Introduction
The corneal epithelium (CE) is the exfoliated
multilayer epithelium that forms the anterior surface layer of the
cornea (1). Corneal epithelial
cell (CEC) homeostasis is a requirement for both ocular surface
integrity and corneal transparency and visual function (2). Corneal damage, including infection,
trauma and denaturation, have severe consequences for the ocular
surface, which in turn may affect the integrity of the CE and
visual function, ultimately leading to functional blindness
(3). There were approximately
4.9–10 million people with corneal blindness worldwide in 2012
(4). One of the most successful
strategies for ocular surface reconstruction is the transplantation
of epithelial cell sheets engineered from autologous limbal
epithelial cells (1,5,6);
it is effective in animal models and humans. However, a limbal
biopsy is required for the isolation of limbal epithelial cells,
which may pose a risk to the contralateral healthy eye and is not
useful for treating bilateral limbal stem cell deficiency.
Allogeneic limbal epithelium transplantation (7), one optional treatment, has
limitations of immunological rejection, donor scarcity and an
extremely low long‑term success rate. Thus, it is reasonable to
find and evaluate alternative autologous stem cell sources for
in vitro culture and transplantation to avoid the risk of
immunological rejection and to satisfy the demand from patients
with corneal blindness.
Other cell sources, including oral mucosal
epithelial cells (8–10), mesenchymal stem cells (11,12), embryonic stem cells (13) and immature dental pulp stem cells
(14), have been established for
corneal epithelium replacement, but to date, no long-term results
have been reported or achieved. Stem cells residing within the
epidermis have the potential of multi-directional differentiation.
In addition to differentiating into skin cells and appendages, skin
epidermal stem cells (SESCs) can differentiate into other lineages,
including nerve cells (15) and
germ cells (16). Moreover, SESCs
and limbal stem cells are of the same origin, namely, the basal
layer of embryonic skin, and are thus keratinocyte stem cells
(17,18), which express specific types of
cytokeratin protein (K1, K3, K5, K10, K12, K14, K15 and K19) on
their surfaces (19–21). It is hypothesized that SESCs are
ideal seed cells that may be utilized to replace limbal stem cells
for tissue engineered cornea construction. There have been certain
attempts to use hair follicle stem cells (22) and skin-derived precursor cells
(2,23); however, the mechanisms involved,
particularly the regulatory role of miRNAs in this process, have
not been fully elucidated. In the present study, the expression
profile of microRNAs (miRNAs/miRs) was compared in the SESCs and
CECs of sheep. The findings of this study may be useful in the
elucidation of the mechanisms underlying the molecular regulation
of the transdifferentiation process of SESCs into CECs.
Materials and methods
Cells
SESCs and CECs were obtained from 2 male
small-tailed Han sheep (obtained from the Experimental Animal
Center of Shaanxi Center for Stem Cell Engineering and Technology,
Northwest A&F University) following a protocol approved by the
Medical Animal Care and Welfare Committee of Luoyang Normal
University. The isolation of CECs and SESCs were performed as
previously described (24,25).
Small RNA sequencing
Total RNA (3 µg) from the SESCs and CECs was
used as input material for the generation of small RNA libraries.
Sequencing libraries were generated using the NEBNext®
Multiplex Small RNA Library Prep Set for Illumina® (New
England Biolabs, Inc., Ipswich, MA, USA) following the
manufacturer's instructions, and index codes were added to
attribute sequences to each sample. The clustering of the
index-coded samples was performed on a cBot Cluster Generation
System using TruSeq SR Cluster kit v3-cBot-HS (Illumina, Inc., San
Diego, CA, USA), according to the manufacturer's instructions.
Following cluster generation, the library preparations were
sequenced on an Illumina HiSeq 2500/2000 platform (Illumina, Inc.)
and 50-bp single-end reads were generated.
The raw sequence reads were first trimmed from the
adapter sequence using a Perl script and adapter-trimmed reads
longer than 18 nt were used for further analysis as clean reads. To
further parse the clean reads, the Integrative Short Reads
Navigator software (freely accessed at http://omicslab.genetics.ac.cn/ISRNA/) was used based
on the manufacturer's instructions. Bowtie software (version 1.1.2,
open-source software maintained by Johns Hopkins University) was
used to map sequences to sheep genomes (Ovis aries).
Annotation of the short sequence reads was performed using the
BLAST + program (ftp://ftp.ncbi.nlm.nih.gov/blast/executables/blast+/LATEST/)
and the Rfam database (http://rfam.xfam.org/). Known miRNA sequences were
extracted from miRBase (http://www.mirbase.org/). The frequency of miRNA reads
was normalized as transcripts per million, then the normalized
expression levels of miRNAs between two samples were conducted to
calculate fold-changes. The identification of differentially
expressed miRNAs between two different samples was performed based
on the log10 ratio of fold-changes. Hierarchical
clustering was performed using Heatmap.2 (R language gplots
package). The levels of miRNA expression were clustered using
average linkage clustering. The average value of the distance of
each miRNA was used to measure cluster-to-cluster distance.
mRNA microarray analysis
The transcriptional profiles of the SESCs and CECs
were analyzed. Total RNA was extracted and purified using the
miRNeasy mini kit (Qiagen GmBH, Hilden, Germany) following the
manufacturer's instructions and an RIN number was checked for to
determine RNA integrity using an Agilent Bioanalyzer 2100 (Agilent
Technologies, Inc., Santa Clara, CA, USA). Total RNA was amplified
and labeled using a Low Input Quick Amp Labeling kit, One‑Color
(Agilent Technologies, Inc.), following the manufacturer's
instructions. Labeled cRNAs were purified using an RNeasy mini kit
(Qiagen GmBH). Each slide was hybridized with 600 ng Cy3-labeled
cRNA using a gene expression hybridization kit in a hybridization
oven (both Agilent Technologies, Inc.), according to the
manufacturer's instructions. After 17 h of hybridization, slides
were washed in staining dishes (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) with a gene expression wash buffer kit (Agilent
Technologies, Inc.), following the manufacturer's instructions.
Slides were scanned on a Agilent Microarray Scanner (Agilent
Technologies) using the following default settings: Dye channel,
green; scan resolution, 5 µm; PMT, 100 and 10%, 16-bit. Data
were extracted using Feature Extraction 10.7 (Agilent Technologies,
Inc.). Raw data were normalized using a Quantile algorithm, Gene
Spring Software 11.0 (Agilent Technologies, Inc.).
Target prediction and network
construction
As there was no appropriate database or methods for
predicting sheep miRNA target genes, the human orthologs of
differentially expressed sheep miRNA sequences were used to
identify potential target genes by searching the TargetScan
database (http://www.targetscan.org/vert_71/). JAVA (version
1.8.0_101; Oracle Software Systems Ltd., Redwood Shore, CA, USA)
and Cytoscape 2.6.0 (version 2.6.0, open-source software) software
were used to construct regulatory miRNA-target gene networks based
on regulatory interactions between differentially expressed
miRNAs.
Gene Ontology (GO) and Kyoto Encyclopedia
of Genes and Genomes (KEGG) pathway analyses
GO (http://geneon-tology.org/) and KEGG (http://www.kegg.jp/) pathway analyses of
differentially expressed genes were performed using the Database
for Annotation, Visualization and Integrated Discovery (DAVID;
https://david.ncifcrf.gov/); a
hypergeometric test with the Benjamini and Hochberg false discovery
rate (FDR) was performed using the default parameters to adjust the
P-value. A bubble chart was plotted using the OmicShare tools, a
free online platform for data analysis (www.omicshare.com/tools).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was performed to validate the expression
levels of the analyzed miRNAs by sequencing. Reverse transcription
was performed in a 20-µl reaction containing 2 µl
total RNA, 1 µl of 5 µM RT primer, 1 µl of 10
mM dNTPs, 4 µl 5X PrimeScript buffer and 200 units of
PrimeScript RTase (Takara Bio, Inc., Otsu, Japan). The primers used
were as follows: U6 forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′; miR-127 forward,
5′-ACACTCCAGCTGGGCTGAAGCTCAGA GGG-3′; miR-376c forward,
5′-ACACTCCAGCTGGGAACATAGAGGAAA-3′; miR-382 forward,
5′-ACACTCCAGCTGGGAATCATTCACGGAC-3′; miR-758 forward,
5′-ACACTCCAGCTGGGTTTGTGACCTGGTC-3′; miR-154 forward,
5′-ACACTCCAGCTGGGTAGGTTATCCGTGTTG-3′; miR-409 forward,
5′-ACACTCCAGCTGGGGAATGTTGCTCGGTG-3′; miR-10b forward,
5′-ACACTCCAGCTGGGTACCCTGTAGAACCGAA-3′ (all miRs had the same
reverse primer, 5′-TGGTGTCGTGGAGTCG-3′); DOCK9 forward,
5′-TTAAGGATGCATCTGGAAACC-3′ and reverse,
5′-CTTGAGCATGTCTTCATTGGA-3′; NEUROD1 forward,
5′-CTTCGATAGCCATTCACATCAT-3′ and reverse,
5′-TGAAACTGACGTGCCTCTAATC-3′; ALCAM forward,
5′-CTTCAGCAGCCATCACAGTT-3′ and reverse, 5′-AGAGCATCGCCTATCTGCTT-3′;
GAPDH forward, 5′-CTGGCCAAGGTCATCCAT-3′ and reverse,
5′-ACAGTCTTCTGGGTGGCAGT-3′. The reaction was incubated at 42°C for
60 min, and then terminated by heating at 85°C for 5 min. The qPCR
amplification was performed using SYBR-Green I® (Roche
Diagnostics, Indianapolis, IN, USA). The reactions were performed
for 35 cycles at 94°C for 30 sec, 54–60°C for 30 sec, and 72°C for
30 sec. All reactions were run in triplicate. U6 small nuclear RNA
was used as an internal reference control, and the data were
analyzed using the 2−ΔΔCt method (26).
