1. Introduction
Protein phosphorylation is one of the most common
and important post-translational modifications (PTMs) (1,2).
This reversible mechanism occurs through protein kinases and
consists of the addition of a phosphate group (PO4) to
the polar group R of various amino acids. Consequently, this
addition modifies the protein from hydrophobic apolar to
hydrophilic polar, allowing the protein to change conformation when
interacting with other molecules. A phosphorylated amino acid can
bind molecules able to interact with other proteins and
consequently assemble and detach proteic complexes (3).
The interactive capacity of the phosphate group is
mainly due to its components. One of its main elements is
phosphorus. It has five outer electrons able to form a maximum of
five covalent bonds, has three pKas, high water
solubility and it can form, for its versatility, mono, di and
trialkyl and aryl esters with hydroxyl groups, but also acid
anhydrides (4).
In particular, many cellular phosphate esters are
phosphoproteins that form, via a catalytic enzyme and adenosine
triphosphate (ATP), a phosphate anhydride, acting as a donor of a
phosphate group.
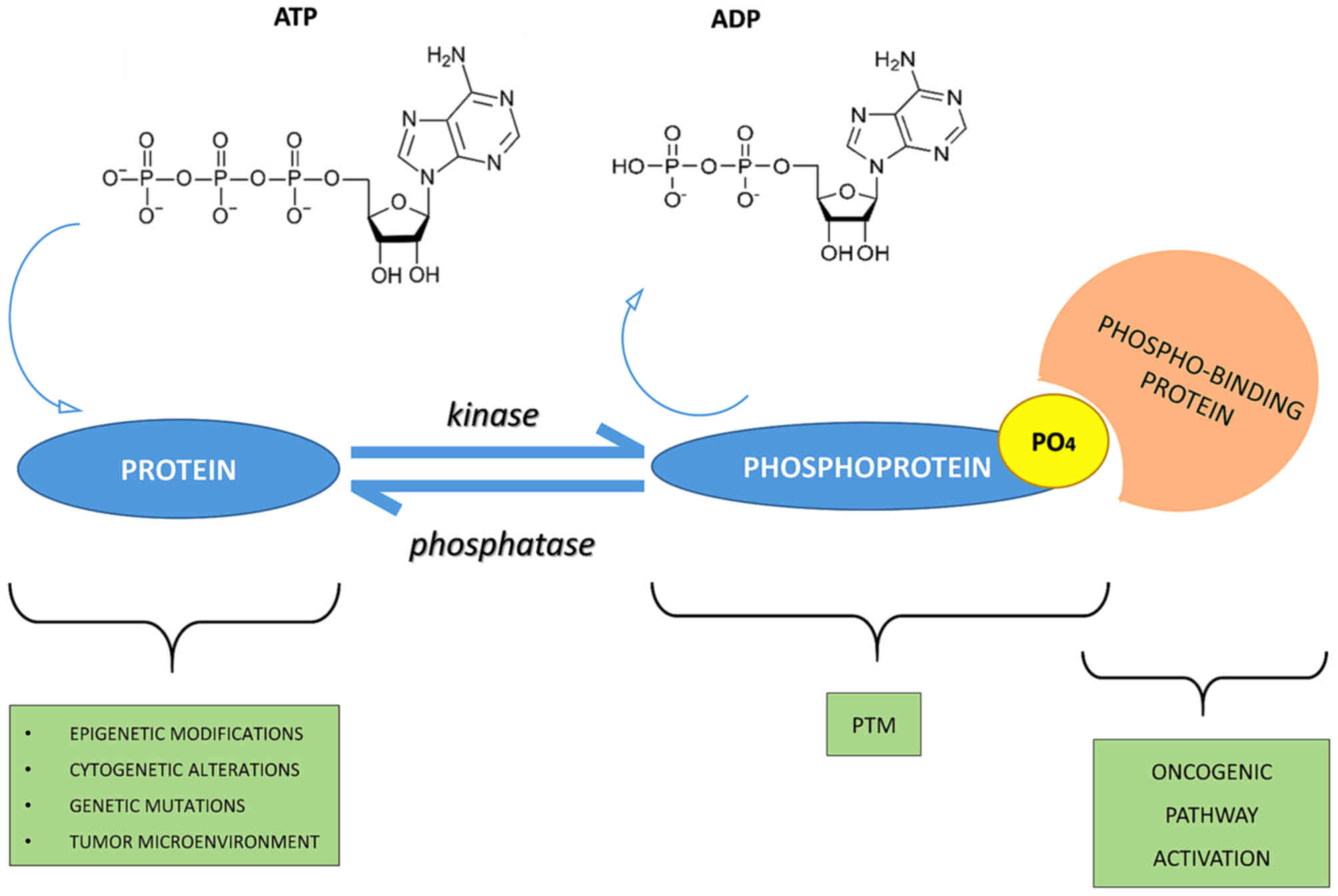
A good energy balance also favors phosphorylation.
Indeed, there is a constant balance between phosphorylation and
dephosphorylation events mediated by kinases, phosphatases, ATP
and/or ADP (protein + ATP ⇄ phosphoprotein + ADP) (5,6)
(Fig. 1).
The Cell Signaling Technology PhosphoSitePlus
(www.phosphosite.org) and the Kinexus
PhosphoNET (www.phosphonet.ca) websites both list
over 200,000 known human phosphosites, and the Kinexus website
predicts another 760,000 additional sites that are likely to be
phosphorylated. More than two-thirds of the 21,000 proteins encoded
by the human genome has been shown to be phosphorylated, and it is
likely that more than 90% are actually subjected to this type of
PTM.
More than one-third of the protein phosphorylation
events occurs on serine (Ser or S), threonine (Thr or T), and
tyrosine residues (Tyr or Y) (O-phosphorylation) (7). In particular, the phosphorylated
residues of serine are 86.4%, followed by residues of threonine
11.8% whereas only 1.8% of tyrosine residues are phosphorylated
(8,9). Tyrosine phosphorylation is
relatively rare compared to the other PTMs and is typical of the
epidermal growth factor receptor (EGFR) family, which owns a domain
called, precisely, tyrosine kinase. Sometimes, phosphorylation of
histidine (His or H) and aspartate residues (Asp or D)
(N-phosphorylation) also occurs, but, in both cases, this
phosphorylation is less stable than others.
Protein phosphorylation is a mechanism of regulation
that is extremely important in most cellular processes such as
protein synthesis, cell division, signal transduction, cell growth,
development and aging as many enzymes and receptors are activated
and deactivated via phosphorylation/dephosphorylation events due to
specific kinases and phosphatases (10).
The human genome, in fact, includes approximately
568 protein kinases and 156 protein phosphatases that regulate
phosphorylation events and, therefore, play an important role in
the control of biological processes such as proliferation,
differentiation and apoptosis.
For instance, p53 protein is activated by
phosphorylation and is then able to stimulate transcription of
genes to inhibit the cell cycle, activate DNA repair and in some
cases lead to apoptosis (13). An
imbalance in the mechanism of phosphorylation/dephosphorylation of
the p53 protein can lead to a chronic inactivation of the protein
itself, which in turn can transform the cell into a cancer
cell.
2. Protein kinases
The protein kinases belong to the great family of
kinases and are responsible for the mechanism of phosphorylation.
They are activated by phosphorylation which in turn activates a
cascade of events leading to the phosphorylation of different amino
acids (3). Activation or
deactivation of kinase occurs in different ways: through the kinase
itself with a cis-phosphorylation/autophosphorylation, by
binding with activator or inhibitor proteins or checking their
localization in the cell in relation to their substrate (7).
The catalytic domain of protein kinase has 2
subdomains, N- and C-terminal (8). Both are connected by a peptidic
stand, which forms an active site with a front pocket (catalytic
residues) and a back pocket. Access to the rear pocket is
controlled by a conserved lysine residue and a residue
'gatekeeper'. The catalytic domain is unavailable when it is active
because propellers of the N- and C-terminal subdomains rotate
inward. The activation of the catalytic domain occurs through
phosphorylation of the activation loop or through an allosteric
mechanism (8). Moreover, the
kinases also have non-catalytic domains allowing the attachment of
substrates and the recruitment of other signaling proteins
(9).
Up to 30% of all human proteins may be modified by
kinase activity, and kinases are known to regulate the majority of
cellular pathways, especially those involved in signal transduction
(10).
In the last few years, kinases have been considered
important not only for their crucial role in signaling but also for
the transduction of the signal, controlling its amplitude (11–13). To facilitate the study of
phospho-signaling networks, different databases have been designed
(2,14,15).
The 518 human protein kinases are classified
according to the amino acid residue that it phosphorylates. Most
kinases act on both serine and threonine (serine/threonine kinases;
STKs), others act on tyrosine (tyrosine kinases; TKs), and a number
act on all three (dual-specificity kinases; DSKs) (16). The latter can phosphorylate STKs
and TKs (17); at least 125 of
the human protein kinases are STKs (18).
