Introduction
Myocardial ischemia-reperfusion injury is defined as
the sudden reintroduction of molecular oxygen due to blood flow
restoration in the ischemic area, and it may cause additional
injury to the myocardium (1).
This pathological process occurs inevitably in a wide range of
patients, such as cardiac arrest survivors, acute myocardial
infarction victims and cardiac surgery patients (2). Although the underlying mechanism has
not been fully elucidated, accumulating evidence indicates that
mitochondrial dysfunction plays a key role in myocardial
ischemia-reperfusion injury (3–7).
Impaired myocardial mitochondrial function leads to diminished
cardiac substrate flexibility, decreased cardiac energy efficiency
and diastolic dysfunction (8–10).
However, there are few effective strategies for preventing the
process of mitochondrial dysfunction in myocardial
ischemia-reperfusion injury. Therefore, identifying potential
therapeutic agents that improve mitochondrial function in
myocardial ischemia-reperfusion has become a field of interest in
research.
Glucagon-like peptide-1 (GLP-1), an endogenous
incretin hormone, has been confirmed to exert potent
insulinotropic, insulinomimetic and glucagonostatic effects;
however, its clinical use is limited by its rapid degradation by
dipeptidyl peptidase-4 (DPP-4) (11). Exenatide, a GLP-1 analogue that is
not susceptible to cleavage by DPP-4, has been developed and is
currently being used as novel antidiabetic drug (11). Exenatide shares 53% homology with
native GLP-1, but still binds to GLP-1 receptors on pancreatic
β-cells to exert its insulin-releasing and glucose-lowering effects
(12). GLP-1 receptors have been
found in extrapancreatic tissues, particularly in the heart
(13,14), and numerous studies have reported
that GLP-1 and its analogues exert cardioprotective effects in
myocardial ischemia-reperfusion injury, as well as in other
pathologies that are associated with myocardial remodeling and
heart failure (15–19). Recent evidence demonstrated that
such cytoprotection appears to rely on direct mitochondrial
preservation by modulating oxidative phosphorylation and inhibiting
oxidative stress (20,21). However, there is little
information on the role of mitochondrial function in this
cardioprotection. In this sense, this commonality in the beneficial
effects on cardiac homeostasis between mitochondrial adjustment and
GLP-1-mediated cardioprotection raises the question whether
mitochondrial function improvement is a component of GLP-1-mediated
cytoprotection against myocardial ischemia-reperfusion injury.
To address this question, in the present study,
hypoxia/reoxygenation (H/R)-treated H9c2 cells, an established
in vitro model resembling ischemia-reperfusion in
vivo, were used to determine the role of mitochondrial function
in GLP-1-mediated cardioprotection. To test this hypothesis,
characteristics of mitochondrial morphology and function, including
ATP synthesis, membrane potential (ΔΨm), mitochondrial permeability
transition pore (mPTP) and activities of mitochondrial ATPases were
investigated, as was mitochondrial oxidative stress at the cellular
level. Furthermore, the underlying mechanism for GLP-1-mediated
cardioprotection was examined by assessing the GLP-1
receptor/cyclic adenosine monophosphate (cAMP)̸protein kinase A
(PKA) signaling pathway.
Materials and methods
Cell culture and H/R treatment
H9c2 cells (rat cardiomyoblast cell line; Chinese
Academy of Medical Sciences, Shanghai, China) were cultured in
Dulbecco's modified Eagle's medium/Nutrient Mixture F-12 (DMEM/F12;
Thermo Fisher Biochemical Products, Beijing, China) containing 10%
(v/v) fetal bovine serum (FBS; Invitrogen Life Technologies; Thermo
Fisher Scientific, Carlsbad, CA, USA) and 100 µg/ml
penicillin/streptomycin (Beyotime Institute of Biotechnology,
Haimen, China).
The H/R model was established according to the
methods previously described, with some modifications (22). In brief, after growing to 80%
confluence, the cells were starved in serum-free DMEM/F12 for 12 h
and were then subjected to hypoxia in a hypoxic incubator (Thermo
Forma, Marietta, OH, USA), saturated with a gas mixture (95%
N2 and 5% CO2) at 37°C. The percent oxygen in
the hypoxic incubator was maintained at 1% to induce simulated
ischemia. After hypoxia treatment, the cells were provided with
fresh 10% FBS DMEM/F12 and rapidly transferred into a normoxic
incubator for reoxygenation. The control group was cultured under
normal incubating conditions for the corresponding times. Exenatide
or the cAMP activator, forskolin (1 µM), was added to the
cells for 30 min prior to exposure to hypoxia. The GLP-1 receptor
antagonist exendin-(9-39) (0.1 µM), the cAMP inhibitor
Rp-Camps (200 µM) and the PKA inhibitor H-89 (5 µM)
were added to the cells for 10 min prior to treatment with
exenatide.
Viability assay
The cell counting kit-8 (CCK-8; Beyotime Institute
of Biotechnology) was employed to examine cell viability as
previously described (23).
Briefly, H9c2 cells (1×104/100 µl) were seeded in
96-well plates for 72 h. The cells were then pretreated with or
without exenatide (0, 0.05, 0.1, 0.2, 0.4 and 0.6 µM) for 30
min prior to being subjected to H/R (4/2, 6/3, 12/4, 14/5, 16/6 and
22/10 h). The cells were provided with fresh media (100 µl)
and CCK-8 solution (10 µl) was added into each well. The
plates were then incubated under normoxic conditions for 2 h. The
optical density values were measured at 450 nm using a microplate
reader (Multiskan MK33; Thermolab Systems, Helsinki, Finland).
Transmission electron microscopy
After the indicated treatment, cells were harvested
by 0.25% trypsinization and centrifugation at 400 × g for 5 min.
