Introduction
As one of the most common causes of end-stage renal
disease, diabetic nephropathy (DN) accounts for the disability and
high mortality rate in patients with diabetes. The incidence of DN
in patients with type 1 or 2 diabetes within 20–25 years of the
onset of the disease is approximately 25–40% (1). Although the prevention and treatment
of DN are attracting increasing attention from researchers, the
current therapeutic options are far from satisfactory as the
intensive therapy of blood glucose and pressure is often difficult
to maintain and may increase the risk of hypoglycemia and/or
hypotension in patients with diabetes (2). Therefore, the development of novel
therapeutic strategies that may specifically target DN is urgently
required.
Podocytes are one of the most important components
of the glomerular filtration barrier which has specific
cytobiological traits and physiological functions. It has been
shown that the loss of podocytes is an early characteristic of DN
that predicts its progressive course (3), and decreased nephrin expression is
observed during the early stages of DN, playing an important role
in accelerating the development of DN (4). DN has been reported to be an
immunological renal disease; the presence of tumor necrosis
factor-α (TNF-α), interleukin (IL)-1β, IL-6, interferon-γ (INF-γ)
and monocyte-chemoattractant protein-1 (MCP-1) has been reported in
patients with diabetes and in animal diabetic models (5–7).
However, the exact role of podocytes in the inflammatory response
has not yet been fully determined.
Various elements, including advanced glycation end
products (AGEs), reactive oxygen species (ROS), protein kinase C
(PKC) and the renin-angiotensin system (RAS), are considered to
play a role in the development and progression of DN (8). AGE formation in the kidneys may
contribute to the progressive alteration in the renal architecture
and loss of renal function in patients, resulting in basement
membrane thickening and mesangial expansion, hallmarks of DN
(9). The activation of AGEs in
podocytes leads to multiple pathophysiological effects, including
hypertrophy with cell cycle arrest and apoptosis, altered migration
and in the generation of pro-inflammatory cytokines (10). Previous studies have reported that
AGEs reduce podocyte adhesion via the upregulation of
integrin-linked kinase (ILK) expression, which occurs partly
through the activation of the RAS in podocytes (11). AGEs are also potent stimulators of
chemokine production, including IL-8 [also known as C-X-C motif
chemokine ligand (CXCL)8], MCP-1 (CCL2), INF-γ inducible protein 10
(IP-10/CXCL10), macrophage inflammatory protein-1α (MIP-1α/CCL3)
and RANTES (CCL5) (7). The
downregulation of CXCL9 and its receptor, CXCR3, suppresses the
loss of renal function and may be a potential therapeutic target
for human immune-mediated nephritis (12). However, the role of CXCL9 in
AGE-induced podocyte injury has not yet been reported to date, at
least to the best of our kowledge.
The activation of the Janus kinase (JAK)/signal
transducers and activators of transcription (STAT) pathway is an
important mechanism through which hyperglycemia contributes to
renal damage (13). Similarly,
JAK/STAT activation has been reported in rat glomerular cells
exposed to high glucose (14) and
may be important in glomerular transforming growth factor-β (TGF-β)
activation in early DN (15).
Moreover, angiotensin-converting-enzyme (ACE) inhibitors and
angiotensin receptor blockers, including valsartan, which prevent
the progression of DN, also prevent JAK/STAT activation in
glomerular cells from diabetic rats (16).
In this study, we examined the effect of AGEs on the
proliferation and apoptosis of podocytes and on the expression of
CXCL9 and the JAK2/STAT3 signaling pathway. We also investigated
whether AGE-induced podocytes injury occurs through the
CXCL9-mediated activation of the JAK2/STAT3 pathway.
Materials and methods
Patient samples
Serum and urine samples were obtained from 45
patients with DN admitted to Tongji Hospital, Shanghai, China and
subjects in the control group were 45 healthy volunteers. Ethical
approval for the study was provided by the independent Ethics
Committee of Tongji Hospital. Written informed consent was obtained
from all participants in this study. All the experimental
procedures were carried out in accordance with the Helsinki
Declaration, 1975.
Cell culture and treatment
Mouse podocytes were obtained from the Shanghai Cell
Bank, Chinese Academy of Sciences (Shanghai, China) and cultured in
RMPI-1640 supplemented with 10% fetal bovine serum (FBS), 100X
penicillin-streptomycin solution and 10 U/ml IFN-γ, and incubated
in a humidified atmosphere at 33°C with 5% CO2.
Following proliferation to 80% confluence, the podocytes were
cultured in the above-mentioned medium without 10 U/ml IFN-γ and
incubated in a humidified atmosphere at 37°C with 5% CO2
for 10–14 days. To establish the injury model, the podocytes were
seeded at 80% confluence in complete medium (Invitrogen Life
Technologies, Carlsbad, CA, USA) containing 10% FBS. After 24 h,
the medium was changed to serum-free medium with AGEs (10, 50, 100
and 150 mg/l) at the indicated time points, respectively. In
addition to AGE (150 mg/l) induction, podocytes were also treated
with 10 µM AG490 or 20 µM valsartan (as a positive
control) for 48 h.
Cell proliferation assay
Mouse podocytes (1×103/well) were plated
in 96-well plates. Following treatment with AGEs for 12, 24, 48 and
72 h, 10% cell counting kit-8 (CCK-8, CK04; Dojindo Molecular
Technologies, Kumamoto, Japan) diluted in serum-free RMPI-1640 was
mixed in each well for a further 1 h. The absorption of each sample
was measured at a 450 nm wavelength using a Labsystems MK3
microplate reader (Thermo Fisher Scientific, Inc., Rockford, IL,
USA) to detect cell viability according to the manufacturer's
instructions. Cells not treated with AGEs served as the control
group.
Transfection with small interfering RNA
(siRNA)
siRNA targeting CXCL9 (siRNA-CXCL9; Sangon Biotech
Co., Ltd., Shanghai, China) was used to knockdown CXCL9 mRNA
expression. The cells were transfected with siRNA (40 nM) using
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) following the
manufacturer's instructions. Non-specific siRNA (Sangon Biotech
Co., Ltd.) was used as a negative control (NC), and the selective
silencing of CXCL9 was confirmed by real-time PCR. The cells were
analyzed at 48 h following transfection.
Cell apoptosis assay
The analysis of cell apoptosis was performed using
flow cytometry and the Annexin V apoptosis detection kit
(eBioscience, San Diego, CA, USA). Briefly, the mouse podocytes
were plated in 6-well plates at a density of 1×105
cells/well and incubated with 195 µl Annexin V and 5
µl propidium iodide (PI) for 15 min in the dark at 4°C. The
early apoptotic cells are represented in the lower right quadrant
of the FACS histogram, and the late apoptotic cells, which were
stained with FITC and PI, emit red-green fluorescence and are
represented in the upper right quadrant of the FACS histogram.
Real-time PCR
Total RNA was extracted using TRIzol reagent
(Invitrogen Life Technologies) according to the manufacturer's
instructions. The complementary DNA was synthesized using a cDNA
synthesis kit (Thermo Fisher Scientific, Inc.). The conditions for
cDNA synthesis were as follows: 37°C for 60 min, followed by 85°C
for 5 min and 4°C for 5 min. Real-time PCR was performed using
SYBR-Green (Takara Biotechnology Co., Ltd., Dalian, China) and data
collection was conducted using an ABI 7500 Real-Time PCR system
(Applied Biosystems Life Technologies, Foster City, CA, USA).
