Introduction
The incidence of Alzheimer's disease (AD) is
gradually increasing, and AD has become a major threat to human
health (1). Unfortunately, an
effective treatment method has not yet been discovered (2,3).
AD is a degenerative disease of the central nervous system. The
deposition of extracellular amyloid plaques and the formation of
intracellular neurofibrillary tangles (NFTs) are its primary
pathological features. β-amyloid (Aβ), a cleavage product of the
amyloid precursor protein, is the main component of amyloid plaques
(4–6). Tau protein is a
microtubule-associated protein closely involved in the maintenance
of microtubule stability (7). Tau
protein has numerous potential phosphorylation sites, which are
mainly serine and threonine residues. The abnormal phosphorylation
of tau protein reduces its affinity for microtubules and damages
its microtubule assembly capacity (8,9).
Furthermore, tau hyperphosphorylation is the dominating cause of
the formation of NFTs (7,10). Although Aβ has been the principal
focus of AD treatments, since tau phosphorylation has been
indicated to be a consequence of Aβ pathology (11), the focus of attention has shifted
from Aβ to tau protein (12).
The hyperphosphorylation of tau protein is mainly
due to an increase in kinase activity and reduction of phosphatase
activity (13,14). Among various kinases associated
with this process, cyclin-dependent kinase 5 (CDK5) is considered
to be particularly relevant (15). Abnormal CDK5 activity leads to the
hyperphosphorylation of tau protein, which contributes to the
formation of NFTs (16). A
previous study indicated that CDK5 silencing decreased the number
of NFTs in transgenic Alzheimer's mice (17). Notably, CDK5 is activated via
subunits p35 or p39, and the cleavage of p35 to form p25 may occur
due to the action of calpain, the activity of which is dependent
upon calcium (18–21). Compared with p35, p25 has a longer
half-life; p25/CDK5-binding prolongs the activity of CDK5 and
further promotes tau protein hyperphosphorylation, which serves an
important role in the development of AD (22). Therefore, blocking the
Ca2+-calpain-p25-CDK5 pathway has considerable
significance for AD. A previous study has shown that A-705253, a
calpain inhibitor, blocks stress-induced tau hyperphosphorylation
(23). The restriction of CDK5
activity has an inhibitory effect on the aggregation of NFTs
(17,23). In addition, a specific calpain
inhibition, calpastatin, has demonstrated the ability to prevent
tauopathy and neurodegeneration and restore a normal lifespan in
tau P301L mice (24).
Quercetin (QUE) is a natural flavonoid compound that
has been shown to exert extensive pharmacological effects,
including antioxidant, antitumor, anti-inflammatory (25–27), anti-chemotherapy-induced fatigue
(28) and anti-aging effects
(29). QUE has been demonstrated
to cross the blood-brain barrier and prevent the progression of
neurodegenerative diseases (30–33). Numerous traditional Chinese
medicines contain QUE, including Japanese pagoda tree flower,
Apocynum venetum and cattail pollen (34,35). A previous study has indicated that
QUE has the ability to reduce Aβ-induced cytotoxicity (36). Additionally, QUE has been revealed
to attenuate tauopathy, although the mechanism has not been
elucidated (37). Furthermore,
QUE has been demonstrated to ameliorate AD pathology and protect
cognitive and emotional functions in vivo (38,39).
The hippocampus, an important brain structure, is
responsible for the strengthening of short-term memories into
long-term memories and is closely relevant to AD (40). Okadaic acid (OA) is widely used to
block protein phosphatase 2A (PP2A) activity (41,42). PP2A serves a vital role in in the
development of neurodegenerative disorders via the
hyperphosphorylation of tau protein (43,44). Therefore, OA-induced HT22 mouse
hippocampal neuronal cells were selected for use in the present
study as a model of AD. The effect of QUE pretreatment on tau
protein hyperphosphorylation in OA-induced HT22 cells was
investigated and the involvement of the
Ca2+-calpain-p25-CDK5 signaling pathway in the
underlying mechanism was evaluated. Natural compounds with fewer
side effects are increasingly favored, which have lower toxicity
and higher efficacy (45). The
present study continues previous studies conducted by the current
research team concerning the neuroprotective effects of other
natural compounds (46,47).
Materials and methods
Materials
QUE (molecular formula,
C15H1007; molecular weight, 302.24
g/mol; purity, >98.5%; CAS no., 117-39-5) was obtained from
Aladdin Industrial Corporation (Shanghai, China). Calpeptin (CALP;
CAS no., 117591-20-5), roscovitine (ROS; CAS no., 186692-46-6) and
OA (molecular formula, C44H68O13;
molecular weight, 805.00 g/mol; purity, >90%; CAS no.,
78111-17-8) were all from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany). Fluo-3 AM (S1056), Super ECL Plus (P1010) and a
bicinchoninic acid (BCA) protein assay kit (P0010) were obtained
from Beyotime Institute of Biotechnology (Jiangsu, China). Primary
antibodies targeting CDK5 (ab40773), calpain-1 (ab28258), tau-5
(ab80579), tau [pS396] (ab32057) and tau [pT231] (ab15559) were
from Abcam (Cambridge, UK). Tau-1 primary antibody (MAB3420) was
from EMD Millipore (Billerica, MA, USA). Primary antibodies for tau
[pS199] (44734G) and tau [pT205] (44738G) were from Invitrogen
(Thermo Fisher Scientific, Inc., Waltham, MA, USA). Primary
antibodies for p35/p25 (C64B10) and β-actin (13E5) were from Cell
Signaling Technology, Inc. (Danvers, MA, USA). The primary antibody
for p-CDK5 (Tyr15) (sc-12918) was from Santa Cruz
Biotechnology, Inc. (Dallas, TX, USA). The horseradish peroxidase
(HRP)-conjugated secondary antibody was from Wuhan Boster
Biological Technology, Ltd. (BA1088; Wuhan, China).
Cell culture
The HT22 cells were a generous gift from Dr Jun Liu
of the Memorial Hospital of Sun Yat-sen University (Guangzhou,
China) (48). The HT22 cells were
grown in a humidified incubator with 5% CO2 and 95% air
at 37°C in Dulbecco's modified Eagle's medium (Hyclone DMEM; GE
Healthcare Life Sciences, Logan, UT, USA) supplemented with 10%
fetal bovine serum, 100 U/ml penicillin and 100 µg/ml
streptomycin (all Gibco; Thermo Fisher Scientific, Inc.).
Cell treatment
The HT22 cells were grown in 6-well plate. OA was
dissolved in DMSO to a concentration of 1 µM as a stock
solution. The stock solution was diluted with DMEM to 80 nM prior
to use. When the cell density reached 80%, the cells were incubated
with QUE (5 or 10 µM), CALP (10 µM) or ROS (0.16
µM) for 24 h prior to exposure to OA (80 nM) for 12 h at
37°C.
Western blotting
HT22 cells were harvested following the treatments
described above and lysed in ice-cold lysis buffer [1X PBS, 1%
NP-40, 0.1% sodium dodecyl sulfate (SDS), 5 mM EDTA, 0.5% sodium
deoxycholate and 1% phenylmethane sulfonyl fluoride] supplemented
with phosphatase inhibitor for 30 min. The lysate was centrifuged
at 14,000 × g for 20 min at 4°C. The supernatant was collected and
its protein content was quantified using the BCA protein assay kit.
