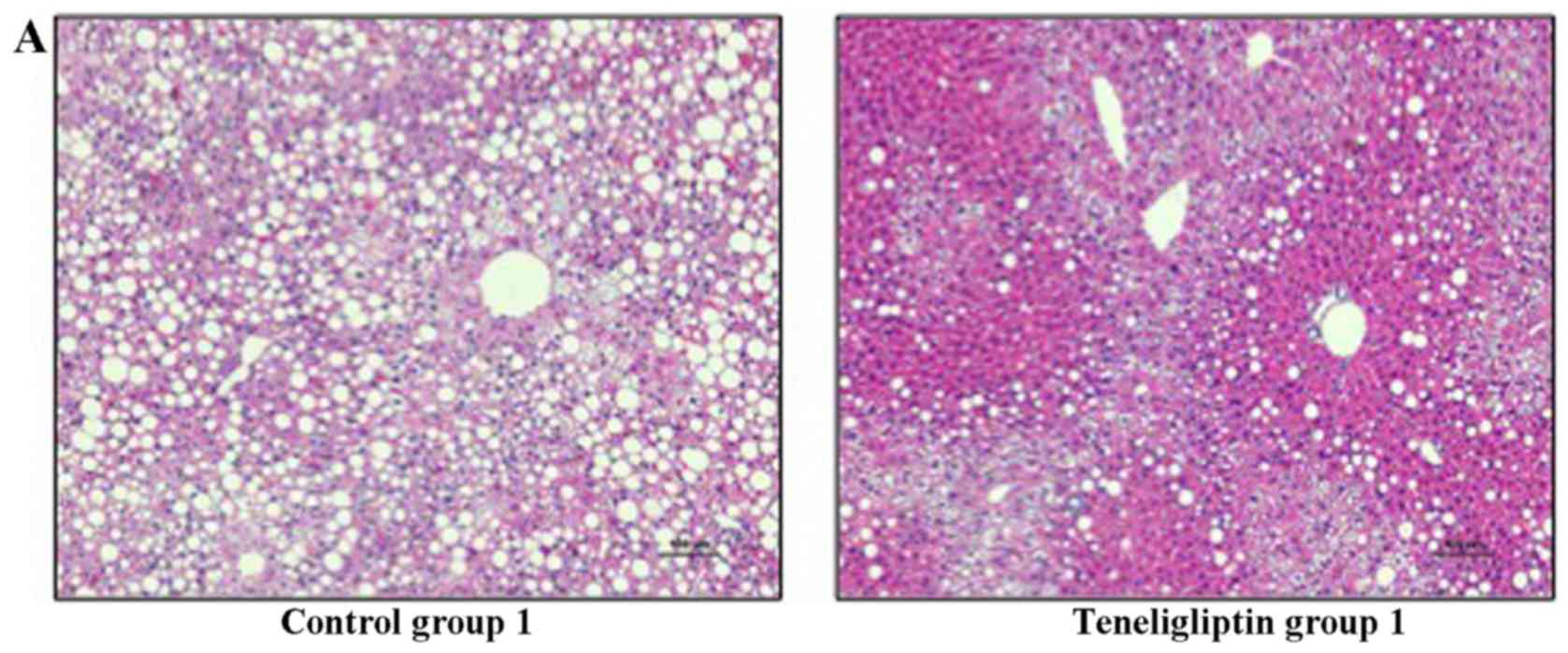
Histological analysis
Hepatic steatosis was observed in all mice used in
the experiment for the prevention of NAFLD in both control group 1
and teneligliptin group 1; however, severe hepatic steatosis was
observed in control group 1 (Fig.
4A and Table III-A).
Furthermore, a great amount of fatty droplets was observed in
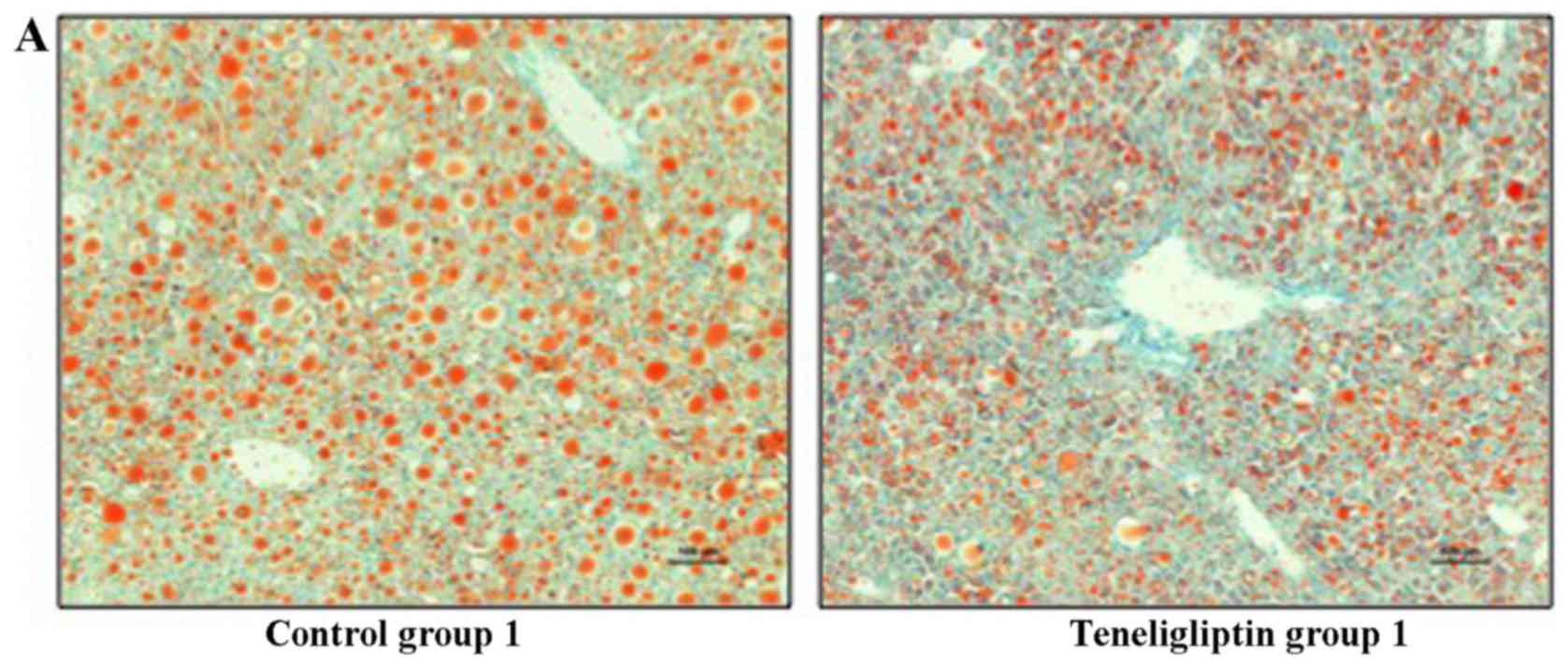
control group 1, as shown by Oil Red O staining (Fig. 5A). The same degree of hepatic
steatosis was observed in all mice used in the experiment for the
treatment of NAFLD (Fig. 4B and
Table III-B). In addition, the
same amount of fatty droplets was observed in both groups (Fig. 5B).
| Table IIIHistological findings of the liver in
mice in both types of experiments. |
Table III
Histological findings of the liver in
mice in both types of experiments.