Primers were list as follows: CXCL9 forward,
5′-CACTTCGCTGCTATCTAATTGG-3′ and reverse,
5′-TAGGCACTGTGGAAGATTTAGG-3′; CXCR3 forward,
5′-ACCATTACTGTGCCTTAGC-3′ a nd reverse,
5′-TATTTGCCTCTCCCTCTTCTC-3′; STAT3 forward,
5′-GACTCAAAGCCACCTCATTC-3′ and reverse, 5′-GCCTTGCCTTCCTAAATACC-3′;
and glyceraldehyde 3-phosphate dehydrogenase (GAPDH) forward,
5′-ATCACTGCCACCCAGAAG-3′ and reverse, 5′-TCCACGACGGACACATTG-3′.
GAPDH was used an internal control for normalization. The real-time
PCR cycling conditions were as follows: 95°C for 10 min, followed
by 40 cycles at 95°C for 15 sec and 60°C for 45 sec, and a final
extension step of 95°C for 15 sec, 60°C for 1 min, 95°C for 15 sec
and 60°C for 15 sec. Gene expression was calculated using the
2−ΔΔCt method.
Western blot analysis
Mouse podocytes were seeded at a density of
5×105 cells/well in 6-well plates, cultured overnight
and then treated with AGEs for 3 or 48 h. Total proteins were
isolated from the mouse podocytes and were subjected to 12% sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
electroblotted onto polyvinylidene fluoride membranes (Roche
Diagnostics, Mannheim, Germany). The membranes were first incubated
with rabbit monoclonal anti-p-STAT3 (ab76315; 1:1,000; Abcam,
Cambridge, MA, USA), anti-JAK2 (#3230; 1:1,000), anti-p-JAK2
(#3771; 1:1,000) (both from Cell Signaling Technology, Danvers, MA,
USA), podocin (ab181143; 1:10,000; Abcam) and anti-GAPDH (#5174;
1:1,500; Cell Signaling Technology) antibodies, and mouse
monoclonal anti-STAT3 (ab119352; 1:1,000) and anti-Bcl-2 (ab117115;
1:400) (both from Abcam) antibodies; as well as rabbit polyclonal
anti-CXCL9 (sc-50302; 1:500; Santa Cruz Biotechnology, Inc.,
Dallas, TX, USA), anti-CXCR3 (ab181013; 1:600; Abcam), anti-Bax
(sc-493; 1:300; Santa Cruz Biotechnology, Inc.), anti-caspase-3
(ab44976; 1:500) and anti-nephrin (ab58968; 1:500) antibodies (both
from Abcam). The blots were then incubated with goat anti-mouse or
anti-rabbit secondary antibodies (A0208 and A0216; 1:1,000;
Beyotime Institute of Biotechnology, Haimen, China) and visualized
using enhanced chemiluminescence (ECL; Thermo Fisher Scientific).
GAPDH antibody was used as an internal control. The blotting bands
were quantified using ImageJ software (National Institutes of
Health, Bethesda, MD, USA).
Enzyme-linked immunosorbent assay
(ELISA)
The TNF-α, IL-6 and CXCL9 levels present in the
mouse podocytes or in the serum and urine of patients with DN were
determined using commercially available murine-specific sandwich
ELISA kit following the manufacturer's instructions.
Statistical analysis
Data are expressed as the means ± SD of triplicate
samples. All results were confirmed in at least 3 time-independent
experiments. Statistical analyses were performed using GraphPad
Prism 5 software (GraphPad Software, Inc., La Jolla, CA, USA).
Statistical analysis was performed using unpaired a two-tailed
Student's t-test and one-way ANOVA tests. A value of P<0.05 was
considered to indicate a statistically significant difference.
Results
CXCL9 levels in the serum and urine of
patients with DN
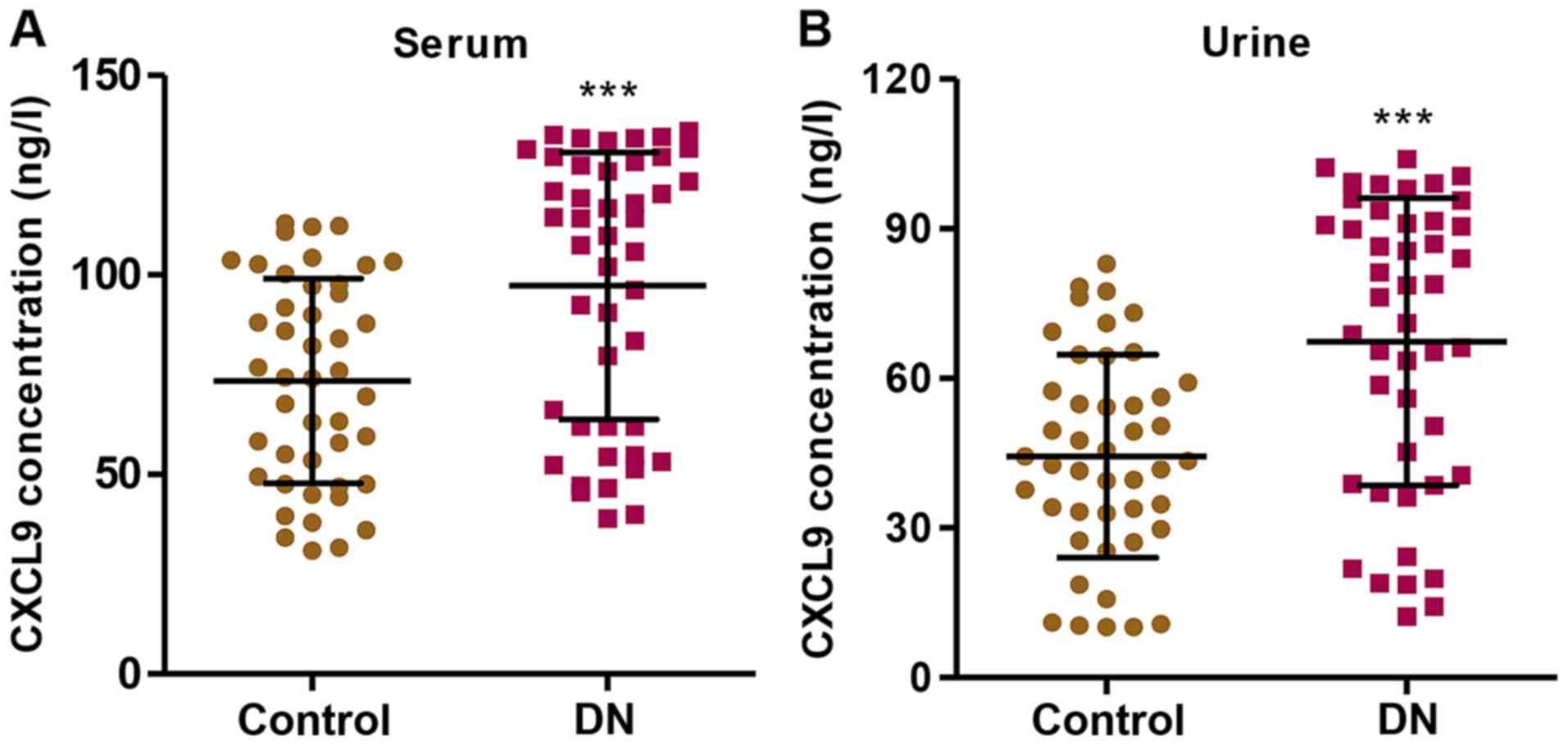
In order to examine the role of CXCL9 in DN, we
first measured the levels of CXCL9 in both the serum and urine of
patients with DN (n=45) and healthy controls (n=45). The CXCL9
concentration was significantly increased in the patients with DN
compared with the healthy controls in both serum and urine
(Fig. 1). Importantly, a higher
concentration of CXCL9 was observed in the serum than in urine.