Statistical analysis
Data were analyzed using SPSS software (version
22.0; SPSS, Inc., Chicago, IL, USA). All measurement data are
presented as the mean ± standard deviation. Statistical
significance analysis of involved measurement data was performed
using paired Student's t-tests. P<0.05 was considered to
indicate a statistically significant difference.
Results
Overview of small RNA sequencing
data
To identify differentially expressed miRNAs in the
SESCs and CECs of sheep, two small RNA libraries were constructed
by deep sequencing. The results of 14.36 million (M) and 13.71 M
total reads were obtained from the cell libraries of SESCs and
CECs, respectively. Subsequent to removing the low-quality and
adaptor sequences, a total of 14.15 and 13.48 M clean reads were
ultimately obtained. Subsequently, all identical sequence reads
were classified as groups, and 0.21 and 0.22 M unique sequences
associated with individual sequence reads, respectively, were
obtained (Table I). The size
distribution of the reads was similar between the two libraries
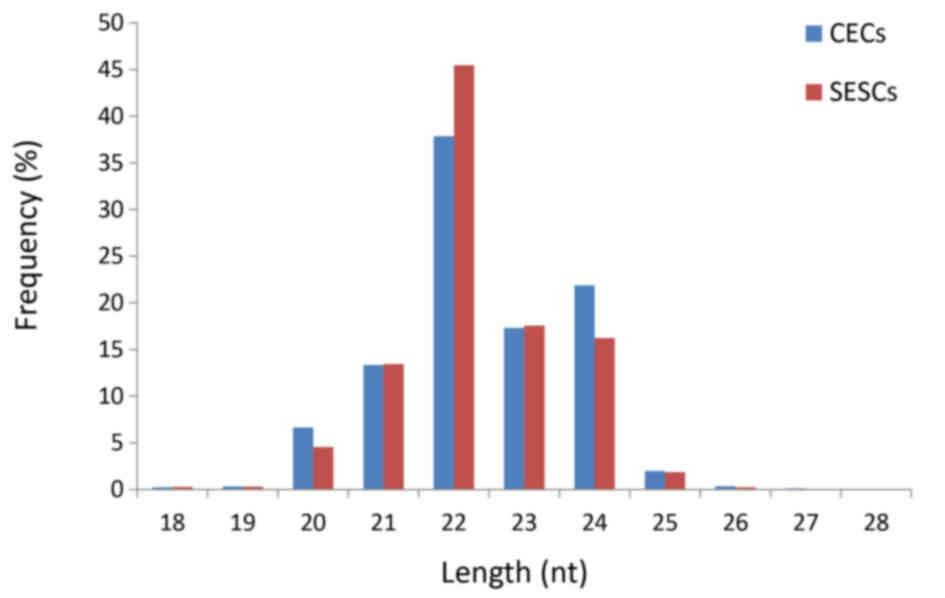
(Fig. 1). The majority of the
small RNAs were 21–24 nt in size. Sequences 22 nt in length, the
typical size of Dicer-derived products, accounted for 45.47 and
35.85% of the total sequence reads in the SESC and CEC libraries,
respectively. The composition of the RNA classes in each library is
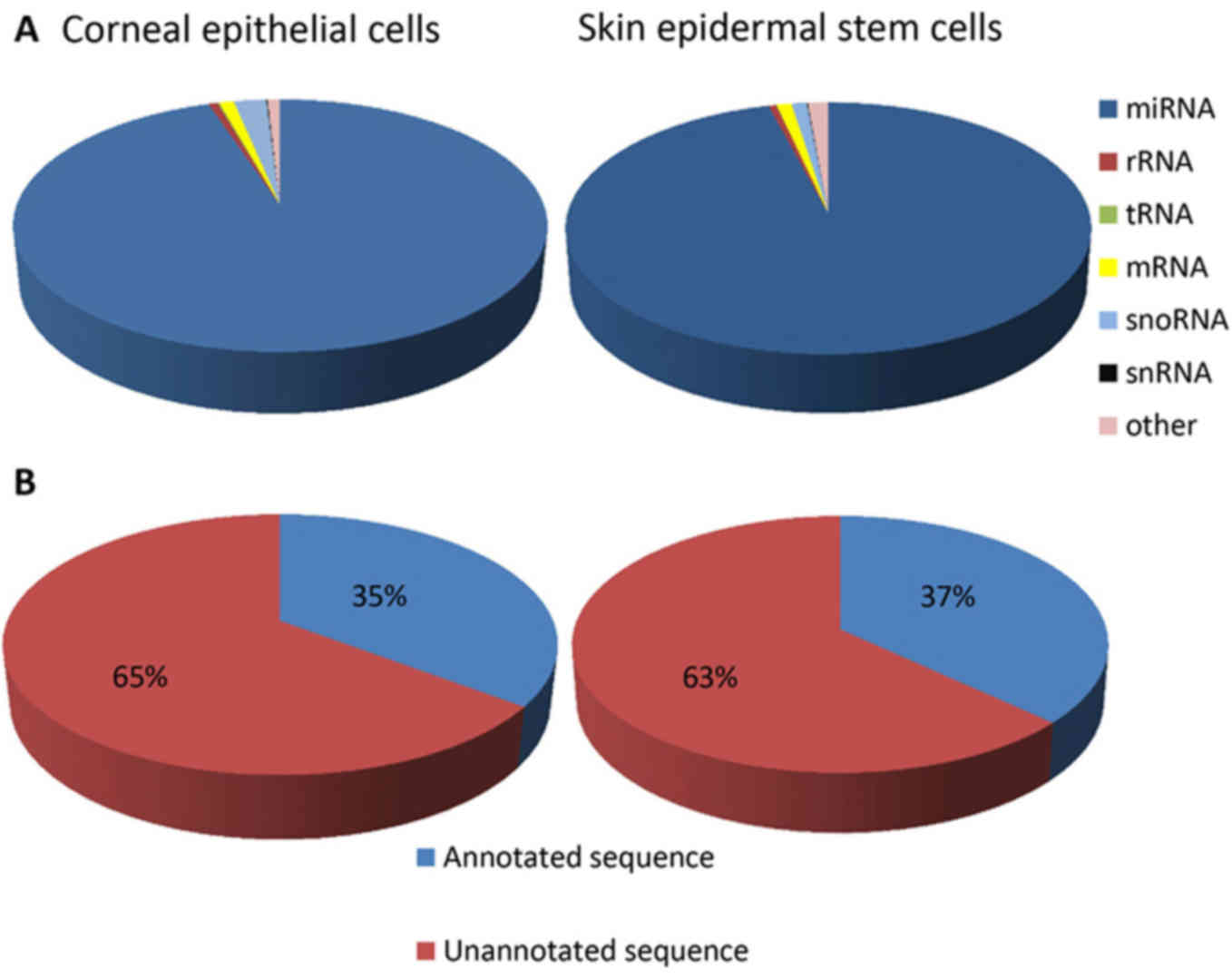
shown in Fig. 2A. Conserved
miRNAs accounted for 95.88 and 95.01% of the total sequence reads
(Fig. 2A) in the SESC and CEC
small RNA libraries, respectively. The detection of a low
proportion of long RNAs, including mRNA (1.02 and 1.03%), rRNA
(0.54 and 0.63%) and snoRNA (1.06 and 2.32%) (Fig. 2A), in the respective SESC and CEC
small RNA libraries indicated that the samples were not
contaminated by degraded RNA. In addition, the highest fraction of
unique reads (63 and 65% in the SESC and CEC libraries,
respectively) was attributed to unannotated sequences (Fig. 2B). A total of 136 and 94 conserved
miRNAs were identified in the SESC and CEC libraries, respectively;
184 and 169 miRNAs were mapped to the sheep genome, with the
extended genome sequences having the potential to form
hairpins.