The cells were then fixed with 2.5% glutaraldehyde for 2 h at 4°C
and post-fixed with 1% osmium tetroxide for 15 min at 4°C. After
dehydration with a graded series of aceton, the cells were washed
by propylene oxide and embedded in Epon 812. Ultrathin sections
were cut with an ultramicrotome, stained with sodium acetate and
lead hydroxide, and examined using a transmission electron
microscope (Hitachi-7500; Hitachi, Tokyo, Japan)
Flow cytometry
Annexin V/propidium iodide (PI) staining was used to
determine cell apoptosis by flow cytometry (24). H9c2 cells (2×104/100
µl) were seeded in 6-well plates for 72 h. After treatment,
the cells were harvested by trypsinization and centrifugation at
400 × g for 5 min, and re-suspended at a density of
1×106/ml. The cells were incubated with 5 µl
Annexin V-fluorescein isothiocyanate (FITC) and PI (10 µl,
20 µg/ml) for 20 min, and then analyzed using a flow
cytometer (BD FACSVantage SE; Beckman Coulter, Brea, CA, USA). The
data on fluorescence intensity were analyzed using the CellQuest™
software (Becton Dickinson and Company, Franklin Lakes, NJ,
USA).
To quantitatively analyze the development of
oxidative stress, the generation of reactive oxygen species (ROS)
and reactive nitrogen species (RNS) was assessed using
2′,7′-dichlorofluorescin diacetate (DCFH-DA) and dihydroethidium
(DHE) (both from Beyotime Institute of Biotechnology) by flow
cytometry, as described previously (24). After the indicated treatment, the
cells were loaded with DCFH-DA (10 µM) for 60 min at 37°C
and DHE (5 µM) for 30 min at 37°C, and then analyzed on a
flow cytometer. DCFH-DA was excited at 488 nm and emitted at 525
nm. DHE was excited at 543 nm and emitted at 560 nm. The data on
fluorescence intensity were analyzed using the CellQuest™
software.
Changes in mitochondrial calcium concentration
[(Ca2+)m] were assessed using a
mitochondrial-permeating calcium fluorophore, Rhod-2AM (Santa Cruz
Biotechnology Inc., Santa Cruz, CA, USA), by flow cytometry as
described previously (24). After
the indicated treatment, the cells were incubated with 2 µM
Rhod-2AM for 30 min at 37°C and were then analyzed on a flow
cytometer. Rhod-2AM was excited at 543 nm and emitted at 560 nm.
The data on fluorescence intensity were analyzed using the
CellQuest™ software.
The opening of mPTP was detected using the
calcein-AM probe (Santa Cruz Biotechnology, Inc.) by flow
cytometry, as described previously (25). The loading of calcein-AM enabled
the localization of fluorescent calcein in mitochondria, and the
calcein signal was reduced when the mPTP opened (26). After the indicated treatment, the
cells were loaded with calcein-AM (1 µM) for 30 min at 37°C
and were then analyzed on a flow cytometer. Calcein-AM was excited
at 488 nm and emitted at 525 nm. The data on fluorescence intensity
were analyzed using the CellQuest™ software.
ΔΨm was measured using a fluorescent, lipophilic and
cationic probe, JC-1 (Beyotime Institute of Biotechnology) by flow
cytometry, as described previously (7). After the indicated treatment, the
cells were loaded with JC-1 (10 µg/ml) for 20 min at 37°C
and were then were analyzed on a flow cytometer under single
excitation (488 nm) and dual emission (530 and 590 nm). The data on
fluorescence intensity were analyzed using the CellQuest™ software.
The fluorescence ratio of red to green was quantitated.
Detection of intracellular ATP
content
Cellular ATP content was measured using the ATP
bioluminescent assay kit (Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. In brief, after the
indicated treatment, the cells were lysed and centrifuged at 12,000
× g for 5 min. The supernatants (100 µl) were mixed with ATP
detection working dilution (100 µl) in a 96-well plate. The
luminance was measured using a microplate reader (Multiskan MK33;
Thermolab Systems). The protein concentration of each group was
determined using the enhanced bicinchoninic acid (BCA) protein
assay kit (Beyotime Institute of Biotechnology). The total ATP
content was expressed as nmol/mg protein.
Mitochondrial isolation
Mitochondria were isolated from H9c2 cells using the
Cell Mitochondria Isolation kit (Beyotime Institute of
Biotechnology) according to the manufacturer's instructions.
Briefly, after the indicated treatment, the cells were collected
and suspended in ice-cold isolation buffer for 15 min. After the
cells were homogenized, the homogenate was centrifuged at 600 × g
for 10 min at 4°C, and then the supernatant was centrifuged at
11,000 × g for 10 min at 4°C. The mitochondria were harvested from
the sediments.
Colorimetry
The activity of lactate dehydrogenase (LDH) in the
culture medium, and the activities of mitochondrial
Na+/K+-ATPase and
Ca2+/Mg2+-ATPase were measured using
commercially available kits (Jiancheng Bioengineering Institute,
Nanjing, China) according to the manufacturer's instructions. In
brief, after the indicated treatment, the cells were lysed and
centrifuged at 1,600 × g for 10 min at 4°C. The mitochondria were
isolated as described above. The supernatants and the mitochondria
were collected and reacted with the respective reagents included in
the kits. Subsequently, the absorbance values at 340 and 660 nm
were measured using a spectrophotometer (721D; Pudong Shanghai
Physical Optical Instrument Factory, Shanghai, China). The protein
concentration of each group was determined using the enhanced BCA
protein assay kit (Beyotime Institute of Biotechnology). The
activity of LDH was expressed as U/l. The activities of
Na+/K+-ATPase and
Ca2+/Mg2+-ATPase were expressed as
µmol Pi/mg protein/h.
Enzyme-linked immunosorbent assay
(ELISA)
The levels of creatine kinase-MB (CK-MB) in the
culture medium and plasma were measured using the CK-MB ELISA assay
(R&D Systems, Minneapolis, MN, USA), according to the
manufacturer's instructions. After the indicated treatment, the
culture medium and plasma was collected and centrifuged at 1,600 ×
g for 10 min at 4°C. The supernatants were collected for the
detection of CK-MB. The supernatants were then incubated with the
reagents in the kits. Finally, the absorbance values were measured
using a microplate reader (Multiskan MK33; Thermolab Systems) at
450 nm. The CK-MB level was expressed as U/l.
Western blot assay
Protein samples were isolated from the H9c2 cells by
homogenization in cell lysis buffer (Beyotime Institute of
Biotechnology). The lysates were kept on ice for 45 min and total
proteins were isolated by centrifugation at 14,000 × g for 10 min
at 4°C. The protein concentration was measured using the enhanced
BCA protein assay kit (Beyotime Institute of Biotechnology).