These results suggest that CXCL9 plays an important role in DN;
thus CXCL9 was further investigated in subsequent experiments.
Treatment with AGEs inhibits the
proliferation of podocytes
To further investigate the association of CXCL9 with
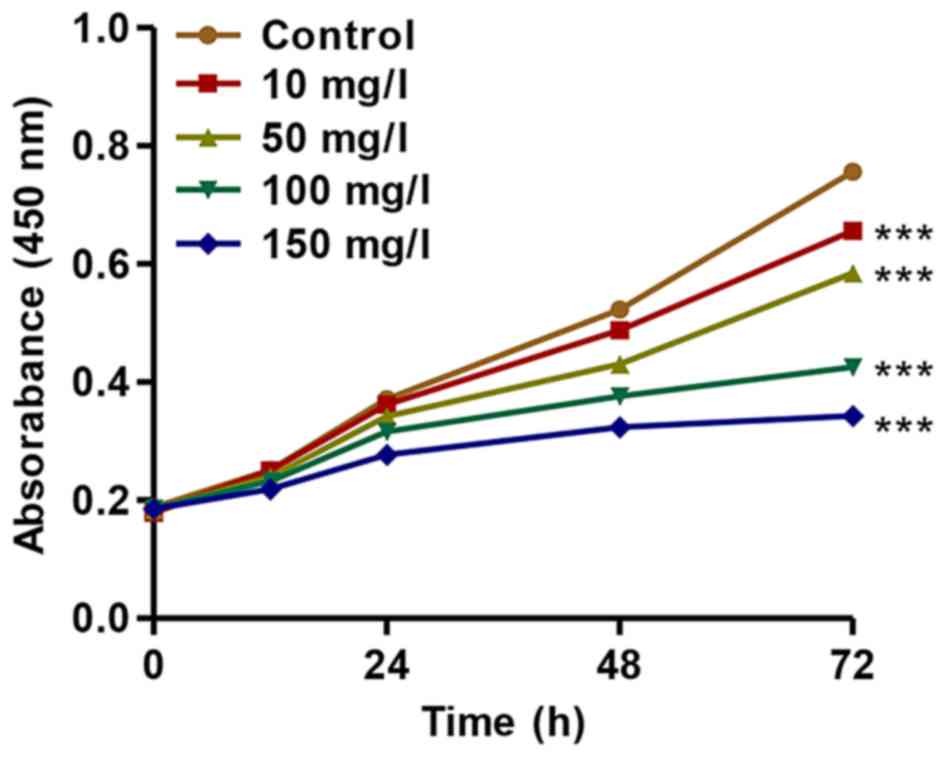
DN, we established an in vitro DN model of mouse podocyte
injury induced by AGEs. We found that AGEs at various
concentrations (10, 50, 100 and 150 mg/l) significantly inhibited
the proliferation of mouse podocytes in a concentration- and
time-dependent manner (Fig. 2).
After 72 h of incubation, the proliferation of podocytes treated
with AGEs (10, 50, 100 and 150 mg/l) was suppressed by 13.31±0.11%,
22.7±0.17%, 43.8±0.25% and 54.6±0.41%, respectively. These findings
indicated that treatment with AGEs inhibited the proliferation of
podocytes in a concentration- and time-dependent manner.
Effect of AGEs on the expression of CXCL9
and CXCR3, and STAT3 activation
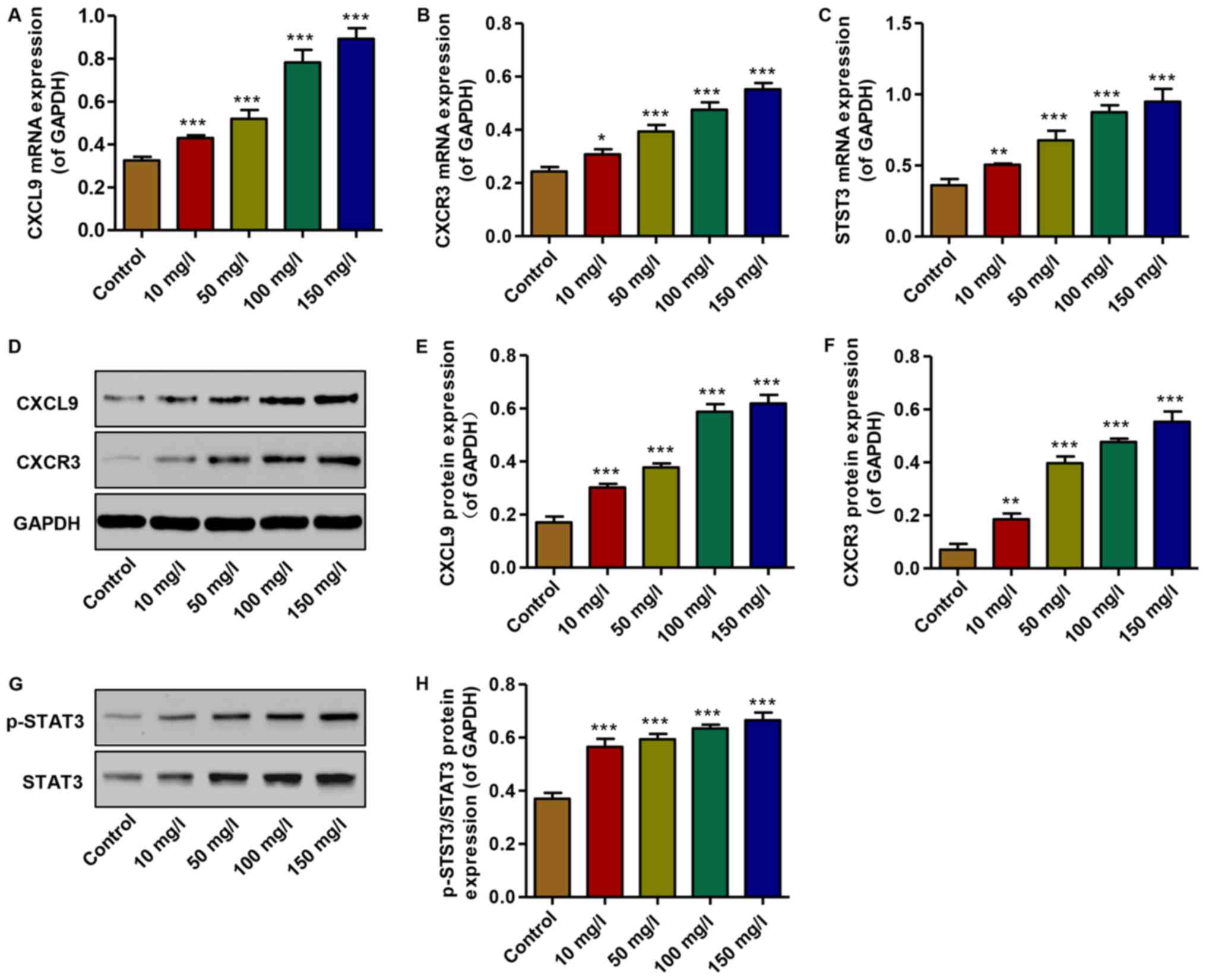
To examine the effects of AGEs on the expression of
CXCL9 and its receptor, and STAT3 activation in vitro,
real-time PCR and western blot analysis were performed. The mRNA
expression levels of CXCL9, CXCR3 and STAT3 were increased in the
podocytes treated with AGEs (10, 50, 100 and 150 mg/l) in a
concentration-dependent manner compared with the controls (Fig. 3A–C). Similarly, the protein
expression levels of CXCL9 and CXCR3 were increased, accompanied by
the activation of STAT3; an increase in the levels of p-STAT3/STAT3
was observed in the AGE-treated podocytes in a
concentration-dependent manner compared with the controls (Fig. 3D–H). Thus, AGEs at 150 mg/l were
therefore used in the subsequent experiments. These data
demonstrate that increased levels of CXCL9 and CXCR3, and STAT3
activation may contribute to the AGE-induced inhibition of podocyte
proliferation.