A, Histological
findings of the liver in mice in the experiment for NAFLD
prevention
|
|---|
| Histological
parameter | Control group 1
(n=8) | Teneligliptin group
1 (n=8) |
|---|
| Hepatic
steatosis | 8/8 (100) | 8/8 (100) |
| Grade 1 | 0 (0) | 7 (83.3) |
| Grade 2 | 6 (83.3) | 1 (16.7) |
| Grade 3 | 2 ((16.7) | 0 (0) |
|
Necroinflammation | 0/8 (0) | 0/8 (0) |
| Grade 1 | 0 (0) | 0 (0) |
| Grade 2 | 0 (0) | 0 (0) |
| Grade 3 | 0 (0) | 0 (0) |
| Ballooning
degeneration | 0/8 (0) | 0/8 (0) |
| Fibrosis | 0/8 (0) | 0/8 (0) |
B, Histological
findings of the liver in mice in the experiment for the treatment
of NAFLD
|
|---|
| Histological
parameter | Control group 2
(n=8) | Teneligliptin group
2 (n=8) |
|---|
| Hepatic
steatosis | 8/8 (100) | 8/8 (100) |
| Grade 1 | 0 (0) | 0 (0) |
| Grade 2 | 3 (37.5) | 4 (50.0) |
| Grade 3 | 5 (62.5) | 4 (50.0) |
|
Necroinflammation | 0/8 (0) | 0/8 (0) |
| Grade 1 | 0 (0) | 0 (0) |
| Grade 2 | 0 (0) | 0 (0) |
| Grade 3 | 0 (0) | 0 (0) |
| Ballooning
degeneration | 0/8 (0) | 0/8 (0) |
| Fibrosis | 0/8 (0) | 0/8 (0) |
GTT
Mice in control group 1 exhibited glucose
intolerance during a GTT, compared with those in teneligliptin
group 1 (Fig. 6). Mice in
teneligliptin group 1 exhibited a significant difference in insulin
content during the GTT compared to the controls; however, mice in
teneligliptin group 2 did not exhibit a significant difference in
insulin content compared to their respective controls (Fig. 7).
Hepatic pro-inflammatory mRNA
expression
A previous study demonstrated that several
pro-inflammatory cytokines are associated with the development of
NASH (17). Since biochemical
parameters, such as AST and ALT in the mice used in the experiment
for the prevention of NAFLD differed significantly between the two
groups, we examined the relative expression levels of hepatic
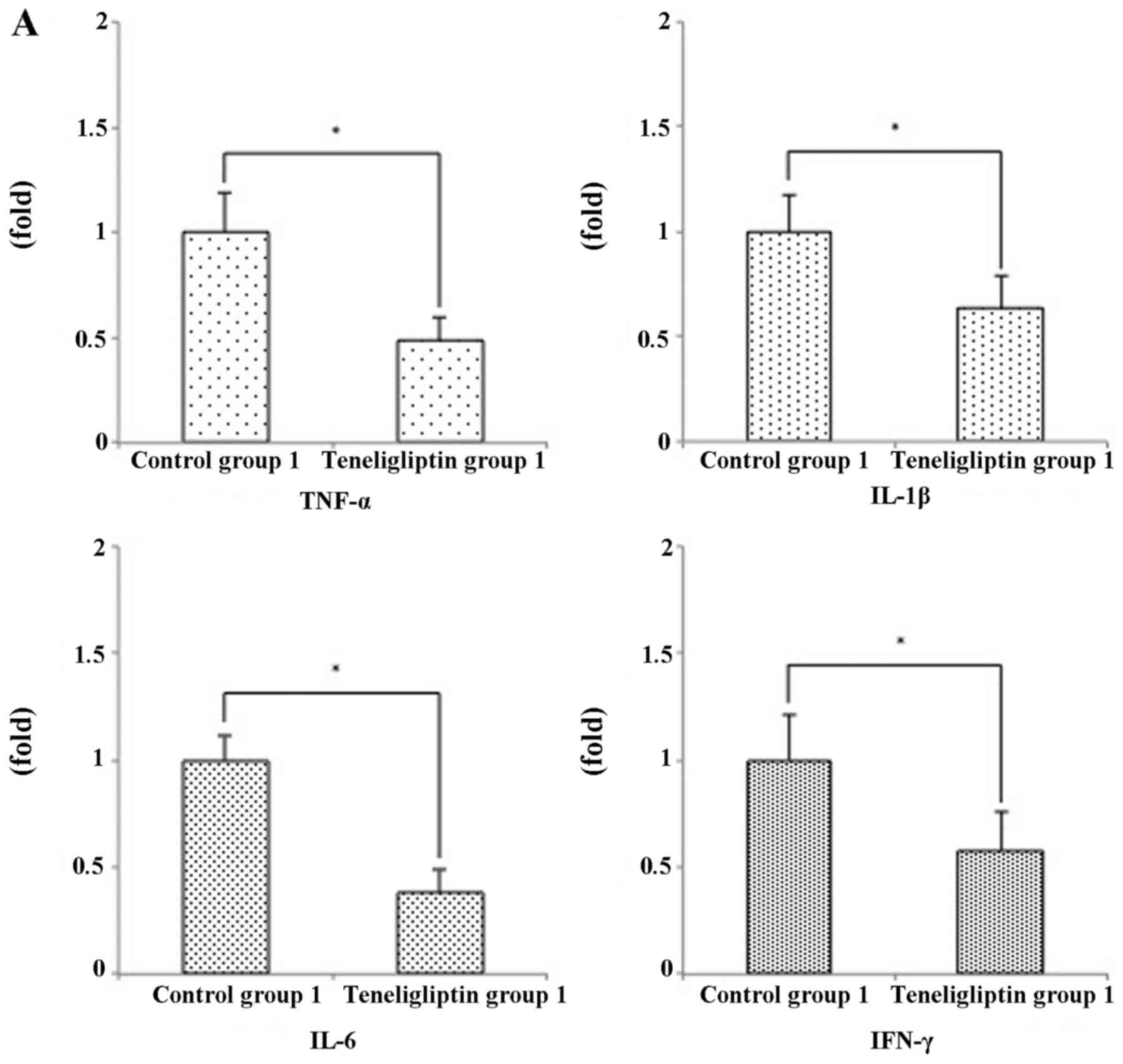
pro-inflammatory cytokines. The mRNA expression levels of tumor
necrosis factor (TNF)-α, interferon (IFN)-γ, interleukin (IL)-1β
and IL-6 were significantly decreased in teligliptin group 1
(respectively, p<0.05) (Fig.