The STKs are enzymes that phosphorylate the OH group
of serine or threonine, and are activated by different events such
as DNA damage or chemical signals mediated for instance by
Ca2+/calmodulin, cyclic-adenosine
monophosphate/cyclic-guanosine monophosphate (cAMP/cGMP) and
diacylglycerol.
There are also the following subfamilies of protein
kinases: AGC, CaMK, CK1, CMGC, STE, TK and TKL (19–27) (Table
I).
 | Table ISubfamilies of protein kinases. |
Table I
Subfamilies of protein kinases.
| Protein kinase
family | Origin of the
name | Description | Refs. |
|---|
| AGC | Named after the
protein kinase A, G and C families (PKA, PKC,
PKG) | Subgroup of Ser/Thr
protein kinases that, based on sequence alignments of their
catalytic kinase domain, are related to cAMP-dependent protein
kinase 1 (PKA; also known as PKAC), cGMP-dependent protein kinase
(PKG; also known as CGK1α) and protein kinase C (PKC) | (20) |
| CaMK | Ca2+/calmodulin-dependent
protein kinases | CaMKs transfer
phosphates from ATP to serine or threonine residues in proteins in
response to increase in concentration of intracellular calcium
ions. They are important for expression of various genes because
after activation, CAMKs phosphorylate several transcription
factors. Members of this enzyme class include: CaMK I, CaMK II,
CaMK III, CaMK IV and CaMK V | (21) |
| CK1 | Originally known as
casein
kinase
1 and now
renamed cell
kinase
1 | CK1 family of
monomeric serine-threonine protein kinases. This family has seven
members and are serine/threonine-selective enzymes that function as
regulators of signal transduction pathways. CK1 isoforms are
involved in Wnt signaling, circadian rhythms, nucleocytoplasmic
shuttling of transcription factors, DNA repair and DNA
transcription | (22) |
| CMGC | Named after another
set of families (CDK, MAPK, GSK3 and CLK) | CDKs regulate cell
progression through the different phases of the cell cycle | |
| MAP kinases are
signal transduction molecules and they play a key role in the
regulation of many cellular processes such as proliferation,
differentiation and death. Abnormalities in MAP kinase cascades are
tightly linked to oncogenic transformation | (23) |
| GSK3, initially
described as a key enzyme involved in glycogen metabolism, is now
known to regulate a diverse array of functions. GSK3 is a
well-established component of the Wnt pathway, which is essential
for establishing the entire body pattern during embryonic
development | (28) |
| CLK encodes
phosphorylation of serine/arginine-rich proteins involved in
pre-mRNA processing, releasing them into the nucleoplasm and it may
play an indirect role in governing splice site selection | (25) |
| STE | Sterile kinase | The STE group
consists of three main families, which sequentially activate each
other to then activate the MAPK family. The Ste7 family directly
phosphorylates MAPKs, while many Ste20 members (MAP4K) act on Ste11
kinases. The Ste20 (MAP4K) family is the largest of the three and
is divided into many subfamilies. Some are implicated in MAPK
cascades, while others are not and may have completely distinct
functions | (26) |
| TK | Tyrosine kinase | Members of the TK
group specifically phosphorylate tyrosine residues and are
therefore distinct from dual specificity kinases, which
phosphorylate serine/threonine in addition to tyrosine. TKs are
cell surface receptors (RTKs) and many of the others function close
to the surface of the cell | (19) |
| TKL | Tyrosine kinase-like | Tyrosine
kinase-like kinases are serine-threonine protein kinases named so
because of their close sequence similarity to tyrosine kinases.
Members of this family include MLK, RAF, STKR, LRRK, LISK, IRAK and
RIPK | (27) |
3. Protein phosphatases
Phosphatases have the opposite function of kinases.
They remove the phosphate group from phosphoproteins by hydrolyzing
phosphoric acid monoesters into a phosphate group and a molecule
with a free hydroxyl group (28,29).
Enzymatic removal reverts the protein to a
non-phosphorylated state with a kinetics more rapid than kinases
(30). When working with proteins
in the laboratory, phosphatases are inactivated using denaturation
or inhibitors so phosphorylation inside of a sample is not lost
(31).
The protein phosphatases are considered passive
housekeeping enzymes compared with protein kinases; their different
structure makes them harder to identify and less important than the
protein kinases (32).
Currently, there are approximately 226 known protein
phosphatases (33) which are
classified into 3 families: phosphoprotein phosphatase (PPP)
family, metallo-dependent protein phosphatase (PPM) family and
protein-tyrosine phosphatase (PTP) family (19).
The PPP family includes PP1, PP2A, PP2B and PP4–7
responsible for many dephosphorylation reactions (1) whereas PP2C is one of the most
important members of the PPM family (34). PPP and PMP groups are responsible
for most dephosphorylation reactions of phosphoserine and
phosphotreonine (pSer/pThr) and they can also dephosphorylate
phosphotyrosine (pTyr) (35,36). However, pSer/pThr have different
domain sequences compared with pTyr (37).
In contrast, the phosphatases that belong to the PTP
family have the same catalytic domain but different selectivity for
phosphorylated proteins (38,39). Of all the phosphatases, at least
100 belong to those that dephosphorylate tyrosine residues, such as
the Tyr-specific phosphatase subfamily, the Cdc25 family and
myotubularin-related phosphatase and low molecular weight Tyr
phosphatase (12,14,32,33,39,40). Aspartate-based phosphatases, such
as FCP/SCP (small CTD phosphatase) (41–43) and TAD (haloacid dehalogenase)
family enzymes are part of the PTP group (44).
PTPs are well-known as they can also dephosphorylate
non-protein targets such as carbohydrates, mRNA and
phosphoinositides (12,45–47).
4. Activities and role of protein
phosphorylation under physiological conditions
Protein phosphorylation is one of the initial steps
that is vital for the coordination of cellular and organic
functions such as the regulation of metabolism, proliferation,
apoptosis, subcellular trafficking, inflammation, and other
important physiological processes.
There are several ways in which the phosphorylation
acts to fulfill its role. First, the activity of
phosphorylation/dephosphorylation acts as a molecular switch
(Fig. 1).
For instance, protein kinase B is only activated
follo wing phosphorylation of its Ser and Thr residues and, thus,
is able to regulate cell survival (48); on the other hand, when
proto-oncogene tyrosine-protein kinase (c-Src) is dephosphorylated,
it is turned off causing a block in the regulation of cell growth
(49).
Another mode of phosphorylation is temporary
protein-protein interaction, which allows the adjustment of many
signaling pathways (50,51). An example is glomerular podocyte
protein nephrin 1 (Neph1), an important protein of renal cells,
which once phosphorylated by Src Fyn, interacts with Grb2, an
adapter protein involved in signal transduction and cell
communication (52).
In addition, the phosphorylation of a protein can
regulate the process of signal transduction since it is able to
trigger the subcellular translocation of the protein phosphorylated
by the mechanism itself.
Moreover, phosphorylation on
serine/threonine-protein kinase (Ser350) residue of the
death-associated protein (DAP) leads to the translocation from the
cytoplasm to the nucleus of apoptosis-inducing kinase 2 (DRAK2)
which is able to induce apoptosis in T and B cells (53).
Another example is the membrane translocation of
synaptosomal-associated protein 25 (SNAP25). After being
phosphorylated, SNAP25 has a reduced binding affinity for
syntaxin-1A and thus changes location (54,55).
Furthermore, phosphorylation is mainly involved in
the production and recycling of ATP and, therefore, is important in
biological reactions that require energy (5,56).
Sometimes, protein phosphorylation may promote the
formation or removal of a second PTM (4,57)
(Fig. 1). An example is the
phosphorylation of insulin receptor substrate-1 (IRS-1), a mediator
of insulin signaling pathway, by the ribosomal protein S6 kinase β1
that induces polyubiquitination of IRS-1 due to E3 ligase-CUL7 and
its successive proteasomal degradation (58).
The processes of phosphorylation and
dephosphorylation can be etremely complex, since a single kinase or
phosphatase may simultaneously have more substrates and may
function in various cell signaling pathways. One of the signaling
pathways known for this function is mitogen activated protein
kinase (MAPK/ERK). It is activated through phosphorylation of MAPK
which in turn phosphorylates many substrates, including 40S
ribosomal protein S6 kinase (RSK, which then phosphorylates
ribosomal protein S6), c-Myc and MNK (which then phosphorylates
CREB in this cascade) (59,60).
MAPK is a known protein involved in a signaling
pathway activated by a cascade effect of phosphorylation events
(61). The binding of
interferon-γ (IFN-γ) to its receptor induces phosphorylation of the
Tyr-440 receptor, which promotes the formation of a complex with
the tyrosine kinase JAK1 and JAK2. This complex phosphorylates
Stat1, leading to its dimerization and nuclear translocation, where
it regulates gene transcription (62–65).