Knockdown of CXCL9 increases the
proliferation and inhibits the apoptosis of podocytes
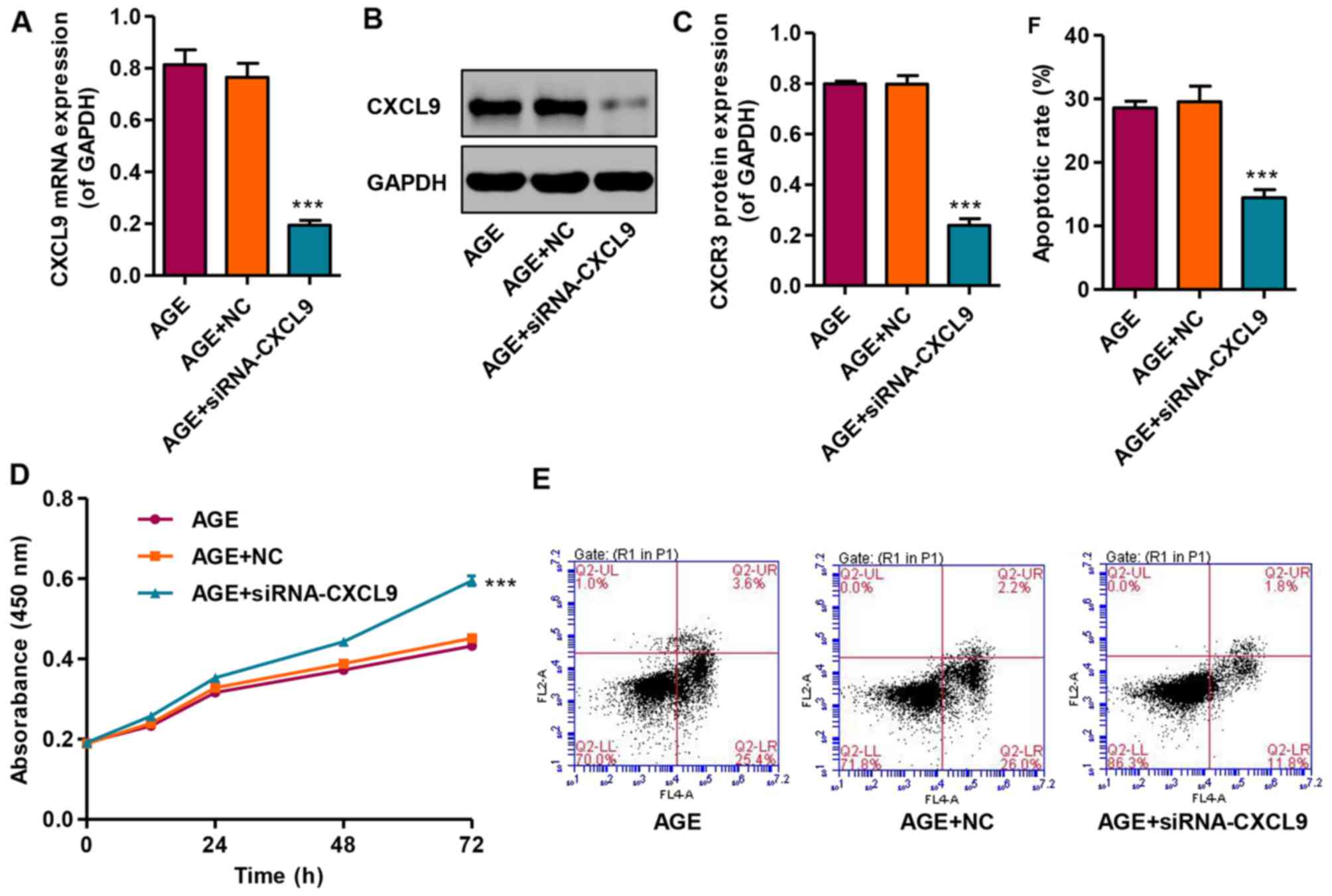
In order to examine the effects of CXCL9 on
podocytes in vitro, CXCCL9 was knocked down by siRNA in
podocytes. The results revealed that transfection with siNRA-CXCL9
significantly decreased the expression of CXCL9 in the AGE-treated
podocytes at both the mRNA and protein level (Fig. 4A–C). In addition, the effects of
CXCL9 on podocyte proliferation and apoptosis were also measured by
CCK-8 assay and flow cytometry, respectively. Transfection with
siNRA-CXCL9 significantly increased the proliferation and decreased
the apoptosis of the AGE-treated podocytes compared with the cells
treated with AGEs alone (Fig.
4D–F). However, the AGE-treated podocytes transfected with
nonspecific siRNA (NC) exhibited no significant changes in CXCL9
expression, proliferation and apoptosis compared with the podocytes
treated with AGEs alone. These results indicate that CXCL9 is
involved in the proliferation and apoptosis of AGE-treated mouse
podocytes.