 | Table IStatistics based on the reads of the
sequencing data. |
Table I
Statistics based on the reads of the
sequencing data.
| Reads | Corneal epithelial
cell count | Skin epidermal stem
cell count |
|---|
| Raw reads | 13,708,257 | 14,358,739 |
| Clean reads | 13,485,770 | 14,152,453 |
| Annotated
reads | 11,289,227 | 11,969,113 |
| with miRBase | 5,774,859 | 6,283,880 |
| with piRNA | 127,061 | 155,238 |
| with Rfam
v.10 | 4,520,579 | 4,752,923 |
| with ncRNA | 866,728 | 777,072 |
| Unannotated
reads | 2,196,543 | 2,183,340 |
| Novel miRNA | 832,45 | 853,42 |
Differential expression of miRNAs and
mRNA in SESCs and CECs
To identify miRNAs that may be involved in the
transdifferentiation process of SESCs to CECs, the miRNA and mRNA
expression profiles were assessed in these two cells using small
RNA deep sequencing and microarray analysis, respectively. The
threshold used to screen upregulated or downregulated miRNAs and
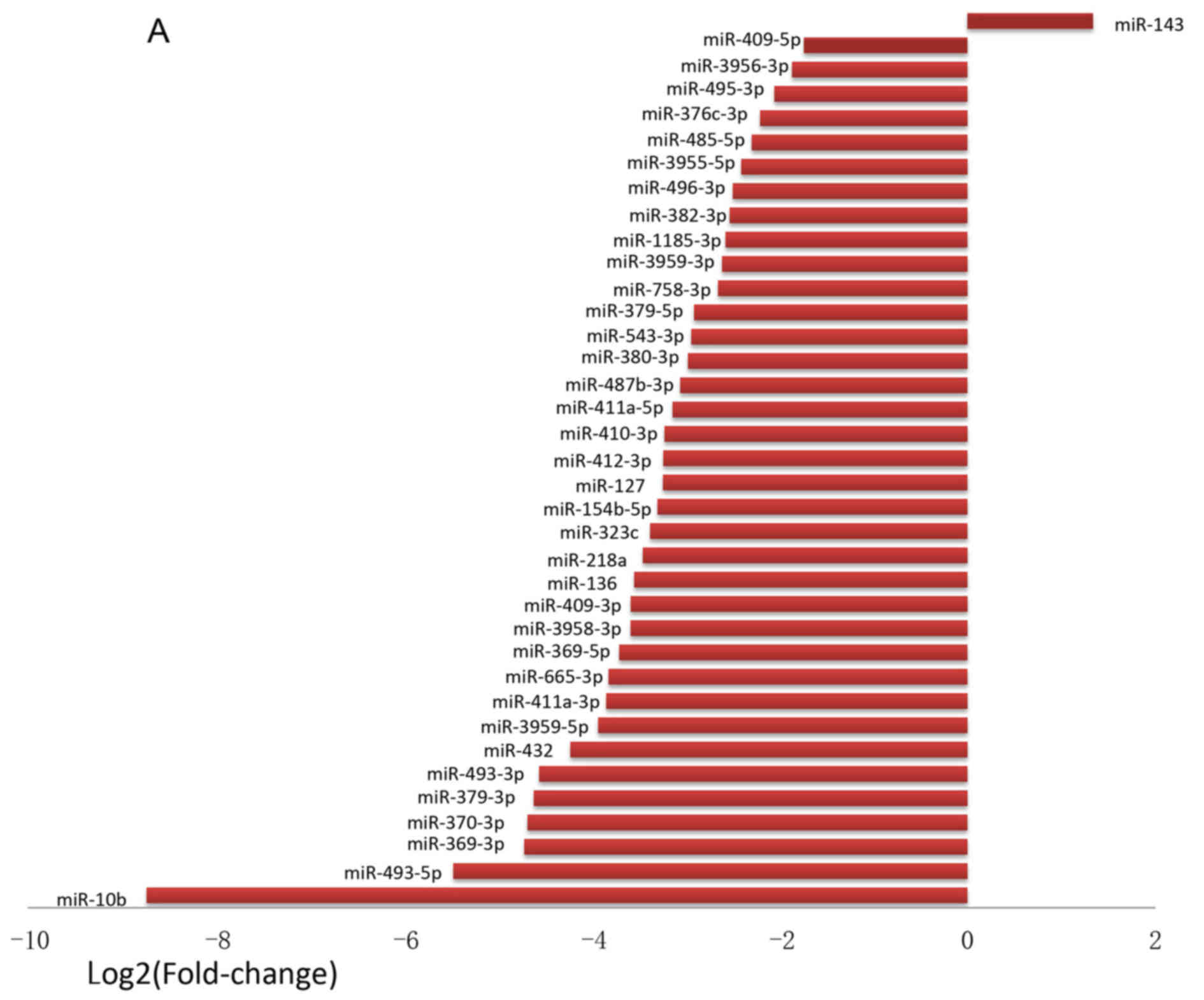
mRNAs was a fold‑change of ≥2.0. A total of 37 miRNAs were found to
be differentially expressed (Fig.
3). Among these miRNAs, 36 were found to be downregulated by
<0.5-fold, and 1 (miR-143) was found to be upregulated
>2.0-fold in SESCs compared with that in CECs (Fig. 3A). The fold-change of
downregulated miRNAs ranged from −425.71 to −3.34, and miR-10b
exhibited the greatest decrease in expression in SESCs compared
with CECs. Hierarchical clustering was performed with normalized
miRNA data (fold-change >2). A total of 37 miRNAs were
identified whose expression was significantly altered in the CECs
and SESCs (Fig. 3B). Differential
expressions of miRNAs and mRNAs were identified as those meeting
the criteria of FDR <0.05 and |log2FoldChange| >1.
A total of 241 mRNAs were found to be differentially
expressed. Among these mRNAs, 123 were downregulated by
<0.5-fold and 118 were upregulated by >2.0-fold in SESCs
compared with that in CECs. The fold-change ranged from −62.50 to
−2.02 and from 2.00 to 155.76 for the downregulated and upregulated
genes in SESCs compared with that in CECs, respectively.
RT-qPCR confirmation of differential
miRNA expression
A total of 7 differentially expressed miRNAs
(Fig. 4) were randomly chosen for
validation by RT-qPCR using the same RNA samples that were applied
to the small RNA sequencing. The expression levels of all 7 miRNAs
determined by RT-qPCR were concordant with their small RNA
sequencing data (Fig. 4),
indicating a strong association between miRNA profiling and the
RT-qPCR data.
