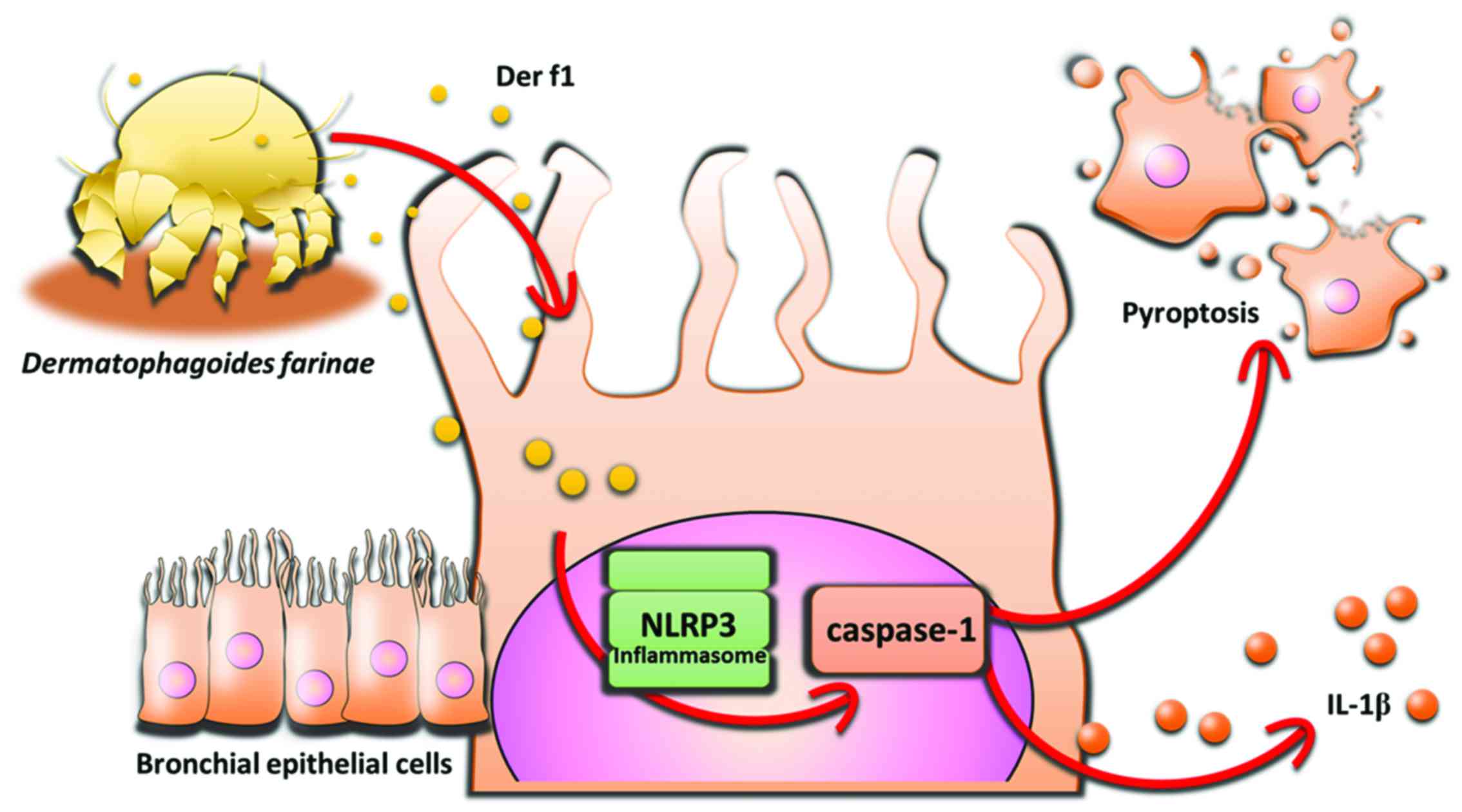
Introduction
Asthma is a chronic inflammatory airway disease that
is defined as reversible airway narrowing, and is characterized by
episodic symptoms of dyspnea, wheezing and coughing (1). Asthma is a major health concern,
affecting more than 300 million individuals worldwide (1). Bronchial epithelial cells (BECs) are
the first line of defence against noxious inhalants, such as
microorganisms, gases and allergens that may cause asthma (2). BECs not only constitute a physical
barrier, but also play a key role in inflammatory, immune and
regenerative processes in response to these noxious inhalants
(3). Loss of BEC integrity is a
hallmark of asthma pathogenesis (4), and sloughing of BECs has been found
in bronchial biopsy samples from patients with mild to severe
asthma (5,6). A number of mechanisms may explain
the loss of epithelial integrity, such as cell death (7) and disrupted cell-cell and
cell-extracellular matrix interactions (7). Repetitive cycles of injury and
repair of BECs is a major factor in airway structural changes
leading to remodelling of the airways (3,8,9).
Therefore, understanding epithelial damage is a valuable approach
to identify optimal treatments for chronic airway diseases.
Pyroptosis is a process of host cell death caused by
microbial infections and non-infectious stimuli (stroke,
chemotherapy and inflammation) (10,11). Pyroptosis differs morphologically
and mechanistically from other types of cell death, such as
apoptosis and necrosis. The unique aspect of pyroptosis is that it
is dependent on caspase-1 activation, which mediates cell death and
cleaves and secretes proinflammatory cytokines, such as interleukin
(IL)-1β and IL-18 (10,12). Both cell death and proinflammatory
signals cause tissue damage, which may lead to permanent structural
changes (13). Therefore, we
hypothesized that pyroptosis may act as a pathogenic mechanism
contributing to inflammatory injury of airway epithelia.
Allergic sensitization to inhaled allergens, such as
house dust mites (HDMs), predisposes susceptible individuals to
developing allergic hypersensitivity reactions and producing IgE
(2), which are key to the
pathogenesis of allergic airway diseases (14). Among HDMs, Dermatophagoides
pteronyssinus (Der p) and Dermatophagoides farinae (Der
f), are the most important triggers of bronchial asthma, rhinitis
and atopic dermatitis (15,16). In addition to the allergic
response, HDMs may also disrupt cell adhesion, induce cell death
and increase the permeability of lung epithelia (17). Furthermore, Dermatophagoides sp.
peptidases have been shown to elicit apoptosis in a bronchial cell
model (15). Der f1 (D.
farina allergen 1), which displays 82% homology to Der p1
(D. pteronyssinus allergen 1), possesses cysteine
protease activity, and it has been shown to enhance tissue damage
and immune activation via downregulation of anti-protease-based
lung defences (18,19). However, little is known on the
effect of HDMs on BEC injury, particularly with regards to
inflammation-mediated cell death. The aim of the present study was
to investigate whether the common inhaled allergen, Der f1, induces
pyroptosis in BECs, suggesting a connection between BEC injury and
an asthmatic inflammatory microenvironment.