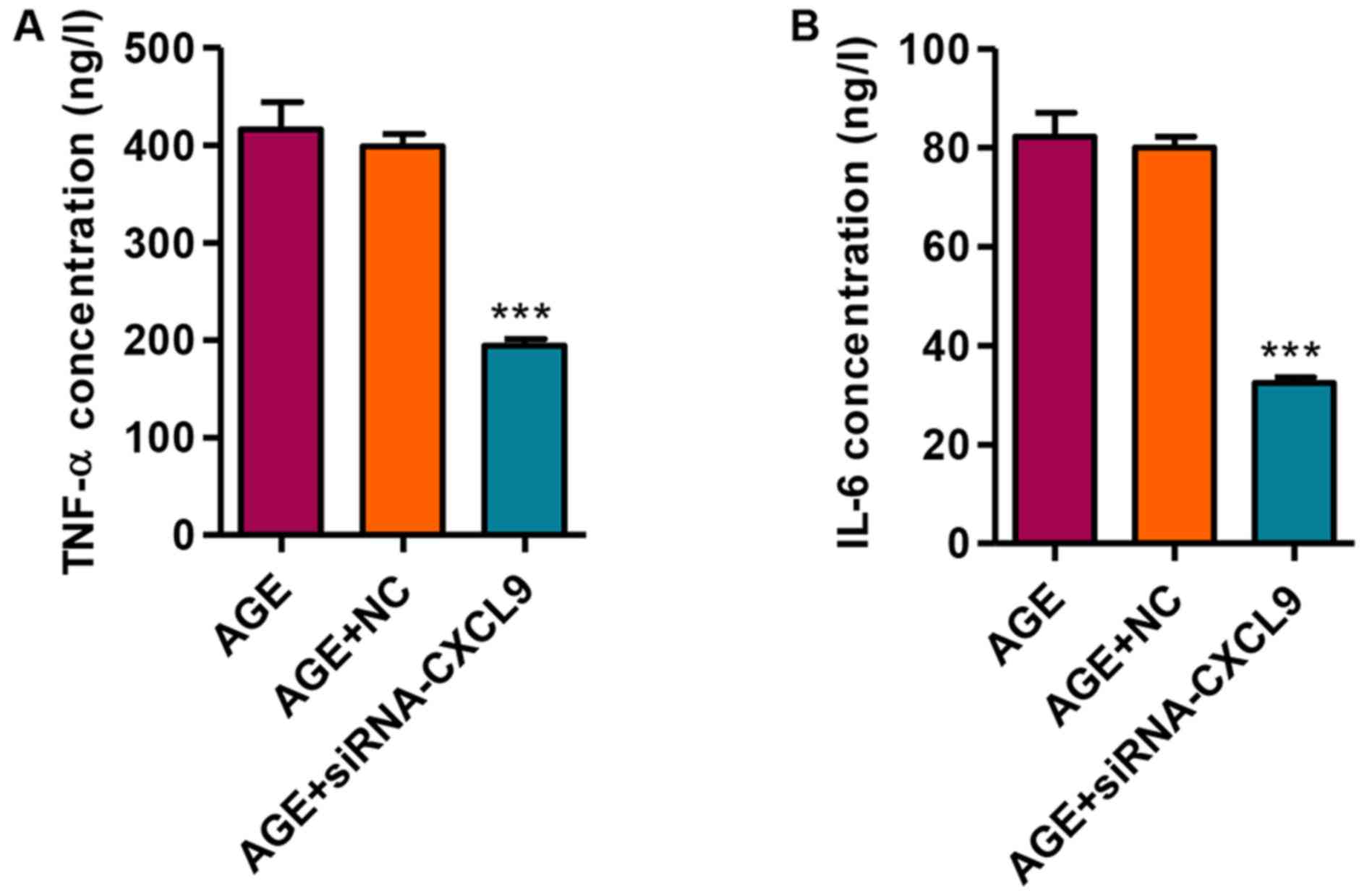
Knockdown of CXCL9 inhibits the release
of the TNF-α and IL-6 inflammatory factors
We then measured the secretion levels of TNF-α and
IL-6 in response to transfection with siRNA-CXCL9 in the
AGE-treated podocytes. Following the transfection of the
AGE-treated podocytes with siRNA-CXCL9 for 48 h, the secretion
levels of TNF-α and IL-6 were significantly decreased (Fig. 5). These findings thus suggest that
the downregulation of CXCL9 exerts an anti-inflammatory effect in
AGE-damaged podocytes.
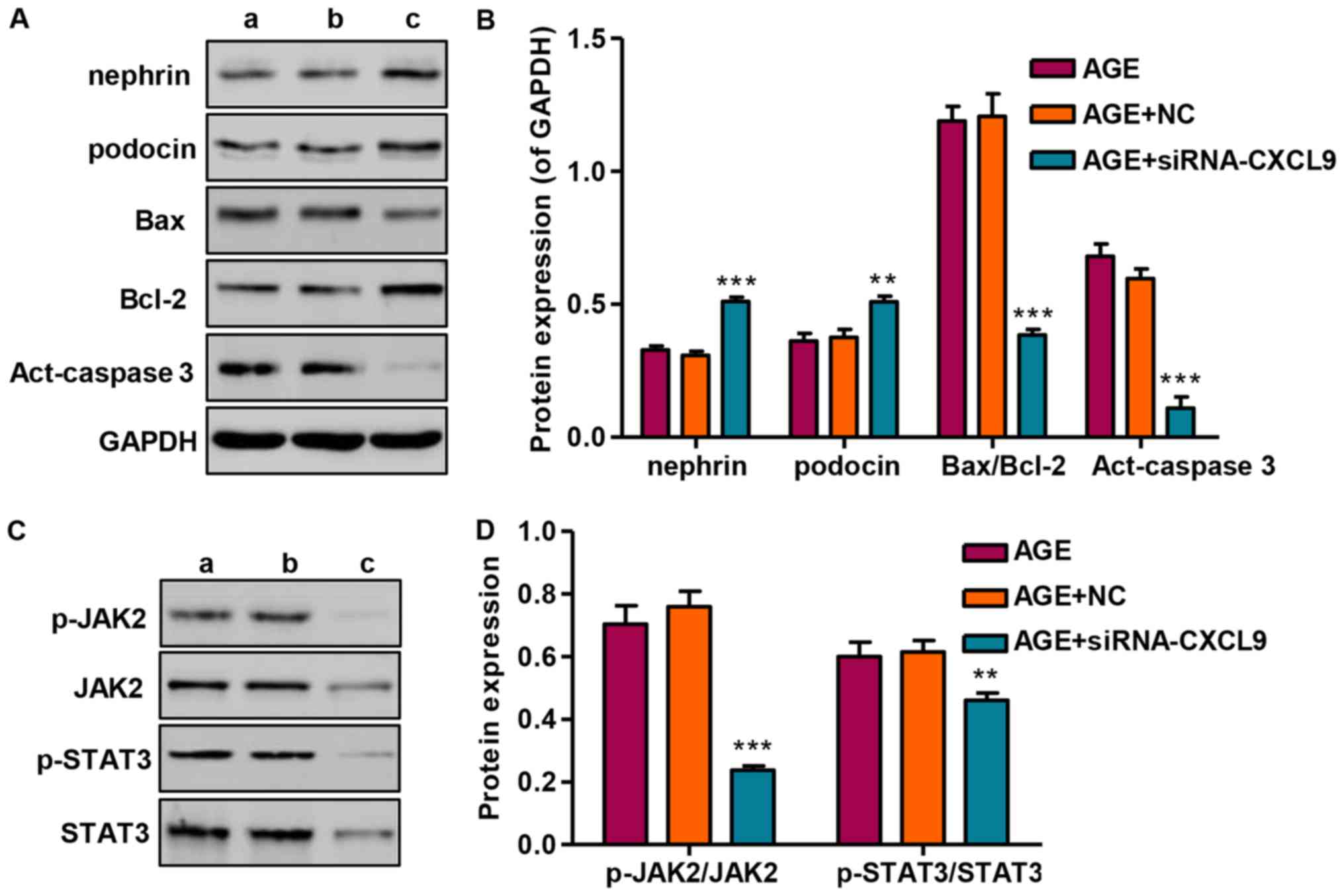
Effect of CXCL9 knockdown on protein
expression and JAK2/STAT3 activation
Following transfection of the podocytes with
siRNA-CXCL9 for 48 h, the expression levels of nephrin and podocin
were significantly increased, while the Bax/Bcl-2 ratio and
activated caspase-3 (act-caspase-3) were significantly suppressed
(Fig. 6A and B). As we had found
that STAT3 was activated in the AGE-treated podocytes, the levels
of STAT3 and upstream JAK2 signaling were also measured by western
blot analysis in the podocytes transfected wtih siRNA-CXCL9. Our
results revealed that siRNA-CXCL9 significantly suppressed the
activation of JAK2 and STAT3, as evidenced by decreased levels of
p-JAK2/JAK2 and p-STAT3/STAT3, in the AGE-treated podocytes
transfected with siRNA-CXCL9 (Fig. 6C
and D). These results thus suggest that the protective effects
against AGE-induced damage to podocytes exerted by the
downregulation CXCL9 are partially associated with the inhibition
of JAK2/STAT3 activation in podocytes.