Samples with equal amounts of protein (40 µg) were separated
using 10% SDS-PAGE. The proteins were then transferred to
polyvinylidene fluoride membranes. the membranes were blocked with
5% bovine serum albumin (Sigma-Aldrich) dissolved in 20 ml TBS with
1 ml Tween-20 buffer for 1 h at room temperature. Subsequently, the
membranes were incubated overnight at 4°C with primary antibodies
against CDK5 (1:2,000), p-CDK5 (1:1,000), tau-5 (1:1,000), tau-1
(1:10,000), tau [pS199] (1:1,000), tau [pT205] (1:1,000), tau
[pS396] (1:1,000), tau [pT231] (1:1,000), calpain-1 (1:1,000),
p35/p25 (1:1,000) and β-actin (1:1,000). Following this, the
membranes were incubated with secondary HRP-conjugated antibody
(1:10,000) at room temperature for 1 h. Immunoreactive proteins
were detected using Super ECL Plus and exposed to X-ray films.
ImageJ 1.410 software (National Institutes of Health, Bethesda, MD,
USA) was used to quantitatively analyze the expression levels of
the target proteins.
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from the HT22 cells in
6-well plates using TRIzol reagent (Thermo Fisher Scientific,
Inc.). Spectrophotometry at 260 nm was conducted to determine the
amount of RNA extracted. The primers used were as follows: p35
forward, 5′-GCA ACG GTC CCA AAA GGC TT-3′ and reverse, 5′-ACA GCA
AGA ACG CCA AGG ACA-3′; CDK5 forward, 5′-GCA TTG AGT TTG GGC ACG
ACA-3′ and reverse, 5′-AAA ACC GGG AAA CCC ATG AGA-3′; β-actin
forward, 5′-ATG CCA TCC TGC GTC TGG ACC TGGC-3′ and reverse, 5′-AGC
ATT TGC GGT GCA CGA TGG AGGG-3′. These primers were described
previously by Chen et al (49). Samples were reverse-transcribed in
RT reaction buffer using 400 ng total RNA according to the
manufacturer's protocol. The PCR was conducted with template,
primers, The RT kit was from Takara Biotechnology Co., Ltd.
(Dalian, China). GoTaq Green Master mix and RNase-free
dH2O added to a volume of 50 µl. Amplification
was carried out under the following conditions: Initial
denaturation at 95°C for 2 min, denaturation at 95°C for 45 sec,
annealing at 59°C for 45 sec, extension at 72°C for 1 min, and a
final extension step of 72°C for 10 min. The number of PCR cycles
was 35. The products were detected in 2% agarose/Tris-acetate-EDTA
gels and stained with ethidium bromide for visualization by Syngene
G:BOX F2 and GeneTools software.
Intracellular calcium measurement
Intracellular calcium was determined by means of
flow cytometry following the staining of the cells with the calcium
indicator Fluo-3 AM. Following the various treatments, the cells
were collected and cultured with 5 µM Fluo-3 AM at 37°C in
the absence of light for 45 min. Cell fluorescence was then
measured by flow cytometry. When the cell density reached 80%, the
cells were incubated with QUE (5 or 10 µM), for 24 h prior
to exposure to OA (80 nM) for 12 h at 37°C.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Statistical significance was determined by one-way
analysis of variance followed by Duncan's multiple range test using
a computerized statistical package (SPSS 16.0). P<0.05 was
considered to indicate a statistically significant difference. All
experiments were repeated at least three times.
Results
Effects of QUE on OA-induced toxicity in
HT22 cells
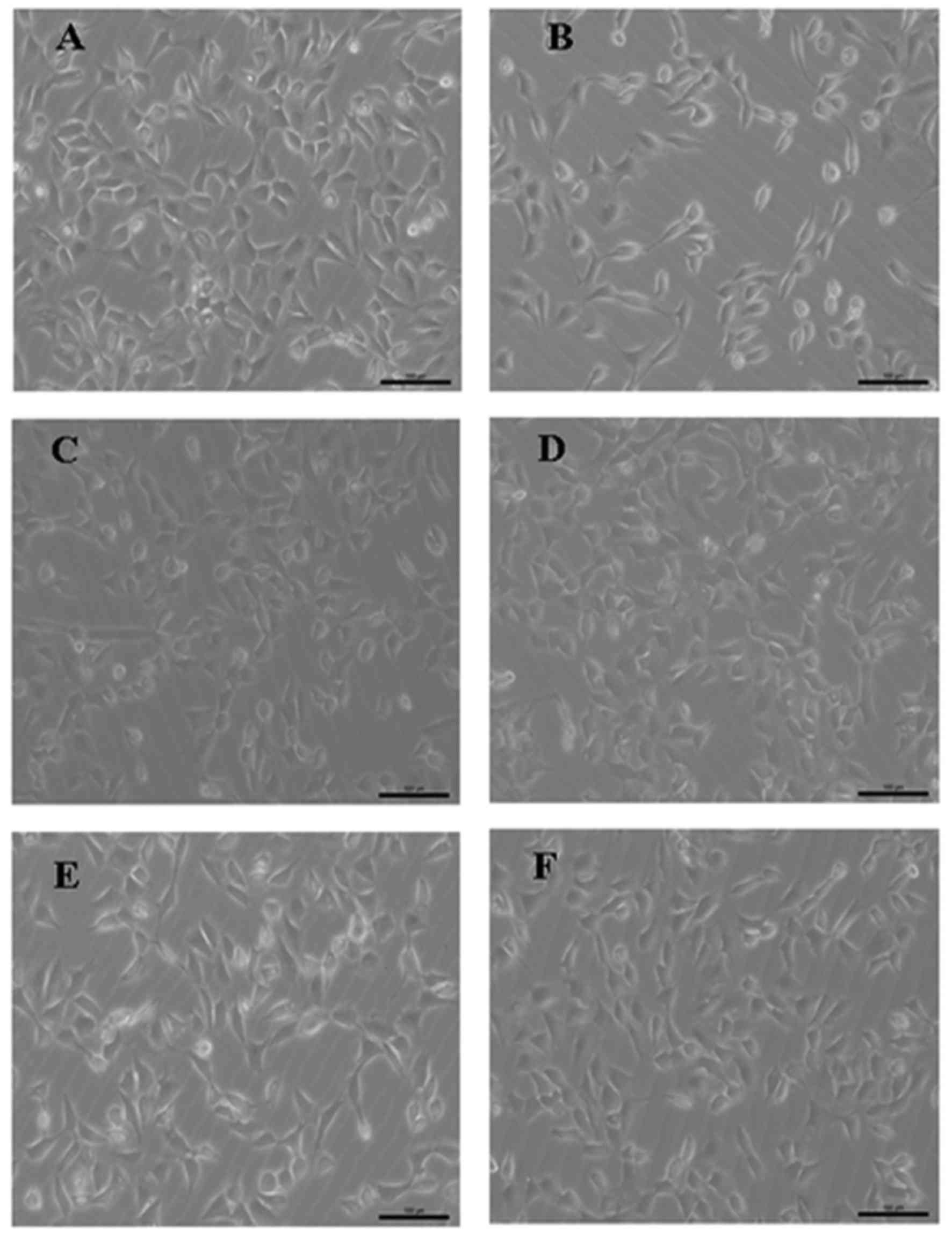
The control HT22 cells grew in a healthy condition,
with the cell bodies exhibiting good translucency and clear
boundaries (Fig. 1A). However,
following the exposure of the cells to 80 nM OA for 12 h, cell
growth was inhibited, the number of cells was markedly reduced, and
the intercellular spaces appeared to be widened (Fig. 1B). Pretreatment with QUE (5 or 10
µM), the calpain inhibitor CALP (10 µM) or the CDK5
inhibitor ROS (0.16 µM) improved the cell morphology and
increase the numbers of the OA-treated HT22 cells (Fig. 1C–F).