Materials and methods
Cell culture
Primary human BECs (pHBECs) were obtained and
maintained in BECs growth medium (Lonza, Walkersville, MD, USA).
HBE135-E6E7 human bronchial epithelial cells (HBE-135, CRL-2741)
were obtained from American Type Culture Collection (Manassas, VA,
USA) and maintained in keratinocyte-serum free medium with 5 ng/ml
human recombinant epithelial growth factor and 0.05 mg/ml bovine
pituitary extract (Invitrogen; Thermo Fisher Scientific, Carlsbad,
CA, USA) supplemented with 0.005 mg/ml insulin and 500 ng/ml
hydrocortisone (Sigma-Aldrich; Merck KGaA, St. Louis, MO, USA).
Both cell lines were cultured at 37°C, with 90% relative humidity
and 5% CO2.
Cell viability
Cells were plated in 96-well culture plates
(5×103/well) and treated with common inhaled allergens,
including recombinant Der f1 (rDer f1), deglycosylated rDer p1
(rDer p1 DG), natural Fel d1 (nFel d1), nCan f1 and nDer p1 (Indoor
Biotechnologies Inc., Charlottesville, VA, USA) for 24 h. BEC
viability was determined by premixed WST-1 cell proliferation
reagent (Clontech Laboratories, Mountain View, CA, USA), according
to the manufacturer's instructions.
TdT-mediated dUTP nick end labeling
(TUNEL) assays and propidium iodide (PI) staining
Cells were treated with rDer f1 for 24 h and then
assayed for apoptotic cell death using a commercial TUNEL assay kit
(BD Biosciences, San Diego, CA, USA). Cells were harvested and
fixed in 4% formaldehyde, then stained using TUNEL reaction mixture
(45 µl labeling solution and 5 µl enzyme solution),
followed by incubation in the dark for 1 h at 37°C. The percentage
of apoptotic cells was assessed by flow cytometry (BD
Biosciences).
Cells were collected by trypsinization and then
stained with PI for 30 min at room temperature. After washing with
phosphate-buffered saline, the penetration of the PI dye was
analyzed by flow cytometry.
Lactate dehydrogenase (LDH) release assay
and cytokine measure
The release of LDH was measured in the cell
supernatants of the BECs after rDer f1 treatment using a LDH assay
kit (BD Biosciences). Cell supernatants (100 µl) were then
transferred to a 96-well microplate and mixed with 100 µl of
the reaction solution provided in the kit. Optical density was
measured at 492 nm using an enzyme-linked immunosorbent assay
(ELISA) reader. The level of IL-1β was determined using an
ELISA-based kit (R&D Systems Europe, Ltd., Abingdon, UK). ELISA
was performed according to the manufacturer's instructions.
Caspase-1 activity analysis
Caspase-1 activity in the cell lysate was determined
with a caspase-1 colorimetric kit (BioVision, Inc., Milpitas, CA,
USA) by cleavage of 5 µl of the caspase-1 substrate
Ac-YVAD-pNA. The cleavage of pNA was monitored by changes in
absorbance at 405 nm.
Immunoblot/co-immunoprecipitation
(co-IP)
The cells were lysed on ice with M-PER lysis reagent
(Pierce Chemical Co., Rockford, IL, USA) and then centrifuged at
14,000 × g for 15 min. The supernatant fraction was collected for
immunoblot analysis. Equal amounts of protein were resolved by
SDS-PAGE (8–12%) and transferred to a polyvinylidene difluoride
membrane. After blocking, the membrane was incubated with the
desired primary antibody for 1–16 h. The membrane was then treated
with the appropriate concentrations of peroxidase-conjugated
secondary antibody, and the immunoreactive proteins were detected
using an enhanced chemiluminescence kit (Millipore Corp.,
Billerica, MA, USA) according to the manufacturer's instructions.
The interaction of NOD-like receptor family pyrin domain-containing
3 (NLRP3) with caspase-1 was assessed by co-IP. Antibodies against
human IL-1β (pro-IL-1β) (rabbit, D3U3E, 1:1000, 12703S), NLRP3
[rabbit, D2P5E, 1:1000 (in IP, 1:100] and caspase-1 (rabbit, D7F10,
1:1000, 3866S) were obtained from Cell Signaling Technology
(Beverly, MA, USA). Monoclonal antibody anti-human
glyceraldehyde-3-phosphate dehydrogenase (GAPDH; rabbit,
polyclonal, 1:5000, ABS16) was obtained from Millipore Corp.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RNA isolation was performed using a TRIzol reagent
(Invitrogen; Thermo Fisher Scientific). cDNA was prepared using an
oligo (dT) primer and RT (Takara Bio, Inc., Otsu, Japan) following
standard protocols. RT-qPCR was performed using SYBR-green on an
ABI 7500 Real-Time PCR system (Applied Biosystems Life
Technologies, Foster City, CA, USA). Each PCR reaction mixture
contained 200 nM of each primer, 10 µl of 2x SYBR-green PCR
master mix (Applied Biosystems Life Technologies), 5 µl cDNA
and RNase-free water in a final volume of 20 µl. The PCR
reaction was performed with a denaturation step at 95°C for 10 min,
followed by 40 cycles at 95°C for 15 sec and 60°C for 1 min. All
PCRs were performed in triplicate and normalized by the internal
control GAPDH. The relative expression was calculated using the
2−ΔΔCq method.
siRNA knockdown
BECs were transfected with 25 nM ON-TARGET plus
control or NLRP3 siRNA by DharmaFECT formulation 4 reagent (Thermo
Fisher Scientific, Waltham, MA, USA) according to the
manufacturer's instructions. After 24 h of transfection, the medium
was changed to whole medium and the cells were treated with rDer f1
for an additional 24 h. The changes in NLRP3 were measured by
RT-qPCR, as described above.
Statistical analysis
Data are presented as mean ± standard deviation.
One-way analysis of variance test with Tukey's multiple comparison
post hoc test was employed for comparison of all data with the
control. p<0.05 was considered to indicate statistically
significant differences.