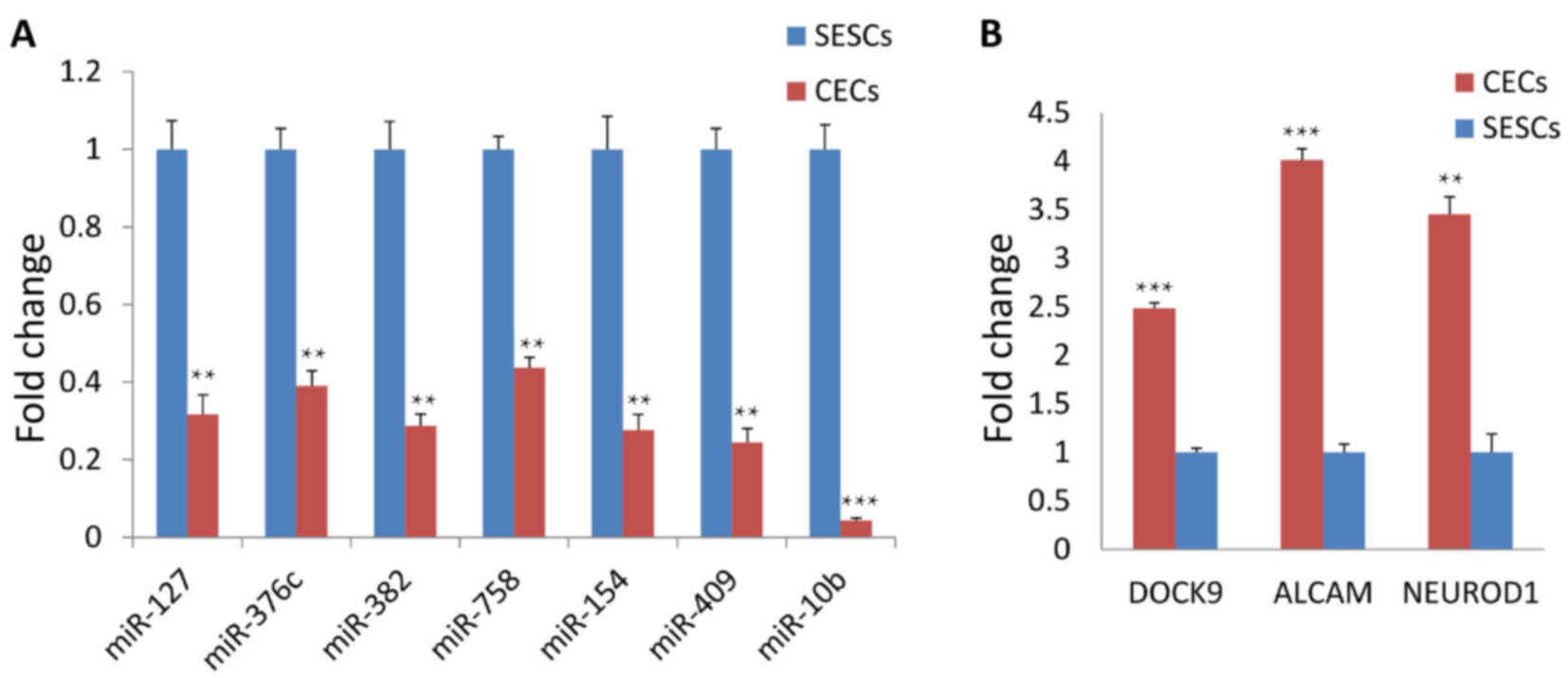 | Figure 4Validation of selected miRNAs and
mRNAs by reverse transcription-quantitative polymerase chain
reaction. (A) The results showed that the expression of miR-127,
376c, 382, 758, 154, 409 and 10b was markedly downregulated in CECs
compared with that in SESCs. (B) Meanwhile, DOCK9, ALCAM and
NEUROD1 expression was upregulated in CECs compared with that in
SESCs. CECs, corneal epithelial cells; SESCs, skin epidermal stem
cells; miR/miRNA, microRNA; DOCK9, dedicator of cytokinesis 9;
NEUROD1, neuronal differentiation 1; ALCAM, activated leukocyte
cell adhesion molecule. |
Target prediction and functional analysis
of differentially expressed miRNAs
As aforementioned, 36 miRNAs were found to be
downregulated in the SESCs compared those in the CECs, and
candidate target genes for all downregulated miRNAs were identified
using the commonly used prediction algorithm, TargetScan. These
targets were sorted by the enrichment of GO categories based on
DAVID and mainly clustered into different functional groups
(Fig. 5A). Targets were then
functionally analyzed using the KEGG pathways database. Pathway
analyses showed that these target genes were apparently associated
with several pathways, including the 'PI3K-Akt signaling pathway',
the 'Wnt signaling pathway', 'focal adhesion', the 'ErbB signaling
pathway' and the 'MAPK signaling pathway' (Fig. 5B).
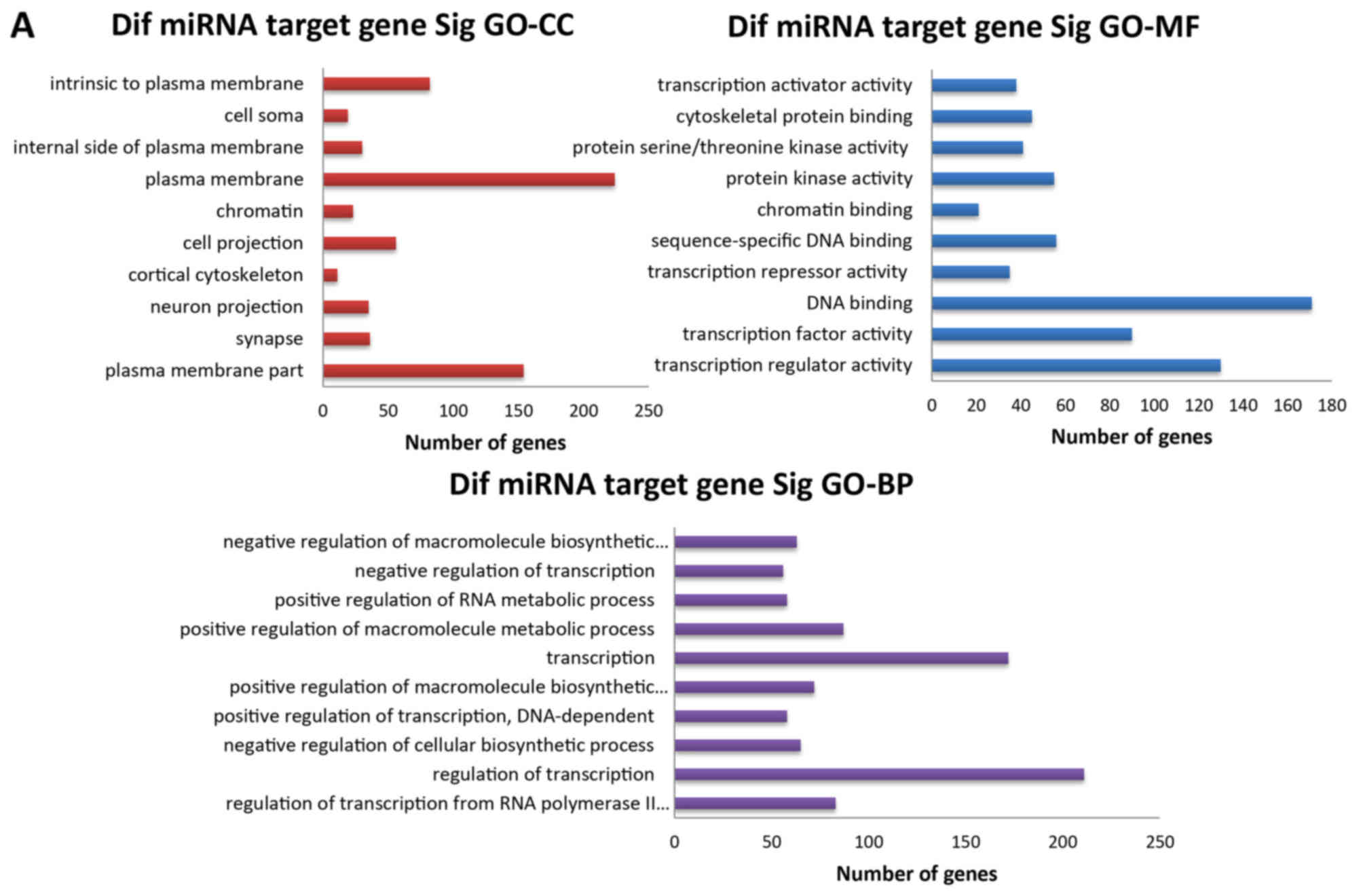 | Figure 5GO and KEGG pathway analyses of
differentially expressed miRNAs. (A) The three GO classifications,
namely, MF, BP and CC, were evaluated separately and (B) the top 10
significant terms of all KEGG pathways are shown. GO, Gene
Ontology; KEGG, Kyoto Encyclopedia of Genes and Genomes; miRNA,
microRNA; MF, molecular function; BP, biological process; CC,
cellular component; Sig, significant; Dif, differentially
expressed. |
Intersection genes between differentially
expressed miRNA target genes and differentially expressed
genes
As previously described, a total of 9,660 genes were
predicted by TargetScan for 36 downregulated miRNAs, and microarray
analysis determined that 123 genes were expressed at higher levels
in CECs than in SESCs. These two set of genes were compared and 43
intersection genes were detected. Notably, 24 miRNAs of the 36
downregulated miRNAs were associated with these 43 intersection
genes. These findings suggested that these miRNAs likely regulate
the same biological process.
Function analysis of differentially
expressed miRNA target genes and intersection genes
GO enrichment and KEGG pathway
analyses
The intersection genes were sorted by the enrichment
of GO categories based on DAVID and mainly clustered into different
functional groups (Fig. 6A), and
then were functionally analyzed using the KEGG pathways database.
Fig. 6B shows 10 of the most
significant pathways.