Proteins were separated by sodium dodecyl sulfate-polyacrylamide
gel electrophoresis and transferred onto PVDF membranes. The
membranes were blocked in 5% non-fat milk and incubated with
primary antibodies to uncoupling protein (UCP)-3 (1:1,000; rabbit,
polyclonal; C19359), nuclear respiratory factor (Nrf)-1 (1:1,000;
rabbit, polyclonal; C20962) (both from Anbo Biotechnology Co.,
Ltd., San Francisco, CA, USA), and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) (1:1,000; Beyotime Institute of
Biotechnology). The membranes were then incubated with horse radish
peroxidase-goat anti-rabbit immunoglobulin G secondary antibody
(cat. no. ZDR 5306; 1:1,000; Zhongshan Goldenbridge Biotechnology
Corporation, Beijing, China). Signals were detected with the ECL
system (Beyotime Institute of Biotechnology). Blots were scanned
using Bio-Rad gel imaging system (Bio-Rad Laboratories, Hercules,
CA, USA) and bands were quantified with Quantity One software.
Statistical analysis
The SPSS 17.0 software (SPSS, Inc., Chicago, IL,
USA) was used for statistical analysis. Data are presented as mean
± standard deviation. Grouped data were analyzed using a one-way
analysis of variance followed by the Student-Newman-Keuls test.
When the equal variance test failed, a Mann-Whitney rank-sum test
was used. A P-value of <0.05 was considered to indicate
statistically significant differences.
Results
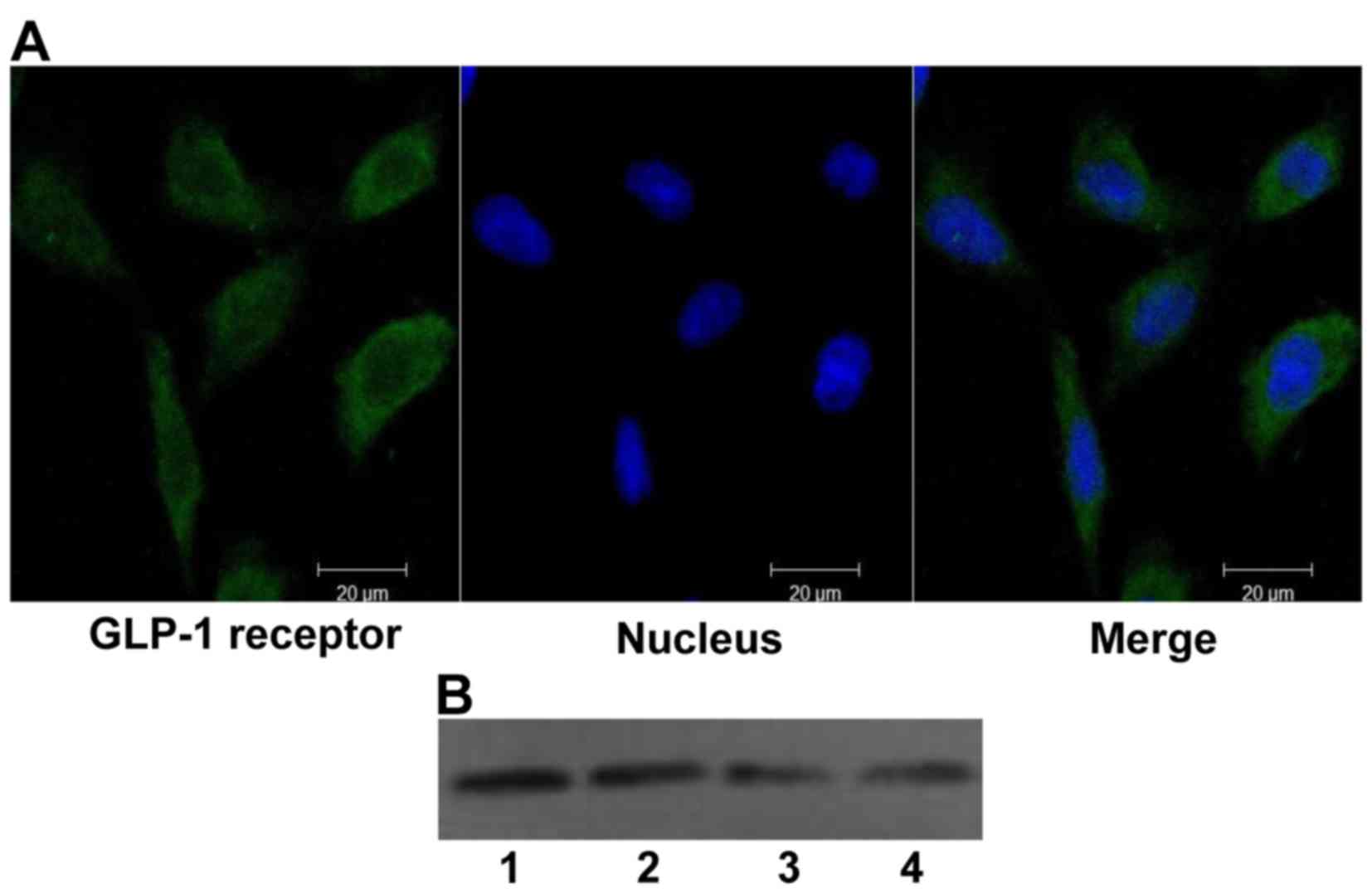
The GLP-1 receptor is expressed in H9c2
cells
Although the GLP-1 receptor has been found in the
hearts of mammals, no information is available regarding its
expression in H9c2 cells. Therefore, the expression of GLP-1
receptor was first tested in H9c2 cells using confocal laser
scanning microscopy and western blot analysis (Fig. 1), and the expression of the GLP-1
receptor in H9c2 cells was confirmed.