Results
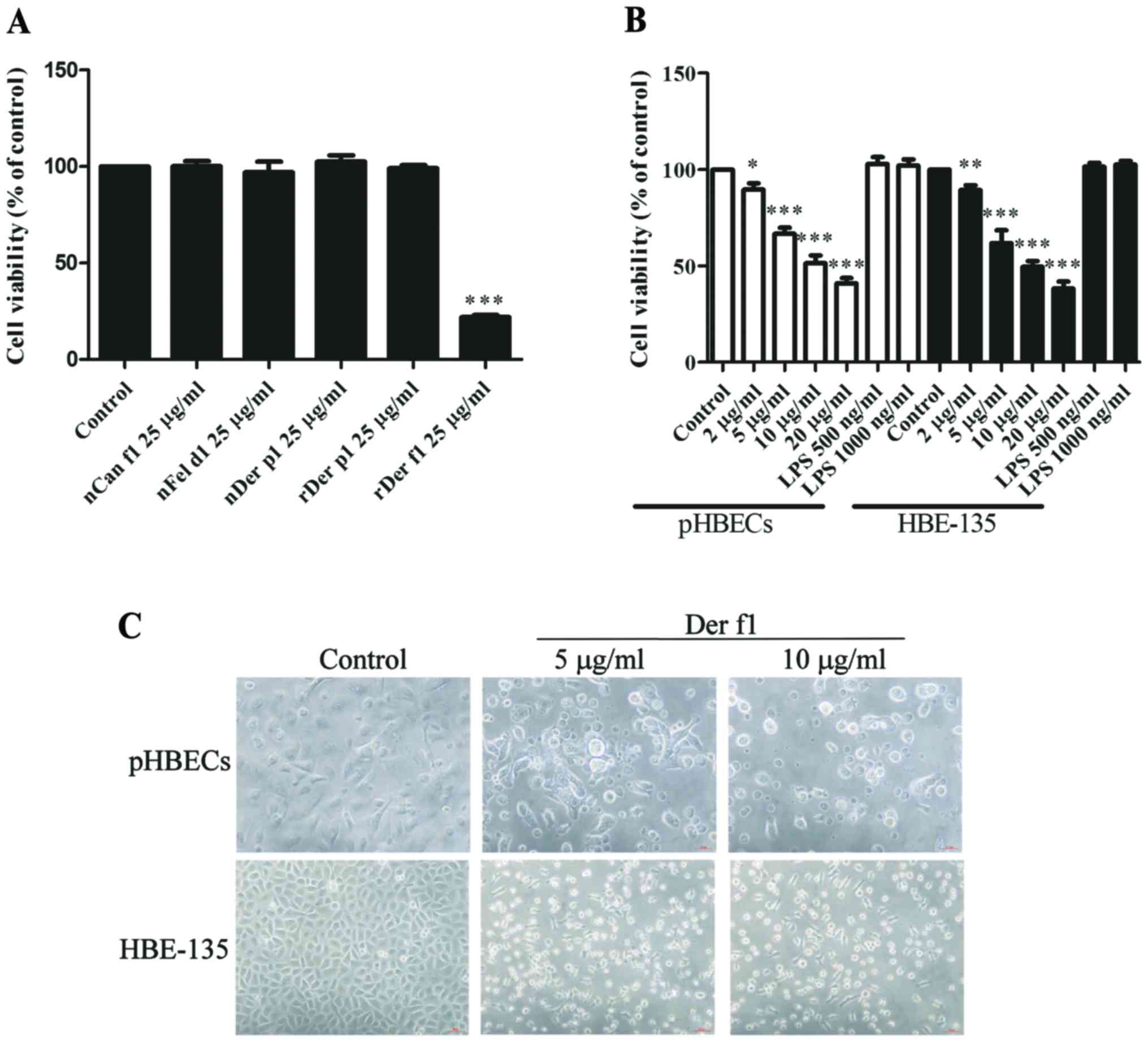
Exposure to rDer f1 decreases BEC
viability by inducing cell death
Exposure to the most common allergens may result in
inflammatory and allergic reactions in susceptible hosts (2). In addition, BEC damage is a critical
pathogenic characteristic of airway diseases (3). Therefore, the effects of common
allergens, including cat, dog and dust mites, on the viability of
BECs were first assessed. HBE-135 cells were treated with a series
of common inhaled allergens, including nCan f1, nFel d1, nDer p1,
rDer p1 Dg and rDer f1 at 25 µg/ml for 24 h. Only rDer f1
treatment exerted a prominent inhibitory effect on the viability of
HBE-135 cells (Fig. 1A). This
effect was dose-dependent in pHBECs and HBE-135 cells (Fig. 1B). Inverted microscopy revealed
that numerous cells had assumed a round shape and had detached from
the culture plates, suggesting that rDer f1 caused BEC death, but
not a reduction in proliferation (Fig. 1C).
rDer f1 induces BEC pyroptosis but not
apoptosis
Whether apoptosis or another means of cell death
caused the decreased cell viability after rDer f1 treatment was
next investigated. Apoptosis was first evaluated by the TUNEL
assay. However, 5 and 10 µg/ml of rDer f1 treatment failed
to induce apoptosis in either pHBECs or HBE-135 cells (Fig. 2A).