Effects of QUE on OA-induced tau protein
hyperphosphorylation in HT22 cells
Tau protein hyperphosphorylation serves a very
important role in AD and is a typical pathological feature of this
disease (50). Therefore, the
effects of QUE on OA-induced tau protein hyperphosphorylation were
investigated. Western blotting demonstrated that tau protein
hyperphosphorylation at four sites (S199, T205, T231 and S396) was
significantly increased following the exposure of HT22 cells to OA
(80 nM) for 12 h. However, these increases in phosphorylation were
significantly attenuated to varying degrees by pretreatment with 5
or 10 µM QUE for 24 h (Fig. 2A
and B). In addition, total tau protein and non-phosphorylated
tau protein were also investigated. As shown in Fig. 2C and D, total tau protein (tau-5)
did not vary significantly among the control, OA and OA plus QUE
treatment groups, and the changes in the levels of
non-phosphorylated tau (tau-1) were the converse of those for
phosphorylated tau, which confirms the consistency of the
results.
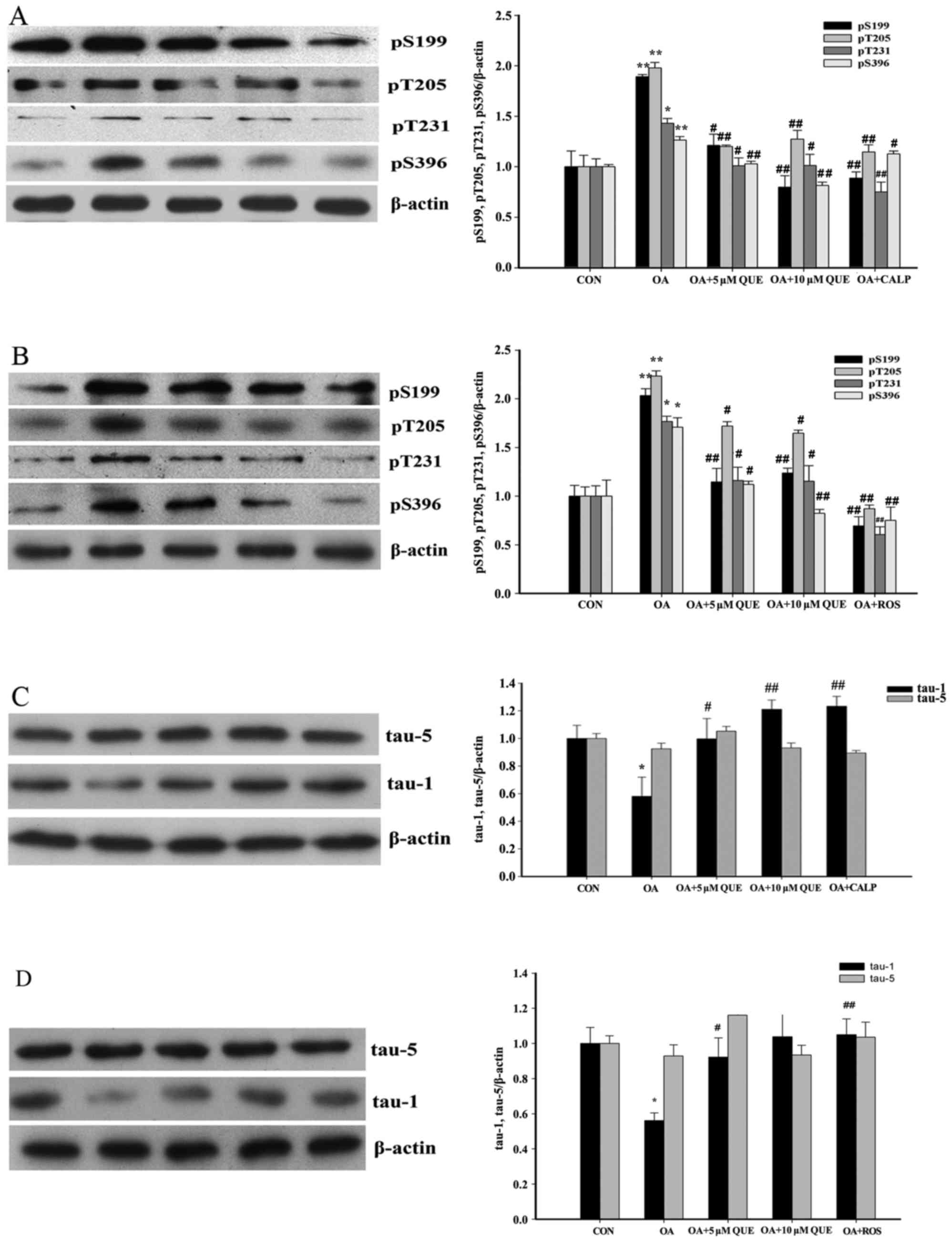 | Figure 2Effects of QUE on OA-induced tau
protein hyperphosphorylation in HT22 cells. QUE decreased the
OA-induced hyperphosphorylation of tau. HT22 cells were pretreated
with QUE (5 and 10 µM), CALP (10 µM) or ROS (0.16
µM) for 24 h prior to OA (80 nM) exposure for 12 h. Western
blotting revealed that treatment with OA alone augmented tau
hyperphosphorylation at Ser396, Ser199, Thr231 and Thr205 sites.
However, pretreatment with QUE, (A) CALP and (B) ROS significantly
decreased tau hyperphosphorylation. Total tau protein (tau-5) was
not altered among the various groups, whereas the changes in
non-phosphorylated tau protein (tau-1) expression induced by OA,
QUE, (C) CALP and (D) ROS were the opposite of those on tau
hyperphosphorylation. Western blotting data were quantified as
densitometry values, normalized using β-actin. Data are expressed
as the mean ± standard error of the mean. a–d,#P<0.05
and A–D,##P<0.01 vs. the OA-treated group;
*P<0.05 and **P<0.01 vs. the CON group.
CON, control; QUE, quercetin; OA, okadaic acid; CALP, calpeptin;
ROS, roscovitine. (E and F) Treatment with QUE alone had no effect
on tau phosphorylation. Western blotting data were quantified as
densitometry values, normalized using β-actin. Data are expressed
as the mean ± standard error of the mean. **P<0.01
vs. CON group; ##P<0.01 vs. OA-treated group. CON,
control; QUE, quercetin; OA, okadaic acid. |
In order to study the effect of QUE on baseline tau
protein phosphorylation, tau phosphorylation levels in HT22 cells
cultured with QUE alone were detected (Fig. 2E and F). However, treatment with
QUE culture exhibited no significant effect on tau phosphorylation
compared with that in the untreated control group.
CDK5 is vital for tau protein hyperphosphorylation;
the augmentation of intracellular Ca2+ levels leads to
increased CDK5 activity, which consequently results in tau protein
hyperphosphorylation (51). As
shown in Fig. 2A and B, tau
protein hyperphosphorylation was significantly reduced by
pretreatment with 10 µM CALP or 0.16 µM ROS for 24 h.
Furthermore, as shown in Fig. 2C and
D, the effects of CALP and ROS on unphosphorylated tau protein
in OA-treated HT22 cells were the converse of those on
phosphorylated tau, and no significant changes in total tau protein
were detected.
These results indicate that QUE markedly attenuated
tau protein hyperphosphorylation in OA-induced HT22 cells via the
Ca2+-calpain-p25-CDK5 signaling pathway, and indicate
that QUE may exhibit a neuroprotective effect. However, QUE
exhibited no effect on normal HT22 cells.