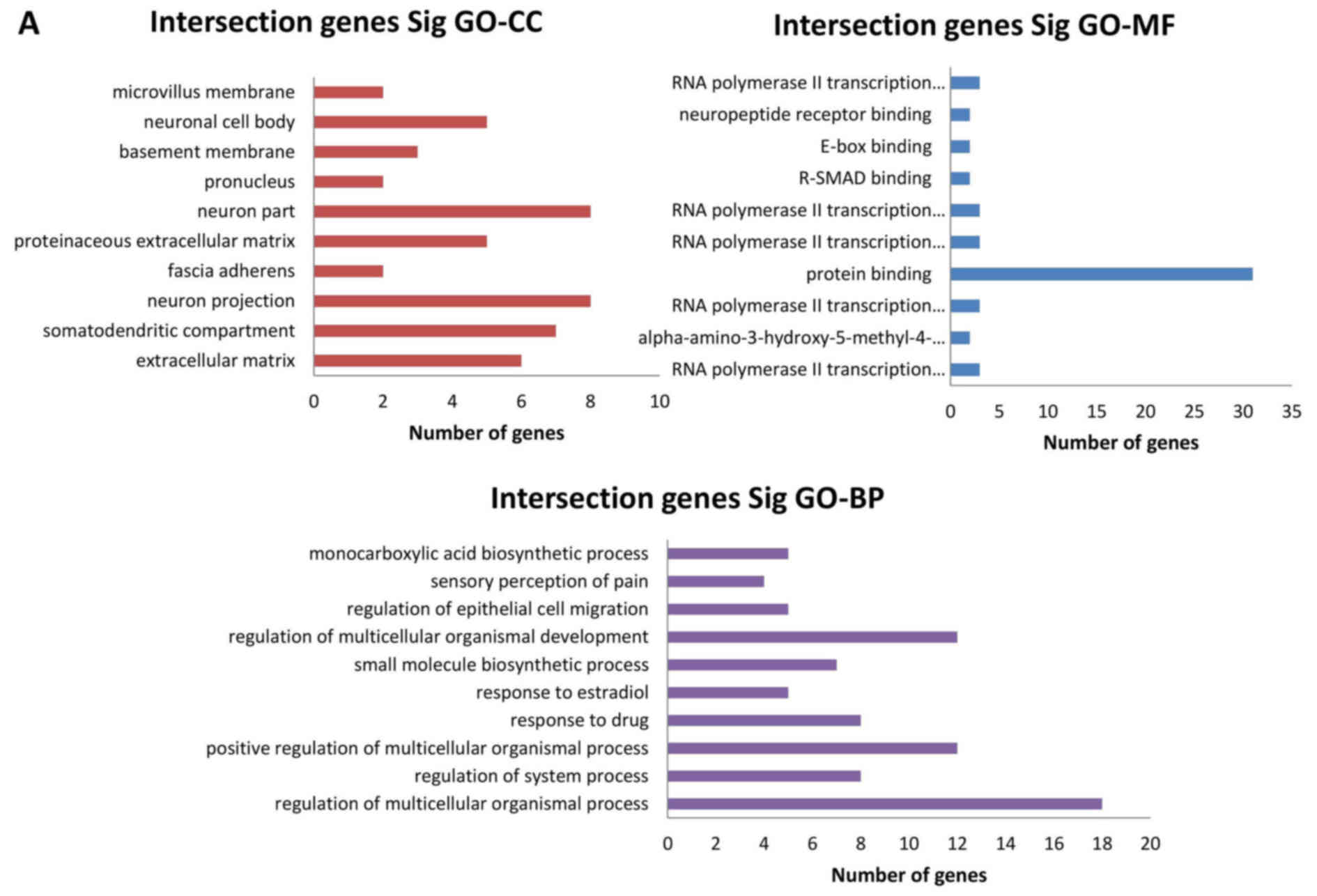 | Figure 6GO and KEGG pathway analyses of
intersection genes. (A) The three GO classifications, namely MF, BP
and CC, were separately evaluated and (B) the 10 most significant
terms of all KEGG pathways are shown. GO, Gene Ontology; KEGG,
Kyoto Encyclopedia of Genes and Genomes; miRNA, microRNA; MF,
molecular function; BP, biological process; CC, cellular component;
Sig, significant; Dif, differentially expressed. |
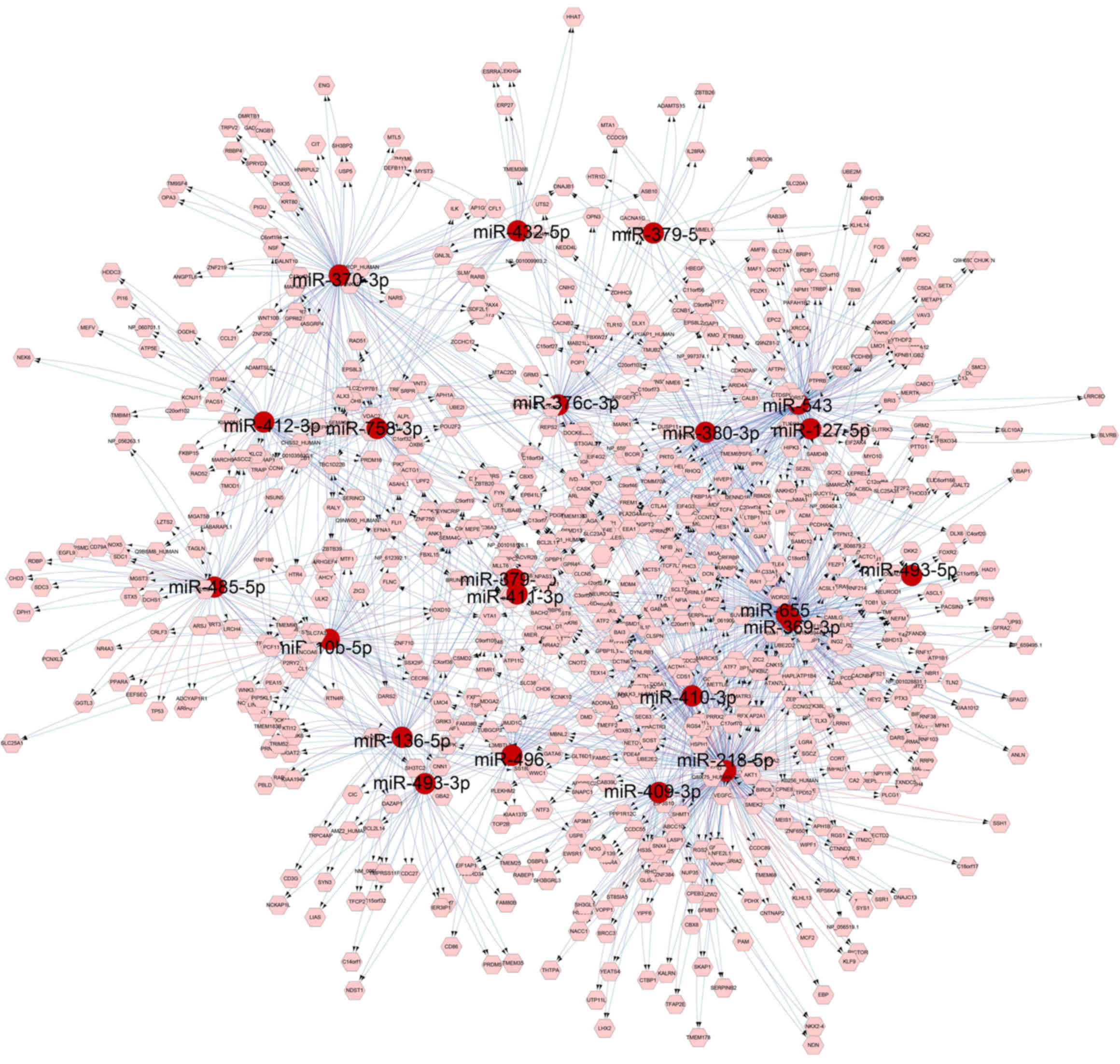
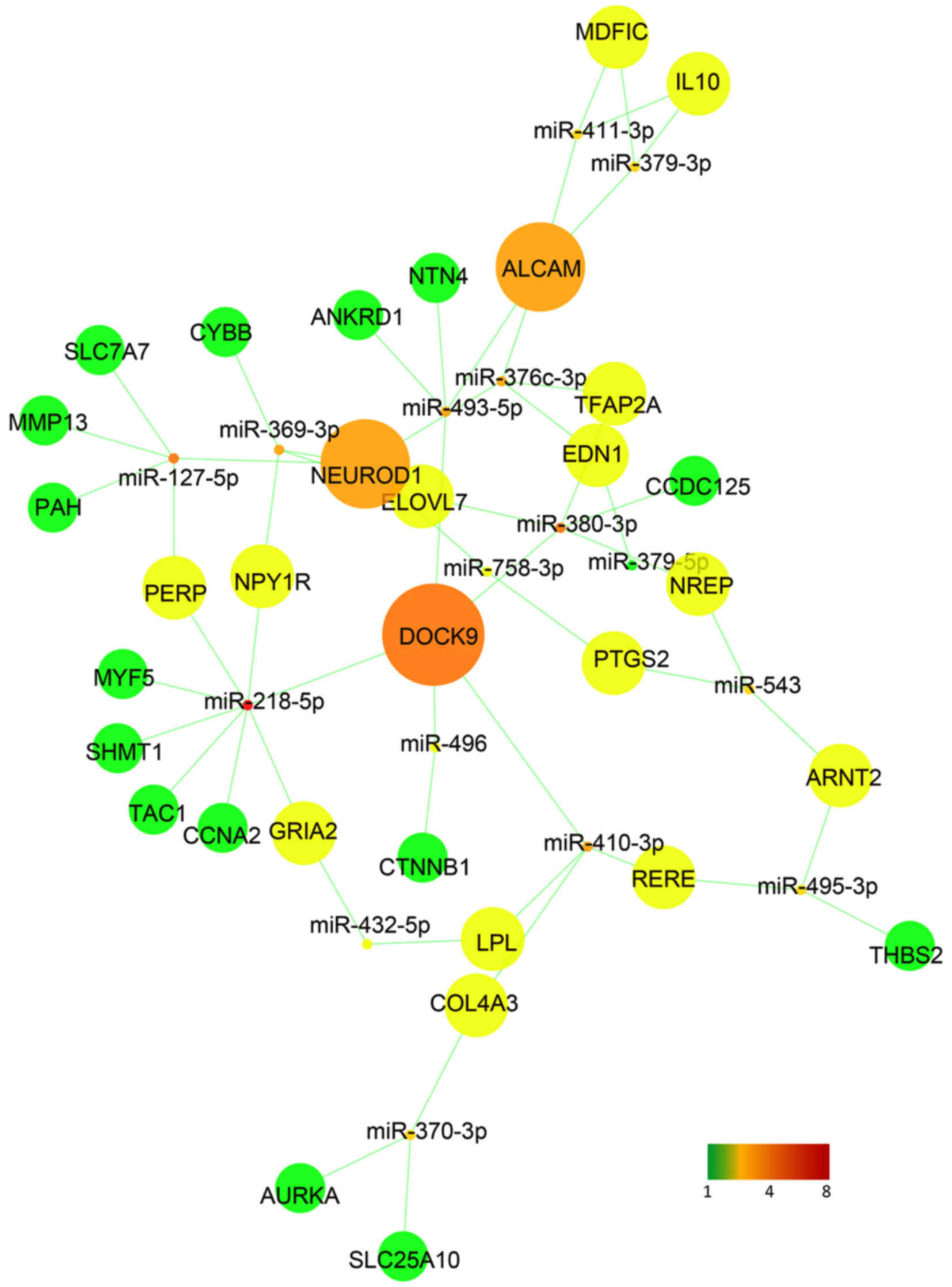
Construction of a regulatory and
interaction network
A regulatory network was performed using the
differentially expressed miRNAs and its target genes (Fig. 7). As previously described, 24
miRNAs of the 36 downregulated miRNAs were associated with these 43
intersection genes. A regulatory network of these 24 miRNAs and 43
intersection genes was then constructed using Cytoscape (Fig. 8). It was observed that dedicator
of cytokinesis 9 (DOCK9), neuronal differentiation 1 (NEUROD1) and
activated leukocyte cell adhesion molecule (ALCAM) had more
interaction with the miRNAs, and RT-qPCR confirmed that these 3
genes were upregulated in CECs compared with that in SESCs
(Fig. 4B). DOCK9, NEUROD1 and
ALCAM may thus serve an important role in CEC homeostasis and in
SESC to CEC transdifferentiation.
Discussion
The present study elucidated a coordinated
miRNA/mRNA expression profile for SESCs and CECs. miRNAs, although
non-coding, play a critical role by binding to the 3′-untranslated
region (3′UTR) of target genes for post-transcriptional regulation
(27). When the expression
profile of miRNAs was analyzed in the present study, it was found
that miR-10b exhibited the greatest decrease in expression in SESCs
compared with that in CECs (Figs.
3 and 4A). miR-10b is
considered as a marker of cell pathological transformation,
particularly the transformation and migration of malignant tumors
(28). A growing number of
studies have indicated that miR-10b expression is markedly
increased in malignant tumors, including breast cancer (29), pancreatic cancer (30), esophageal cancer (31) and glioblastoma (32). Thus, miR-10b may serve a critical
role in cell pathological transformation, and is considered as a
target for cancer therapy of, for example, breast cancer and
neurofibromatosis (33,34). In pathological conditions, CECs
convert into skin-like epithelial cells, which in turn may lead to
a loss in corneal transparency (2,3);
however, the underlying mechanism remains largely unknown. In this
process, the expression of paired box protein Pax-6 (PAX6), a
critical transcription factor during eye development, is markedly
decreased (3). Analysis of the
3′UTR region of PAX6 indicated that this gene is one of the
potential targets of miR-10b (data not shown), and as a tumor
suppressor, is suppressed by miR-10b in gliomagenesis (35). Thus, miR-10b may serve an
important role in homeostasis and the pathogenesis of CECs through
the suppression of PAX6 expression.
KEGG analysis indicated that the Wnt signaling
pathway is one of the most significant pathways involved in the
differential expression of miRNA target genes. SESCs and limbal
stem cells originate from the basal layer of embryonic skin
(17,18), which develops into different cell