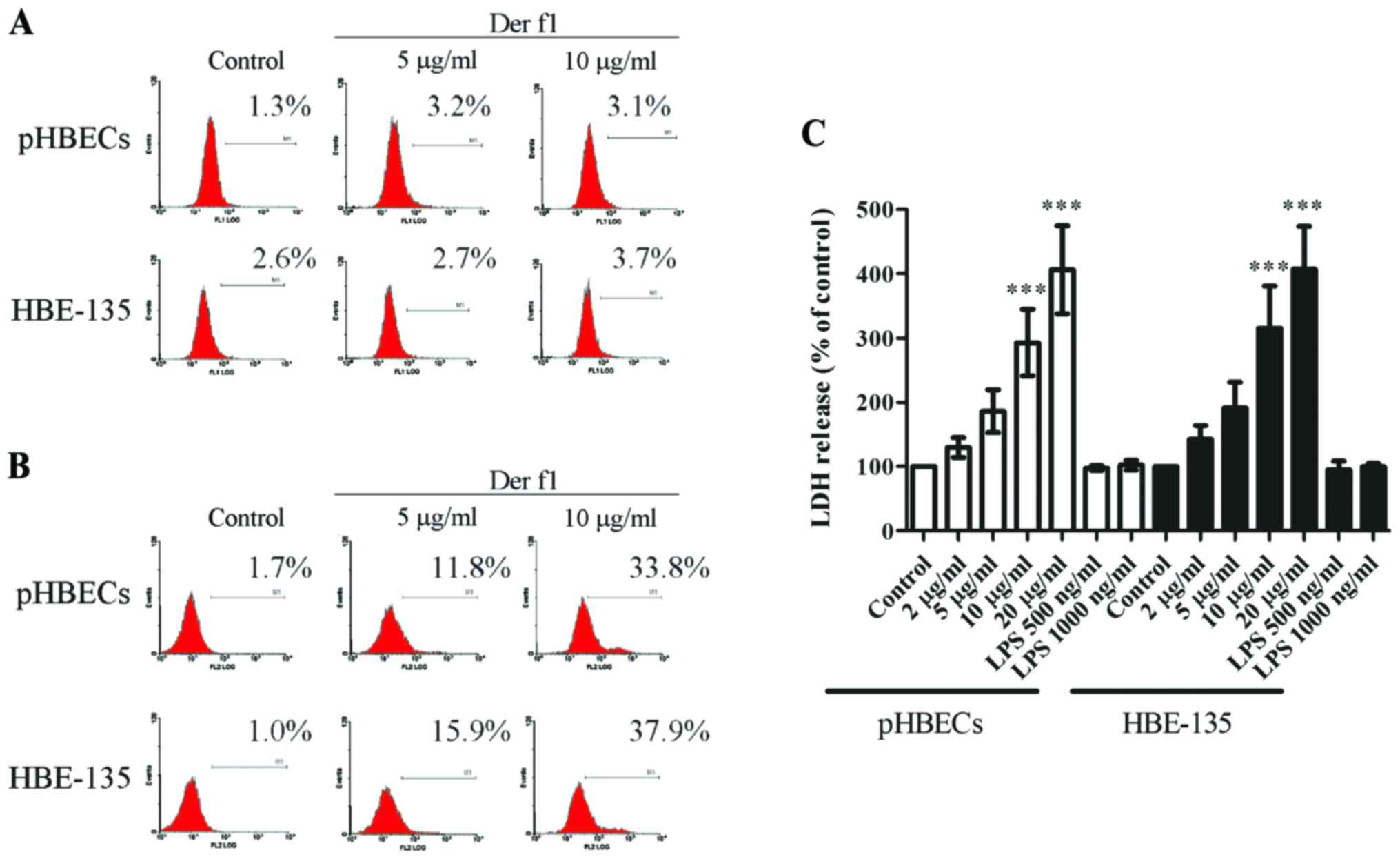 | Figure 2rDer f1 induced pyroptosis in BECs.
(A) rDer f1 did not trigger cell apoptosis. rDer f1 induced
pyroptosis in BECs, as determined by (B) PI staining and (C) LDH
release analysis in pHBECs and HBE-135 cells treated with various
concentrations of rDer f1 for 24 h. The percentage of apoptosis was
assessed by terminal deoxynucleotidyl TdT-mediated duTP nick end
labeling (TUNEL) analysis to determine cellular DNA fragments. For
pyroptosis analysis, the supernatants of rDer f1-treated pHBECs and
HBE-135 cells were collected by centrifugation, and the levels of
LDH were then determined using a cytotoxicity detection kit. rDer
f1-treated BECs were harvested by trypsinization and
centrifugation. The cells were then stained by PI and assessed by
flow cytometry. Data are expressed as the mean of three independent
experiments ± standard deviation, ***p<0.001 vs.
control. LDH, lactate dehydrogenase; BECs, bronchial epithelial
cells; pHBECs, primary human BECs; Der f1, Dermatophagoides
farina allergen 1; PI, propidium iodide; LPS,
lipopolysaccharide. |
It was then investigated whether rDer f1 induced
pyroptosis in BECs. Pyroptosis is characterized by the formation of
pores in the cell membrane, which may be detected by LDH release
and PI staining without cell membrane permeabilization (10,12). Flow cytometric analysis revealed
that an increase in PI-positive cells was well-correlated with an
increase in rDer f1 concentration (Fig. 2B). In addition, with increasing
concentrations of rDer f1, more LDH was released (Fig. 2C). This finding suggested that
cell death was associated with the loss of cellular membrane
integrity and release of LDH, further indicating that there was
pore formation during rDer f1 treatment. Lipopolysaccharide (LPS)
treatment (up to 1,000 ng/ml) did not affect cell viability,
suggesting that LPS contamination may not be a major concern
(Fig. 2B and C). Taken together,
these findings confirmed that rDer f1 induced epithelial cell death
by pyroptosis, but not apoptosis.