Exenatide increases the viability of H9c2
cells subjected to H/R
After H9c2 cells were exposed to various durations
of H/R (4/2, 6/3, 12/4, 14/5, 16/6 and 22/10 h), cell viability was
assessed with the CCK-8 kit and was found to be significantly
decreased in a time-dependent manner compared with the control
group (Fig. 2A). Cell viability
after 4/2 and 6/3 h H/R was reduced to 0.96 and 0.92 (% of
control), respectively, compared with that in the control group
(P<0.05), while cell viability was reduced to 0.71, 0.61, 0.53
and 0.36 after 12/4, 14/5, 16/6 and 22/10 h H/R, respectively
(P<0.01). H/R 12/4 h was selected to investigate the potential
protective effects of exenatide on cardiomyocytes, as it was the
earliest time-point when cell viability exhibited a statistically
significant difference (P<0.01).
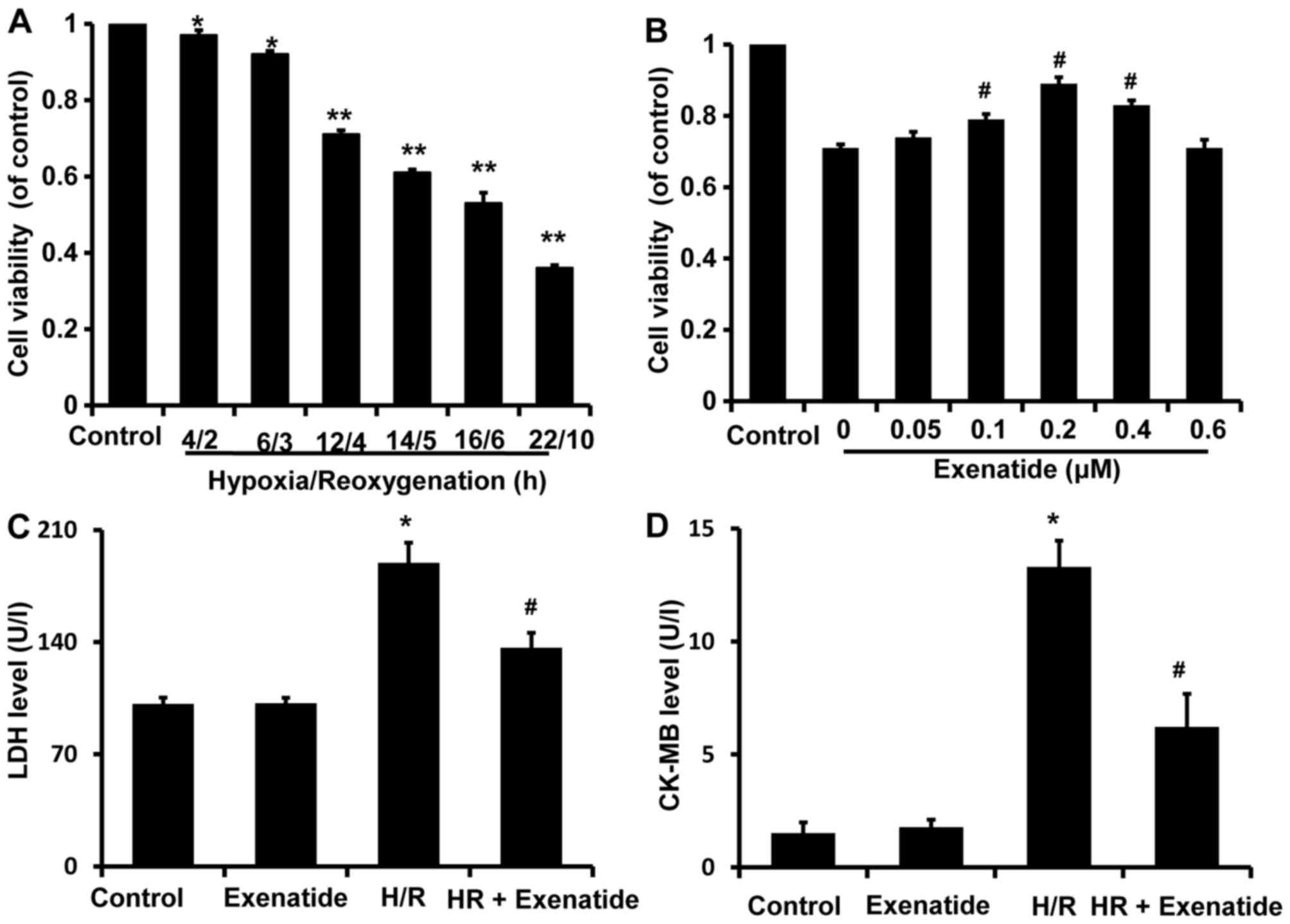 | Figure 2Effects of hypoxia/reoxygenation
(H/R) on the viability of H9c2 cells and the protective effects of
exenatide in H/R-injury. (A) H9c2 cells were exposed to H/R
conditions for different times (4/2, 6/3, 12/4, 14/5, 16/6 and
22/10 h). After treatment, cell viability was assessed using the
cell counting kit-8 (CCK-8). Data are expressed as percentage of
control and represented as mean ± SD; n=6. *P<0.05;
**P<0.01 vs. control group. (B) H9c2 cells were
pretreated with exenatide (0, 0.05, 0.1, 0.2, 0.4 and 0.6
µM) for 30 min and underwent 12 h hypoxia followed by 4 h
reoxygenation. After treatment, cell viability was assessed using
the CCK-8 kit. Data are expressed as percentage of control and
represented as mean ± SD; n=6. #P<0.05 vs. the 0
group. (C) Effects of exenatide on the lactate dehydrogenase (LDH)
levels in the culture medium. H9c2 cells were pretreated with
exenatide (0.2 µM) for 30 min and underwent 12 h hypoxia
followed by 4 h reoxygenation. After treatment, the LDH levels in
the culture medium were measured by colorimetry and expressed as
U/l. (D) Effects of exenatide on the creatine kinase-MB (CK-MB)
levels in the culture medium. H9c2 cells were pretreated with
exenatide (0.2 µM) for 30 min and underwent 12 h hypoxia
followed by 4 h reoxygenation. After treatment, the CK-MB levels in
the culture medium were measured by ELISA and expressed as U/l.