Therefore, phospho-signaling networks represent the
basis of many cellular processes. They consist mainly of protein
kinases, phosphatases, and their respective substrates
phospho-binding proteins (66)
(Fig. 1).
There is also a mechanism of competition for kinases
and phosphatases at the level of protein sites to adjust the states
of phosphorylation of common substrates (67,68).
PhosphoNET and PhosphoSitePlus websites document the
inhibition or activation of human protein with more than 850
different phosphosites with predictions for over 1,000 additional
sites.
Therefore, it is customary to classify
phosphorylation into two categories: one refers to functional
changes (stable) and the other one, transitory, has no effect on
regulatory functions. For this reason, it is thought that all
stable phosphosites are functional and those not stable, are not
functional (69–71).
In addition, the functional effects of phosphosites
within a protein are site-dependent (72), and this means that they are
functional only if phosphorylation takes place on a specific site
and not random. This endorses the view that the detailed study of
phosphorylation networks may help to understand the physiological
and pathological mechanisms (72–76).
5. Protein phosphorylation and cancer
Phosphorylation is one of the most common PTMs
involved in the regulation of multiple biological processes and
overexpression of kinase. Mutations or defects in regulatory
mechanisms can lead to aberrant activation or dysregulation of
kinase signaling pathways (77)
and this is the basis of oncogenesis for multiple tumors (78–80).
Cancer is not only considered a disease that arises
from genetic mutations, but also a disease that results from
epigenetic changes (81–83) that mainly lead to a deregulation
of signal transduction pathways with subsequent changes in normal
cellular mechanisms (84).
Many key regulatory proteins controlling gene
expression are targets of kinases. The addition of a phosphate
group to a protein by a kinase can alter the activity of the
protein and this action is often exploited as a switch on or off
(85,86).
In chronic myeloid leukemia, a particular
chromosomal translocation (Philadelphia chromosome) was identified
that generates a novel kinase that is always active, the
retinoblastoma, pRb. The process normally controlled by this kinase
is stuck in the 'on' position. This leads to the proliferation of
tumor cells (87).
D. Stehelin was one of the first researchers to
understand the direct involvement of protein kinases in tumors,
with the study of the oncogene v-SRC (88). This tyrosine kinase with the
phosphate group of Tyr-527 has a key role in tumor cell
proliferation, and has been studied extensively in Rous sarcoma
virus (RSV) as the main cause of sarcoma in chickens (89–92). Its carcinogenic action is due to a
mutation of the carboxyl terminal of the molecule able to eliminate
the tyrosine residue, which causes conformational changes and also
an irregular unregulated autophosphorylation, leading to a signal
of increased growth (93,94).
Aberrations of kinases have been reported in
different types of cancer. An example is the amplification of
Her2/neu observed in tumor cells of invasive breast cancer
(95,96).
Phosphorylation plays a key role even in oral
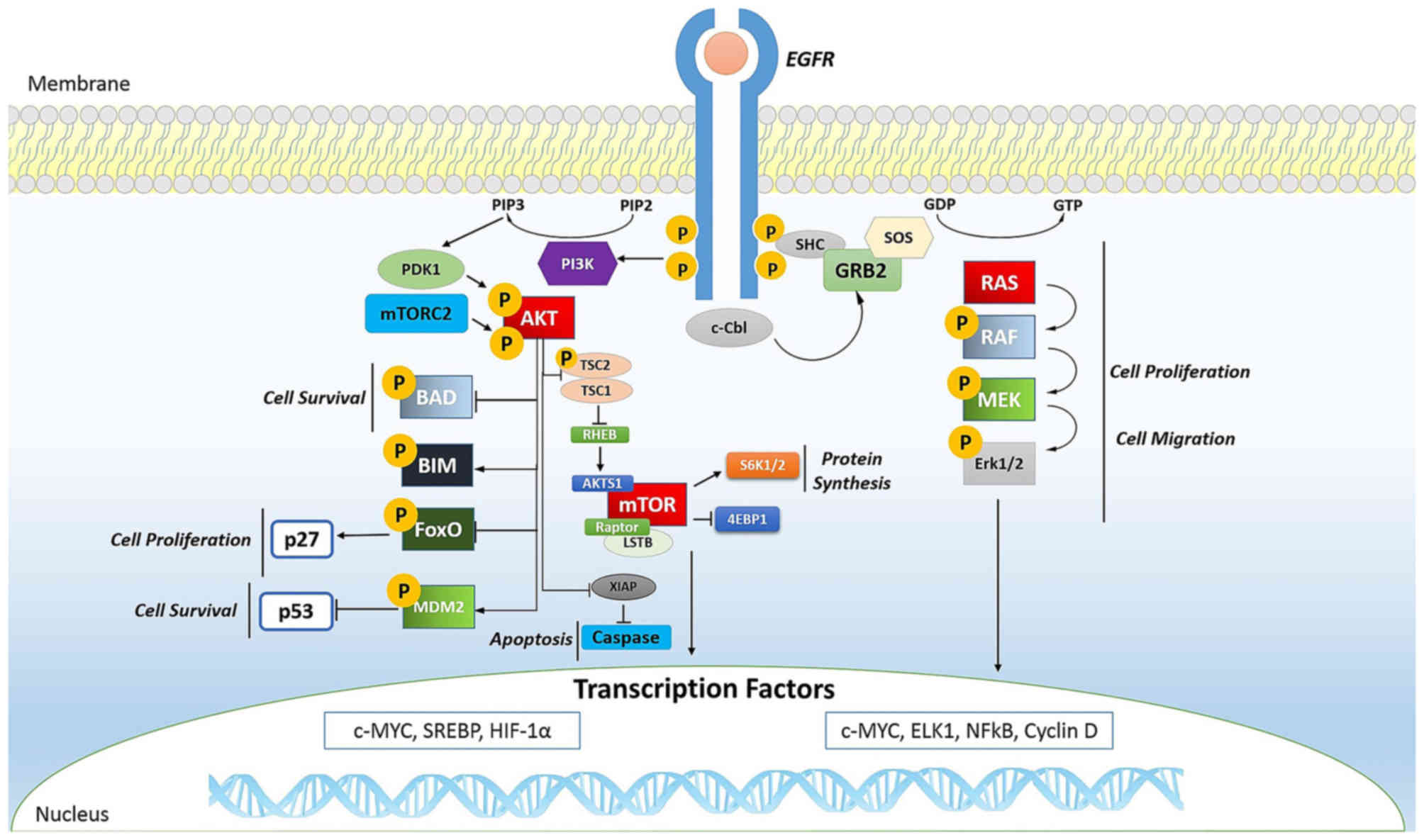
cancer. In fact, the phosphorylation of EGFR (Fig. 2) is important for transactivation
of interleukin (IL)-1β through the CXCL1 and CXCR2 axis (97).
Alteration of the phosphoproteome also affects
gastrointestinal stromal tumors (GISTs) (98,99), lung cancer (73,100), hematologic malignancies
(101,102), breast cancer (103,104), pancreatic cancer (105,106) and prostate cancer (103,107).
To date, more than 1,000 variations in the
expression of protein kinases have been detected in human tumors
(108–111). Many of these kinases are now
considered cancer biomarkers, such as EGFR for colon cancer, cKIT
for GISTs, and human EGFR-related gene (Her2) for breast cancer
(112).
In tumors, mTOR (Fig.
2) is a kinase and, when activated, is able to induce
activation of its downstream effectors and consequently increases
the synthesis of cell cycle proteins such as hypoxia-inducible
factor-1 (HIF-1) (cyclin D and HIF-1α) which in turn stimulates
vascular endothelial growth factor (VEGF) (113,114). mTOR is particularly active in
renal cancer by promoting angiogenesis (115,116).
The Ras oncogene (Fig.
2) is the most common in human tumors (117) and its activation is very
complex, characterized by countless phosphorylation events. It
begins with the binding of a ligand to a receptor tyrosine kinase
(RTK) loca ted on the plasma membrane. This receptor is activated
only if it dimerizes with another RTK. They then phosphorylate each
other and become activated. The activated receptor binds to the SH2
domain of the adapter protein Grb2, which plays its role without
being phosphorylated. In fact, its SH3 domain binds to the protein
activating SOS, without any phosphorylation reactions. SOS then
moves close to the plasma membrane where it can bind Ras, replacing
GDP with GTP and then becomes activated; SOS therefore acts as a
nucleotide exchange factor (GEF). It is known that activated Ras
binds to the N-terminus of a protein Ser/Thr kinase called c-Raf,
activating them (118).
6. Protein kinases as drug targets
The signaling pathways regulated by protein kinases
contribute to the onset and progression of almost all types of
cancer. Consequently, research of the signaling pathways mediated
by kinase and therefore the possibility of blocking them with
targeted treatment could have major clinical-therapeutic utility
especially since many of these proteins act as oncogenes (78,119,120).