types in response to different micro-environments. The Wnt signal
has emerged as the predominant pathway in the dermal condensation
message that determines whether the basal layer of embryonic skin
should differentiate into skin and its appendages (36), as well as subsequently controlling
the function of differentiated skin cells (37). CECs transdifferentiate into
skin-like epithelial cells when situated in a pathological
condition or induced by dermal developmental signals (2,38).
The expression level of β-catenin then sharply increases,
indicating that the Wnt signaling pathway has been activated
(2,38). Moreover, knockdown of the Wnt
signaling inhibitor dickkopf-related protein 2 promotes the
differentiation of the corneal epithelia into skin-like epithelial
cells and the development of appendages such as hair follicles
(39). Thus, the Wnt signaling
pathway triggers the pathological transdifferentiation of corneal
epithelia into skin-like epithelia and the development, formation
and maintenance of the corneal epithelium, which is required to
inhibit Wnt signals. It is also possible that the inhibition of a
Wnt signal promotes the trans-differentiation of SESCs into
CECs.
The present study found 3 intersection genes, DOCK9,
NEUROD1 and ALCAM, which had more interactions with miRNAs. DOCK9
(OMIM 607325) encodes a DOCK protein family member that has GTP/GDP
exchange factor activity and specifically activates G-protein cell
division control protein 42 homolog (40). Furthermore, it has been reported
as the most functionally significant gene for keratoconus (41). DOCK3, one of the members of the
DOCK protein family, was found as a negative regulator of Wnt
signaling, as it inhibited the nuclear expression of β-catenin
(42). Therefore, DOCK9 may play
a role in corneal epithelium homeostasis by inhibiting Wnt
signaling. NEUROD1 is a member of the NeuroD family of basic
helix-loop-helix transcription factors and activates the
transcription of genes that contain a specific DNA sequence known
as the E-box (43,44). There is evidence that PAX6, a
critical transcription factor that defines corneal epithelium
homeostasis and pathogenesis (2),
is activated by NeuroD1 by binding the E-box in the PAX6 promoter
(44). The present study found
that NeuroD1 was upregulated in CECs, and thus may act as the
upstream activator of PAX6 and maintain the characteristics of
corneal epithelia. ALCAM, also known as cluster of differentiation
(CD)166, serves an important role in mediating adhesion
interactions in epithelial cells. In CD166-deficient cells, mRNA
expression of the epithelial-mesenchymal transition (EMT) activator
zinc finger E-box-binding homeobox 1 is overexpressed (45), which implies that CD166 deficiency
may cause EMT in epithelial cells. MET is one of the pathological
changes when homeostasis of the corneal epithelia is disturbed by
infection, injury, epithelial hypertrophy or stem cell deficiency
(46). Thus, ALCAM may maintain
the homeostasis of corneal epithelium by regulating adhesion
interactions in corneal epithelial cells. In conclusion, DOCK9,
NEUROD1 and ALCAM, which are upregulated in CECs, may serve an
important role in maintaining the homeostasis of corneal epithelia,
and their overexpression induces the transdifferentiation of SESCs
into corneal epithelial cells.