Values are expressed as means ± SD; n=6. *P<0.05 vs.
control group; #P<0.05 vs. H/R group. SD, standard
deviation. |
To investigate the possible cardioprotective effects
of exenatide against H/R injury, H9c2 cells were pretreated with
exenatide (0, 0.05, 0.1, 0.2, 0.4 and 0.6 µM) for 30 min
prior to undergoing 12/4 h H/R. It was observed that pretreatment
with exenatide (0.1, 0.2 and 0.4 µM) successfully alleviated
the decrease of cell viability induced by H/R injury (P<0.05)
(Fig. 2B). Exenatide at 0.2
µM exhibited the best efficiency in preserving cell
viability. Thus, the concentration of 0.2 µM was selected to
treat H9c2 cells in the following experiment.
LDH and CK-MB release are two well-known markers of
cardiomyocyte injury. To further investigate the cardioprotection
of exenatide against H/R injury, LDH and CK-MB release in the
culture medium was further examined (Fig. 2C and D). LDH and CK-MB release was
significantly increased in the H/R group compared with the control
group (P<0.05), while pretreatment with 0.2 µM exenatide
significantly decreased LDH and CK-MB release induced by H/R
(P<0.05). These results strongly suggest that exenatide exerted
cardioprotective effects against H/R injury in H9c2 cells.
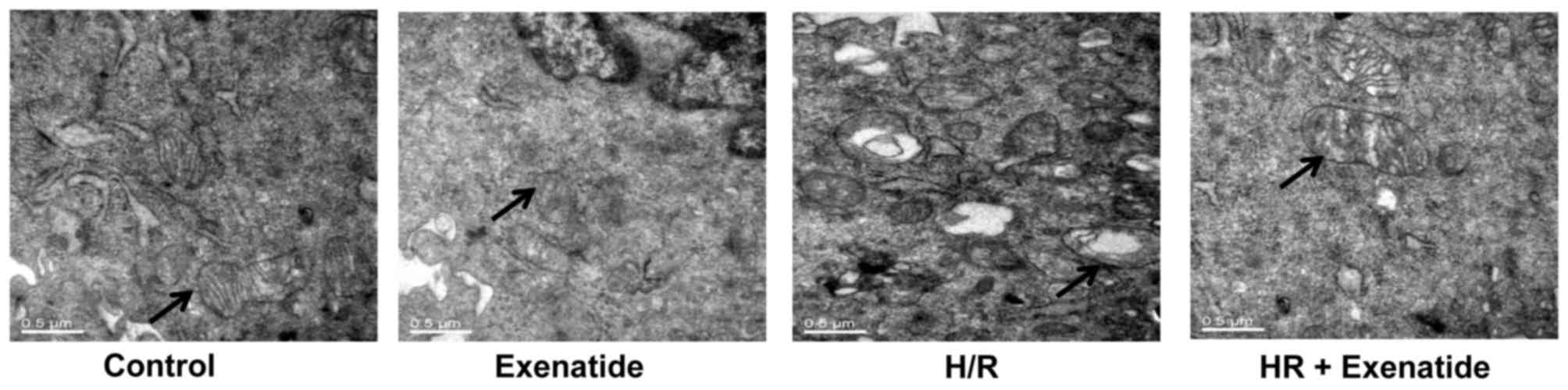
Exenatide inhibits structural changes in
mitochondria
Transmission electron microscopy was used to detect
mitochondrial structural changes. As shown in Fig. 3, mitochondria in the control cells
presented as integrated structures with numerous transversely
orientated cristae enveloped by an intact outer membrane. However,
H/R injury resulted in swollen mitochondria, appearing as spherical
structures with disarrayed cristae, disorganized matrix and more
cytosolic vacuoles. Exenatide treatment attenuated mitochondrial
swelling, cristae disarray and membrane rupture in H9c2 cells
following H/R.
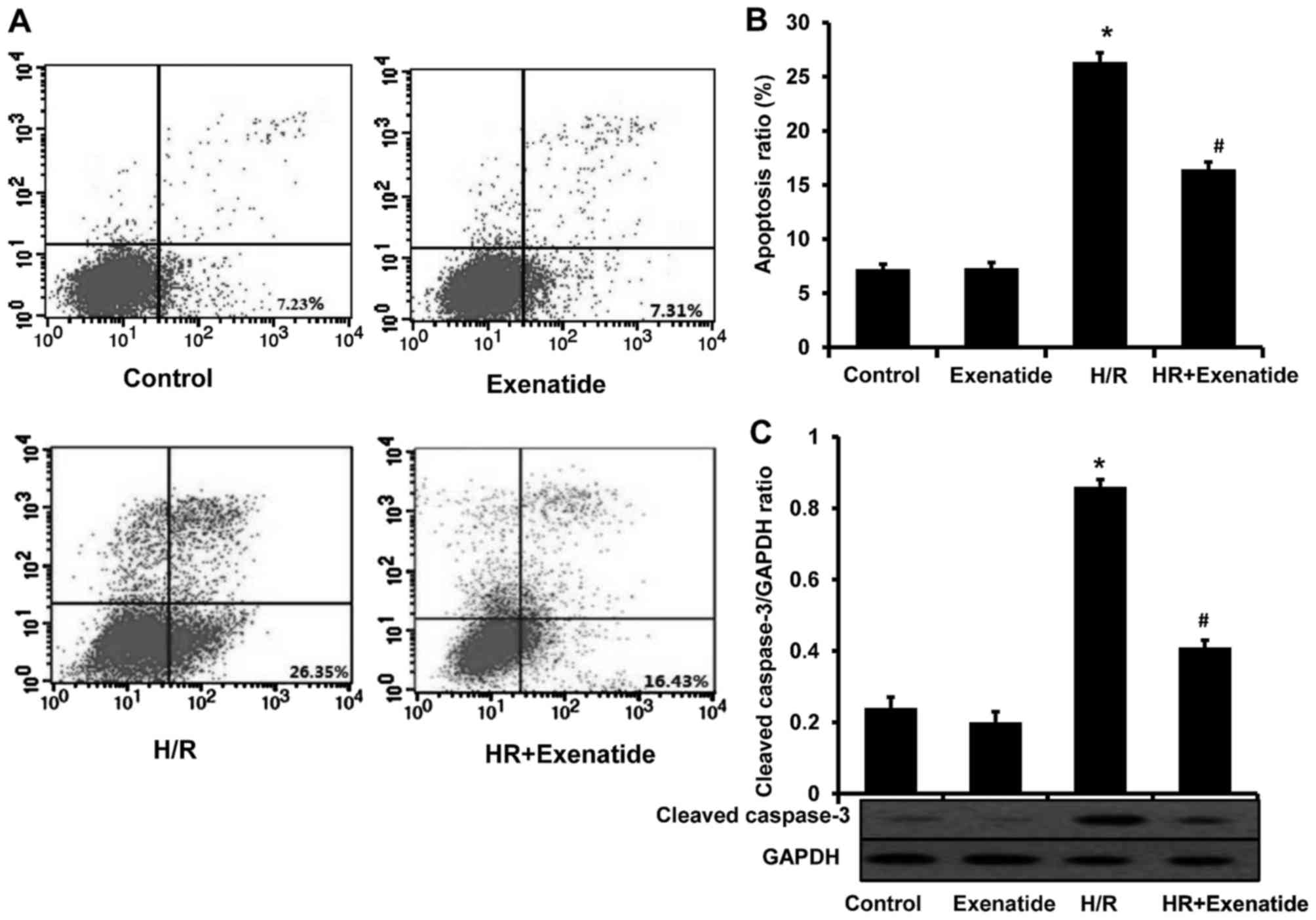
Exenatide protects H9c2 cells from
apoptosis
Considering the anti-apoptotic effect of exenatide
in several studies, this effect was investigated in the H/R model.