Considerable advances have led to the identification
of inhibitors directed against activated tyrosine kinases in
cancer, 17 of which are already used for the treatment of several
cancers and more than 390 molecules are being tested (121).
Imatinib (Glivec®) is a known inhibitor
that blocks the action of the tyrosine kinase BCR-Abl in patients
with chronic myeloid leukemia (CML) (122,123). This drug also targets against
PI3K in solid tumors (124,125), serine/threonine kinase BRAF to
treat melanoma (126–128), the receptor tyrosine kinase EGFR
for lung cancer (129,130), and serine/threonine kinase mTOR
for the treatment of renal tumors (131).
Gefitinib/erlotinib (Tarceva®) is
targeted against EGFR in lung tumors (132) with a success rate of 71.2%
(129), whereas crizotinib
(Xalkori®) acts in the same tumor against EML4-ALK
(133).
Vemurafenib (Zelboraf®) in melanoma is
directed against mutations of BRAF V600E (134) with a successful response of 48%
during treatment (127).
If overexpressed, HER2 is a protein tyrosine kinase
which enhances the proliferation of cancer cells, and enhances the
formation of blood vessels thereby increasing the invasiveness of
breast cancer. Currently, improvements in the prognosis of this
cancer are due to the use of trastuzumab (Herceptin®) a
monoclonal antibody targeted against this protein (135).
Sorafenib (Nexavar®) is another kinase
inhibitor which blocks the action of Raf kinase in kidney and liver
tumors (136).
Sunitinib (Sutent®) is a multi-targeted
receptor tyrosine kinase inhibitor that acts against
platelet-derived growth factor receptors (PDGFRs) and VEGF
receptors (VEGFRs) (137). The
simultaneous inhibition of these targets induces a reduction in
tumor vascularization and triggers cancer cell apoptosis. It has
been recommended as a drug in renal cell carcinoma and in GISTs
(138,139). Furthermore, since sunitinib
targets many different receptors, it leads to dermatologic toxic
side effects such as hand-foot syndrome (140).
Temsirolimus (Torisel®) is a drug used
for the treatment of renal cell carcinoma and it is a specific
inhibitor of mTOR (141), a
cellular kinase enzyme that may favor tumor growth. Temsirolimus
leads to cell cycle arrest in the G1 phase, and also inhibits tumor
angiogenesis by reducing synthesis of VEGF (142).
The success of therapies based on kinase inhibitors
relies on different aspects: the clinical targeted kinase, the
structure of the signaling network and the mechanisms of innate or
acquired resistance.
First of all, both the patients and the therapeutic
approach functions must be appropriately selected (119). For instance, in CML, this
therapy only works with BCR-Abl-positive patients as the main
targets of therapy are BCR and ABL (Philadelphia chromosome genes)
fused together by means of an activated protein tyrosine
kinase.
In the same way, the response rate of Herceptin was
34% in patients whose tumors had amplified HER2 compared with 7% in
those whose tumors did not (143).
However, not all tumors respond to inhibitors of
kinases and often patients with the same cancer respond differently
to the same therapy. For this reason, patients should be further
stratified using biomarkers (144) and further studies are warranted
to investigate the signaling pathways (145–147).
In this respect, we know that changes in the
signaling pathways, caused by several factors (genetic and
epigenetic mutations, alterations of the microenvironment), lead to
the formation of oncogenes and, very often, there is a release of
tumoral molecules that can be tracked and used as biomarkers. For
example, hepatocyte growth factor (HGF) released in stromal cells
of melanoma, influences the way BRAF inhibitors act (148).
The signaling networks of cancer cells can also
develop innate or acquired resistance, since they are able to
create the most common or rare oncogenic mutations different from
tumor to tumor (the so-called polygenic tumor biology) (149).
There are two main types of resistance to a drug
treatment based on kinase inhibitors. Intrinsic or innate
resistance (on target) occurs when the drug target protein has
changed due to steric hindrance to inhibitor binding (150), altered active site topography
(151), disruption of favorable
inhibitor interactions (152),
altered protein dynamics (153),
increased oncogenicity (151),
and alteration of ATP affinity (154). In this way, this resistance is
not inhibited by the drug and continues to perform its normal
activity in the tumor cell.
In extrinsic or acquired resistance (off target),
the signaling network is able to restore the function of oncogenic
mutation in the signaling network (155,156) or via bypass/compensatory
signaling or even through feedback loops to adjust the signal. The
cancer cells are able in fact to exploit and reactivate the
mechanism of signaling that the drug would inhibit (157). In addition, during treatment
acquired resistance can occur and the tumors can develop subclones
which foster even relapse.
New studies of the signaling network of tumors with
particular attention to the mechanism of action of drug inhibitors
of protein kinases are therefore needed.
7. Conclusions
Phosphoproteomics has a critical relevance for many
aspects of biology and has a significant role for understanding the
molecular mechanisms, especially those that lead to the genesis and
growth of tumors (77–79). Signaling networks in which protein
kinases operate are highly complex, but we believe that
understanding the regulatory functions of kinases may be a valid
means to identify more effective therapies against cancer (146,147). Many drug kinase inhibitors are
already on the market (122,133–137) but, often, their effectiveness is
reduced due to the development of complex mechanisms of drug
resistance (149).
However, great progress has been made in recent
years thanks to the numerous techniques of proteomics. Proteomics
is the most important way by which to study the sites and behavior
of phosphoprotein and phosphosite in tumor biology. The
identification of biomarkers that aid in the selection of the most
appropriate therapy for individual patients remains a major
challenge (144).
Glossary
Abbreviations
Abbreviations:
|
ADP
|
adenosine diphosphate
|
|
AGC kinases
|
protein kinase A, G and C families
|
|
Asp or D
|
aspartate residues
|
|
ATP
|
adenosine triphosphate
|
|
CaMK
|
Ca2+/calmodulin-dependent
protein kinase class
|
|
cAMP/cGMP
|
cyclic-adenosine
monophosphate/cyclic-guanosine monophosphate
|
|
Cdc25
|
cell division cycle 25 phosphatase
|
|
CK1
|
casein kinase 1
|
|
CML
|
chronic myeloid leukemia
|
|
c-Src
|
proto-oncogene tyrosine-protein
kinase
|
|
DAP protein
|
death-associated protein
|
|
DRAK2
|
apoptosis-inducing kinase 2
|
|
DSKs
|
dual-specificity kinases
|
|
EGF
|
epidermal growth factor
|
|
GISTs
|
gatrointestinal stromal tumors
|
|
Grb2
|
growth factor receptor-bound protein
2
|
|
HGF
|
hepatocyte growth factor
|
|
HIF-1α
|
hypoxia-inducible factor-1α
|
|
His or H
|
histidine residues and
N-phosphorylation
|
|
IFN-γ
|
interferon-γ
|
|
IRS-1
|
insulin receptor substrate-1
|
|
mRNA
|
messenger ribonucleic acid
|
|
Neph1
|
glomerular podocyte protein nephrin
1
|
|
PDGFRs
|
platelet-derived growth factor
receptors
|
|
PO4
|
phosphate group
|
|
PP1
|
phosphoprotein phosphatase 1
|
|
PP2A
|
phosphoprotein phosphatase 2A
|
|
PP2B
|
phosphoprotein phosphatase 2B
|
|
PP2C
|
phosphoprotein phosphatase 2C
|
|
PP4–7
|
phosphoprotein phosphatase 4–7
|
|
PPM
|
metallo-dependent protein phosphatase
family
|
|
PPP
|
phosphoprotein phosphatase family
|
|
pSer/pThr
|
phosphoserine and
phosphothreonine
|
|
PTMs
|
post-translational modifications
|
|
PTP
|
protein-tyrosine phosphatase
family
|
|
pTyr
|
phosphotyrosine
|
|
Ser or S
|
serine residues
|
|
Ser350
|
serine/threonine-protein kinase
|
|
SNAP25
|
synaptosomal-associated protein
25
|
|
STKs
|
serine/threonine kinases
|
|
Thr or T
|
threonine residues
|
|
TKs
|
tyrosine kinases
|
|
TKLs
|
tyrosine kinase-like kinases
|
|
Tyr or Y
|
tyrosine residues
|
|
VEGFRs
|
vascular endothelial growth factor
receptors
|
References
|
1
|
Li X, Wilmanns M, Thornton J and Köhn M:
Elucidating human phosphatase-substrate networks. Sci Signal.
6:rs102013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sacco F, Perfetto L, Castagnoli L and
Cesareni G: The human phosphatase interactome: an intricate family
portrait. FEBS Lett. 586:2732–2739. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Alberts B, Johnson A, Lewis J, Raff M,
Roberts K and Walter P: Molecular Biology of the Cell. Anderson M
and Granum S: 5th edition. Garland Science; New York, NY: pp.