In conclusion (Fig.
9), the homeostasis of CECs is maintained by the sustained
upregulation of PAX6, which can inhibit the Wnt/β-catenin signaling
pathway, and this dermal condensation message causes epidermal
changes in the CECs. The inhibitory effect of PAX6 may be enhanced
by NEUROD1 and DOCK9. In SESCs, the expression of PAX6 is inhibited
by miR-10b and the Wnt/β-catenin signaling pathway. Therefore, the
transdifferentiation from SESCs to CECs may be realized by the
following approach: i) Forced expression of PAX6 in SESCs; this
approach has previously been demonstrated to be feasible (2). ii) Forced expression of DOCK9,
NEUROD1, and ALCAM. iii) Inhibition of miR-10b expression in SESCs.
iv) Inhibition of the Wnt/β-catenin signaling pathway in SESCs.
PAX6, miR-10b and the Wnt/β-catenin signaling pathway are the
critical factors that predominate the transdifferentiation
process.
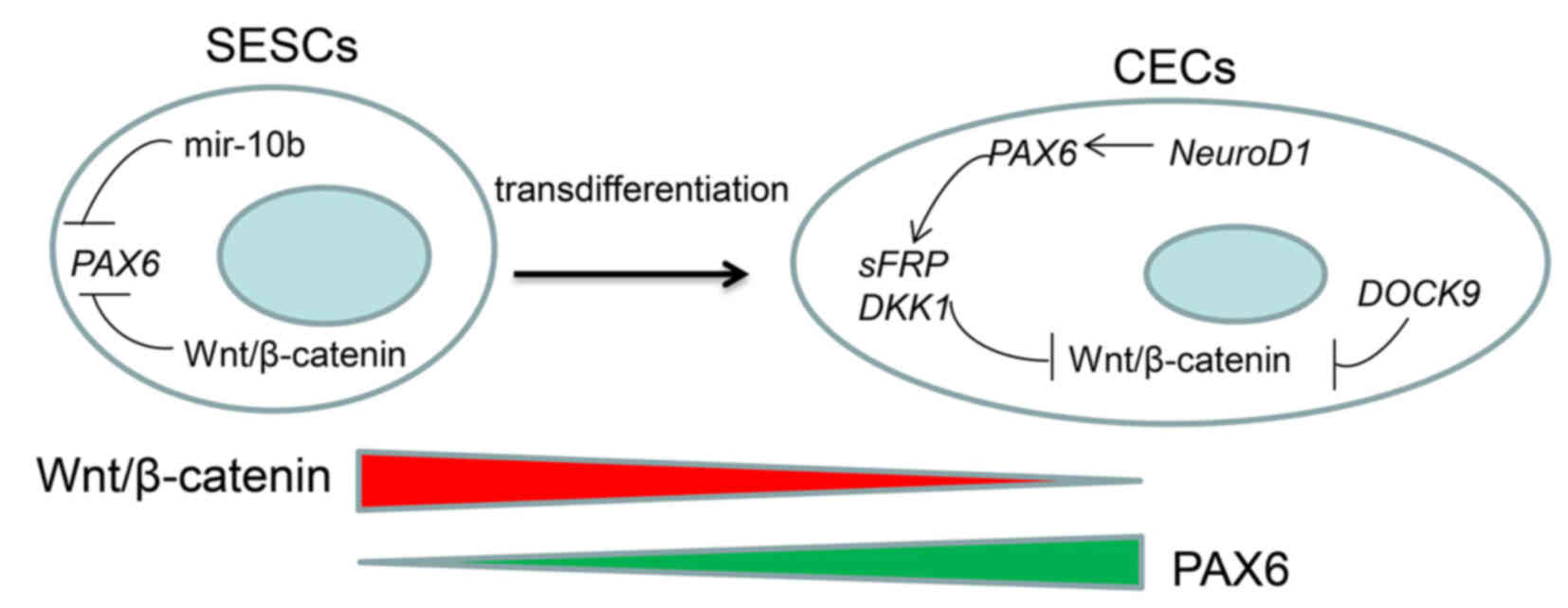 | Figure 9Schematic representation of the
putative mechanism involved in the transdifferentiation from SESCs
to CECs. The homeostasis of CECs is maintained by stabilized high
PAX6 expression, which can inhibit the Wnt/β-catenin signaling
pathway. The inhibitory effect of PAX6 may be enhanced by NEUROD1
and DOCK9. In SESCs, the expression of PAX6 is inhibited by miR-10b
and the Wnt/β-catenin signaling pathway together. PAX6, miR-10b and
the Wnt/β-catenin signaling pathway are the critical factors that
predominate the transdifferentiate process. CECs, corneal
epithelial cells;SESCs, skin epidermal stem cells; DOCK9, dedicator
of cytokinesis 9; NEUROD1, neuronal differentiation 1; miR/miRNA,
microRNA; PAX6, paired box protein Pax-6; DKK2, Dickkopf-related
protein 2; sFRP, secreted frizzled-related protein. |
The present study investigated the miRNA and mRNA
expression profiles of sheep SESCs and CECs. Coordinated miRNA/mRNA
expression analysis revealed that miR-10b, DOCK9, NEUROD1 and the
Wnt/β-catenin signaling pathway may be critical factors in the
construction of a regulatory network to regulate corneal
homeostasis and transdifferentiation from SESCs to CECs.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (nos. 31240089 and 31701121)
and the Key Project of Higher Education Institutions in Henan
Province (nos. 15A180008 and 16A180009).
References
|
1
|
Pellegrini G, Traverso CE, Franzi AT,
Zingirian M, Cancedda R and De Luca M: Long-term restoration of
damaged corneal surfaces with autologous cultivated corneal
epithelium. Lancet. 349:990–993. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ouyang H, Xue Y, Lin Y, Zhang X, Xi L,
Patel S, Cai H, Luo J, Zhang M, Zhang M, et al: WNT7A and PAX6
define corneal epithelium homeostasis and pathogenesis. Nature.
511:358–361. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Li W, Chen YT, Hayashida Y, Blanco G,
Kheirkah A, He H, Chen SY, Liu CY and Tseng SC: Down-regulation of
Pax6 is associated with abnormal differentiation of corneal
epithelial cells in severe ocular surface diseases. J Pathol.
214:114–122. 2008. View Article : Google Scholar
|
|
4
|
Lamm V, Hara H, Mammen A, Dhaliwal D and
Cooper DK: Corneal blindness and xenotransplantation.
Xenotransplantation. 21:99–114. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yang X, Moldovan I, Zhao Q, Mi S, Zhou Z,
Chen D, Gao Z, Tong D and Dou Z: Reconstruction of damaged cornea
by autologous transplantation of epidermal adult stem cells. Mol
Vis. 14:1064–1070. 2008.PubMed/NCBI
|
|
6
|
Shortt AJ, Secker GA, Notara MD, Limb GA,
Khaw PT, Tuft SJ and Daniels JT: Transplantation of ex vivo
cultured limbal epithelial stem cells: A review of techniques and
clinical results. Surv Ophthalmol. 52:483–502. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tseng SC, Prabhasawat P, Barton K, Gray T
and Meller D: Amniotic membrane transplantation with or without
limbal allografts for corneal surface reconstruction in patients
with limbal stem cell deficiency. Arch Ophthalmol. 116:431–441.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakamura T and Kinoshita S: Ocular surface
reconstruction using cultivated mucosal epithelial stem cells.
Cornea. 22(Suppl 7): S75–S80. 2003. View Article : Google Scholar
|
|
9
|
Utheim TP, Utheim OA, Khan QE and Sehic A:
Culture of oral mucosal epithelial cells for the purpose of
treating limbal stem cell deficiency. J Funct Biomater. 7:52016.
View Article : Google Scholar
|
|
10
|
Nishida K, Yamato M, Hayashida Y, Watanabe
K, Yamamoto K, Adachi E, Nagai S, Kikuchi A, Maeda N, Watanabe H,
et al: Corneal reconstruction with tissue-engineered cell sheets
composed of autologous oral mucosal epithelium. N Engl J Med.
351:1187–1196. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Katikireddy KR, Dana R and Jurkunas UV:
Differentiation potential of limbal fibroblasts and bone marrow
mesenchymal stem cells to corneal epithelial cells. Stem Cells.
32:717–729. 2014. View Article : Google Scholar
|
|
12
|
Gu S, Xing C, Han J, Tso MO and Hong J:
Differentiation of rabbit bone marrow mesenchymal stem cells into
corneal epithelial cells in vivo and ex vivo. Mol Vis. 15:99–107.