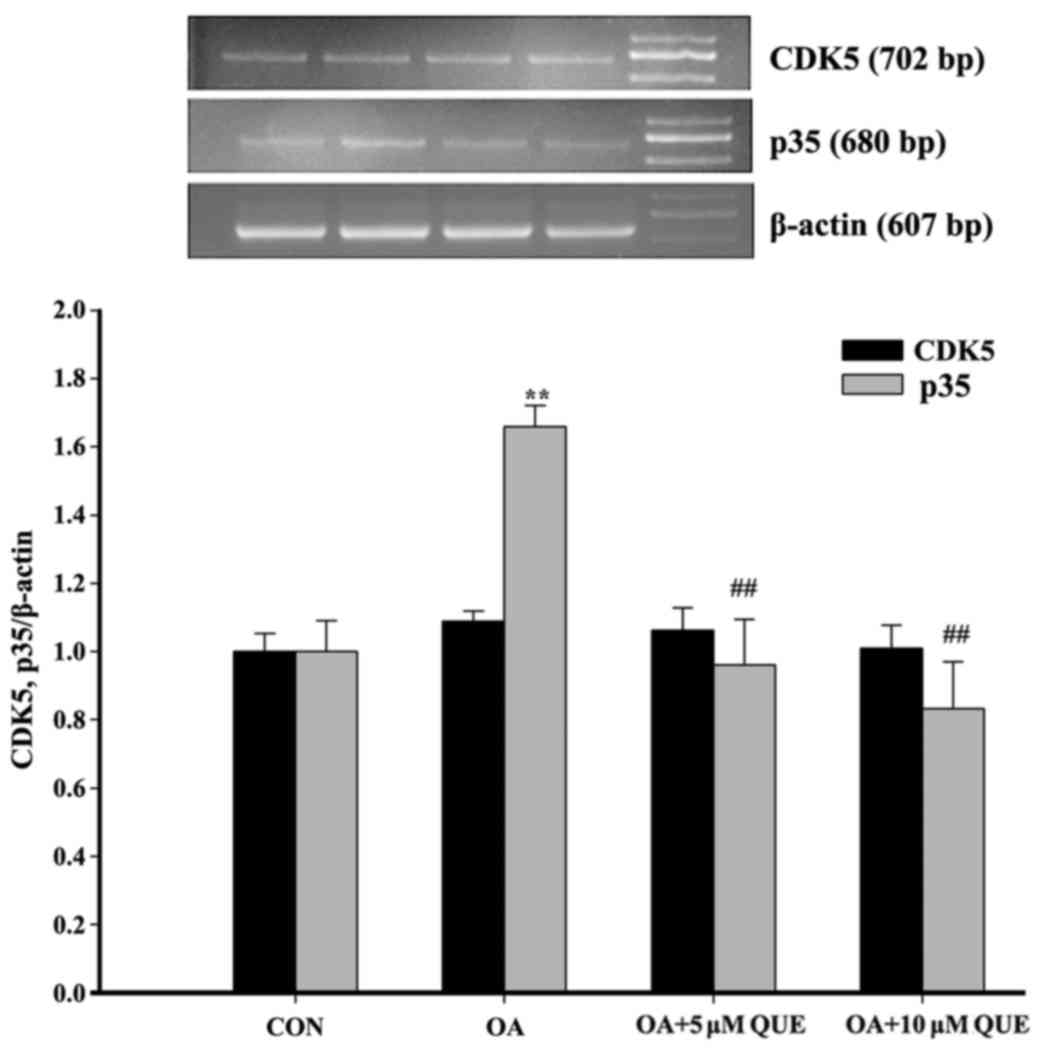
Effects of QUE on the OA-induced cleavage
of p35
To investigate whether the
Ca2+-calpain-p25-CDK5 signaling pathway is associated
with the effects of QUE on OA-induced tau protein
hyperphosphorylation, the changes of p25, p35 and CDK5 in
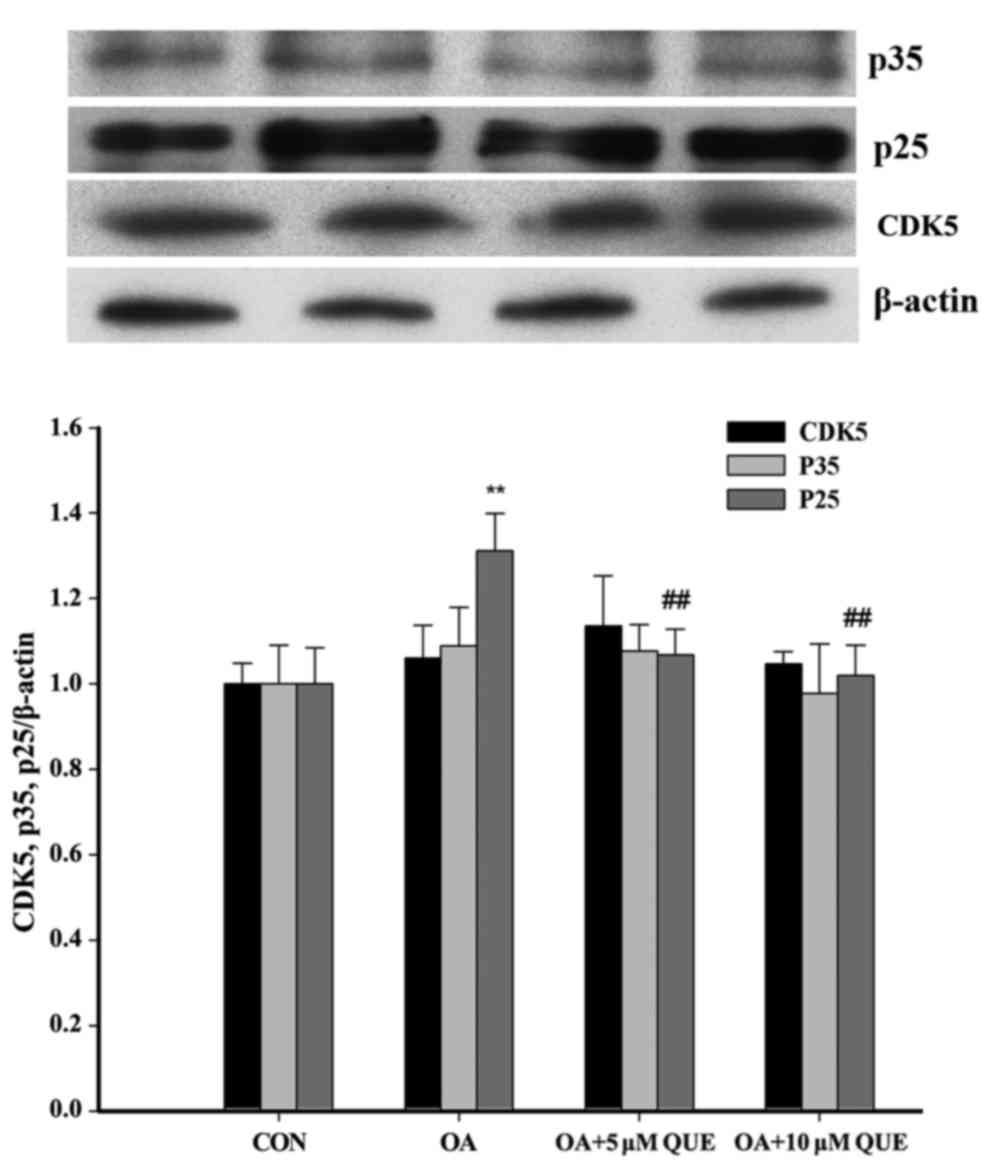
OA-induced HT22 cells were explored. As shown in Fig. 3, p35 and CDK5 levels exhibited no
significant difference among the groups. However, p25 was
significantly increased following treatment with OA for 12 h, and
this effect was significantly attenuated via pretreatment with QUE.
These results indicate that QUE decreased the conversion of p35
into p25.