1752007
|
|
4
|
Hunter T: Why nature chose phosphate to
modify proteins. Philos Trans R Soc Lond B Biol Sci. 367:2513–2516.
2009. View Article : Google Scholar
|
|
5
|
Fukami Y and Lipmann F: Reversal of Rous
sarcoma-specific immunoglobulin phosphorylation on tyrosine (ADP as
phosphate acceptor) catalyzed by the src gene kinase. Proc Natl
Acad Sci USA. 80:1872–1876. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kole HK, Abdel-Ghany M and Racker E:
Specific dephosphorylation of phosphoproteins by protein-serine and
-tyrosine kinases. Proc Natl Acad Sci USA. 85:5849–5853. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roskoski R Jr: ERK1/2 MAP kinases:
structure, function, and regulation. Pharmacol Res. 66:105–143.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schwartz PA and Murray BW: Protein kinase
biochemistry and drug discovery. Bioorg Chem. 39:192–210. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nishi H, Shaytan A and Panchenko AR:
Physicochemical mechanisms of protein regulation by
phosphorylation. Front Genet. 5:2702014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
McCance KL and Huether SE:
Pathophysiology: The Biologic Basis for Disease in Adults and
Children. Brashers VL and Rote NS: 7th edition. Elsevier; 2014
|
|
11
|
Hornberg JJ, Bruggeman FJ, Binder B, Geest
CR, de Vaate AJ, Lankelma J, Heinrich R and Westerhoff HV:
Principles behind the multifarious control of signal transduction.
ERK phosphorylation and kinase/phosphatase control. FEBS J.
272:244–258. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tonks NK: Protein tyrosine phosphatases:
from genes, to function, to disease. Nat Rev Mol Cell Biol.
7:833–846. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Heinrich R, Neel BG and Rapoport TA:
Mathematical models of protein kinase signal transduction. Mol
Cell. 9:957–970. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liberti S, Sacco F, Calderone A, Perfetto
L, Iannuccelli M, Panni S, Santonico E, Palma A, Nardozza AP,
Castagnoli L, et al: HuPho: the human phosphatase portal. FEBS J.
280:379–387. 2013. View Article : Google Scholar
|
|
15
|
Hatzihristidis T, Liu S, Pryszcz L,
Hutchins AP, Gabaldón T, Tremblay ML and Miranda-Saavedra D:
PTP-central: a comprehensive resource of protein tyrosine
phosphatases in eukaryotic genomes. Methods. 65:156–164. 2014.
View Article : Google Scholar
|
|
16
|
Miller ML, Jensen LJ, Diella F, Jørgensen
C, Tinti M, Li L, Hsiung M, Parker SA, Bordeaux J, Sicheritz-Ponten
T, et al: Linear motif atlas for phosphorylation-dependent
signaling. Sci Signal. 1:–ra2. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hunter T: Tyrosine phosphorylation: thirty
years and counting. Curr Opin Cell Biol. 21:140–146. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Capra M, Nuciforo PG, Confalonieri S,
Quarto M, Bianchi M, Nebuloni M, Boldorini R, Pallotti F, Viale G,
Gishizky ML, et al: Frequent alterations in the expression of
serine/threonine kinases in human cancers. Cancer Res.
66:8147–8154. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin J and Pawson T: Modular evolution of
phosphorylation-based signalling systems. Philos Trans R Soc Lond B
Biol Sci. 367:2540–2555. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Pearce LR, Komander D and Alessi DR: The
nuts and bolts of AGC protein kinases. Nat Rev Mol Cell Biol.
11:9–22. 2010. View Article : Google Scholar
|
|
21
|
Wayman GA, Tokumitsu H, Davare MA and
Soderling TR: Analysis of CaM-kinase signaling in cells. Cell
Calcium. 50:1–8. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Eide EJ and Virshup DM: Casein kinase I:
another cog in the circadian clockworks. Chronobiol Int.
18:389–398. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sundaram MV: RTK/Ras/MAPK signaling.
WormBook: pp. 1–19. 2006, View Article : Google Scholar
|
|
24
|
Cohen P and Goedert M: GSK3 inhibitors:
development and therapeutic potential. Nat Rev Drug Discov.
3:479–487. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Moeslein FM, Myers MP and Landreth GE: The
CLK family kinases, CLK1 and CLK2, phosphorylate and activate the
tyrosine phosphatase, PTP-1B. J Biol Chem. 274:26697–26704. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Müller-Taubenberger A, Ishikawa-Ankerhold
HC, Kastner PM, Burghardt E and Gerisch G: The STE group kinase
SepA controls cleavage furrow formation in Dictyostelium. Cell
Motil Cytoskeleton. 66:929–939. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Abdi AI, Carvalho TG, Wilkes JM and Doerig
C: A secreted Plasmodium falciparum kinase reveals a signature
motif for classification of tyrosine kinase-like kinases.
Microbiology. 159:2533–2547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Barford D: Molecular mechanisms of the
protein serine/thre-onine phosphatases. Trends Biochem Sci.
21:407–412. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang ZY: Protein tyrosine phosphatases:
structure and function, substrate specificity, and inhibitor
development. Annu Rev Pharmacol Toxicol. 42:209–234. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mackintosh C: Protein Phosphorylation: A
Practical Approach. Hardie GD: IRL Press; New York, NY: pp.
3281993
|
|
31
|
Thingholm TE, Larsen MR, Ingrell CR,
Kassem M and Jensen ON: TiO(2)-based phosphoproteomic analysis of
the plasma membrane and the effects of phosphatase inhibitor
treatment. J Proteome Res. 7:3304–3313. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Stern DF: Phosphoproteomics for oncology
discovery and treatment. Expert Opin Ther Targets. 9:851–860. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Liu Y and Chance MR: Integrating
phosphoproteomics in systems biology. Comput Struct Biotechnol J.
10:90–97. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Moorhead GB, De Wever V, Templeton G and
Kerk D: Evolution of protein phosphatases in plants and animals.
Biochem J. 417:401–409. 2009. View Article : Google Scholar
|
|
35
|
Das AK, Helps NR, Cohen PT and Barford D:
Crystal structure of the protein serine/threonine phosphatase 2C at
2.0 A resolution. EMBO J. 15:6798–6809. 1996.PubMed/NCBI
|
|
36
|
Shi Y: Serine/threonine phosphatases:
mechanism through structure. Cell. 139:468–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Virshup DM and Shenolikar S: From
promiscuity to precision: protein phosphatases get a makeover. Mol
Cell. 33:537–545. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Guan KL and Dixon JE: Evidence for
protein-tyrosine-phosphatase catalysis proceeding via a
cysteine-phosphate intermediate. J Biol Chem. 266:17026–17030.
1991.PubMed/NCBI
|
|
39
|
Alonso A, Sasin J, Bottini N, Friedberg I,
Friedberg I, Osterman A, Godzik A, Hunter T, Dixon J and Mustelin
T: Protein tyrosine phosphatases in the human genome. Cell.
117:699–711. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Tonks NK and Neel BG: Combinatorial
control of the specificity of protein tyrosine phosphatases. Curr
Opin Cell Biol. 13:182–195. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wrighton KH, Willis D, Long J, Liu F, Lin
X and Feng XH: Small C-terminal domain phosphatases dephosphorylate
the regulatory linker regions of Smad2 and Smad3 to enhance
transforming growth factor-beta signaling. J Biol Chem.
281:38365–38375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Archambault J, Pan G, Dahmus GK, Cartier
M, Marshall N, Zhang S, Dahmus ME and Greenblatt J: FCP1, the
RAP74-interacting subunit of a human protein phosphatase that
dephosphorylates the carboxyl-terminal domain of RNA polymerase
IIO. J Biol Chem. 273:27593–27601. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Salton SR: Teaching resources. Protein
phosphatases. Sci STKE. 2005:tr82005.PubMed/NCBI
|
|
44
|
Tootle TL, Silver SJ, Davies EL, Newman V,
Latek RR, Mills IA, Selengut JD, Parlikar BE and Rebay I: The
transcription factor eyes absent is a protein tyrosine phosphatase.
Nature. 426:299–302. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Gentry MS, Dixon JE and Worby CA: Lafora
disease: insights into neurodegeneration from plant metabolism.
Trends Biochem Sci. 34:628–639. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Gentry MS, Dowen RH III, Worby CA, Mattoo
S, Ecker JR and Dixon JE: The phosphatase laforin crosses
evolutionary boundaries and links carbohydrate metabolism to
neuronal disease. J Cell Biol. 178:477–488. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Niittylä T, Comparot-Moss S, Lue WL,
Messerli G, Trevisan M, Seymour MD, Gatehouse JA, Villadsen D,
Smith SM, Chen J, et al: Similar protein phosphatases control
starch metabolism in plants and glycogen metabolism in mammals. J
Biol Chem. 281:11815–11818. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Case N, Thomas J, Sen B, Styner M, Xie Z,
Galior K and Rubin J: Mechanical regulation of glycogen synthase
kinase 3β (GSK3β) in mesenchymal stem cells is dependent on Akt
protein serine 473 phosphorylation via mTORC2 protein. J Biol Chem.