As shown in Fig. 4A and B,
H/R-treated cells exhibited a significant increase in apoptosis
(P<0.05). Compared with cells treated with H/R, the H/R +
exenatide group exhibited a significant decrease in the proportion
of apoptotic cells (P<0.05). The expression of cleaved caspase-3
was also detected (Fig. 4C); it
was observed that exenatide statistically significantly decreased
the expression of cleaved caspase-3 in H/R-treated H9c2 cells
(P<0.05). These findings demonstrated that exenatide exerts
anti-apoptotic effects on H/R-treated H9c2 cells.
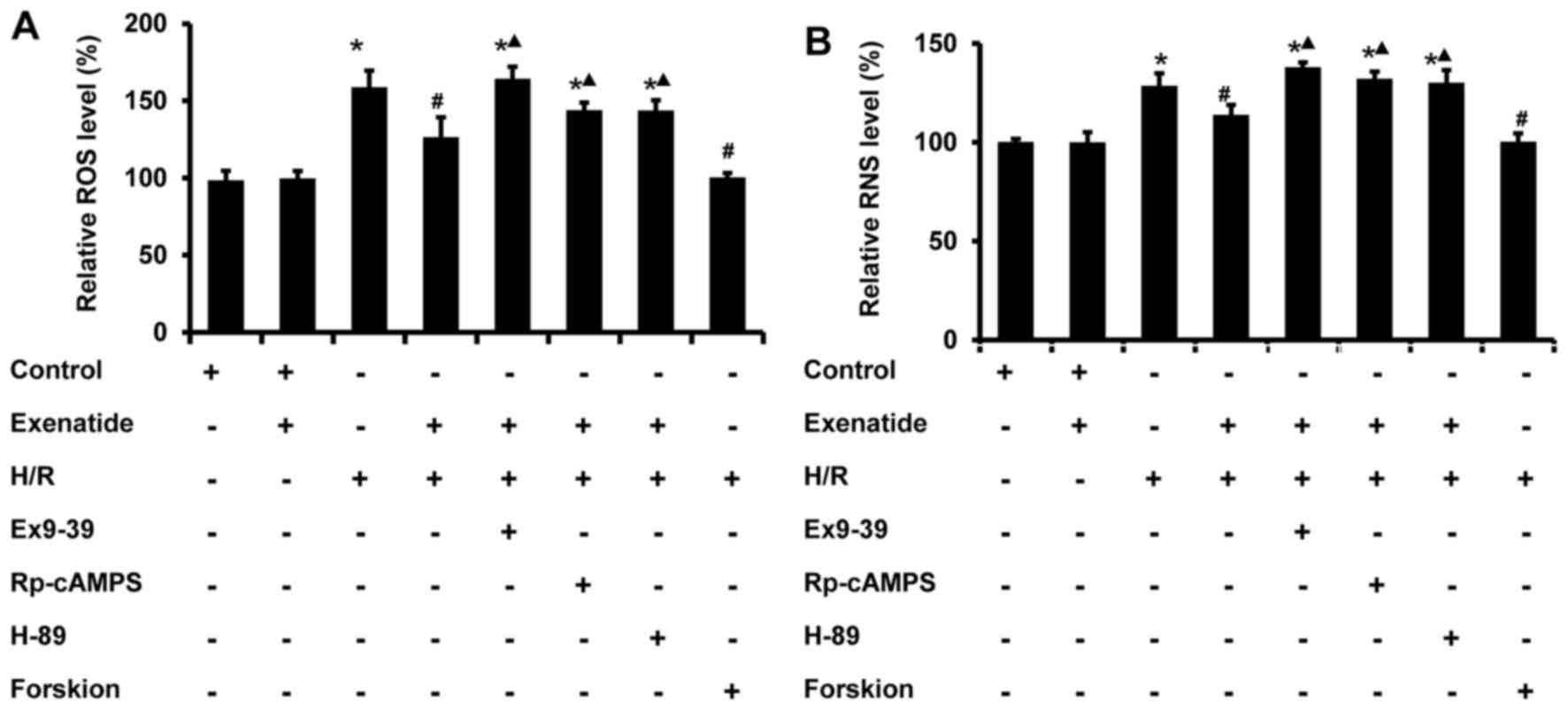
Exenatide reduces the H/R-induced
oxidative stress via the GLP-1 receptor/cAMP/PKA pathway in H9c2
cells
Mitochondria are one of the major cellular sources
of oxidative stress, and play a crucial role in oxidative injury
during H/R; thus, the effects of exenatide on the generation of ROS
and RNS induced by H/R were determined in H9c2 cells. As shown in
Fig. 5, ROS and RNS were
significantly increased in H9c2 cells subjected to H/R (P<0.05),
whereas exenatide reduced ROS and RNS generation in H/R-treated
H9c2 cells (P<0.05). The results indicated that exenatide
reduced H/R-induced oxidative stress in H9c2 cells.