Effects of QUE on OA-induced p-CDK5
expression
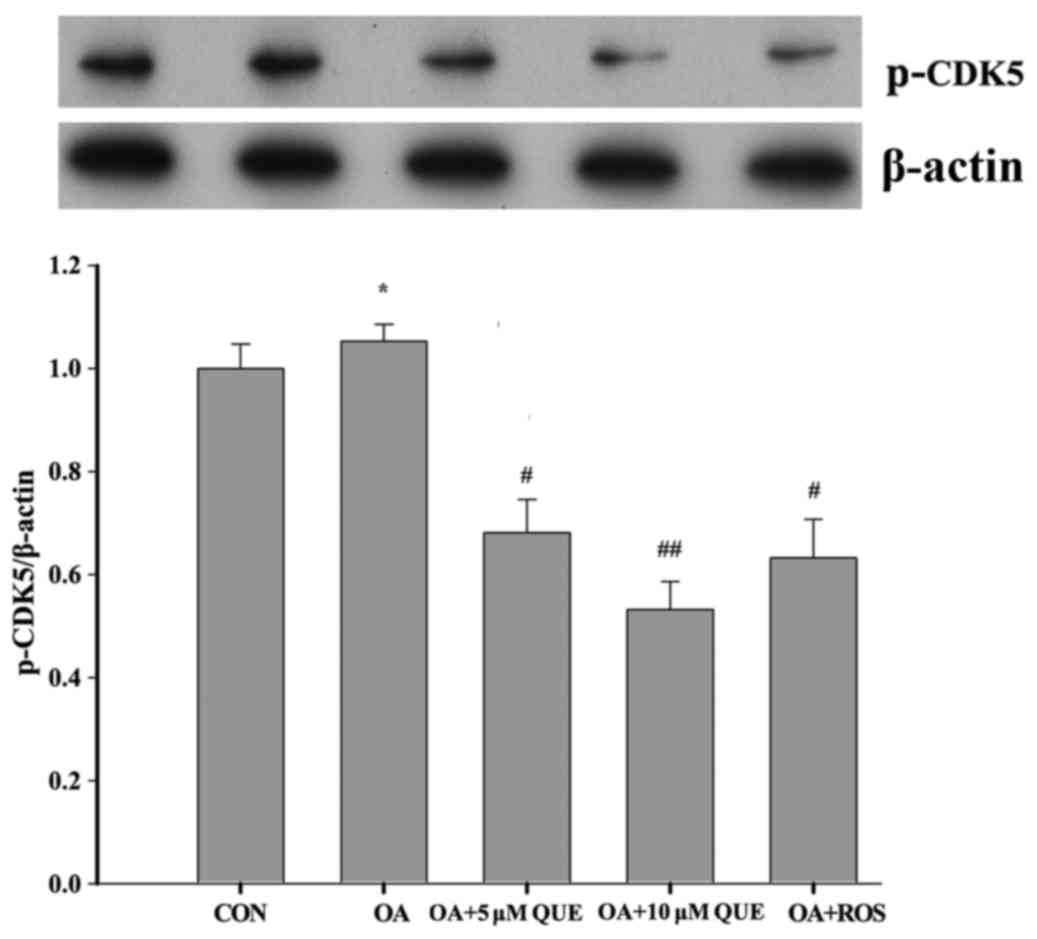
p-CDK5, the active form of CDK5, is involved in the
aggregation of phosphorylated tau protein (52). Therefore, the expression of p-CDK5
in the OA-induced HT22 cells was investigated. As shown in Fig. 4, p-CDK5 expression was increased
significantly following exposure to OA for 12 h. However, this
increase was significantly attenuated by pretreatment with 5 or 10
µM QUE, or 0.16 µM ROS for 24 h. These results are in
accordance with the variations in p25 expression, and suggest that
QUE reduced tau protein hyperphosphorylation by attenuating the
cleavage of p35 and downregulating CDK5 activity.
Effects of QUE on OA-induced calpain
expression
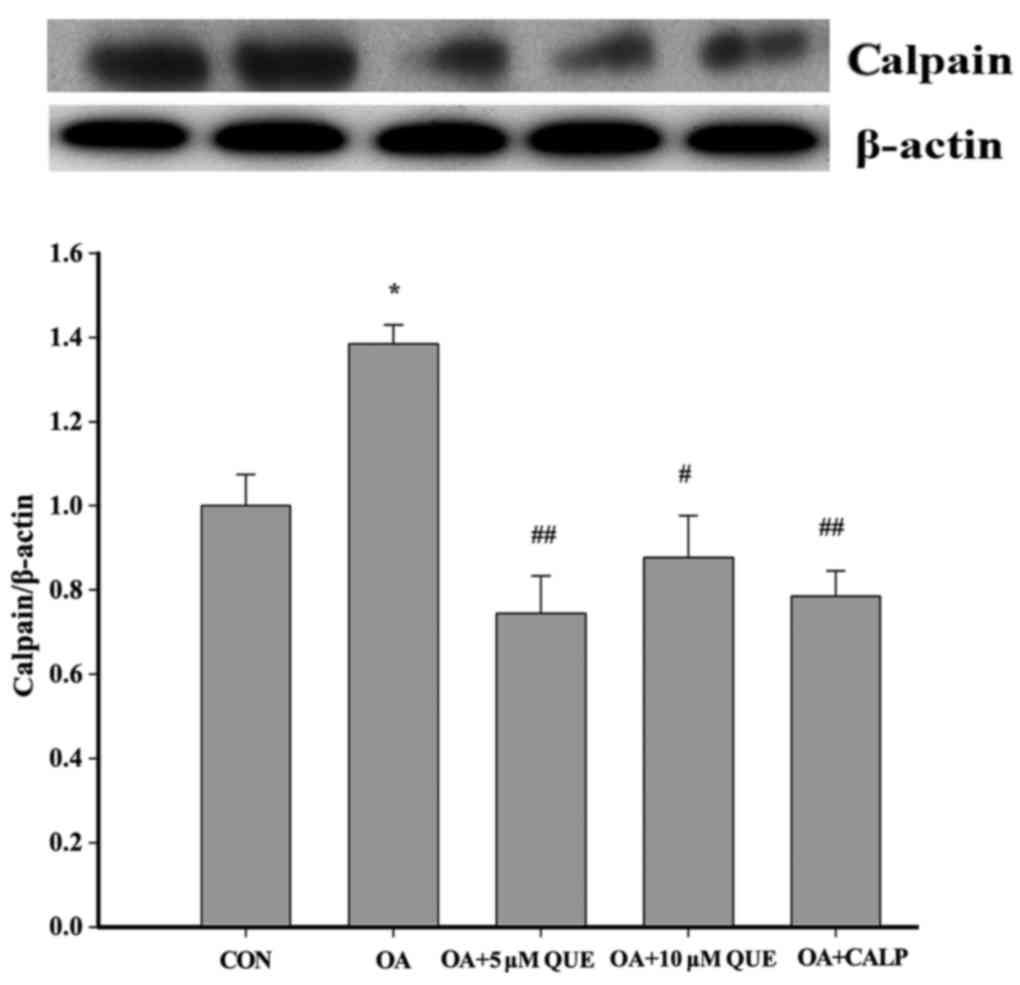
As p35 is converted to p25 by calpain, a
calcium-dependent protease, the possibility that QUE may affect
calpain expression was investigated. As shown in Fig. 5, calpain expression was
significantly augmented following treatment with OA for 12 h.
However, this increase was significantly attenuated by pretreatment
with 5 or 10 µM QUE or 10 µM CALP for 24 h. These
results indicate that QUE inhibited tau protein
hyperphosphorylation, which was associated with a reduction of
calpain expression.