286:39450–39456. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cole PA, Shen K, Qiao Y and Wang D:
Protein tyrosine kinases Src and Csk: a tail's tale. Curr Opin Chem
Biol. 7:580–585. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nishi H, Hashimoto K and Panchenko AR:
Phosphorylation in protein-protein binding: effect on stability and
function. Structure. 19:1807–1815. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nishi H, Fong JH, Chang C, Teichmann SA
and Panchenko AR: Regulation of protein-protein binding by coupling
between phosphorylation and intrinsic disorder: analysis of human
protein complexes. Mol Biosyst. 9:1620–1626. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Harita Y, Kurihara H, Kosako H, Tezuka T,
Sekine T, Igarashi T and Hattori S: Neph1, a component of the
kidney slit diaphragm, is tyrosine-phosphorylated by the Src family
tyrosine kinase and modulates intracellular signaling by binding to
Grb2. J Biol Chem. 283:9177–9186. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kuwahara H, Nishizaki M and Kanazawa H:
Nuclear localization signal and phosphorylation of Serine350
specify intracellular localization of DRAK2. J Biochem.
143:349–358. 2008. View Article : Google Scholar
|
|
54
|
Shimazaki Y, Nishiki T, Omori A, Sekiguchi
M, Kamata Y, Kozaki S and Takahashi M: Phosphorylation of 25-kDa
synaptosome-associated protein. Possible involvement in protein
kinase C-mediated regulation of neurotransmitter release. J Biol
Chem. 271:14548–14553. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kataoka M, Kuwahara R, Iwasaki S,
Shoji-Kasai Y and Takahashi M: Nerve growth factor-induced
phosphorylation of SNAP-25 in PC12 cells: a possible involvement in
the regulation of SNAP-25 localization. J Neurochem. 74:2058–2066.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Rosen OM and Erlichman J: Reversible
autophosphorylation of a cyclic 3′:5′-AMP-dependent protein kinase
from bovine cardiac muscle. J Biol Chem. 250:7788–7794.
1975.PubMed/NCBI
|
|
57
|
Hunter T: The age of crosstalk:
phosphorylation, ubiquitination, and beyond. Mol Cell. 28:730–738.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Xu X, Sarikas A, Dias-Santagata DC, Dolios
G, Lafontant PJ, Tsai SC, Zhu W, Nakajima H, Nakajima HO, Field LJ,
et al: The CUL7 E3 ubiquitin ligase targets insulin receptor
substrate 1 for ubiquitin-dependent degradation. Mol Cell.
30:403–414. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Pende M, Um SH, Mieulet V, Sticker M, Goss
VL, Mestan J, Mueller M, Fumagalli S, Kozma SC and Thomas G:
S6K1(−/−)/S6K2(−/−) mice exhibit perinatal lethality and
rapamycin-sensitive 5′-terminal oligopyrimidine mRNA translation
and reveal a mitogen-activated protein kinase-dependent S6 kinase
pathway. Mol Cell Biol. 24:3112–3124. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ferrer I, Blanco R, Carmona M, Puig B,
Domínguez I and Viñals F: Active, phosphorylation-dependent MAP
kinases, MAPK/ERK, SAPK/JNK and p38, and specific transcription
factor substrates are differentially expressed following systemic
administration of kainic acid to the adult rat. Acta Neuropathol.
103:391–407. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Darnell JE Jr, Kerr IM and Stark GR:
Jak-STAT pathways and transcriptional activation in response to
IFNs and other extracellular signaling proteins. Science.
264:1415–1421. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Greenlund AC, Farrar MA, Viviano BL and
Schreiber RD: Ligand-induced IFN gamma receptor tyrosine
phosphorylation couples the receptor to its signal transduction
system (p91). EMBO J. 13:1591–1600. 1994.PubMed/NCBI
|
|
64
|
Igarashi K, Garotta G, Ozmen L, Ziemiecki
A, Wilks AF, Harpur AG, Larner AC and Finbloom DS: Interferon-gamma
induces tyrosine phosphorylation of interferon-gamma receptor and
regulated association of protein tyrosine kinases, Jak1 and Jak2,
with its receptor. J Biol Chem. 269:14333–14336. 1994.PubMed/NCBI
|
|
65
|
Decker T and Kovarik P: Serine
phosphorylation of STATs. Oncogene. 19:2628–2637. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Liu Z, Wang Y and Xue Y:
Phosphoproteomics-based network medicine. FEBS J. 280:5696–5704.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Hirschi A, Cecchini M, Steinhardt RC,
Schamber MR, Dick FA and Rubin SM: An overlapping kinase and
phosphatase docking site regulates activity of the retinoblastoma
protein. Nat Struct Mol Biol. 17:1051–1057. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Salazar C and Höfer T: Competition effects
shape the response sensitivity and kinetics of phosphorylation
cycles in cell signaling. Ann NY Acad Sci. 1091:517–530. 2006.
View Article : Google Scholar
|
|
69
|
Lienhard GE: Non-functional
phosphorylations? Trends Biochem Sci. 33:351–352. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Landry CR, Levy ED and Michnick SW: Weak
functional constraints on phosphoproteomes. Trends Genet.
25:193–197. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Levy ED, Michnick SW and Landry CR:
Protein abundance is key to distinguish promiscuous from functional
phosphorylation based on evolutionary information. Philos Trans R
Soc Lond B Biol Sci. 367:2594–2606. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Olsen JV, Blagoev B, Gnad F, Macek B,
Kumar C, Mortensen P and Mann M: Global, in vivo, and site-specific
phosphorylation dynamics in signaling networks. Cell. 127:635–648.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Rikova K, Guo A, Zeng Q, Possemato A, Yu
J, Haack H, Nardone J, Lee K, Reeves C, Li Y, et al: Global survey
of phosphotyrosine signaling identifies oncogenic kinases in lung
cancer. Cell. 131:1190–1203. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Linding R, Jensen LJ, Ostheimer GJ, van
Vugt MA, Jørgensen C, Miron IM, Diella F, Colwill K, Taylor L,
Elder K, et al: Systematic discovery of in vivo phosphorylation
networks. Cell. 129:1415–1426. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Newman RH, Hu J, Rho HS, Xie Z, Woodard C,
Neiswinger J, Cooper C, Shirley M, Clark HM, Hu S, et al:
Construction of human activity-based phosphorylation networks. Mol
Syst Biol. 9:6552013. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Drake JM, Graham NA, Stoyanova T, Sedghi
A, Goldstein AS, Cai H, Smith DA, Zhang H, Komisopoulou E, Huang J,
et al: Oncogene-specific activation of tyrosine kinase networks
during prostate cancer progression. Proc Natl Acad Sci USA.
109:1643–1648. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Harsha HC and Pandey A: Phosphoproteomics
in cancer. Mol Oncol. 4:482–495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Hanahan D and Weinberg RA: Hallmarks of
cancer: the next generation. Cell. 144:646–674. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Hynes NE and MacDonald G: ErbB receptors
and signaling pathways in cancer. Curr Opin Cell Biol. 21:177–184.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Sharma A, Tan TH, Cheetham G, Scott HS and
Brown MP: Rare and novel epidermal growth factor receptor mutations
in non-small-cell lung cancer and lack of clinical response to
gefitinib in two cases. J Thorac Oncol. 7:941–942. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Vogelstein B and Kinzler KW: Cancer genes
and the pathways they control. Nat Med. 10:789–799. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Petricoin EF, Zoon KC, Kohn EC, Barrett JC
and Liotta LA: Clinical proteomics: translating benchside promise
into bedside reality. Nat Rev Drug Discov. 1:683–695. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Jones PA and Baylin SB: The fundamental
role of epigenetic events in cancer. Nat Rev Genet. 3:415–428.
2002.PubMed/NCBI
|
|
84
|
Hanahan D and Weinberg RA: The hallmarks
of cancer. Cell. 100:57–70. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Tarrant MK and Cole PA: The chemical
biology of protein phosphorylation. Annu Rev Biochem. 78:797–825.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Paul MK and Mukhopadhyay AK: Tyrosine
kinase - role and significance in cancer. Int J Med Sci. 1:101–115.
2004. View Article : Google Scholar
|
|
87
|
Murphree AL and Benedict WF:
Retinoblastoma: clues to human oncogenesis. Science. 223:1028–1033.
1984. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Stehelin D, Guntaka RV, Varmus HE and
Bishop JM: Purification of DNA complementary to nucleotide
sequences required for neoplastic transformation of fibroblasts by
avian sarcoma viruses. J Mol Biol. 101:349–365. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Hunter T and Cooper JA: Protein-tyrosine
kinases. Annu Rev Biochem. 54:897–930. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Sefton BM and Hunter T: From c-src to
v-src, or the case of the missing C terminus. Cancer Surv.