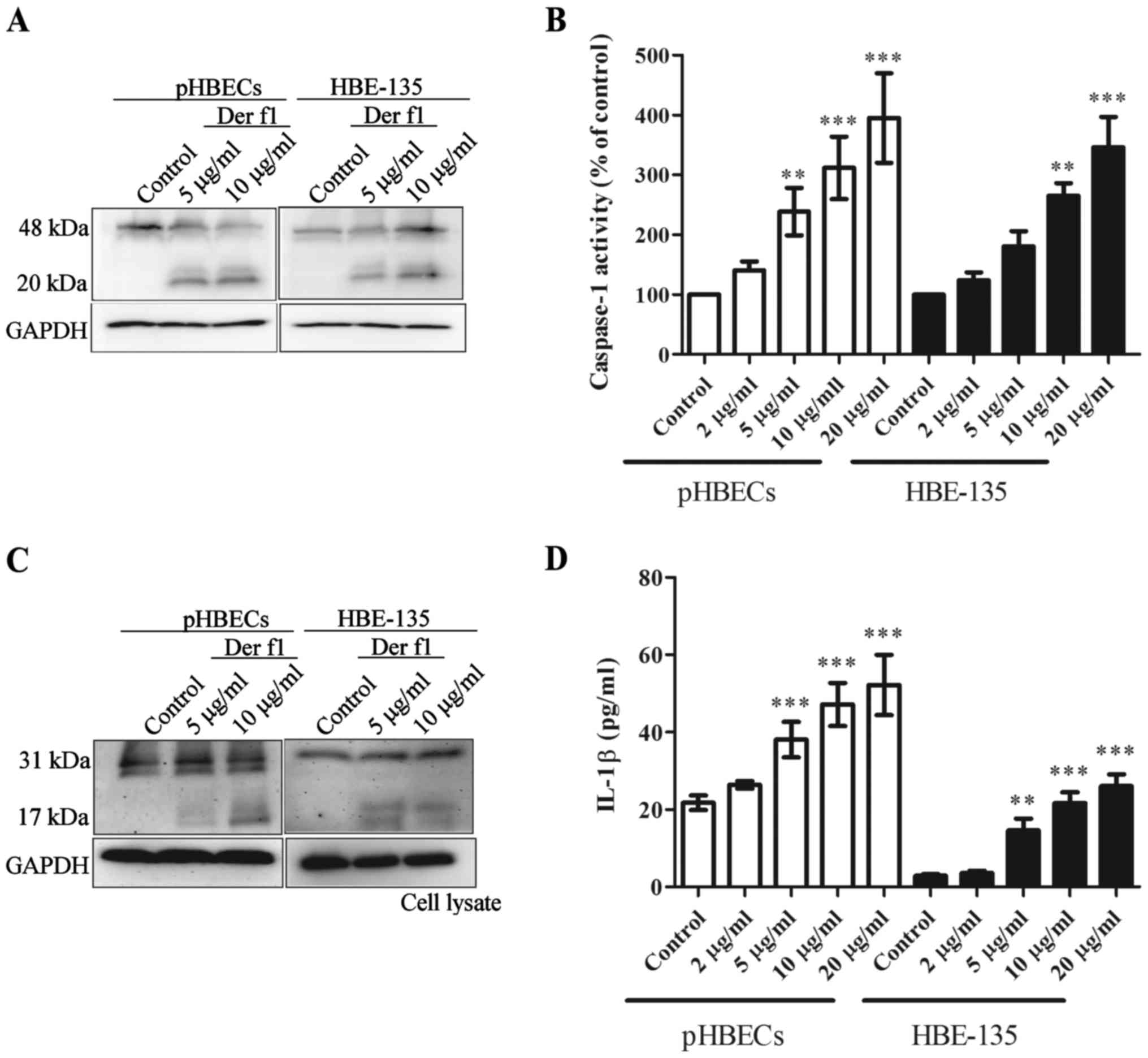
rDer f1 triggers caspase-1 activation and
IL-1β release in BECs
Pyroptosis is characterised by caspase-1 activation
and IL-1β release (10,12). To confirm caspase-1 activation and
IL-1β release, the pHBECs and HBE-135 cells were treated with
various concentrations of rDer f1 for 12 h, and whole-cell lysates
were assessed to confirm caspase-1 cleavage and activity by
immunoblot analysis and caspase-1 activity kits, respectively.
Fig. 3A shows that rDer f1
treatment caused caspase-1 activation, as demonstrated by the
presence of cleaved caspase-1 (20 kDa). Increased caspase-1
activity was observed in the whole-cell lysates of rDer f1-treated
BECs, compared with the controls (Fig. 3B).
In addition, rDer f1 treatment led to conversion of
pro-IL-1β to mature IL-1β, as demonstrated by the presence of
cleaved IL-1β (17 kDa) (Fig. 3C).
Compared with controls, increased concentrations of IL-1β were
detected in the supernatant of BECs following rDer F1 treatment
(Fig. 3D). These findings suggest
that rDer f1 increases caspase-1 activation and IL-1β release in
BECs.
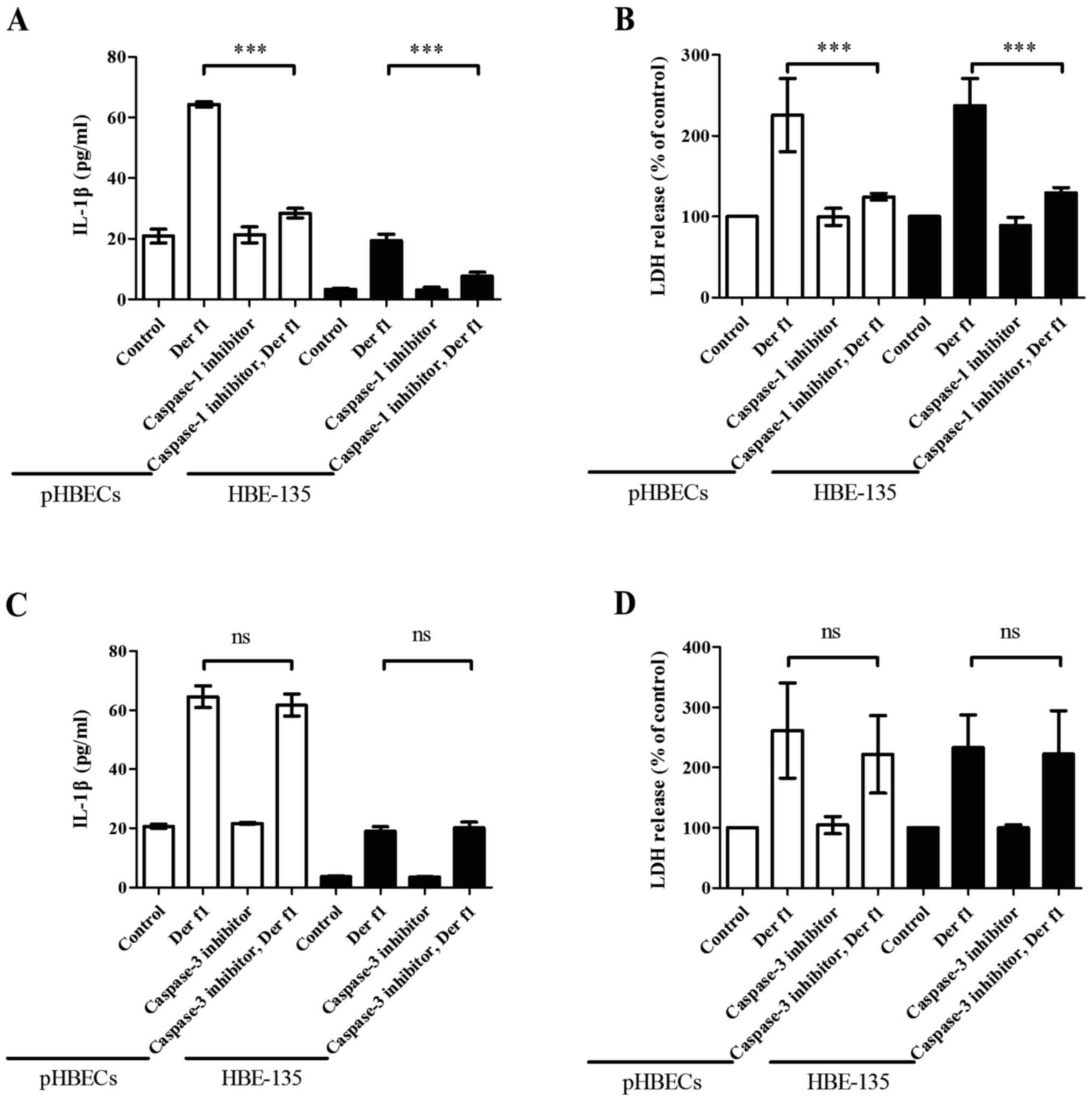
rDer f1-induces IL-1β release and
pyroptosis in BECs in a caspase-1-dependent manner
Our findings indicated that exposure to rDer f1
increased IL-1β production and caused pyroptosis in BECs. To
confirm that both phenomena were associated with caspase-1
activation, a specific caspase-1 inhibitor was used to assess the
involvement of caspase-1. pHBECs and HBE-135 cells were pre-treated
with a caspase-1 inhibitor (Z-YVAD-FMK; 20 µM) or a
caspase-3 inhibitor (20 µM) for 1 h, followed by rDer f1 (10
µg/ml) for 12 h for IL-1β and 24 h for LDH analysis. In the
presence of the caspase-1 inhibitor, IL-1β and LDH release caused
by rDer f1 decreased to nearly the same level as that in control
pHBECs and HBE-135 cells (Fig. 4A and
B). However, in the presence of caspase-3 inhibitor, there was
no inhibitory effect on either IL-1β secretion or LDH release
(Fig. 4C and D), suggesting that
the secretion of IL-1β and triggering of pyroptosis by rDer f1 are
dependent on caspase-1.