8A). On the other hand, the ALT levels in the mice used in the
experiment for the treatment of NAFLD was significantly increased
in teneligliptin group 2 (Table
II-B). As expected, the expression levels of TNF-α and IL-6
were also significantly increased in the mice in teneligliptin
group 2 (Fig. 8B). However, the
mRNA expression levels of IL-1β were significantly decreased in the
mice in teneligliptin group 2, while the levels of IFN-γ exhibited
no significant difference (Fig.
8B).
Hepatic lipogenic-related mRNA
expression
The differences in the histological findings and
hepatic TG content observed between the mice used in the experiment
for the prevention of NAFLD and those used in the experiment for
the treatment of NAFLD suggest that the expression of cytokines is
involved in the development of NAFLD. The expression of lipogenic
enzymes is mainly regulated at the transcriptional level in a
hyperinsulinemic and hyperglycemic state. Sterol regulatory element
binding protein-1c (SREBP-1c) and carbohydrate response element
binding protein (ChREBP), are well known to be involved in these
states (18). The induction of
lipogenic genes, such as fatty acid synthase (FAS) is under the
concerned action of ChREBP and of the transcription factor SREBP-1c
in response to glucose and insulin (18). In particular, the expression of
SREBP-1c is stimulated by insulin (19,20). In addition to FAS, stearoyl-CoA
desaturase-1 (SCD-1) may be critical to the role of triglyceride
accumulation in hepatocytes (21). Peroxisome proliferator-activated
receptors (PPARs) are nuclear transcription factors that include
three subtypes: α, β and γ. PPAR-γ agonists, such as
thiazolidinediones improve insulin action in peripheral tissues and
are effective in the treatment of patients with NAFLD (22). Unlike PPAR-γ, PPAR-α mediates the
expression of genes that regulate lipid oxidation (23). PPAR-α agonists, such as fibrates,
have been used in the treatment of hypertriglyceridemia and to
reduce cardiovascular risk (24).
In the present study, the hepatic TG content in the
teneligliptin group 1 was reduced, compared with that in control
group 1 (Table II-A). On the
other hand, there was no significant difference in the hepatic TG
between control group 2 and teneligliptin group 2 (Table II-B). These results coincided
with the result that the relative mRNA expression levels of
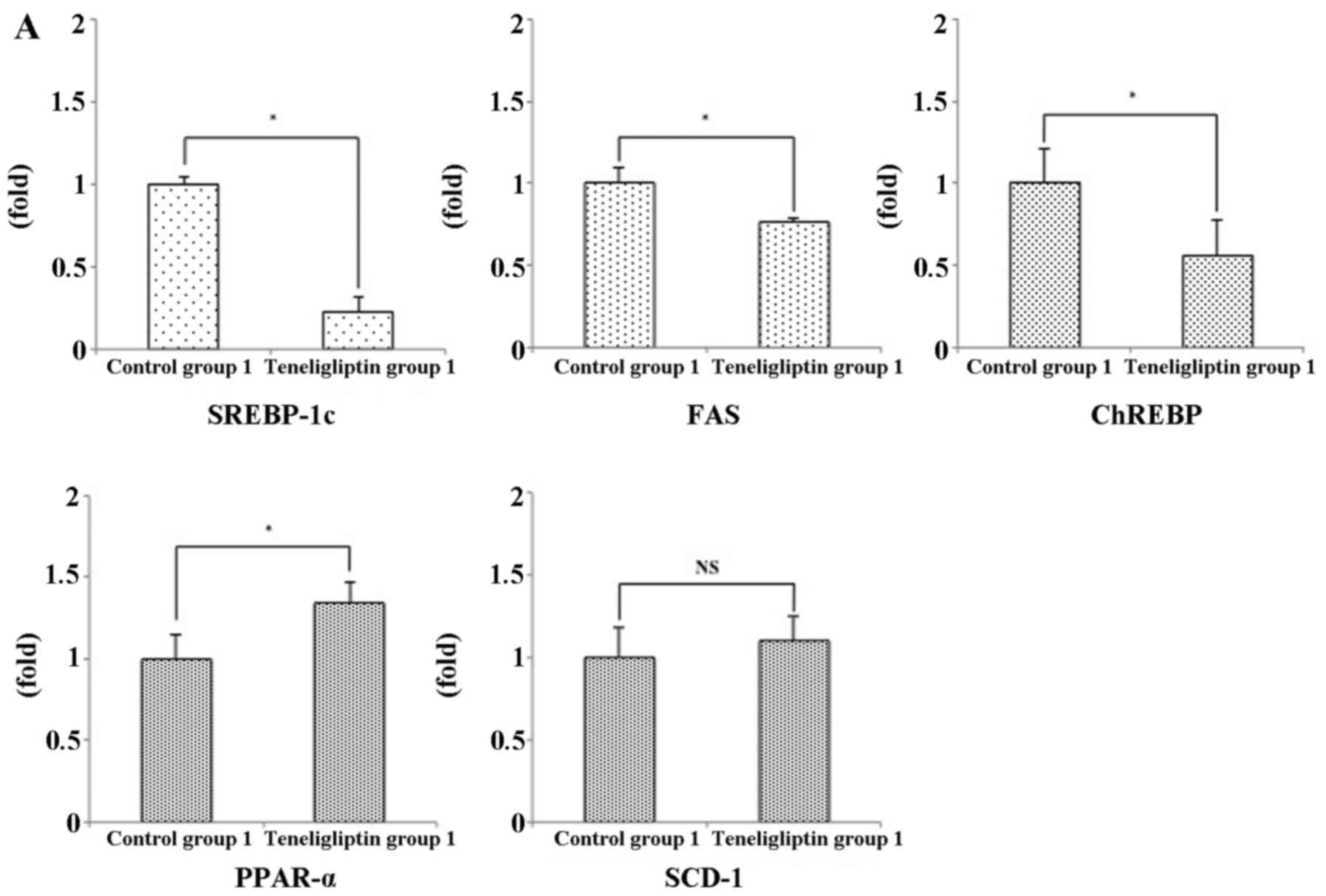
SREBP-1c and ChREBP were reduced in teneligliptin group 1, but were
not reduced in teneligliptin group 2 compared with control group 2
(Fig. 9). On the other hand, the
mRNA expression levels of FAS were significantly decreased in
teneligliptin group 1 (Fig. 9A);
however, there was no significant difference in the mRNA expression
levels of SCD-1 between control group 2 and teneligliptin group 2
(Fig. 9B). In addition, the
expression of PPAR-α, a key element in the β-oxidation of FFAs,
differed significantly between control group 1 and teneligliptin
group (Fig. 9A). However, the
expression of PPAR-α in control group 2 and teneligliptin group 2
did not exhibit a significant difference (Fig. 9B). The mRNA expressions of PPAR-γ
was below the lower limit of detection in all groups (data not
shown).