Effects of QUE on OA-induced p35 and CDK5
mRNA
To further study the mechanism by which QUE
decreased tau protein hyperphosphorylation via the CDK5 signaling
pathway, the expression of p35 and CDK5 mRNA was examined. The
exposure of HT22 cells to OA caused a significant increase in p35
mRNA, which was significantly blocked by pretreatment with 5 or 10
µM QUE. However, treatment with OA alone or with QUE
pretreatment exhibited no significant effect on CDK5 mRNA (Fig. 6).
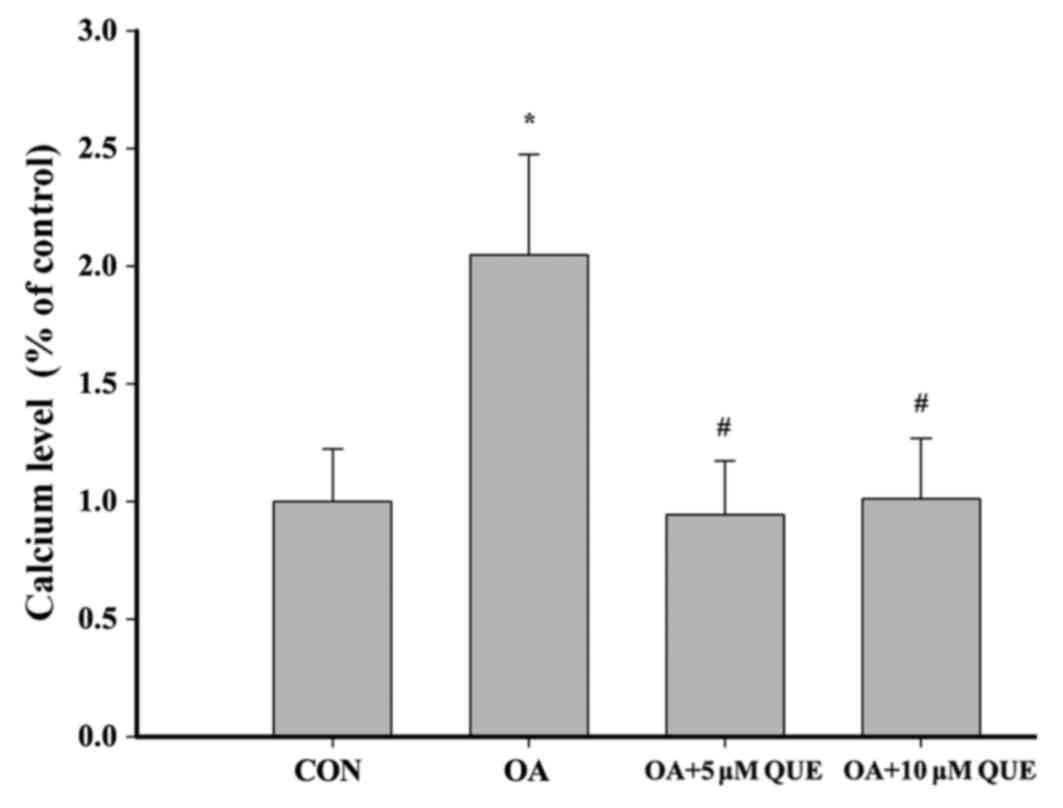
Effects of QUE on the OA-induced
intracellular Ca2+ level
Calcium influx activates calpain, which has been
reported as a factor in AD pathogenesis (18). Fig.
7 indicates that exposing HT22 cells to OA (80 nM) for 12 h
caused a significant increase in intracellular Ca2+
levels. This increase was significantly attenuated by pretreatment
with 5 or 10 µM QUE.
Discussion
Hyperphosphorylated tau protein is an essential
component of NFTs, which are a major pathological factor in AD
(12). Thus, tau protein
hyperphosphorylation is a potential therapeutic target.
Furthermore, it has been reported that tau pathology has a greater
effect than amyloid burden on the clinical symptoms associated with
AD; magnetic resonance imaging and electroencephalography indicate
that Aβ deposition is relevant to functional network destruction
whereas hyperphosphorylated tau directly affects memory deficits
and cognition (53). Therefore,
the inhibition of tau protein hyperphosphorylation is an important
aim in AD. The present study provides evidence that QUE defended
HT22 cells from OA-induced neurotoxicity by reducing tau protein
hyperphosphorylation. During the study, it was observed that QUE
reduced OA-induced tau protein hyperphosphorylation at Ser396,
Ser199, Thr231 and Thr205 sites. However, no evident difference
between 5 and 10 µM QUE was observed, with the exception of
the Thr231 site, where QUE (10 µM) exhibited a stronger
effect. Additionally, CALP and ROS, which are specific inhibitors
of calpain and CDK5, respectively, decreased OA-induced tau protein
hyperphosphorylation. Furthermore, none of the treatments affected
the total tau levels. However, unphosphorylated tau levels were
reduced by treatment with OA alone, and the reduction was
attenuated by pretreatment with QUE, CALP or ROS. These results
indicate that QUE effectively reduced tau hyperphosphorylation
without changing the levels of total tau protein.
The aberrant phosphorylation of tau due to
overactivated CDK5 activity is considered a major pathological
mechanism in the development of AD (54). CDK5 serves as an upstream
signaling molecule in the regulation of tau protein
hyperphosphorylation, and is an important determinant of the state
of tau protein (20). Therefore,
downregulating CDK5 kinase activity is a potential target for AD.
In the present study, whether the effect of QUE on
hyperphosphorylated tau is attributable to downregulated CDK5
kinase activity was investigated. CDK5 activation was further
examined by detecting the phosphorylation of CDK5 at
Tyr15, which represents the activity of CDK5. Western
blotting results demonstrated that the phosphorylation of
CDK5-Tyr15 was attenuated by pretreatment with QUE or
ROS. This indicates that QUE affected the activity of CDK5, and
demonstrates the potential of QUE as a CDK5 inhibitor for use in
AD.
As mentioned above, CDK5 activity is dependent on
the p35 or p39 subunits of the enzyme, and the former can be
cleaved to form p25. Therefore, CDK5 activity may be blocked by
decreasing p35 or p25, which should consequently reduce tau protein
hyperphosphorylation. Thus, the expression of p35, p25 and CDK5 was
investigated in HT22 cells following exposure to OA. OA caused a
significant increase in p25 levels, and pretreatment with QUE
blocked the conversion of p35 to p25, leading to a reduction of
hyperphosphorylated tau protein levels. Notably, the expression of
CDK5 exhibited no difference among the groups. Based on these
results, it may be concluded that QUE restrained OA-induced tau
hyperphosphorylation by inhibiting the cleavage of p35 to p25
without affecting CDK5 expression. Additionally, the cleavage of
p35 to p25 requires calpain, which is activated by intracellular
calcium (55,56). To better explain the molecular
mechanism by which QUE attenuated tau protein hyperphosphorylation,
calpain expression was explored in the present study. Calpain
expression was significantly increased following exposure to OA.
However, this increase was attenuated by pretreatment with QUE. The
positive control, CALP, also reduced calpain expression.
Furthermore, intracellular calcium levels were evaluated, and the
changes observed were in accordance with those for calpain. These
findings demonstrate that QUE inhibited the OA-induced increases in
calpain expression and intracellular calcium levels. Furthermore,
the results indicate that QUE blocked the
Ca2+-calpain-p25-CDK5 signaling pathway, ,and thus may
be an effective treatment for AD.
To further elucidate the molecular mechanism by
which QUE modulates the Ca2+-calpain-p25-CDK5 signaling
pathway, p35 and CDK5 mRNA levels were detected. Notably, only p35
mRNA exhibited any changes when different treatments were applied.