2009.PubMed/NCBI
|
|
13
|
Ahmad S, Stewart R, Yung S, Kolli S,
Armstrong L, Stojkovic M, Figueiredo F and Lako M: Differentiation
of human embryonic stem cells into corneal epithelial-like cells by
in vitro replication of the corneal epithelial stem cell niche.
Stem Cells. 25:1145–1155. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gomes JA, Geraldes Monteiro B, Melo GB,
Smith RL, Cavenaghi Pereira da Silva M, Lizier NF, Kerkis A,
Cerruti H and Kerkis I: Corneal reconstruction with
tissue-engineered cell sheets composed of human immature dental
pulp stem cells. Invest Ophthalmol Vis Sci. 51:1408–1414. 2010.
View Article : Google Scholar
|
|
15
|
Toma JG, Akhavan M, Fernandes KJ,
Barnabé-Heider F, Sadikot A, Kaplan DR and Miller FD: Isolation of
multipotent adult stem cells from the dermis of mammalian skin. Nat
Cell Biol. 3:778–784. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dyce PW, Wen L and Li J: In vitro germline
potential of stem cells derived from fetal porcine skin. Nat Cell
Biol. 8:384–390. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chee KY, Kicic A and Wiffen SJ: Limbal
stem cells: The search for a marker. Clin Experiment Ophthalmol.
34:64–73. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lavker RM and Sun TT: Epidermal stem
cells: Properties, markers, and location. Proc Natl Acad Sci USA.
97:13473–13475. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bose A, Teh MT, Mackenzie IC and Waseem A:
Keratin k15 as a biomarker of epidermal stem cells. Int J Mol Sci.
14:19385–19398. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Forni MF, Trombetta-Lima M and Sogayar MC:
Stem cells in embryonic skin development. Biol Res. 45:215–222.
2012. View Article : Google Scholar
|
|
21
|
Ghadially R: 25 years of epidermal stem
cell research. J Invest Dermatol. 132:797–810. 2012. View Article : Google Scholar
|
|
22
|
Blazejewska EA, Schlötzer-Schrehardt U,
Zenkel M, Bachmann B, Chankiewitz E, Jacobi C and Kruse FE: Corneal
limbal microenvironment can induce transdifferentiation of hair
follicle stem cells into corneal epithelial-like cells. Stem Cells.
27:642–652. 2009. View Article : Google Scholar :
|
|
23
|
Saichanma S, Bunyaratvej A and Sila-Asna
M: In vitro trans-differentiation of corneal epithelial-like cells
from human skin-derived precursor cells. Int J Ophthalmol.
5:158–163. 2012.
|
|
24
|
Araki K, Ohashi Y, Sasabe T, Kinoshita S,
Hayashi K, Yang XZ, Hosaka Y, Aizawa S and Handa H: Immortalization
of rabbit corneal epithelial cells by a recombinant SV40-adenovirus
vector. Invest Ophthalmol Vis Sci. 34:2665–2671. 1993.PubMed/NCBI
|
|
25
|
Yang X, Qu L, Wang X, Zhao M, Li W, Hua J,
Shi M, Moldovan N, Wang H and Dou Z: Plasticity of epidermal adult
stem cells derived from adult goat ear skin. Mol Reprod Dev.
74:386–396. 2007. View Article : Google Scholar
|
|
26
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
27
|
Shukla GC, Singh J and Barik S: MicroRNAs:
Processing, maturation, target recognition and regulatory
functions. Mol Cell Pharmacol. 3:83–92. 2011.PubMed/NCBI
|
|
28
|
Baffa R, Fassan M, Volinia S, O'Hara B,
Liu CG, Palazzo JP, Gardiman M, Rugge M, Gomella LG, Croce CM, et
al: MicroRNA expression profiling of human metastatic cancers
identifies cancer gene targets. J Pathol. 219:214–221. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ma L, Teruya-Feldstein J and Weinberg RA:
Tumour invasion and metastasis initiated by microRNA-10b in breast
cancer. Nature. 449:682–688. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bloomston M, Frankel WL, Petrocca F,
Volinia S, Alder H, Hagan JP, Liu CG, Bhatt D, Taccioli C and Croce
CM: MicroRNA expression patterns to differentiate pancreatic
adenocarcinoma from normal pancreas and chronic pancreatitis. JAMA.
297:1901–1908. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Tian Y, Luo A, Cai Y, Su Q, Ding F, Chen H
and Liu Z: MicroRNA-10b promotes migration and invasion through
KLF4 in human esophageal cancer cell lines. J Biol Chem.
285:7986–7994. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ciafrè SA, Galardi S, Mangiola A, Ferracin
M, Liu CG, Sabatino G, Negrini M, Maira G, Croce CM and Farace MG:
Extensive modulation of a set of microRNAs in primary glioblastoma.
Biochem Biophys Res Commun. 334:1351–1358. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ma L, Reinhardt F, Pan E, Soutschek J,
Bhat B, Marcusson EG, Teruya-Feldstein J, Bell GW and Weinberg RA:
Therapeutic silencing of miR-10b inhibits metastasis in a mouse
mammary tumor model. Nat Biotechnol. 28:341–347. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chai G, Liu N, Ma J, Li H, Oblinger JL,
Prahalad AK, Gong M, Chang LS, Wallace M, Muir D, et al:
MicroRNA-10b regulates tumorigenesis in neurofibromatosis type 1.
Cancer Sci. 101:1997–2004. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin J, Teo S, Lam DH, Jeyaseelan K and
Wang S: MicroRNA-10b pleiotropically regulates invasion,
angiogenicity and apoptosis of tumor cells resembling mesenchymal
subtype of glioblastoma multiforme. Cell Death Dis. 3:e3982012.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Widelitz RB: Wnt signaling in skin
organogenesis. Organogenesis. 4:123–133. 2008. View Article : Google Scholar
|
|
37
|
Lim X and Nusse R: Wnt signaling in skin
development, homeostasis, and disease. Cold Spring Harb Perspect
Biol. 5:52013. View Article : Google Scholar
|
|
38
|
Pearton DJ, Yang Y and Dhouailly D:
Transdifferentiation of corneal epithelium into epidermis occurs by
means of a multistep process triggered by dermal developmental
signals. Proc Natl Acad Sci USA. 102:3714–3719. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mukhopadhyay M, Gorivodsky M, Shtrom S,
Grinberg A, Niehrs C, Morasso MI and Westphal H: Dkk2 plays an
essential role in the corneal fate of the ocular surface
epithelium. Development. 133:2149–2154. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kwofie MA and Skowronski J: Specific
recognition of Rac2 and Cdc42 by DOCK2 and DOCK9 guanine nucleotide
exchange factors. J Biol Chem. 283:3088–3096. 2008. View Article : Google Scholar
|
|
41
|
Czugala M, Karolak JA, Nowak DM,
Polakowski P, Pitarque J, Molinari A, Rydzanicz M, Bejjani BA, Yue
BY, Szaflik JP, et al: Novel mutation and three other sequence
variants segregating with phenotype at keratoconus 13q32
susceptibility locus. Eur J Hum Genet. 20:389–397. 2012. View Article : Google Scholar :
|
|
42
|
Caspi E and Rosin-Arbesfeld R: A novel
functional screen in human cells identifies MOCA as a negative
regulator of Wnt signaling. Mol Biol Cell. 19:4660–4674. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Poulin G, Turgeon B and Drouin J:
NeuroD1/beta2 contributes to cell-specific transcription of the
proopiomelanocortin gene. Mol Cell Biol. 17:6673–6682. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Marsich E, Vetere A, Di Piazza M, Tell G
and Paoletti S: The PAX6 gene is activated by the basic
helix-loop-helix transcription factor NeuroD/BETA2. Biochem J.
376:707–715. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Fujiwara K, Ohuchida K, Sada M, Horioka K,
Ulrich CD III, Shindo K, Ohtsuka T, Takahata S, Mizumoto K, Oda Y,
et al: CD166/ALCAM expression is characteristic of tumorigenicity
and invasive and migratory activities of pancreatic cancer cells.
PLoS One. 9:e1072472014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Park GB, Kim D, Kim YS, Kim S, Lee HK,
Yang JW and Hur DY: The Epstein-Barr virus causes
epithelial-mesenchymal transition in human corneal epithelial cells
via Syk/src and Akt/Erk signaling pathways. Invest Ophthalmol Vis
Sci. 55:1770–1779. 2014. View Article : Google Scholar : PubMed/NCBI
|