Hepatic steatosis develops as result of abnormally
enhanced de novo lipid synthesis and fat delivery (25). ATP-citrate lyase (ACL) is an
important lipogenic enzyme that regulates the flow of glucose
carbons to cytosolic acetyl-coenzyme A (CoA) (26). Acetyl-CoA carboxylase (ACC) is a
biotinylated enzyme that catalyzes the ATP-dependent carboxylation
of acetyl-CoA to produce malonyl-CoA. Animals have two ACC genes
(ACC1 and ACC2). In particular, ACC1 is a protein that is mainly
expressed in liver and adipose tissue (27). The phosphorylation of ACC by
AMP-activated protein kinase (AMPK) results in its inactivation and
inability to inhibit carnitine palmitoyltransferase-1 (CPT-1).
CPT-1 is responsible for fatty acid transport into the
mitochondria. The liver isoform (CPT1A) is localized in the outer
mitochondrial membrane and exposes its active site at the cytosolic
face of the mitochondrion (28).
Insulin can regulate the sensitivity of CPT1A for malonyl-CoA in
the liver (29). In this study,
we examined the relative expression levels of these hepatic
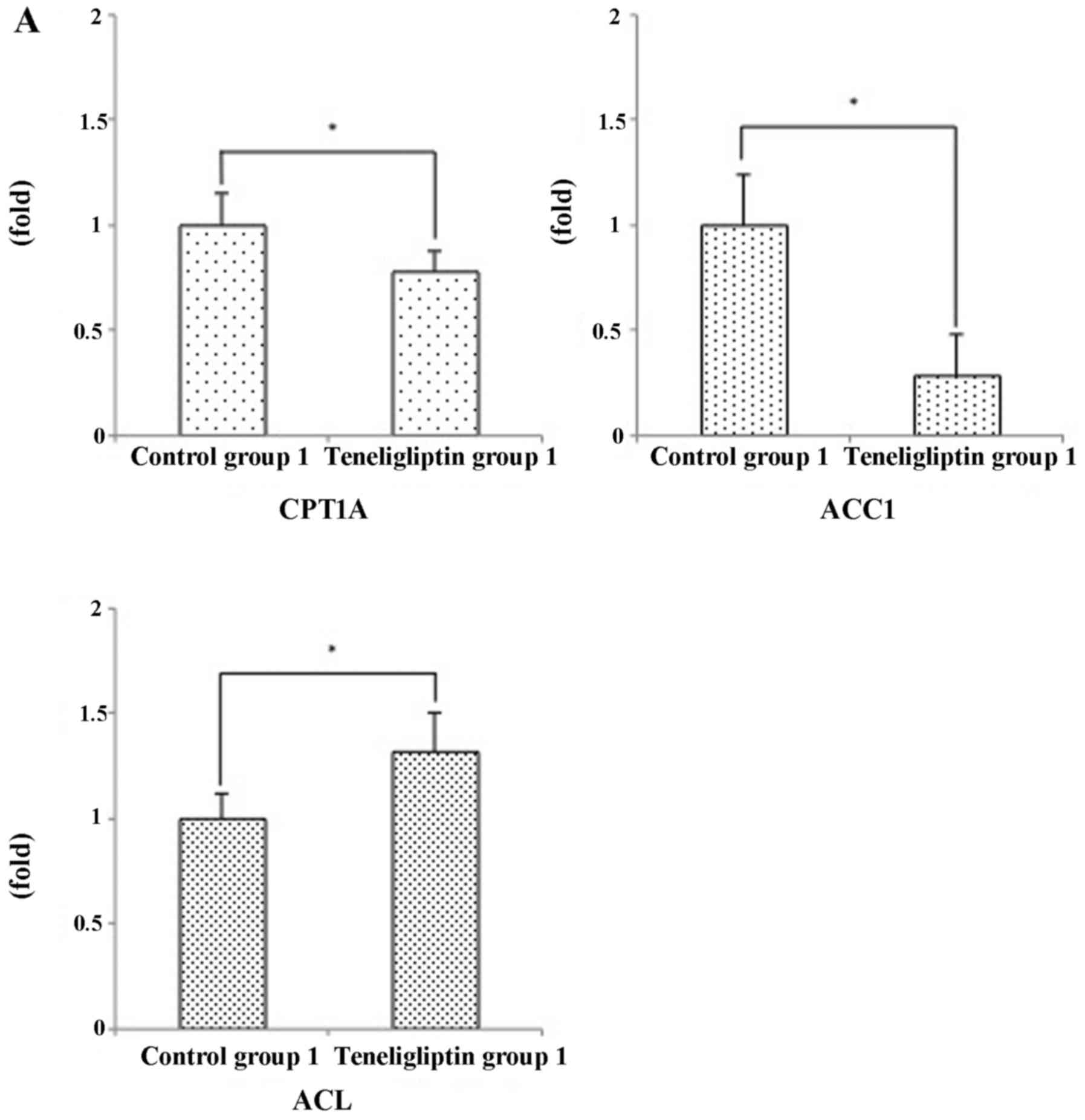
lipogenic-related enzymes. Against our prediction, the expression
levels of ACC1 were significantly decreased in teneligliptin group
1 (Fig. 10A), but were
significantly increased in teneligliptin group 2 (Fig. 10B). Furthermore, the expression
levels of ACL were significantly increased in teneligliptin group 1
(Fig. 10A); however these
expression levels were significantly decreased in teneligliptin
group 2 (Fig. 10B). In addition,
the expression levels of CPT1A were significantly decreased in
teneligliptin group 1; however, the expression levels of CPT1A were
not significantly different between control group 2 and
teneligliptin group 2 (Fig.
10B).
Mitochondrial oxidation of FFA-related
mRNA expression
It is known that once the fatty acid is inside the
mitochondrial matrix, β-oxidation can then begin. It has 4 steps
(30). First, acyl-CoA is
oxidized by acyl-CoA dehydrogenase to yield a trans-2-enoyl-CoA.