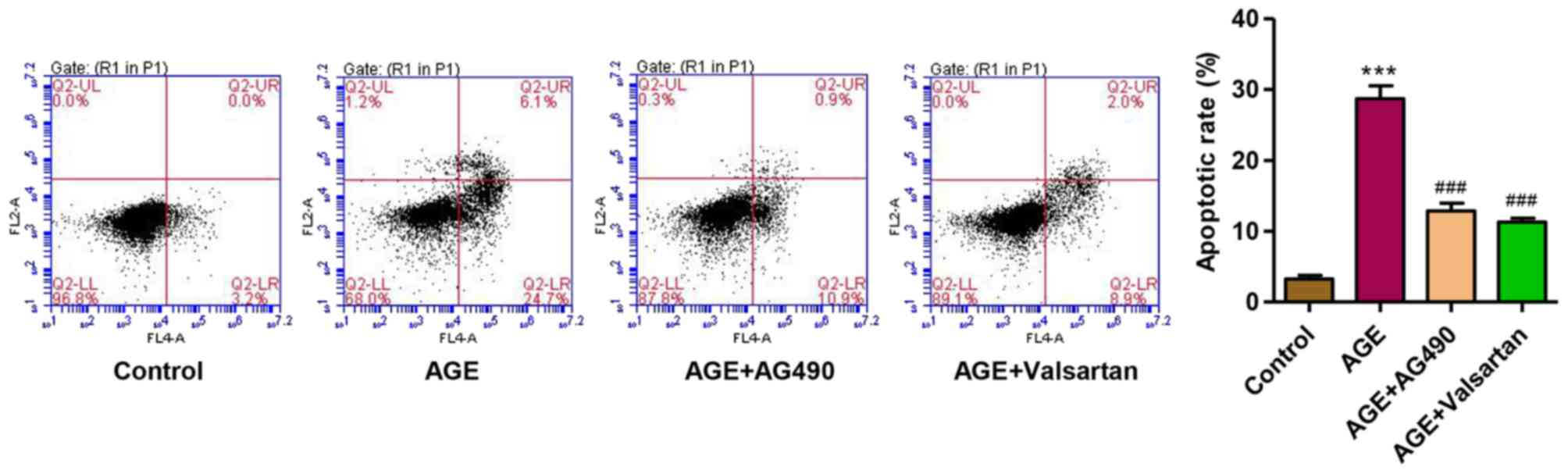
AG490 and valsartan attenuate the
AGE-induced apoptosis of podocytes
Considering the role of JAK2/STAT3 signaling in
AGE-induced damage to podocytes, the JAK2 inhibitor, AG490, was
used. Valsartan, an angiotensin II receptor antagonist, has long
been proven to exert an anti-proteinuria effect and to decrease
podocytes damage in DN (37).
Therefore, with valsartan as a control, we observed that following
treatment of the podocytes with AGEs for 48 h, the apoptotic rate
was significantly increased. However, treatment with AG490 and
valsartan markedly attenuated the apoptosis of the podocytes
induced by treatment with AGEs (Fig.
7). These findings suggest that the activation of JAK2/STAT3 is
implicated in AGE-induced podocyte apoptosis.
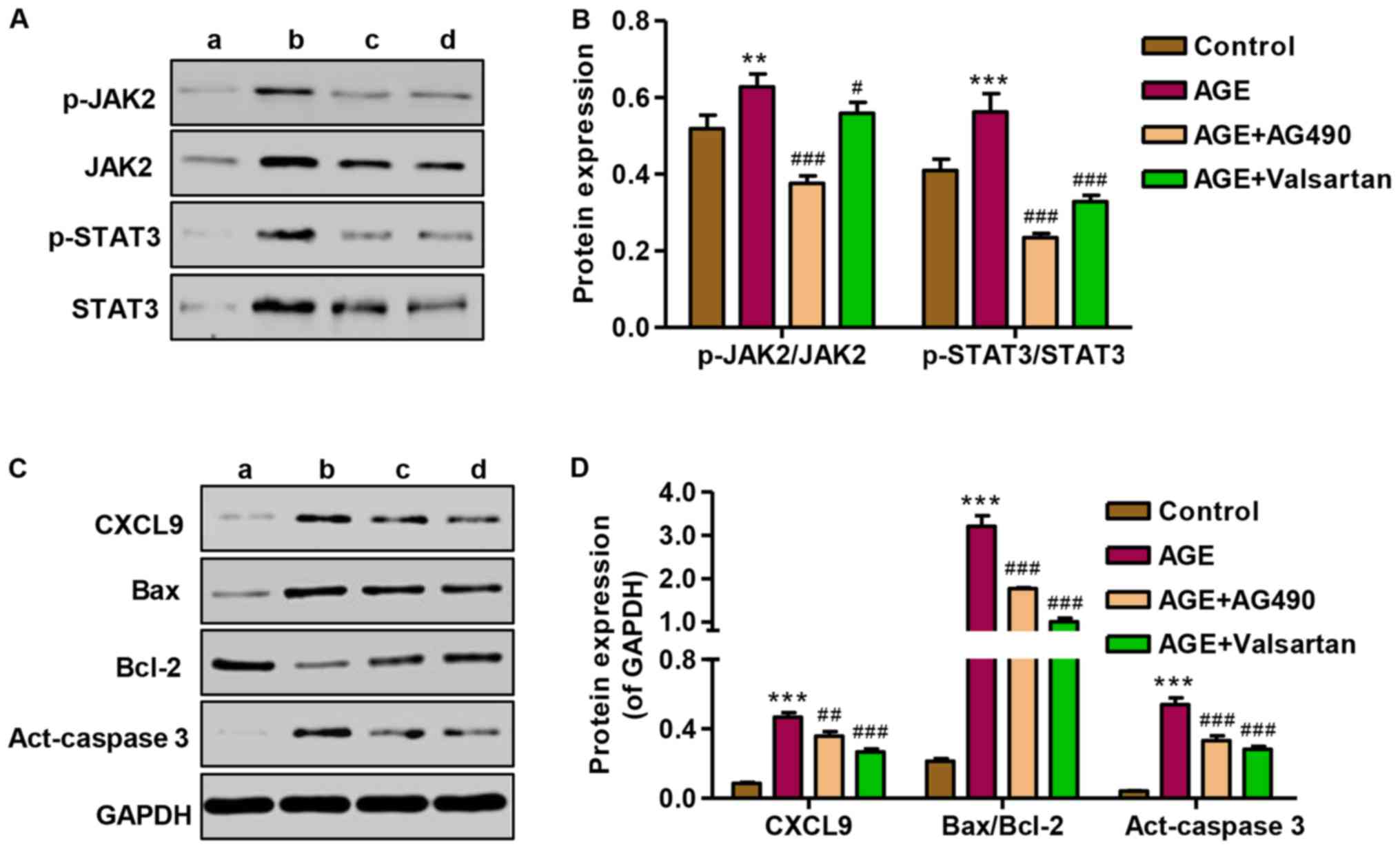
Effect of AG490 and valsartan on
JAK2/STAT3 activation and protein expression
To investigate the role of AG490 in the apoptosis of
podocytes induced by AGEs, the expression of CXCL9, Bax/Bcl-2 and
activated caspase-3, as well as JAK2/STAT3 signaling was measured
by western blot analysis. Following treatment of the podocytes with
AGEs, the promoting effect of AGEs on JAK2 and STAT3 signaling in
podocytes was markedly attenuated by treatment with AG490 and
valsartan (Fig. 8A and B). The
expression levels of CXCL9, Bax/Bcl-2 and activated caspase-3 were
significantly increased following treatment with AGE; however,
treatment with AG490 and valsartan markedly attenuated the
overexpression of CXCL9, Bax/Bcl-2 and activated caspase-3 induced
by AGE treatment (Fig. 8C and D).
These results indicate that the AGE-induced apoptosis of podocytes
is associated with the CXCL9-mediated activation of JAK2/STAT3
signaling.