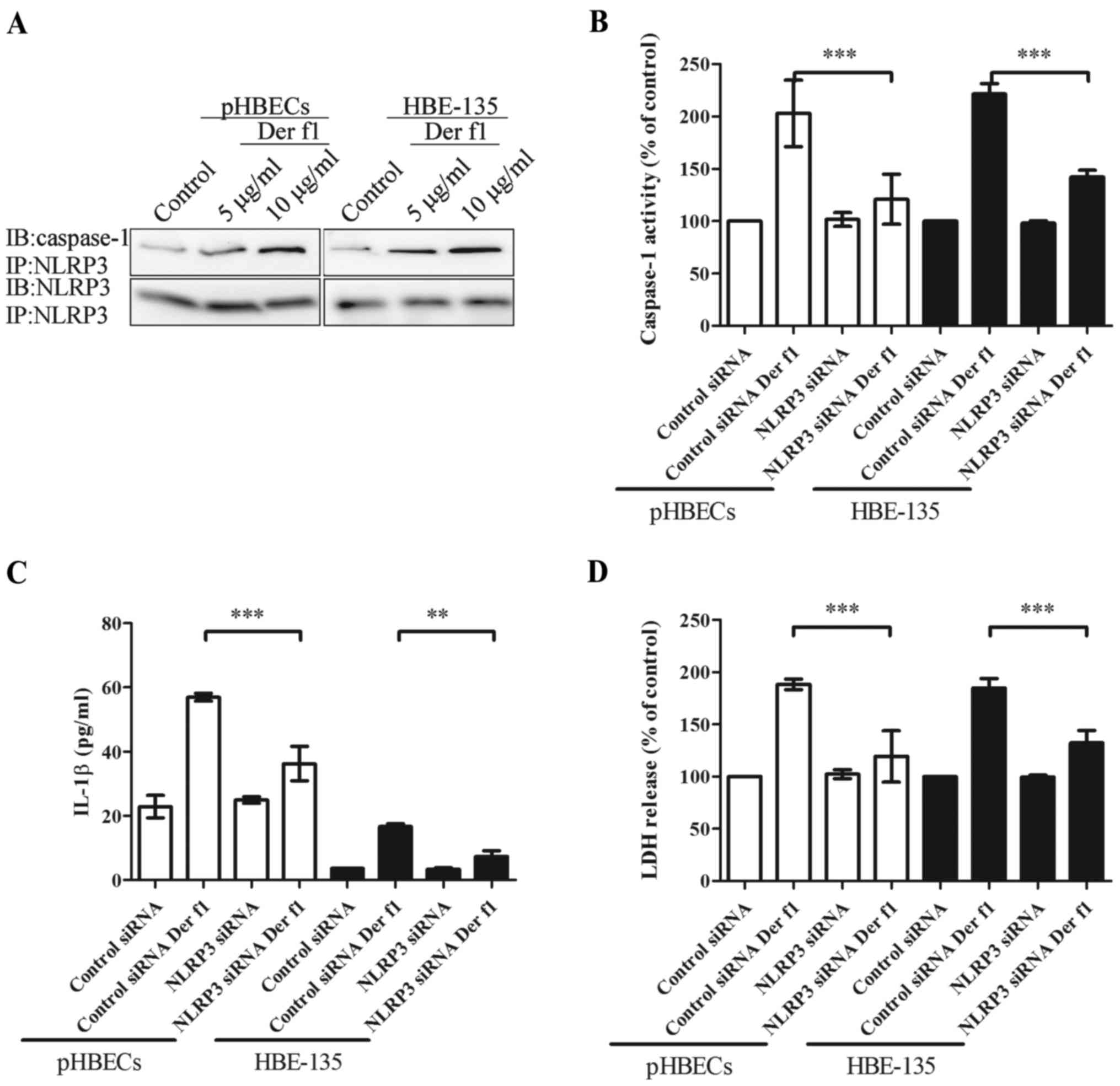
rDer f1-induced pyroptosis is due to the
NLRP3 inflammasome
To further elucidate the possible role of NLRP3 in
rDer f1-mediated caspase-1 activation, the association of NLRP3 and
caspase-1 was assessed. rDer f1 was applied to pHBECs and HBE-135
cells; subsequently, co-IP was performed to confirm the association
between NLRP3 and caspase-1 (Fig.
5A). Increased concentrations of rDer f1 were well-correlated
with an upregulated interaction of NLRP3 and caspase-1.