This step is followed by hydratation of the double bond. The
resulting L-3-hydroxy-acyl-CoA is again oxidized into
3-keto-acyl-CoA in the third step. Finally, the thiolytic cleavage
of 3-keto-acyl-CoA produces a 2-carbon chain-shortened acyl-CoA
plus acetyl-CoA. In this study, we examined the expression of
mitochondrial oxidation of fatty acid-related genes, such as
medium-chain acyl-CoA dehydrogenase (MCAD), long-chain acyl-CoA
dehydrogenase (LCAD), enoyl-CoA hydratase (ECAH), 3-hydroxyacyl-CoA
dehydrogenase (HACDH) and 3-ketoacyl-CoA thiolase (bKACTB). The
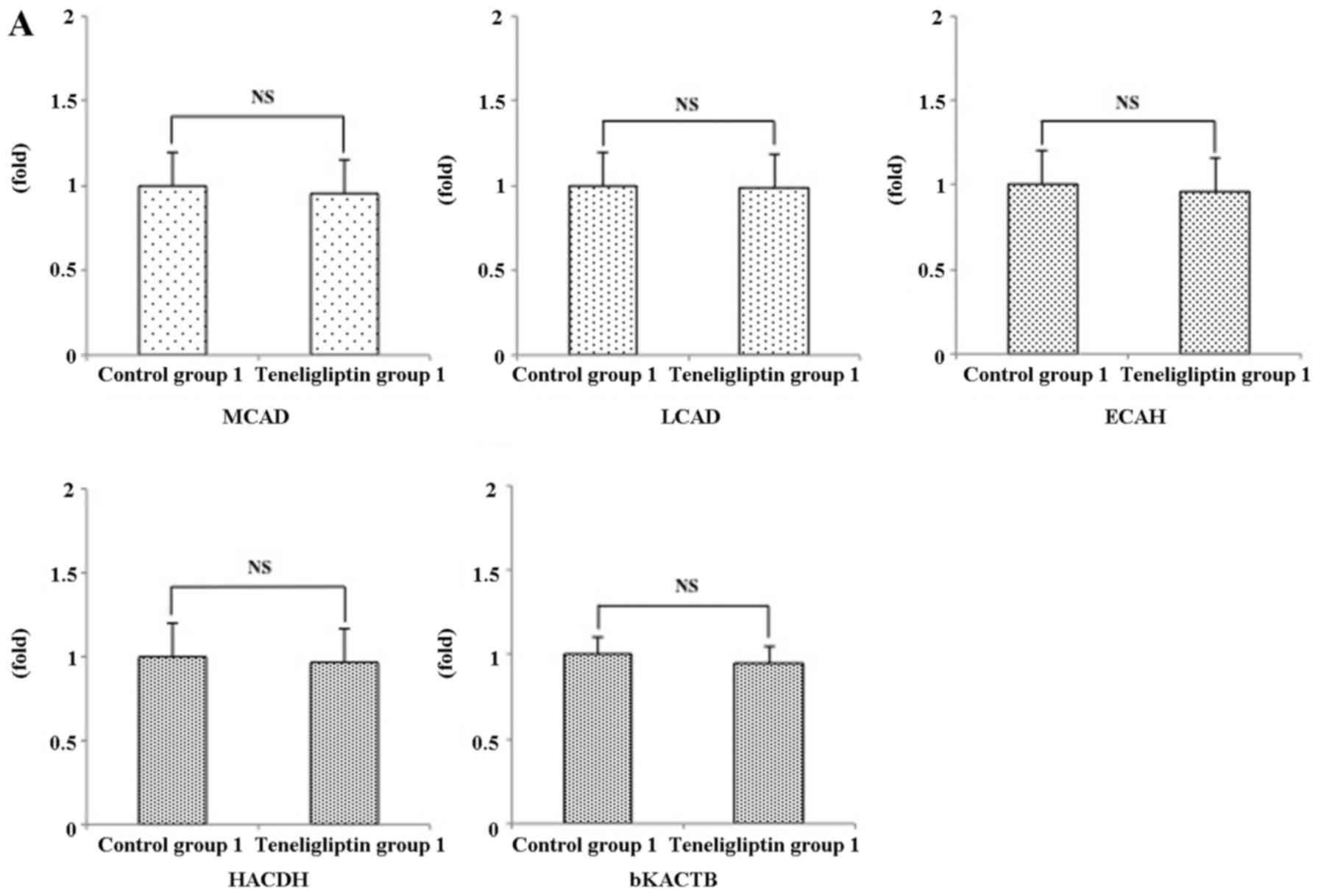
relative expression levels of these genes, such as MCAD, LCAD,
ECAH, HACDH and bKACTB were not significantly different between
control group 1 and teneligliptin group 1 (Fig. 11A). In addition, the expression
levels of these same genes were not significantly different between
control group 2 and teneligliptin group 2 (Fig. 11B).
Insulin resistance-related mRNA
expression
Insulin signaling is initiated when insulin binds to
its receptor expressed on the cell membrane. The insulin receptor
is a receptor tyrosine kinase upon the binding of insulin, is
autophosphorylated and activated. Once activated, the receptor can
phosphorylate tyrosine residues on the insulin receptor substrate
(IRS) molecules. IRS proteins bind the
phosphatidylinositol-3-kinase (PI3K) and activate it. PI3K
eventually leads to many of the effects of insulin on glucose,
lipid and protein metabolism. IRS-1 and IRS-2 exhibit high
structural homology, are abundantly expressed in the liver. In
addition, AMPK promotes glucose uptake into skeletal muscle and
suppresses glucose output from the liver via insulin-independent
mechanisms (31). These receptors
are thought to be responsible for transducing insulin signals from
the insulin receptor to the intracellular effectors in the
regulation of glucose and lipid homeostasis (32,33). IRS-2 mainly functions during the
fasting state and immediately after re-feeding, while IRS-1
functions primarily after re-feeding (34). Moreover, IRS-1 has been observed
to play a dominant role under states of nutrient excess (35). In this study, the relative
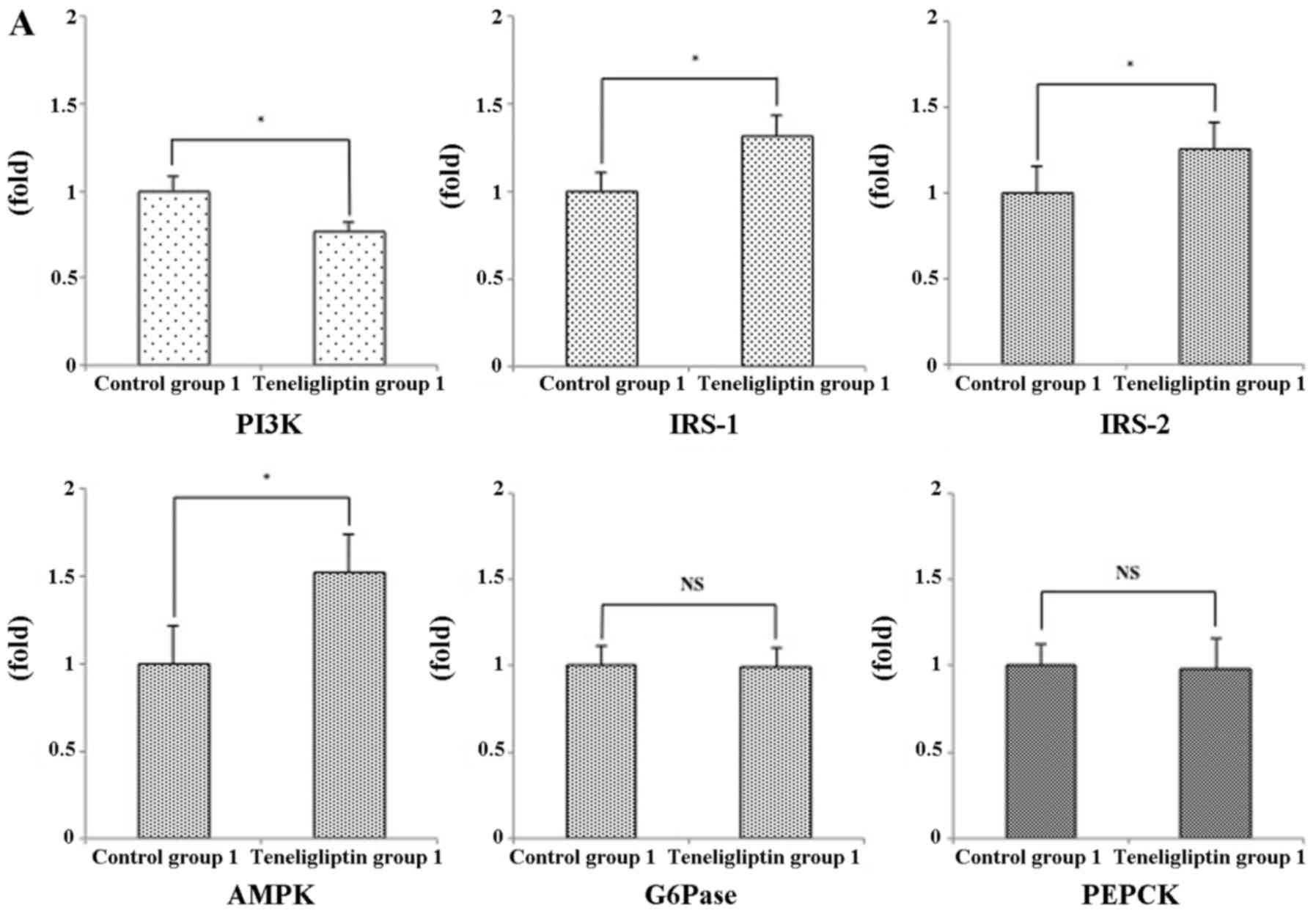
expression levels of these genes, such as PI3K, IRS-1, IRS-2 and
AMPK were significantly different between teneligliptin group 1 and
control group 1 (Fig. 12A).
However, the expression levels of PI3K and AMPK were not
significantly different between control group 2 and teneligliptin
group 2 (Fig. 12B). The levels
of IRS-1 and IRS-2 were significantly decreased in teneligliptin
group 2 compared to control group 2 (Fig. 12B). In addition, gluconeogenic
genes, such as phosphoenolpyruvate carboxykinase (PEPCK) and
glucose-6-phosphatase (G6Pase) contribute to insulin resistance and
glucose intolerance (36). In
this study, the relative expression levels of PEPCK and G6Pase were
not significantly different between control group 1 and
teneligliptin group 1 (Fig.
12A); however, these expression levels were significantly
increased in teneligliptin group 2 compared to control group 2
(Fig. 12B).
Discussion
There are many patients with NAFLD associated with
T2DM and obesity. In the present study, we fed ob/ob mice, animal
models of the disease, a HCD containing fructose as a main
component to evaluate the efficacy of teneligliptin, a diabetes
drug, for the treatment of HCD-induced NAFLD.
In the experiment for the prevention of NAFLD, a
comparison of mice fed the HCD supplemented with teneligliptin from
the initial stage of the study with mice fed the HCD alone revealed
no significant difference in weight gain between the two groups.
However, histological finding in the liver revealed that the
occurrence of hepatic steatosis was suppressed in the mice fed the
HCD supplemented with teneligliptin. In the following experiment
for the treatment of NAFLD, we compared mice fed the HCD
supplemented with teneligliptin after the occurrence of a certain