5:159–172. 1986.PubMed/NCBI
|
|
91
|
Sefton BM, Hunter T and Raschke WC:
Evidence that the Abelson virus protein functions in vivo as a
protein kinase that phosphorylates tyrosine. Proc Natl Acad Sci
USA. 78:1552–1556. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Sefton BM, Hunter T, Beemon K and Eckhart
W: Evidence that the phosphorylation of tyrosine is essential for
cellular transformation by Rous sarcoma virus. Cell. 20:807–816.
1980. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Xu W, Doshi A, Lei M, Eck MJ and Harrison
SC: Crystal structures of c-Src reveal features of its
autoinhibitory mechanism. Mol Cell. 3:629–638. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Young MA, Gonfloni S, Superti-Furga G,
Roux B and Kuriyan J: Dynamic coupling between the SH2 and SH3
domains of c-Src and Hck underlies their inactivation by C-terminal
tyrosine phosphorylation. Cell. 105:115–126. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Shawver LK, Slamon D and Ullrich A: Smart
drugs: tyrosine kinase inhibitors in cancer therapy. Cancer Cell.
1:117–123. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Slamon DJ, Clark GM, Wong SG, Levin WJ,
Ullrich A and McGuire WL: Human breast cancer: correlation of
relapse and survival with amplification of the HER-2/neu oncogene.
Science. 235:177–182. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Lee CH, Syu SH, Liu KJ, Chu PY, Yang WC,
Lin P and Shieh WY: Interleukin-1 beta transactivates epidermal
growth factor receptor via the CXCL1-CXCR2 axis in oral cancer.
Oncotarget. 6:38866–38880. 2015.PubMed/NCBI
|
|
98
|
Corless CL, Fletcher JA and Heinrich MC:
Biology of gastrointestinal stromal tumors. J Clin Oncol.
22:3813–3825. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Javidi-Sharifi N, Traer E, Martinez J,
Gupta A, Taguchi T, Dunlap J, Heinrich MC, Corless CL, Rubin BP,
Druker BJ, et al: Crosstalk between KIT and FGFR3 promotes
gastrointestinal stromal tumor cell growth and drug resistance.
Cancer Res. 75:880–891. 2015. View Article : Google Scholar :
|
|
100
|
Sharma SV, Bell DW, Settleman J and Haber
DA: Epidermal growth factor receptor mutations in lung cancer. Nat
Rev Cancer. 7:169–181. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zhu N, Xiao H, Wang LM, Fu S, Zhao C and
Huang H: Mutations in tyrosine kinase and tyrosine phosphatase and
their relevance to the target therapy in hematologic malignancies.
Future Oncol. 11:659–673. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
102
|
Kraus J, Kraus M, Liu N, Besse L, Bader J,
Geurink PP, de Bruin G, Kisselev AF, Overkleeft H and Driessen C:
The novel β2-selective proteasome inhibitor LU-102 decreases
phosphorylation of I kappa B and induces highly synergistic
cytotoxicity in combination with ibrutinib in multiple myeloma
cells. Cancer Chemother Pharmacol. 76:383–396. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
103
|
Jagarlamudi KK, Hansson LO and Eriksson S:
Breast and prostate cancer patients differ significantly in their
serum thymidine kinase 1 (TK1) specific activities compared with
those hematological malignancies and blood donors: implications of
using serum TK1 as a biomarker. BMC Cancer. 15:662015. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Hou S, Isaji T, Hang Q, Im S, Fukuda T and
Gu J: Distinct effects of β1 integrin on cell proliferation and
cellular signaling in MDA-MB-231 breast cancer cells. Sci Rep.
6:184302016. View Article : Google Scholar
|
|
105
|
Paladino D, Yue P, Furuya H, Acoba J,
Rosser CJ and Turkson J: A novel nuclear Src and p300 signaling
axis controls migratory and invasive behavior in pancreatic cancer.
Oncotarget. 7:7253–7267. 2016.
|
|
106
|
Li Z, Lin P, Gao C, Peng C, Liu S, Gao H,
Wang B, Wang J, Niu J and Niu W: Integrin β6 acts as an unfavorable
prognostic indicator and promotes cellular malignant behaviors via
ERK-ETS1 pathway in pancreatic ductal adenocarcinoma (PDAC). Tumour
Biol. 37:5117–5131. 2016. View Article : Google Scholar
|
|
107
|
Meh raein-Ghom i F, Chu rch DR, Sch reiber
CL, Weichmann AM, Basu HS and Wilding G: Inhibitor of p52 NF-κB
subunit and androgen receptor (AR) interaction reduces growth of
human prostate cancer cells by abrogating nuclear translocation of
p52 and phosphorylated AR(ser81). Genes Cancer. 6:428–444.
2015.
|
|
108
|
Barber TD, Vogelstein B, Kinzler KW and
Velculescu VE: Somatic mutations of EGFR in colorectal cancers and
glioblastomas. N Engl J Med. 351:28832004. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Blume-Jensen P and Hunter T: Oncogenic
kinase signalling. Nature. 411:355–365. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Parsons DW, Wang TL, Samuels Y, Bardelli
A, Cummins JM, DeLong L, Silliman N, Ptak J, Szabo S, Willson JK,
et al: Colorectal cancer: mutations in a signalling pathway.
Nature. 436:7922005. View Article : Google Scholar : PubMed/NCBI
|
|
111
|
Stephens P, Edkins S, Davies H, Greenman
C, Cox C, Hunter C, Bignell G, Teague J, Smith R, Stevens C, et al:
A screen of the complete protein kinase gene family identifies
diverse patterns of somatic mutations in human breast cancer. Nat
Genet. 37:590–592. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
112
|
Ludwig JA and Weinstein JN: Biomarkers in
cancer staging, prognosis and treatment selection. Nat Rev Cancer.
5:845–856. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
113
|
Rubio-Viqueira B and Hidalgo M: Targeting
mTOR for cancer treatment. Adv Exp Med Biol. 587:309–327. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Hudson CC, Liu M, Chiang GG, Otterness DM,
Loomis DC, Kaper F, Giaccia AJ and Abraham RT: Regulation of
hypoxia-inducible factor 1alpha expression and function by the
mammalian target of rapamycin. Mol Cell Biol. 22:7004–7014. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
115
|
Dancey JE: Therapeutic targets: MTOR and
related pathways. Cancer Biol Ther. 5:1065–1073. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Thomas GV, Tran C, Mellinghoff IK, Welsbie
DS, Chan E, Fueger B, Czernin J and Sawyers CL: Hypoxia-inducible
factor determines sensitivity to inhibitors of mTOR in kidney
cancer. Nat Med. 12:122–127. 2006. View
Article : Google Scholar
|
|
117
|
Ahmadian MR: Prospects for anti-ras drugs.
Br J Haematol. 116:511–518. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
118
|
Goodsell DS: The molecular perspective:
the ras oncogene. Oncologist. 4:263–264. 1999.PubMed/NCBI
|
|
119
|
Sawyers CL: Shifting paradigms: the seeds
of oncogene addiction. Nat Med. 15:1158–1161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
120
|
Hainaut P and Plymoth A: Targeting the
hallmarks of cancer: towards a rational approach to next-generation
cancer therapy. Curr Opin Oncol. 25:50–51. 2013. View Article : Google Scholar
|
|
121
|
Gonzalez de Castro D, Clarke PA,
Al-Lazikani B and Workman P: Personalized cancer medicine:
molecular diagnostics, predictive biomarkers, and drug resistance.
Clin Pharmacol Ther. 93:252–259. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
122
|
Druker BJ: Imatinib mesylate in the
treatment of chronic myeloid leukaemia. Expert Opin Pharmacother.
4:963–971. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
123
|
Stegmeier F, Warmuth M, Sellers WR and
Dorsch M: Targeted cancer therapies in the twenty-first century:
lessons from imatinib. Clin Pharmacol Ther. 87:543–552. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
124
|
Bachman KE, Argani P, Samuels Y, Silliman
N, Ptak J, Szabo S, Konishi H, Karakas B, Blair BG, Lin C, et al:
The IK3CA gene is mutated with high frequency in human breast
cancers. Cancer Biol Ther. 3:772–775. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
125
|
Serra V, Markman B, Scaltriti M, Eichhorn
PJ, Valero V, Guzman M, Botero ML, Llonch E, Atzori F, Di Cosimo S,
et al: NVP-BEZ235, a dual PI3K/mTOR inhibitor, prevents PI3K
signaling and inhibits the growth of cancer cells with activating
PI3K mutations. Cancer Res. 68:8022–8030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
126
|
Brose MS, Volpe P, Feldman M, Kumar M,
Rishi I, Gerrero R, Einhorn E, Herlyn M, Minna J, Nicholson A, et
al: BRAF and RAS mutations in human lung cancer and melanoma.