To confirm that NLRP3 mediated rDer f1-induced
pyroptosis of BECs, cells were transfected with NLRP3 siRNA to
inhibit its expression. Compared with control siRNA transfection,
transfection of pHBECs and HBE-135 cells with NLRP3 siRNA led to
~70% inhibition of NLRP3 expression (data not shown). Transfection
of BECs with NLRP3 siRNA inhibited rDer f1-mediated caspase-1
activation and IL-1β release (Fig. 5B
and C). Furthermore, rDer f1-induced pyroptosis was also
inhibited by NLRP3 siRNA (Fig.
5D). These findings confirmed that rDer f1 acts through
NLRP3-activated caspase-1, which in turn induces IL-1β release and
pyroptosis.
Discussion
Asthma is a major public health concern worldwide.
The most well-known mechanism of asthma pathogenesis is
allergen-induced airway inflammation (20,21). The subsequent reactions include
bronchospasm, oedema and epithelial damage. Epithelial damage may
enhance susceptibility to allergens and sensitivity of the airways,
leading to persistent asthma and airway remodelling (2,4,6).
The novel finding of the present study is that Der f1, an allergen
of D. farinae, was found to induce cell death in
BECs. The increased LDH release and intracellular PI, but not
TUNEL, suggested that this cell death process was due to
pyroptosis. In addition, Der f1 led to inflammasome formation
characterized by caspase-1 activation and IL-1β secretion. This
whole process occurred via the NLRP3 inflammasome pathway. These
findings suggest that Der f1-induced epithelial pyroptosis plays a
major role in asthma pathogenesis and airway remodelling.
BECs are the first line of defence against external
antigens, and are considered to play an important role in the
development of airway allergic inflammation (17). Intact epithelia provide
mucociliary clearance and remove noxious agents, thereby avoiding
allergen sensitization and airway hyperresponsiveness. Sloughing of
BECs has been found in the majority of asthmatic patients,
regardless of the severity (5,6).
In asthmatic patients, clumps of epithelial cells in the sputum and
increased epithelial cells in bronchoalveolar lavage suggest that
epithelial cell sloughing is a pathological characteristic of
asthma (22). The exact mechanism
underlying epithelial cell sloughing in asthma remains a matter of
debate. Pyroptosis, a type of programmed cell death, is triggered
by microbial infections and non-infectious stimuli (10,11). Once activated,
inflammasome-mediated caspase-1 activation leads to membrane
rupture followed by the secretion of pro-inflammatory cytokines,
including IL-1β. Pyroptosis plays a key role in several clinically
relevant conditions, including respiratory diseases. Reisetter
et al demonstrated that carbon black nanoparticles mimicking
particulate ambient pollution also led to pyroptosis of lung
alveolar macrophages (23). Acute
respiratory distress syndrome has been shown to be due to the
induction of NLRP1-dependent pyroptosis (24). In contrast to apoptosis of
bronchial epithelial cells, which lessens inflammation inside the
airway, pyroptosis induces intense airway inflammation, which may
lead to permanent structural changes (13). In the present study, BECs were
exposed to several common allergens, but only rDer f1 treatment led
to cell death. TUNEL analysis failed to provide evidence that
apoptosis played a major role in Der f1-mediated cell death. By
contrast, the LDH release assay as well as PI staining favoured Der
f1-induced cell death by pyroptosis. To evaluate whether LPS
contamination affected the results, treatment with LPS ≤1,000 ng/ml
was performed. The results revealed that LPS treatment did not
exert any effect on cell viability and LDH release, suggesting that
Der f1-mediated pyroptosis is not due to LPS contamination.
Caspase-1 catalyses the conversion of pro-IL-1β to
mature IL-1β, a key inflammatory mediator that controls both local
and systemic immune responses (25). IL-1β drives diverse biological
processes, including extravasation, cell proliferation and
differentiation, cytokine secretion, angiogenesis, wound healing
and pyrexia (26,27). Unregulated and sustained release
of IL-1β has been shown to lead to a number of chronic inflammatory
diseases, such as psoriasis and inflammatory bowel disease.
Moreover, IL-1β has also been shown to play a role in the early
phase of asthma pathogenesis and to modulate airway constriction
and relaxation responses directly on the airway smooth muscles
(26). Our results demonstrated
that Der f1 increased the proteolytic activation and activity of
caspase-1, which in turn induced the secretion of IL-1β from BECs.
Moreover, only caspase-1 inhibition, and not caspase-3 inhibition,
prevented Der f1-mediated IL-1β release and pyroptosis, suggesting
that caspase-1 plays a critical role in Der f1-mediated cell death
in BECs.
The NLRs play important roles in the recognition of
exogenous microbial components or endogenous destructive cellular
factors in innate immunity (28).
Various inflammasomes, such as NLRP1, NLRP3/ASC and IPAF, are
associated with pyroptosis and the secretion of pro-inflammatory
cytokines (29). The NLRP3
inflammasome may be triggered by various pathogens, toxins,
bacterial RNA and uric acid through two sensors of danger signals,
toll-like receptors and P2X7 receptors (28). Following endogenous or exogenous
stimulation, oligomerised NLRP3 interacts with apoptosis-associated
speck-like protein containing a CARD (ASC) through homotypic
protein-protein interactions of the pyrin domains. The interactions
between ASC and the CARD domain of pro-caspase-1 are considered to
activate caspase-1 (28,30). Our results demonstrated that Der
f1 increased the association of NLRP3 and caspase-1. Following
NLRP3 knockdown, the Der f1-induced increased activity of caspase-1
was shown to decrease in pHBECs and HBE-135 cells compared with
that in controls. In addition, the results revealed that Der
f1-induced IL-1β release and pyroptosis were reduced in pHBECs and
HBE-135 cells compared with the controls. These results indicate
that the NLRP-3-related inflammasome contributes to Der f1-mediated
pyroptosis in BECs.
The novel finding of the present study is that Der
f1 induces pyroptosis in BECs via the NLRP3 inflammasome (Fig. 6). The ensuing caspase-1 activation
leads to BEC death and release of the pro-inflammatory cytokine
IL-1β. Der f1 may not act solely as an allergen, and Der
f1-mediated pyroptosis may represent a pathogenic mechanism
contributing to inflammatory injury of airway epithelia. Further
studies are required to elucidate the connection between Der f1 and
NLRP3 inflammasome and Der f1-mediated pyroptosis of BECs in
asthma. In conclusion, the results of the present study suggest
that Der f1-induced epithelial cell pyroptosis plays a major role
in the pathogenesis of asthma, providing novel evidence on the
effect of HDMs on BEC injury and offering novel insight into the
pathobiology of asthma and other respiratory diseases.