degree of steatosis with the mice continuously fed HCD alone. The
results revealed no significant difference in weight gain between
the two groups, and a similar degree of steatosis was observed in
both groups. In addition, our results revealed that the plasma
level of GLP-1 was significantly increased in both teneligliptin
groups. Furthermore, both teneligliptin groups exhibited the same
degree of DPP-4 activity. Therefore, we examined whether some
factors, apart from GLP-1 and DPP-4 activity, are associated with
these histological changes. To investigate the mechanism
contributing to the differences in histological findings observed
between these two experiments, we measured plasma parameters
related to both hepatic disorders and lipids and the hepatic TG
levels, which were likely to yield some differences. The results
revealed a decrease in the plasma AST and ALT levels, the
parameters indicative of hepatic disorder, in the mice fed the
teneligliptin-supplemented HCD from the initial stage of the study.
In addition, decreased liver TG levels, which are hallmarks of
steatosis, were observed in the mice fed the
teneligliptin-supplemented HCD from the initial stage of the study.
The plasma glucose and insulin levels were also decreased in these
mice. In the mice fed the HCD supplemented with teneligliptin after
the occurrence of steatosis, the ALT levels increased, and no
significant difference was observed in the plasma glucose and
insulin levels between the two groups. In contrast to the mice
between control group 1 and teneligliptin group 1, the mice in
teneligliptin group 2 did not exhibit a significant difference in
glucose tolerance and insulin content during the GTT between
control group 2 and teneligliptin group 2. These results indicate
that teneligliptin does not always improve insulin resistance.
These biochemistry results were mostly consistent with the
histological findings.
Most importantly, the T2DM drug, teneligliptin,
showed resistance to the occurrence of hepatic steatosis induced by
a diet containing fructose as a main component when the diet was
fed from the initial stage of the study, but the drug exerted a
poor therapeutic effect, as determined by histological analysis, in
the presence of a certain degree of steatosis. To elucidate the
mechanisms behind these results, we examined the liver expression
of genes associated with the progression of NAFLD, various
lipid-related genes including factors related to mitochondrial
β-oxidation, and genes associated with insulin resistance. A
comparison of the liver expression of pro-inflammatory cytokines
(TNF-α, IL-6, IL-1β and IFN-γ) revealed the decreased expression of
these cytokines in the mice used in the experiment for the
prevention of NAFLD. By contrast, the expression of TNF-α and IL-6
increased in the mice used in the experiment for the treatment of
NAFLD. Next, the expression of lipid synthesis-related genes, such
as SREBP-1c, FAS and ChREBP was decreased in the mice used in the
experiment for the prevention of NAFLD. By contrast, the expression
of FAS increased in the mice used in the experiment for the
treatment of NAFLD. These results were consistent with the
histological findings. We further studied genes associated with
fatty acid oxidation and mitochondrial β-oxidation. The expression
of CPT1A and ACC1 decreased and the expression of ACL increased in
the mice used in the experiment for the prevention of NAFLD. In the
mice fed the diet after the occurrence of steatosis, however, the
expression of CPT1A did not differ significantly from that of the
controls, but the expression of ACC1 increased while and the
expression of ACL decreased. As for the expression of factors
associated with other stages of β-oxidation, the expression of
PPAR-α increased in the mice used in the experiment for the
prevention of NAFLD. These results suggest that the early
administration of teneligliptin also affects fatty acid
oxidation.