Next, the role of GLP-1 receptor/cAMP/PKA signaling
pathway in the anti-oxidative effects of exenatide on H/R injury
was further evaluated. The GLP-1 receptor antagonist
exendin-(9-39), the cAMP inhibitor Rp-cAMPS and the PKA inhibitor
H-89 were employed. As shown in Fig.
5, the inhibitory effects of exenatide on H/R-induced ROS and
RNS accumulation were significantly attenuated by treatment with
exendin-(9-39), Rp-cAMPS and H-89 (P<0.05). Furthermore, in line
with the results following exenatide pretreatment, pretreatment
with the cAMP activator forskolin also reduced the production of
ROS and RNS in H9c2 cells subjected to H/R, suggesting that the
reduction of the H/R-induced oxidative stress by exenatide is
dependent on the GLP-1 receptor/cAMP/PKA pathway. Taken together,
these results suggest that exenatide reduces the H/R-induced
oxidative stress via activating the GLP-1 receptor/cAMP/PKA pathway
in H9c2 cells.
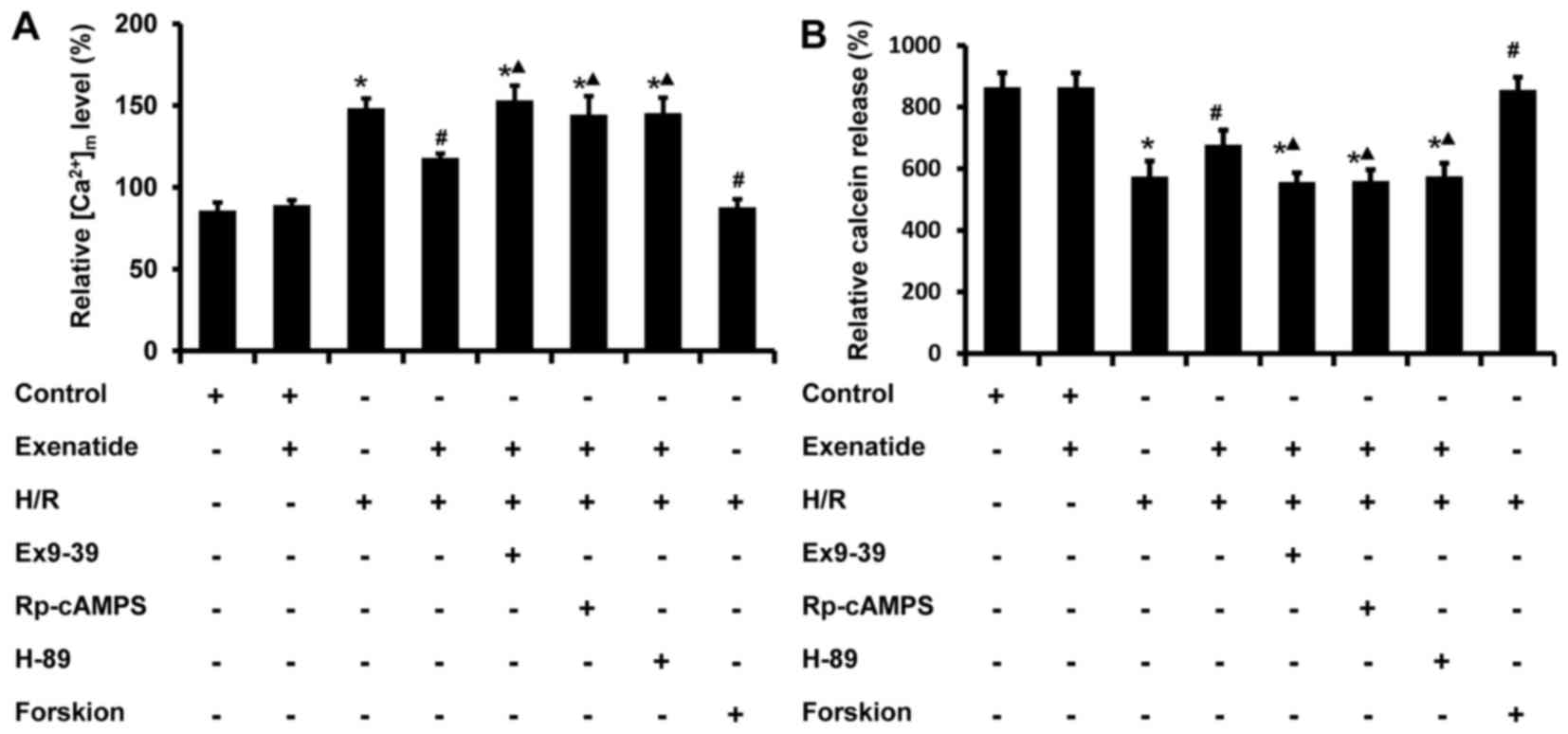
Exenatide reduces the H/R-induced
(Ca2+)m overload and the opening of mPTP via
the GLP-1 receptor/cAMP/PKA pathway in H9c2 cells
It is well-known that an increase in
(Ca2+)m impairs mitochondrial function; thus,
the effects of exenatide on (Ca2+)m changes
induced by H/R in H9c2 cells were tested using flow cytometry. As
shown in Fig. 6A, the
(Ca2+)m level in the H/R group was
statistically significantly increased compared with that in the
control group (P<0.05), while pretreatment with 0.2 µM
exenatide inhibited the increase of (Ca2+)m
induced by H/R (P<0.05). Similar to exenatide, forskolin (0.1
µM) pretreatment also inhibited the increase of
(Ca2+)m. However, incubation of cells with
exendin-(9-39), Rp-cAMPS and H-89 abrogated the normalizing effect
of exenatide on (Ca2+)m (P<0.05). These
results indicated that exenatide attenuates the H/R-induced
(Ca2+)m overload via activating the GLP-1
receptor/cAMP/PKA pathway in H9c2 cells.