Cancer Res. 62:6997–7000. 2002.PubMed/NCBI
|
|
127
|
Chapman PB, Hauschild A, Robert C, Haanen
JB, Ascierto P, Larkin J, Dummer R, Garbe C, Testori A, Maio M, et
al BRIM-3 Study Group: Improved survival with vemurafenib in
melanoma with BRAF V600E mutation. N Engl J Med. 364:2507–2516.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
128
|
Young K, Minchom A and Larkin J: BRIM-1,
-2 and -3 trials: improved survival with vemurafenib in metastatic
melanoma patients with a BRAF(V600E) mutation. Future Oncol.
8:499–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
129
|
Mok TS, Wu YL, Thongprasert S, Yang CH,
Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, et
al: Gefitinib or carboplatin-paclitaxel in pulmonary
adenocarcinoma. N Engl J Med. 361:947–957. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
130
|
Paez JG, Jänne PA, Lee JC, Tracy S,
Greulich H, Gabriel S, Herman P, Kaye FJ, Lindeman N, Boggon TJ, et
al: EGFR mutations in lung cancer: correlation with clinical
response to gefitinib therapy. Science. 304:1497–1500. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
131
|
Motzer RJ, Escudier B, Oudard S, Hutson
TE, Porta C, Bracarda S, Grünwald V, Thompson JA, Figlin RA,
Hollaender N, et al RECORD-1 Study Group: Efficacy of everolimus in
advanced renal cell carcinoma: a double-blind, randomised,
placebo-controlled phase III trial. Lancet. 372:449–456. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
132
|
Abdurahman A, Anwar J, Turghun A, Niyaz M,
Zhang L and Awut I: Epidermal growth factor receptor gene mutation
status and its association with clinical characteristics and tumor
markers in non-small-cell lung cancer patients in Northwest China.
Mol Clin Oncol. 3:847–850. 2015.PubMed/NCBI
|
|
133
|
Ulivi P, Chiadini E, Dazzi C, Dubini A,
Costantini M, Medri L, Puccetti M, Capelli L, Calistri D, Verlicchi
A, et al: Nonsquamous, non-small-cell lung cancer patients who
carry a double mutation of EGFR, EML4-ALK or KRAS: frequency,
clinical-pathological characteristics, and response to therapy.
Clin Lung Cancer. 17:384–390. 2016. View Article : Google Scholar
|
|
134
|
Larkin J, Ascierto PA, Dréno B, Atkinson
V, Liszkay G, Maio M, Mandalà M, Demidov L, Stroyakovskiy D, Thomas
L, et al: Combined vemurafenib and cobimetinib in BRAF-mutated
melanoma. N Engl J Med. 371:1867–1876. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
135
|
Carvajal-Hausdorf DE, Schalper KA, Pusztai
L, Psyrri A, Kalogeras KT, Kotoula V, Fountzilas G and Rimm DL:
Measurement of domain-specific HER2 (ERBB2) expression may classify
benefit from trastuzumab in breast cancer. J Natl Cancer Inst.
107:djv1362015. View Article : Google Scholar : PubMed/NCBI
|
|
136
|
Hasskarl J: Sorafenib: targeting multiple
tyrosine kinases in cancer. Recent Results Cancer Res. 201:145–164.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
137
|
Aita Y, Ishii KA, Saito Y, Ikeda T,
Kawakami Y, Shimano H, Hara H and Takekoshi K: Sunitinib inhibits
catecholamine synthesis and secretion in pheochromocytoma tumor
cells by blocking VEGF receptor 2 via PLC-γ-related pathways. Am J
Physiol Endocrinol Metab. 303:E1006–E1014. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
138
|
Demetri GD, van Oosterom AT, Garrett CR,
Blackstein ME, Shah MH, Verweij J, McArthur G, Judson IR, Heinrich
MC, Morgan JA, et al: Efficacy and safety of sunitinib in patients
with advanced gastrointestinal stromal tumour after failure of
imatinib: a randomised controlled trial. Lancet. 368:1329–1338.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
139
|
Czarnecka AM, Solarek W, Kornakiewicz A
and Szczylik C: Tyrosine kinase inhibitors target cancer stem cells
in renal cell cancer. Oncol Rep. 35:1433–1442. 2016.
|
|
140
|
Lankheet NA, Huitema AD, Mallo H,
Adriaansz S, Haanen JB, Schellens JH, Beijnen JH and Blank CU: The
effect of seasonal variation and secretion of sunitinib in sweat on
the development of hand-foot syndrome. Eur J Clin Pharmacol.
69:2065–2072. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
141
|
Axelsson J, Rippe A and Rippe B: mTOR
inhibition with temsirolimus causes acute increases in glomerular
permeability, but inhibits the dynamic permeability actions of
puromycin aminonucleoside. Am J Physiol Renal Physiol.
308:F1056–F1064. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
142
|
Wan X, Shen N, Mendoza A, Khanna C and
Helman LJ: CCI-779 inhibits rhabdomyosarcoma xenograft growth by an
antiangiogenic mechanism linked to the targeting of
mTOR/Hif-1alpha/VEGF signaling. Neoplasia. 8:394–401. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
143
|
Vogel CL, Cobleigh MA, Tripathy D, Gutheil
JC, Harris LN, Fehrenbacher L, Slamon DJ, Murphy M, Novotny WF,
Burchmore M, et al: Efficacy and safety of trastuzumab as a single
agent in first-line treatment of HER2-overexpressing metastatic
breast cancer. J Clin Oncol. 20:719–726. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
144
|
Cutillas PR: Role of phosphoproteomics in
the development of personalized cancer therapies. Proteomics Clin
Appl. 9:383–395. 2015. View Article : Google Scholar
|
|
145
|
Klempner SJ, Myers AP and Cantley LC: What
a tangled web we weave: emerging resistance mechanisms to
inhibition of the phosphoinositide 3-kinase pathway. Cancer Discov.
3:1345–1354. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
146
|
Robin X, Creixell P, Radetskaya O, Santini
CC, Longden J and Linding R: Personalized network-based treatments
in oncology. Clin Pharmacol Ther. 94:646–650. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
147
|
Elkabets M, Vora S, Juric D, Morse N,
Mino-Kenudson M, Muranen T, Tao J, Campos AB, Rodon J, Ibrahim YH,
et al: mTORC1 inhibition is required for sensitivity to PI3K110α
inhibitors in PIK3CA-mutant breast cancer. Sci Transl Med.
5:196ra992013. View Article : Google Scholar
|
|
148
|
Straussman R, Morikawa T, Shee K,
Barzily-Rokni M, Qian ZR, Du J, Davis A, Mongare MM, Gould J,
Frederick DT, et al: Tumour micro-environment elicits innate
resistance to RAF inhibitors through HGF secretion. Nature.
487:500–504. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
149
|
Murray BW and Miller N: Durability of
kinase-directed therapies - a network perspective on response and
resistance. Mol Cancer Ther. 14:1975–1984. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
150
|
Daub H, Specht K and Ullrich A: Strategies
to overcome resistance to targeted protein kinase inhibitors. Nat
Rev Drug Discov. 3:1001–1010. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
151
|
Yoshida T, Zhang G and Haura EB: Targeting
epidermal growth factor receptor: central signaling kinase in lung
cancer. Biochem Pharmacol. 80:613–623. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
152
|
Balius TE and Rizzo RC: Quantitative
prediction of fold resistance for inhibitors of EGFR. Biochemistry.
48:8435–8448. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
153
|
Dixit A and Verkhivker GM: Hierarchical
modeling of activation mechanisms in the ABL and EGFR kinase
domains: thermodynamic and mechanistic catalysts of kinase
activation by cancer mutations. PLOS Comput Biol. 5:e10004872009.
View Article : Google Scholar : PubMed/NCBI
|
|
154
|
Yun CH, Mengwasser KE, Toms AV, Woo MS,
Greulich H, Wong KK, Meyerson M and Eck MJ: The T790M mutation in
EGFR kinase causes drug resistance by increasing the affinity for
ATP. Proc Natl Acad Sci USA. 105:2070–2075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
155
|
Solit DB, Garraway LA, Pratilas CA, Sawai
A, Getz G, Basso A, Ye Q, Lobo JM, She Y, Osman I, et al: BRAF
mutation predicts sensitivity to MEK inhibition. Nature.
439:358–362. 2006. View Article : Google Scholar
|
|
156
|
Denis MG, Vallée A and Théoleyre S: EGFR
T790M resistance mutation in non small-cell lung carcinoma. Clin
Chim Acta. 444:81–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
157
|
Niederst MJ and Engelman JA: Bypass
mechanisms of resistance to receptor tyrosine kinase inhibition in
lung cancer. Sci Signal. 6:re62013. View Article : Google Scholar : PubMed/NCBI
|