Acknowledgments
The present study was supported by the Ministry of
Science and Technology (grant nos. MOST 104-2314-B-037-053-MY4 and
MOST 103-2320-B-037-006-MY3), the 'KMu-KMuH Co-Project of Key
Research' (grant no. KMu-DK 105002 from Kaohsiung Medical
university), the Kaohsiung Medical university Hospital Research
Foundation (grant no. KMuH104-4R72), the Kaohsiung Municipal
Ta-Tung Hospital Research Foundation (grant no. kmtth-104-025), and
the Chi-Mei Medical Center and Kaohsiung Medical university
Research Foundation (grant no. 105 CM-KMu-12).
References
|
1
|
Bel EH: Clinical Practice. Mild asthma. N
Engl J Med. 369:549–557. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lambrecht BN and Hammad H: The airway
epithelium in asthma. Nat Med. 18:684–692. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Holgate ST, Roberts G, Arshad HS, Howarth
PH and Davies DE: The role of the airway epithelium and its
interaction with environmental factors in asthma pathogenesis. Proc
Am Thorac Soc. 6:655–659. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Lloyd CM and Saglani S: Asthma and
allergy: The emerging epithelium. Nat Med. 16:273–274. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Martínez-Girón R and van Woerden HC:
Disruption of airway epithelium in asthma pathogenesis: Are
protozoa responsible. Proc Am Thorac So. 7:161author reply 161.
2010.
|
|
6
|
Chanez P: Severe asthma is an epithelial
disease. Eur Respir J. 25:945–946. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Holgate ST: The sentinel role of the
airway epithelium in asthma pathogenesis. Immunol Rev. 242:205–219.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Holgate ST: Epithelial damage and
response. Clin Exp Allerg. 30(Suppl 1): 37–41. 2000. View Article : Google Scholar
|
|
9
|
Davies DE: The role of the epithelium in
airway remodeling in asthma. Proc Am Thorac Soc. 6:678–682. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bergsbaken T, Fink SL and Cookson BT:
Pyroptosis: Host cell death and inflammation. Nat Rev Microbiol.
7:99–109. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yeretssian G, Labbé K and Saleh M:
Molecular regulation of inflammation and cell death. Cytokine.
43:380–390. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
LaRock CN and Cookson BT: Burning down the
house: Cellular actions during pyroptosis. PLoS Pathog. 9:pp.
e10037932013, View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tanaka H, Miyazaki N, Oashi K, Teramoto S,
Shiratori M, Hashimoto M, Ohmichi M and Abe S: IL-18 might reflect
disease activity in mild and moderate asthma exacerbation. J
Allergy Clin Immunol. 107:331–336. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Maezawa Y, Nakajima H, Kumano K, Kubo S,
Karasuyama H and Iwamoto I: Role of IgE in Th2 cell-mediated
allergic airway inflammation. Int Arch Allergy Immuno. 131(Suppl
1): pp. 2–6. 2003, View Article : Google Scholar
|
|
15
|
Jacquet A: Innate immune responses in
house dust mite allergy. ISRN Allergy. 735031:2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Baker SF, Yin Y, Runswick SK, Stewart GA,
Thompson PJ, Garrod DR and Robinson C: Peptidase allergen Der
1initiates apoptosis of epithelial cells independently of tight
junction proteolysis. Mol Membr Biol. 20:71–81. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Gandhi VD, Davidson C, Asaduzzaman M,
Nahirney D and Vliagoftis H: House dust mite interactions with
airway epithelium: Role in allergic airway inflammation. Curr
Allergy Asthma Rep. 13:262–270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang JY: The innate immune response in
house dust mite-induced allergic inflammation. Allergy Asthma
Immunol Res. 5:68–74. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Takai T, Kato T, Sakata Y, Yasueda H,
Izuhara K, Okumura K and Ogawa H: Recombinant Der 1 and Der f 1
exhibit cysteine protease activity but no serine protease activity.
Biochem Biophys Res Commun. 328:944–952. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Holgate ST: Pathogenesis of asthma. Clin
Exp Allergy. 38:872–897. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Holt PG and Sly PD: Viral infections and
atopy in asthma pathogenesis: New rationales for asthma prevention
and treatment. Nat Med. 18:726–735. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Fahy JV: Remodeling of the airway
epithelium in asthma. Am J Respir Crit Care Me. 164(Suppl 2):
S46–S51. 2001. View Article : Google Scholar
|
|
23
|
Reisetter AC, Stebounova LV, Baltrusaitis
J, Powers L, gupta A, grassian VH and Monick MM: Induction of
inflammasome-dependent pyroptosis by carbon black nanoparticles. J
Biol Chem. 286:21844–21852. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kovarova M, Hesker PR, Jania L, Nguyen M,
Snouwaert JN, Xiang Z, Lommatzsch SE, Huang MT, Ting JP and Koller
BH: NLRP1-dependent pyroptosis leads to acute lung injury and
morbidity in mice. J Immunol. 189:2006–2016. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Denes A, Lopez-Castejon G and Brough D:
Caspase-1: Is IL-1 just the tip of the ICEberg. Cell Death Di.
3:e3382012. View Article : Google Scholar
|
|
26
|
Whelan R, Kim C, Chen M, Leiter J,
Grunstein MM and Hakonarson H: Role and regulation of interleukin-1
molecules in pro-asthmatic sensitised airway smooth muscle. Eur
Respir. 24:559–567. 2004. View Article : Google Scholar
|
|
27
|
Delaleu N and Bickel M: Interleukin-1 beta
and interleukin-18: Regulation and activity in local inflammation.
Periodontol. 35:42–52. 2004. View Article : Google Scholar
|
|
28
|
Mariathasan S and Monack DM: Inflammasome
adaptors and sensors: Intracellular regulators of infection and
inflammation. Nat Rev Immunol. 7:31–40. 2007. View Article : Google Scholar
|
|
29
|
Aachoui Y, Sagulenko V, Miao EA and Stacey
KJ: Inflammasome-mediated pyroptotic and apoptotic cell death, and
defense against infection. Curr Opin Microbiol. 16:319–326. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miao EA, Rajan JV and Aderem A:
Caspase-1-induced pyroptotic cell death. Immunol Rev. 243:206–214.
2011. View Article : Google Scholar : PubMed/NCBI
|