Discussion
CXC chemokines are particularly important for
leukocyte infiltration in inflammatory diseases. It has been
demonstrated that inflammation is one of the potential pathogenic
mechanisms responsible for the development of DN (17). However, to date, there are limited
data available on inflammation related to CXC chemokines in human
DN. In this study, we measured the serum and urine levels of the
CXC chemokine, CXCL9, in 45 patients wth DN and 45 healthy controls
by ELISA. The serum and urine levels of CXCL9 in the patients with
DN were significantly elevated compared with those in the controls.
In agreement with our findings, a previous study found that the
levels of 3 CXC chemokines, CXCL5, CXCL8 and CXCL9, were
significantly increased in the urine and serum of patients with DN
(18). These results suggest
CXCL9 may be involved in the progression of DN.
In patients with type 1 and 2 diabetes, the serum
and tissue levels of AGEs have been shown to be significantly
increased compared with the healthy control subjects (19), and the tissue levels of AGEs in
diabetic renal disease have been shown to be twice those of
patients with only diabetes and no renal disease (20), suggesting that AGEs are
responsible for the progression of DN. A reduction in podocyte
density is an important determinant of progressive DN and precedes
the development of renal dysfunction and albuminuria in diabetic
patients and in animal models of diabetes (21,22). Chen et al (23) reported that AGEs induced podocytes
apoptosis in a dose-dependent manner and Chuang et al
(24) showed that AGEs activated
FOXO4, leading to the apoptosis of podocytes, which was similar to
our findings in that AGEs inhibited the proliferation of mouse
podocytes in a dose- and time-dependent manner.
Accumulating evidence from animal models supports
the notion that CXCL9 and its receptor, CXCR3, which is highly
expressed in Th1 CD4+ cells, play a critical role in the
recruitment of T cells, macrophages and dendritic cells during the
development of chronic renal injury (25). An increase in CXCL9 protein levels
was detected in streptozotocin-injected mice and showed its
pronociceptive properties (26).
Activated and resting CXCR3 macrophages express CXCR3 during kidney
disease and are therefore central to inducing renal injury
(12). In the present study, we
found that CXCL9, as well as CXCR3, was significantly increased in
response to AGEs in podocytes in a dose-dependent manner.
Furthermore, STAT3 signaling was also activated by AGE treatment.
After binding to their receptors, CXCL9 activates JAKs, which in
turn leads to the tyrosine phosphorylation of STAT3 (27). Previous studies have reported that
the increased expression of STAT3 reduces the IFN-α induction of
CXCL9 mRNA in myeloid cells (28); in vitro, in human bronchial
epithelial cells, IL-13 augmented IL-27-induced CXCL9 expression,
which appeared to be due to augmented STAT1 activation and reduced
STAT3 activation (29). The
different stimulators, including AGEs, IL-27 and IFN-α may
interpret the contradictory findings.
In this study, to further investigate the role of
CXCL9 in AGE-induced podocyte damage, siRNA-CXCL9 was used to
transfected into the podocytes. We found that CXCL9 downregulation
significantly increased the proliferation and decreased the
apoptosis of podocytes, and decreased the levels of the
inflammatory factors, TNF-α and IL-6. In line with the experimental
data, patients with type 2 diabetes have 3–4-fold greater serum
levels of TNF-α compared with non-diabetic patients, and that
urinary TNF-α excretion correlates well with the clinical markers
of DN and the progression of the disease (30). Experimental studies have indicated
that IL-6 overexpression in diabetic kidneys correlates with kidney
hypertrophy and albumin excretion (31,32). Nephrin and podocin are two
recently discovered podocyte-specific proteins, pivotal in
establishing podocyte slit membrane structure and in the
maintenance of an intact filtration barrier (33). In the present study, nephrin and
podocin expression levels were markedly increased following
transfection of the AGEs-treated podocytes with siRNA-CXCL9.x
Furthermore, the apoptosis-associated protein
expression of Bax/Bcl-2 and activated caspase-3 was decreased,
which is consistent with our apoptosis analysis measured by flow
cytometry. In the present study, siRNA-CXCL9 triggered an effect
opposite to that of the AGEs, whereby JAK2 and STAT3 activation was
significantly suppressed in podocytes. Similar data by other
investigators have indicated that increases in JAK2 and STAT3 gene
expression occur in patients with DN compared with healthy controls
(34). Lu et al (35) reported a mouse with reduced
capacity of STAT3 activation showing less proteinuria, macrophage
infiltration and inflammation at an early stage of DN. Total
glucosides of paeony (TGP) significantly inhibited DN progression
and these protective effects are associated with the ability of TGP
to inhibit the JAK2/STAT3 pathway (13). AG490, a JAK2 specific inhibitor,
was used in this study to determine the role of JAK2/STAT3
signaling in AGE-induced podocyte damage. Our results revealed that
AG490 significantly inhibited JAK2 and STAT3 activation and the
apoptosis of podocytes and the expression of CXCL9, Bax/Bcl-2, and
activated caspase-3. Podocyte STAT3 activation can result in more
severe nephropathy independent of upstream JAK signaling, or at
least in changes in upstream JAK signaling. Thus, the activation of
JAK2 and STAT3 in podocytes is important in the pathogenesis of DN
(36). In addition, valsartan has
been shown to exert protective effects against the progression of
DN (37) and exerts a similar
effect as AG490, suggesting that JAK2/STAT3 signaling is also
implicated in the protective effects of valsartan against
AGE-induced podocyte damage.
In conclusion, our study demonstrated that CXCL9
synthesis was upregulated in patients with DN and in AGE-treated
mouse podocytes, and both CXCL9 downregulation and JAK2 inhibitor
treatment significantly inhibited the decrease in proliferation and
apoptosis induced by AGEs, as well as inflammatory factor secretion
and JAK2/STAT3 activation. Our data may aid in the understanding of
the multifactorial nature of DN, and suggest that CXCL9 may be a
novel therapeutic target in the treatment of DN.
References
|
1
|
Remuzzi G, Schieppati A and Ruggenenti P:
Clinical practice. Nephropathy in patients with type 2 diabetes. N
Engl J Med. 346:1145–1151. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yamagishi S and Matsui T: Advanced
glycation end products, oxidative stress and diabetic nephropathy.
Oxid Med Cell Longev. 3:101–108. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Susztak K, Raff AC, Schiffer M and
Böttinger EP: Glucose-induced reactive oxygen species cause
apoptosis of podocytes and podocyte depletion at the onset of
diabetic nephropathy. Diabetes. 55:225–233. 2006. View Article : Google Scholar
|
|
4
|
Navarro-González JF, Mora-Fernández C,
Muros de Fuentes M and García-Pérez J: Inflammatory molecules and
pathways in the pathogenesis of diabetic nephropathy. Nat Rev
Nephrol. 7:327–340. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koike N, Takamura T and Kaneko S:
Induction of reactive oxygen species from isolated rat glomeruli by
protein kinase C activation and TNF-α stimulation, and effects of a
phosphodiesterase inhibitor. Life Sci. 80:1721–1728. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dalla Vestra M, Mussap M, Gallina P,
Bruseghin M, Cernigoi AM, Saller A, Plebani M and Fioretto P:
Acute-phase markers of inflammation and glomerular structure in
patients with type 2 diabetes. J Am Soc Nephrol. 16(Suppl 1):
S78–S82. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lim AK and Tesch GH: Inflammation in
diabetic nephropathy. Mediators Inflamm. 2012:1461542012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yamagishi S and Imaizumi T: Diabetic
vascular complications: Pathophysiology, biochemical basis and
potential therapeutic strategy. Curr Pharm Des. 11:2279–2299. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bohlender JM, Franke S, Stein G and Wolf
G: Advanced glycation end products and the kidney. Am J Physiol
Renal Physiol. 289:F645–F659. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Busch M, Franke S, Rüster C and Wolf G:
Advanced glycation end-products and the kidney. Eur J Clin Invest.