Thus, it appears that QUE reduced CDK5 activity by inhibiting the
expression of p35, thereby decreasing the conversion of p35 to p25.
This demonstrates that p35 mRNA is an effective target for QUE.
Together, the results indicate that QUE acted on various targets in
the Ca2+-calpain-p25-CDK5 signaling pathway to
downregulate tau protein hyperphosphorylation.
In conclusion, the results of the present study
suggest that QUE exhibited a marked neuroprotective effect on
OA-induced HT22 cells by inhibiting the hyperphosphorylation of tau
protein, and this effect may have been mediated via inhibition of
the Ca2+-calpain-p25-CDK5 signaling pathway. This
demonstrates that QUE is a potential therapeutic candidate for the
prevention of tau protein hyperphosphorylation. Collectively, these
findings expand our knowledge of the neuroprotective mechanism of
QUE. However, the present study was restricted to in vitro
experiments, and future studies to investigate the effect of QUE on
transgenic mouse are planned. These may support the use of QUE as
an effective therapeutic agent for AD and other tauopathies.
Acknowledgments
The present study was supported by the National Key
Development Program for Basic Research of China (grant no.
2006cb500700), Medical Scientific Research Foundation of Guangdong
Province (grant no. A2015032), Fundamental Research Funds for the
Central Universities of China (grant no. 14ykpy03), National
Natural Science Foundation of China (grant nos. 81774099 and
81173577) National Natural Science Foundation of China (grant no.
81501093), Natural Science Foundation of Guangdong Province (grant
nos. 2015A030313066 and 2015A030310251) and Science and Technology
Planning Project of Guangdong Province (grant no.
2014A020212622).
Glossary
Abbreviations
Abbreviations:
|
AD
|
Alzheimer's disease
|
|
NFTs
|
neurofibrillary tangles
|
|
QUE
|
quercetin
|
|
CDK5
|
cyclin-dependent kinase 5
|
|
Aβ
|
β-amyloid
|
|
OA
|
okadaic acid
|
|
CALP
|
calpeptin
|
|
ROS
|
roscovitine
|
References
|
1
|
Prince M, Bryce R and Ferri CA: World
Alzheimer Report 2011: The Benefits of Early Diagnosis and
Intervention. Alzheimer's Disease International; London: pp. 1–72.
2011
|
|
2
|
Bassil N and Grossberg GT: Novel regimens
and delivery systems in the pharmacological treatment of
Alzheimer's disease. CNS Drugs. 23:293–307. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Neugroschl J and Sano M: An update on
treatment and prevention strategies for Alzheimer's disease. Curr
Neurol Neurosci Rep. 9:368–376. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ballatore C, Lee VM and Trojanowski JQ:
Tau-mediated neurodegeneration in Alzheimer's disease and related
disorders. Nat Rev Neurosci. 8:663–672. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Giacobini E and Becker RE: One hundred
years after the discovery of Alzheimer's disease. A turning point
for therapy? J Alzheimers Dis. 12:37–52. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pallàs M and Camins A: Molecular and
biochemical features in Alzheimer's disease. Curr Pharm Des.
12:4389–4408. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Takashima A: Tau aggregation is a
therapeutic target for Alzheimer's disease. Curr Alzheimer Res.
7:665–669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Gong CX and Iqbal K: Hyperphosphorylation
of micro-tubule-associated protein tau: A promising therapeutic
target for Alzheimer disease. Curr Med Chem. 15:2321–2328. 2008.
View Article : Google Scholar
|
|
9
|
Iqbal K, Liu F, Gong CX and Grundke-Iqbal
I: Tau in Alzheimer disease and related tauopathies. Curr Alzheimer
Res. 7:656–664. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Alonso AC, Mederlyova A, Novak M,
Grundke-Iqbal I and Iqbal K: Promotion of hyperphosphorylation by
frontotemporal dementia tau mutations. J Biol Chem.
279:34873–34881. 2004. View Article : Google Scholar
|
|
11
|
Zheng WH, Bastianetto S, Mennicken F, Ma W
and Kar S: Amyloid beta peptide induces tau phosphorylation and
loss of cholinergic neurons in rat primary septal cultures.
Neuroscience. 115:201–211. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Giacobini E and Gold G: Alzheimer disease
therapy - moving from amyloid-β to tau. Nat Rev Neurol. 9:677–686.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Stoothoff WH and Johnson GV: Tau
phosphorylation: physiological and pathological consequences.
Biochim Biophys Acta. 1739:280–297. 2005. View Article : Google Scholar
|
|
14
|
Wang JZ, Grundke-Iqbal I and Iqbal K:
Kinases and phosphatases and tau sites involved in Alzheimer
neurofibrillary degeneration. Eur J Neurosci. 25:59–68. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Engmann O and Giese KP: Crosstalk between
Cdk5 and GSK3β: Implications for Alzheimer's disease. Front Mol
Neurosci. 2:22009. View Article : Google Scholar
|
|
16
|
Tsai LH, Lee MS and Cruz J: Cdk5, a
therapeutic target for Alzheimer's disease? Biochim Biophys Acta.
1697:137–142. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Piedrahita D, Hernández I, López-Tobón A,
Fedorov D, Obara B, Manjunath BS, Boudreau RL, Davidson B, Laferla
F, Gallego-Gómez JC, et al: Silencing of CDK5 reduces
neurofibrillary tangles in transgenic Alzheimer's mice. J Neurosci.
30:13966–13976. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Angelo M, Plattner F and Giese KP:
Cyclin-dependent kinase 5 in synaptic plasticity, learning and
memory. J Neurochem. 99:353–370. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Alvarez A, Muñoz JP and Maccioni RB: A
Cdk5-p35 stable complex is involved in the beta-amyloid-induced
deregulation of Cdk5 activity in hippocampal neurons. Exp Cell Res.
264:266–274. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Noble W, Olm V, Takata K, Casey E, Mary O,
Meyerson J, Gaynor K, LaFrancois J, Wang L, Kondo T, et al: Cdk5 is
a key factor in tau aggregation and tangle formation in vivo.
Neuron. 38:555–565. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Utreras E, Maccioni R and
González-Billault C: Cycl-in-dependent kinase 5 activator p35
over-expression and amyloid beta synergism increase apoptosis in
cultured neuronal cells. Neuroscience. 161:978–987. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dhavan R and Tsai LH: A decade of CDK5.
Nat Rev Mol Cell Biol. 2:749–759. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nikkel AL, Martino B, Markosyan S,
Brederson JD, Medeiros R, Moeller A and Bitner RS: The novel
calpain inhibitor A-705253 prevents stress-induced tau
hyperphosphorylation in vitro and in vivo. Neuropharmacology.
63:606–612. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rao MV, McBrayer MK, Campbell J, Kumar A,
Hashim A, Sershen H, Stavrides PH, Ohno M, Hutton M and Nixon RA:
Specific calpain inhibition by calpastatin prevents tauopathy and
neurodegeneration and restores normal lifespan in tau P301L mice. J
Neurosci. 34:9222–9234. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Orsolić N, Knezević AH, Sver L, Terzić S
and Basić I: Immunomodulatory and antimetastatic action of propolis
and related polyphenolic compounds. J Ethnopharmacol. 94:307–315.