We also examined the insulin resistance associated
with the occurrence of NAFLD. The biochemistry and GTT results
suggested that the early administration of teneligliptin improved
insulin resistance. Determining the expression of genes associated
with insulin signaling, the expression of IRS-1 and AMPK increased
and the expression of PI3K decreased in the mice of the experiment
for prevention of NAFLD. In the mice fed the
teneligliptin-supplemented diet after the occurrence of steatosis.
However, the expression of IRS-1 and IRS-2 decreased and the
expression of PI3K and AMPK did not significantly differ between
groups in the mice used in the experiment for the treatment of
NAFLD. In addition, the expression levels of gluconeogenic genes,
such as PEPCK and G6Pase were significantly different in the mice
used in the experiment for the treatment of NAFLD. These results
suggest that, whereas the early administration of teneligliptin may
improve insulin resistance, the administration of the drug after
the occurrence of a certain degree of steatosis does little to
improve the insulin resistance. These results are suggested to be
one of the factors contributing to the histological differences
between the two groups.
Since the GLP-1 concentration and DPP-4 activity in
the groups receiving teneligliptin were at similar levels, we
hypothesized that pathways unlike GLP-1 and DPP-4 activity were
responsible for the results in our experiment. An important
difference in the two experimental groups was the existence of
excess adiposity, including hepatic steatosis that was caused by 4
weeks of being fed a HCD. In addition to adipocytokine secreted
from adipose tissue, our results suggested that the DPP-4 inhibitor
aggravatd the expression of certain pro-inflamatory cytokines in
the presence of hepatic steatosis. This result may be associated
with the aggravation of insulin resistance in teneligliptin group
2. We will have to examine the mechanisms of the effect of the
DPP-4 inhibitor apart from DPP-4 activity and GLP-1 in detail in
the future.
On the whole, it was shown that the administration
of the DPP-4 inhibitor, teneligliptin, from the initial stage of
diabetes may not only suppress the expression of a number of
pro-inflammatory cytokines in the liver, but can also inhibit the
expression of lipid synthesis-related genes and, secondarily,
inhibit the occurrence of steatosis and prevent the development of
NAFLD by preventing the expression of pro-inflammatory cytokines,
lipid synthesis-related genes and improving insulin resistance.
In conclusion, the results of the present study
suggest that the DPP-4 inhibitor, teneligliptin, may prevent the
development of NAFLD, which is a hepatic phenotype of metabolic
syndrome, if the drug is administered from the initial stage of
diabetes. Since DPP-4 inhibitors are recommended to be used in the
treatment of early-stage type 2 diabetes (34); it is considered important to use
teneligliptin as rescommended. Further experiments are warranted in
order to determine whether similar effects can be obtained with
other DPP-4 inhibitors.
Abbreviations:
|
NAFLD
|
non-alcoholic fatty liver disease
|
|
T2DM
|
type 2 diabetes mellitus
|
|
NASH
|
non-alcoholic steatohepatitis
|
|
GLP-1
|
glucagon-like peptide-1
|
|
GIP
|
glucose-dependent insulinotropic
polypeptide
|
|
DPP-4
|
dipeptidyl peptidase-4
|
|
HCD
|
high carbohydrate diet
|
|
AST
|
aspartate aminotransferase
|
|
ALT
|
alanine aminotransferase
|
|
GA
|
glico albumin
|
|
T-CHO
|
total cholesterol
|
|
TG
|
triglyceride
|
|
FFA
|
free fatty acid
|
|
H&E
|
hematoxylin and eosin
|
|
GTT
|
glucose tolerance test
|
|
TNF
|
tumor necrosis factor
|
|
IFN
|
interferon
|
|
IL
|
interleukin
|
|
SREBP-1c
|
sterol regulatory element binding
protein-1c
|
|
ChREBP
|
carbohydrate response element binding
protein
|
|
FAS
|
fatty acid synthase
|
|
SCD-1
|
stearoyl-CoA desaturase-1
|
|
PPAR
|
peroxisome proliferator-activated
receptors
|
|
ACL
|
ATP-citrate lyase
|
|
CoA
|
coenzyme A
|
|
ACC
|
acetyl-CoA carboxylase
|
|
AMPK
|
AMP-activated protein kinase
|
|
CPT-1
|
carnitine palmitoyltransferase-1
|
|
MCAD
|
medium-chain acyl-CoA
dehydrogenase
|
|
LCAD
|
long-chain acyl-CoA dehydrogenase
|
|
ECAH
|
enoyl-CoA hydratase
|
|
HACDH
|
3-hydroxyacyl-CoA dehydrogenase
|
|
bKACTB
|
3-ketoacyl-CoA thiolase
|
|
IRS
|
insulin receptor substrate
|
|
PI3K
|
phosphatidylinositol-3-kinase
|
|
PEPCK
|
phosphoenolpyruvate carboxykinase
|
|
G6Pase
|
glucose-6-phosphatase
|
Acknowledgments
The authors would like to thank Yukio Nakahira and
Eiko Koubayashi, at the Osaka Medical College, for providing
technical support.
References
|
1
|
Matsuzawa Y: The role of fat topology in
the risk of disease. Int J Obes. 32(Suppl 7): S83–S92. 2008.
View Article : Google Scholar
|
|
2
|
Ludwig J, Viggiano TR, McGill DB and Oh
BJ: Nonalcoholic steatohepatitis: Mayo Clinic experiences with a
hitherto unnamed disease. Mayo Clin Proc. 55:434–438.
1980.PubMed/NCBI
|
|
3
|
Powell EE, Cooksley WG, Hanson R, Searle
J, Halliday JW and Powell LW: The natural history of nonalcoholic
steatohepatitis: A follow-up study of forty-two patients for up to
21 years. Hepatology. 11:74–80. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yoshiike N and Lwin H: Epidemiological
aspects of obesity and NASH/NAFLD in Japan. Hepatol Res. 33:77–82.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ahrén B: Gut peptides and type 2 diabetes
mellitus treatment. Curr Diab Rep. 3:365–372. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Drucker DJ: Biological actions and
therapeutic potential of the glucagon-like peptides.
Gastroenterology. 122:531–544. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mentlein R, Gallwitz B and Schmidt WE:
Dipeptidyl-peptidase IV hydrolyses gastric inhibitory polypeptide,
glucagon-like peptide-1(7–36)amide, peptide histidine methionine
and is responsible for their degradation in human serum. Eur J
Biochem. 214:829–835. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Deacon CF, Johnsen AH and Holst JJ:
Degradation of glucagon-like peptide-1 by human plasma in vitro
yields an N-terminally truncated peptide that is a major endogenous
metabolite in vivo. J Clin Endocrinol Metab. 80:952–957.