40:742–755. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cheng C, Zheng Z, Shi C, Liu X, Ye Z and
Lou T: Advanced glycation end-products reduce podocyte adhesion by
activating the renin-angiotensin system and increasing
integrin-linked kinase. Exp Ther Med. 6:1494–1498. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Menke J, Zeller GC, Kikawada E, Means TK,
Huang XR, Lan HY, Lu B, Farber J, Luster AD and Kelley VR: CXCL9,
but not CXCL10, promotes CXCR3-dependent immune-mediated kidney
disease. J Am Soc Nephrol. 19:1177–1189. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang K, Wu YG, Su J, Zhang JJ, Zhang P and
Qi XM: Total glucosides of paeony regulates JAK2/STAT3 activation
and macrophage proliferation in diabetic rat kidneys. Am J Chin
Med. 40:521–536. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Banes AK, Shaw S, Jenkins J, Redd H, Amiri
F, Pollock DM and Marrero MB: Angiotensin II blockade prevents
hyperglycemia-induced activation of JAK and STAT proteins in
diabetic rat kidney glomeruli. Am J Physiol Renal Physiol.
286:F653–F659. 2004. View Article : Google Scholar
|
|
15
|
Wang X, Shaw S, Amiri F, Eaton DC and
Marrero MB: Inhibition of the Jak/STAT signaling pathway prevents
the high glucose-induced increase in tgf-beta and fibronectin
synthesis in mesangial cells. Diabetes. 51:3505–3509. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiao B, Wang YS, Cheng YN, Gao JJ and
Zhang QZ: Valsartan attenuated oxidative stress, decreased MCP-1
and TGF-β1 expression in glomerular mesangial and epithelial cells
induced by high-glucose levels. Biosci Trends. 5:173–181. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chung AC and Lan HY: Chemokines in renal
injury. J Am Soc Nephrol. 22:802–809. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Higurashi M, Ohya Y, Joh K, Muraguchi M,
Nishimura M, Terawaki H, Yagui K, Hashimoto N, Saito Y and Yamada
K: Increased urinary levels of CXCL5, CXCL8 and CXCL9 in patients
with type 2 diabetic nephropathy. J Diabetes Complications.
23:178–184. 2009. View Article : Google Scholar
|
|
19
|
Galler A, Müller G, Schinzel R, Kratzsch
J, Kiess W and Münch G: Impact of metabolic control and serum
lipids on the concentration of advanced glycation end products in
the serum of children and adolescents with type 1 diabetes, as
determined by fluorescence spectroscopy and
nepsilon-(carboxymethyl)lysine ELISA. Diabetes Care. 26:2609–2615.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Genuth S, Sun W, Cleary P, Sell DR, Dahms
W, Malone J, Sivitz W and Monnier VM: Glycation and
carboxymethyllysine levels in skin collagen predict the risk of
future 10-year progression of diabetic retinopathy and nephropathy
in the diabetes control and complications trial and epidemiology of
diabetes interventions and complications participants with type 1
diabetes. Diabetes. 54:3103–3111. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Siu B, Saha J, Smoyer WE, Sullivan KA and
Brosius FC III: Reduction in podocyte density as a pathologic
feature in early diabetic nephropathy in rodents: Prevention by
lipoic acid treatment. BMC Nephrol. 7:62006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dai C, Stolz DB, Kiss LP, Monga SP,
Holzman LB and Liu Y: Wnt/β-catenin signaling promotes podocyte
dysfunction and albuminuria. J Am Soc Nephrol. 20:1997–2008. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen Y, Liu CP, Xu KF, Mao XD, Lu YB, Fang
L, Yang JW and Liu C: Effect of taurine-conjugated ursodeoxycholic
acid on endoplasmic reticulum stress and apoptosis induced by
advanced glycation end products in cultured mouse podocytes. Am J
Nephrol. 28:1014–1022. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chuang PY, Yu Q, Fang W, Uribarri J and He
JC: Advanced glycation endproducts induce podocyte apoptosis by
activation of the FOXO4 transcription factor. Kidney Int.
72:965–976. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Holdsworth SR and Tipping PG: Leukocytes
in glomerular injury. Semin Immunopathol. 29:355–374. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zychowska M, Rojewska E, Pilat D and Mika
J: The role of some chemokines from the CXC subfamily in a mouse
model of diabetic neuropathy. J Diabetes Res. 2015:7501822015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang JS, Lee YH, Chuang LY, Guh JY and
Hwang JY: Cinnamaldehyde and nitric oxide attenuate advanced
glycation end products - induced the JAK/STAT signaling in human
renal tubular cells. J Cell Biochem. 116:1028–1038. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ho HH and Ivashkiv LB: Role of STAT3 in
type I interferon responses. Negative regulation of STAT1-dependent
inflammatory gene activation. J Biol Chem. 281:14111–14118. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xie M, Mustovich AT, Jiang Y, Trudeau JB,
Ray A, Ray P, Hu H, Holguin F, Freeman B and Wenzel SE: IL-27 and
type 2 immunity in asthmatic patients: Association with severity,
CXCL9, and signal transducer and activator of transcription
signaling. J Allergy Clin Immunol. 135:386–394. 2015. View Article : Google Scholar
|
|
30
|
Navarro JF, Mora C, Muros M and García J:
Urinary tumour necrosis factor-α excretion independently correlates
with clinical markers of glomerular and tubulointerstitial injury
in type 2 diabetic patients. Nephrol Dial Transplant. 21:3428–3434.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Navarro JF, Milena FJ, Mora C, León C and
García J: Renal pro-inflammatory cytokine gene expression in
diabetic nephropathy: Effect of angiotensin-converting enzyme
inhibition and pentoxifylline administration. Am J Nephrol.
26:562–570. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Thomson SC, Deng A, Bao D, Satriano J,
Blantz RC and Vallon V: Ornithine decarboxylase, kidney size, and
the tubular hypothesis of glomerular hyperfiltration in
experimental diabetes. J Clin Invest. 107:217–224. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saleem MA, Ni L, Witherden I, Tryggvason
K, Ruotsalainen V, Mundel P and Mathieson PW: Co-localization of
nephrin, podocin, and the actin cytoskeleton: Evidence for a role
in podocyte foot process formation. Am J Pathol. 161:1459–1466.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brosius FC III and Alpers CE: New targets
for treatment of diabetic nephropathy: What we have learned from
animal models. Curr Opin Nephrol Hypertens. 22:17–25. 2013.
|
|
35
|
Lu TC, Wang ZH, Feng X, Chuang PY, Fang W,
Shen Y, Levy DE, Xiong H, Chen N and He JC: Knockdown of Stat3
activity in vivo prevents diabetic glomerulopathy. Kidney Int.
76:63–71. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Brosius FC III and He JC: JAK inhibition
and progressive kidney disease. Curr Opin Nephrol Hypertens.
24:88–95. 2015. View Article : Google Scholar :
|
|
37
|
Zhang Y, Chen B, Hou XH, Guan GJ, Liu G,
Liu HY and Li XG: Effects of mycophenolate mofetil, valsartan and
their combined therapy on preventing podocyte loss in early stage
of diabetic nephropathy in rats. Chin Med J (Engl). 120:988–995.
2007.
|