2004. View Article : Google Scholar
|
|
26
|
Gulati N, Laudet B, Zohrabian VM, Murali R
and Jhanwar-Uniyal M: The antiproliferative effect of quercetin in
cancer cells is mediated via inhibition of the PI3K-Akt/PKB
pathway. Anticancer Res. 26(2A): 1177–1181. 2006.PubMed/NCBI
|
|
27
|
Landis-Piwowar KR, Milacic V and Dou QP:
Relationship between the methylation status of dietary flavonoids
and their growth-inhibitory and apoptosis-inducing activities in
human cancer cells. J Cell Biochem. 105:514–523. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mahoney SE, Davis JM, Murphy EA, McClellan
JL and Pena MM: Dietary quercetin reduces chemotherapy-induced
fatigue in mice. Integr Cancer Ther. 13:417–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chondrogianni N, Kapeta S, Chinou I,
Vassilatou K, Papassideri I and Gonos ES: Anti-ageing and
rejuvenating effects of quercetin. Exp Gerontol. 45:763–771. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ferri P, Angelino D, Gennari L, Benedetti
S, Ambrogini P, Del Grande P and Ninfali P: Enhancement of
flavonoid ability to cross the blood-brain barrier of rats by
co-administration with α-tocopherol. Food Funct. 6:394–400. 2015.
View Article : Google Scholar
|
|
31
|
Li Y, Zhou S, Li J, Sun Y, Hasimu H, Liu R
and Zhang T: Quercetin protects human brain microvascular
endothelial cells from fibrillar β-amyloid1-40-induced toxicity.
Acta Pharm Sin B. 5:47–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ishisaka A, Mukai R, Terao J, Shibata N
and Kawai Y: Specific localization of quercetin-3-O-glucuronide in
human brain. Arch Biochem Biophys. 557:11–17. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Faria A, Pestana D, Teixeira D, Azevedo J,
De Freitas V, Mateus N and Calhau C: Flavonoid transport across
RBE4 cells: a blood-brain barrier model. Cell Mol Biol Lett.
15:234–241. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Li H, Liu Y, Yi Y, Miao Q, Liu S, Zhao F,
Cong W, Wang C and Xia C: Purification of quercetin-3-O-sophoroside
and isoquercitrin from Poacynum hendersonii leaves using
macroporous resins followed by Sephadex LH-20 column
chromatography. J Chromatogr B Analyt Technol Biomed Life Sci.
1048:56–63. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang SH, Wang SQ, Liu CH and Yang YH:
Studies on quality standards for Pollen Typhae(puhuang). Zhongguo
Zhong Yao Za Zhi. 25:136–139. 2000.In Chinese.
|
|
36
|
Ansari MA, Abdul HM, Joshi G, Opii WO and
Butterfield DA: Protective effect of quercetin in primary neurons
against Aβ(1-42): relevance to Alzheimer's disease. J Nutr Biochem.
20:269–275. 2009. View Article : Google Scholar
|
|
37
|
Rezai-Zadeh K, Arendash GW, Hou H,
Fernandez F, Jensen M, Runfeldt M, Shytle RD and Tan J: Green tea
epigallo-catechin-3-gallate (EGCG) reduces beta-amyloid mediated
cognitive impairment and modulates tau pathology in Alzheimer
transgenic mice. Brain Res. 1214:177–187. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Devi L and Ohno M: 7,8-Dihydroxyflavone, a
small-molecule TrkB agonist, reverses memory deficits and BACE1
elevation in a mouse model of Alzheimer's disease.
Neuropsychopharmacology. 37:434–444. 2012. View Article : Google Scholar :
|
|
39
|
Spencer JP: The impact of flavonoids on
memory: physiological and molecular considerations. Chem Soc Rev.
38:1152–1161. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Oishi K and Lyketsos CG: Alzheimer's
disease and the fornix. Front Aging Neurosci. 6:2412014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Shi Y: Serine/threonine phosphatases:
Mechanism through structure. Cell. 139:468–484. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sun L, Liu SY, Zhou XW, Wang XC, Liu R,
Wang Q and Wang JZ: Inhibition of protein phosphatase 2A- and
protein phosphatase 1-induced tau hyperphosphorylation and
impairment of spatial memory retention in rats. Neuroscience.
118:1175–1182. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bennecib M, Gong CX, Grundke-Iqbal I and
Iqbal K: Inhibition of PP-2A upregulates CaMKII in rat forebrain
and induces hyperphosphorylation of tau at Ser 262/356. FEBS Lett.
490:15–22. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Martin L, Latypova X, Wilson CM,
Magnaudeix A, Perrin ML and Terro F: Tau protein phosphatases in
Alzheimer's disease: The leading role of PP2A. Ageing Res Rev.
12:39–49. 2013. View Article : Google Scholar
|
|
45
|
Tao T, He C, Deng J, Huang Y, Su Q, Peng
M, Yi M, Darko KO, Zou H and Yang X: A novel synthetic derivative
of quercetin,
8-trifluoromethyl-3,5,7,3′,4′-O-pentamethyl-quercetin, inhibits
bladder cancer growth by targeting the AMPK/mTOR signaling pathway.
Oncotarget. 8:71657–71671. 2017.PubMed/NCBI
|
|
46
|
Luo T, Jiang W, Kong Y, Li S, He F, Xu J
and Wang HQ: The protective effects of jatrorrhizine on
β-amyloid(25–35)-induced neurotoxicity in rat cortical neurons. CNS
Neurol Disord Drug Targets. 11:1030–1037. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Luo T, Zhang H, Zhang WW, Huang JT, Song
EL, Chen SG, He F, Xu J and Wang HQ: Neuroprotective effect of
Jatrorrhizine on hydrogen peroxide-induced cell injury and its
potential mechanisms in PC12 cells. Neurosci Lett. 498:227–231.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu J, Li L and Suo WZ: HT22 hippocampal
neuronal cell line possesses functional cholinergic properties.
Life Sci. 84:267–271. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Chen X, Huang T, Zhang J, Song J, Chen L
and Zhu Y: Involvement of calpain and p25 of CDK5 pathway in
ginsenoside Rb1's attenuation of β-amyloid
peptide25–35-induced tau hyperphosphorylation in
cortical neurons. Brain Res. 1200:99–106. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Wang JZ, Wang ZH and Tian Q: Tau
hyperphosphorylation induces apoptotic escape and triggers
neurodegeneration in Alzheimer's disease. Neurosci Bull.
30:359–366. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shukla V, Seo J, Binukumar BK, Amin ND,
Reddy P, Grant P, Kuntz S, Kesavapany S, Steiner J, Mishra SK, Tsai
LH and Pant HC: TFP5, a peptide inhibitor of aberrant and
hyperactive Cdk5/p25, attenuates pathological phenotypes and
restores synaptic function in CK-p25Tg mice. J Alzheimers Dis.
56:335–349. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Zukerberg LR, Patrick GN, Nikolic M,
Humbert S, Wu CL, Lanier LM, Gertler FB, Vidal M, Van Etten RA and
Tsai LH: Cables links Cdk5 and c-Abl and facilitates Cdk5 tyrosine
phosphorylation, kinase upregulation, and neurite outgrowth.
Neuron. 26:633–646. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Pievani M, de Haan W, Wu T, Seeley WW and
Frisoni GB: Functional network disruption in the degenerative
dementias. Lancet Neurol. 10:829–843. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kimura T, Ishiguro K and Hisanaga S:
Physiological and pathological phosphorylation of tau by Cdk5.
Front Mol Neurosci. 7:652014. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Kawahara M and Kuroda Y: Intracellular
calcium changes in neuronal cells induced by Alzheimer's
beta-amyloid protein are blocked by estradiol and cholesterol. Cell
Mol Neurobiol. 21:1–13. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kusakawa G, Saito T, Onuki R, Ishiguro K,
Kishimoto T and Hisanaga S: Calpain-dependent proteolytic cleavage
of the p35 cyclin-dependent kinase 5 activator to p25. J Biol Chem.
275:17166–17172. 2000. View Article : Google Scholar : PubMed/NCBI
|