1995.PubMed/NCBI
|
|
9
|
Ahrén B, Landin-Olsson M, Jansson PA,
Svensson M, Holmes D and Schweizer A: Inhibition of dipeptidyl
peptidase-4 reduces glycemia, sustains insulin levels, and reduces
glucagon levels in type 2 diabetes. J Clin Endocrinol Metab.
89:2078–2084. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Pratley RE and Salsali A: Inhibition of
DPP-4: A new therapeutic approach for the treatment of type 2
diabetes. Curr Med Res Opin. 23:919–931. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vella A, Bock G, Giesler PD, Burton DB,
Serra DB, Saylan ML, Dunning BE, Foley JE, Rizza RA and Camilleri
M: Effects of dipeptidyl peptidase-4 inhibition on gastrointestinal
function, meal appearance, and glucose metabolism in type 2
diabetes. Diabetes. 56:1475–1480. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maiztegui B, Borelli MI, Madrid VG, Del
Zotto H, Raschia MA, Francini F, Massa ML, Flores LE, Rebolledo OR
and Gagliardino JJ: Sitagliptin prevents the development of
metabolic and hormonal disturbances, increased β-cell apoptosis and
liver steatosis induced by a fructose-rich diet in normal rats.
Clin Sci (Lond). 120:73–80. 2011. View Article : Google Scholar
|
|
13
|
Yoshida T, Akahoshi F, Sakashita H,
Kitajima H, Nakamura M, Sonda S, Takeuchi M, Tanaka Y, Ueda N,
Sekiguchi S, et al: Discovery and preclinical profile of
teneligliptin
(3-[(2S,4S)-4-[4-(3-methyl-1-phenyl-1H-pyrazol-5-yl)piperazin-1-yl]pyrrolidin-2-ylcarbonyl]thiazolidine):
A highly potent, selective, long-lasting and orally active
dipeptidyl peptidase IV inhibitor for the treatment of type 2
diabetes. Bioorg Med Chem. 20:5705–5719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Friedman JM, Leibel RL, Siegel DS, Walsh J
and Bahary N: Molecular mapping of the mouse ob mutation. Genomics.
11:1054–1062. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brunt EM, Janney CG, Di Bisceglie AM,
Neuschwander-Tetri BA and Bacon BR: Nonalcoholic steatohepatitis: A
proposal for grading and staging the histological lesions. Am J
Gastroenterol. 94:2467–2474. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tomas E, Wood JA, Stanojevic V and Habener
JF: Glucagon-like peptide-1 (9–36) amide metabolite inhibits weight
gain and attenuates diabetes and hepatic steatosis in diet-induce
obese.
|
|
17
|
Gao B: Innate immunity and
steatohepatitis: A critical role of another toll (TLR-9).
Gastroenterology. 139:27–30. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dentin R, Girard J and Postic C:
Carbohydrate responsive element binding protein (ChREBP) and sterol
regulatory element binding protein-1c (SREBP-1c): Two key
regulators of glucose metabolism and lipid synthesis in liver.
Biochimie. 87:81–86. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Foretz M, Guichard C, Ferré P and Foufelle
F: Sterol regulatory element binding protein-1c is a major mediator
of insulin action on the hepatic expression of glucokinase and
lipogenesis-related genes. Proc Natl Acad Sci USA. 96:12737–12742.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Foretz M, Pacot C, Dugail I, Lemarchand P,
Guichard C, Le Lièpvre X, Berthelier-Lubrano C, Spiegelman B, Kim
JB, Ferré P and Foufelle F: ADD1/SREBP-1c is required in the
activation of hepatic lipogenic gene expression by glucose. Mol
Cell Biol. 19:3760–3768. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cohen P and Friedman JM: Leptin and the
control of metabolism: Role for stearoyl-CoA desaturase-1 (SCD-1).
J Nutr. 134:2455S–2463S. 2004.PubMed/NCBI
|
|
22
|
Gastaldelli A, Harrison S, Belfort-Aguiar
R, Hardies J, Balas B, Schenker S and Cusi K: Pioglitazone in the
treatment of NASH: The role of adiponectin. Aliment Pharmacol Ther.
32:769–775. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kersten S, Desvergne B and Wahli W: Roles
of PPARs in health and disease. Nature. 405:421–424. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Knopp RH: Drug treatment of lipid
disorders. N Engl J Med. 341:498–511. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Postic C and Girard J: Contribution of de
novo fatty acid synthesis to hepatic steatosis and insulin
resistance: Lessons from genetically engineered mice. J Clin
Invest. 118:829–838. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Srere PA: The citrate cleavage enzyme. I
Distribution and purification. J Biol Chem. 234:2544–2547.
1959.PubMed/NCBI
|
|
27
|
Abu-Elheiga L, Jayakumar A, Baldini A,
Chirala SS and Wakil SJ: Human acetyl-CoA carboxylase:
Characterization, molecular cloning, and evidence for two isoforms.
Proc Natl Acad Sci USA. 92:4011–4015. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
van der Leij FR, Kram AM, Bartelds B,
Roelofsen H, Smid GB, Takens J, Zammit VA and Kuipers JR:
Cytological evidence that the C-terminus of carnitine
palmitoyltransferase I is on the cytosolic face of the
mitochondrial outer membrane. Biochem J. 341:777–784. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park EA, Mynatt RL, Cook GA and Kashfi K:
Insulin regulates enzyme activity, malonyl-CoA sensitivity and mRNA
abundance of hepatic carnitine palmitoyltransferase-I. Biochem J.
310:853–858. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bartlett K and Eaton S: Mitochondrial
beta-oxidation. Eur J Biochem. 271:462–469. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hegarty BD, Turner N, Cooney GJ and
Kraegen EW: Insulin resistance and fuel homeostasis: The role of
AMP-activated protein kinase. Acta Physiol (Oxf). 196:129–145.
2009. View Article : Google Scholar
|
|
32
|
Saltiel AR and Kahn CR: Insulin signalling
and the regulation of glucose and lipid metabolism. Nature.
414:799–806. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kubota N, Kubota T, Itoh S, Kumagai H,
Kozono H, Takamoto I, Mineyama T, Ogata H, Tokuyama K, Ohsugi M, et
al: Dynamic functional relay between insulin receptor substrate 1
and 2 in hepatic insulin signaling during fasting and feeding. Cell
Metab. 8:49–64. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Guo S, Copps KD, Dong X, Park S, Cheng Z,
Pocai A, Rossetti L, Sajan M, Farese RV and White MF: The Irs1
branch of the insulin signaling cascade plays a dominant role in
hepatic nutrient homeostasis. Mol Cell Biol. 29:5070–5083. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cernea S and Raz I: Therapy in the early
stage: Incretins. Diabetes Care. 34(Suppl 2): S264–S271. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Valera A, Pujol A, Pelegrin M and Bosch F:
Transgenic mice overexpressing phosphoenolpyruvate carboxykinase
develop non-insulin-dependent diabetes mellitus. Proc Natl Acad Sci
USA. 91:9151–9154. 1994. View Article : Google Scholar : PubMed/NCBI
|