To further investigate the effects of exenatide on
mitochondrial function, the status of mPTP was determined using
calcein-AM probes by flow cytometry. Previous studies reported that
the loading of calcein-AM enabled the localization of fluorescent
calcein in mitochondria, and that the calcein-AM signal was reduced
when the mPTP opened (26). As
shown in Fig. 6B, H/R treatment
significantly decreased the calcein-AM fluorescence intensity
compared with that of the control group (P<0.05), while
pretreatment with exenatide or forskolin increased the calcein-AM
signal intensity. When cells were pre-incubated with
exendin-(9-39), Rp-cAMPS and H-89, the effects of exenatide on
calcein-AM intensity were inhibited (P<0.05). These results
indicated that exenatide inhibits the H/R-induced opening of mPTP
via activating the GLP-1 receptor/cAMP/PKA pathway in H9c2
cells.
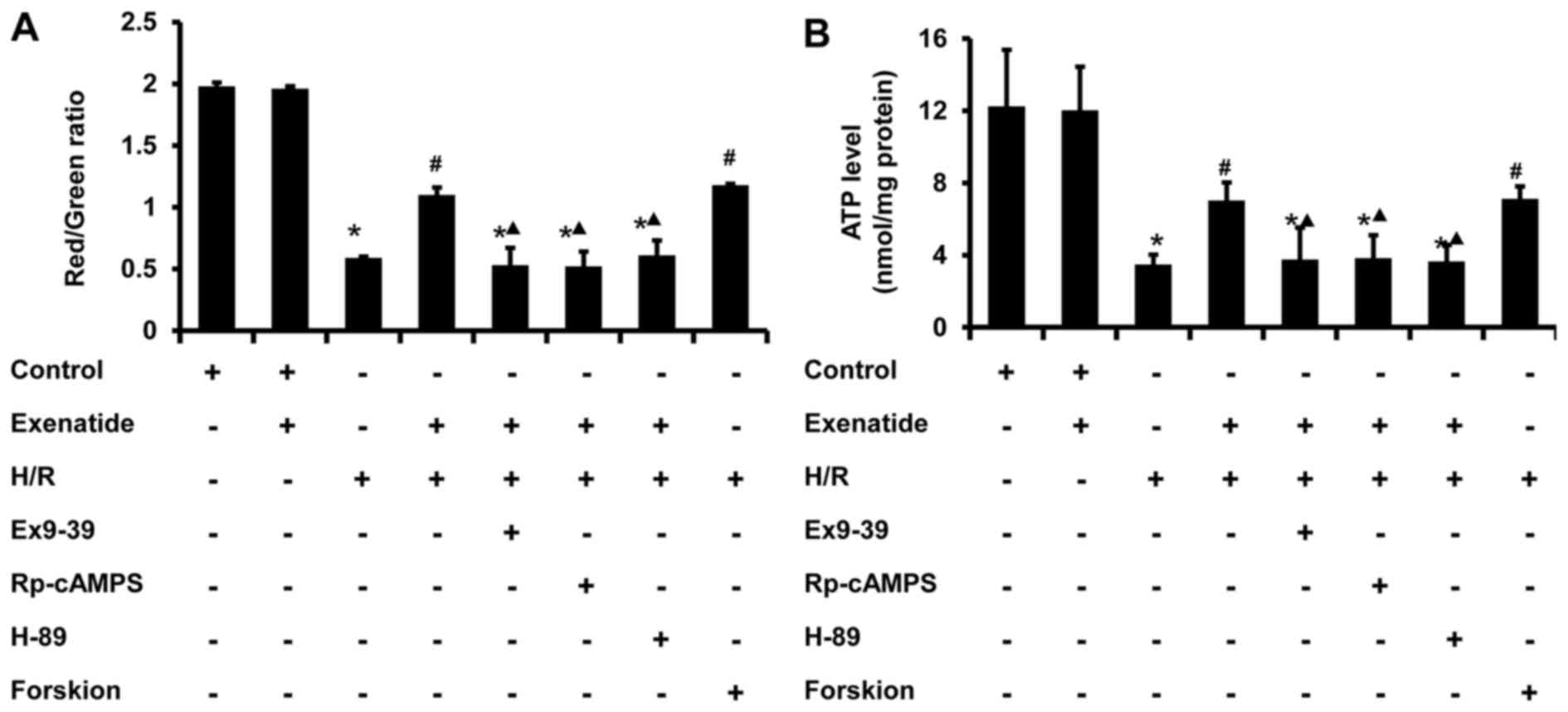
Exenatide inhibits the H/R-induced
depolarization of ΔΨm and the reduction of ATP synthesis in H9c2
cells via the GLP-1 receptor/cAMP/PKA pathway
Since ΔΨm is one of the indicators of mitochondrial
function, the effect of exenatide on ΔΨm was investigated. As shown
in Fig. 7A, H/R treated cells
exhibited a decrease in polarized mitochondria (P<0.05) and an
increase in depolarized mitochondria (P<0.05) compared with the
control group, whereas pretreatment with exenatide or forskolin
reversed these changes (P<0.05); there was no significant
difference between the H/R + exenatide and H/R + forskolin groups
(P>0.05). When the cells were preincubated with exendin-(9-39),
Rp-cAMPS and H-89, the effect of exenatide on ΔΨm was inhibited
(P<0.05). These results suggest that exenatide prevents ΔΨ
depolarization induced by H/R via activating the GLP-1
receptor/cAMP/PKA pathway in H9c2 cells.
Cellular ATP content is also a sensitive indicator
of mitochondrial function. As shown in Fig. 7B, ATP concentration significantly
decreased from 12.25 nmol/mg protein in the control group to 3.49
nmol/mg protein in the H/R group (P<0.05). However, exenatide
pretreatment resulted in an increase of cellular ATP level compared
with that in the H/R group (P<0.05), and these results were
similar to those in the H/R + forskolin group. By contrast,
incubation of cells with exendin-(9-39), Rp-cAMPS and H-89
abrogated the effect of exenatide on cellular ATP content in
H/R-treated cells (P<0.05). These results suggest that exenatide
prevents the reduction of ATP synthesis induced by H/R via
activating the GLP-1 receptor/cAMP/PKA pathway in H9c2 cells.
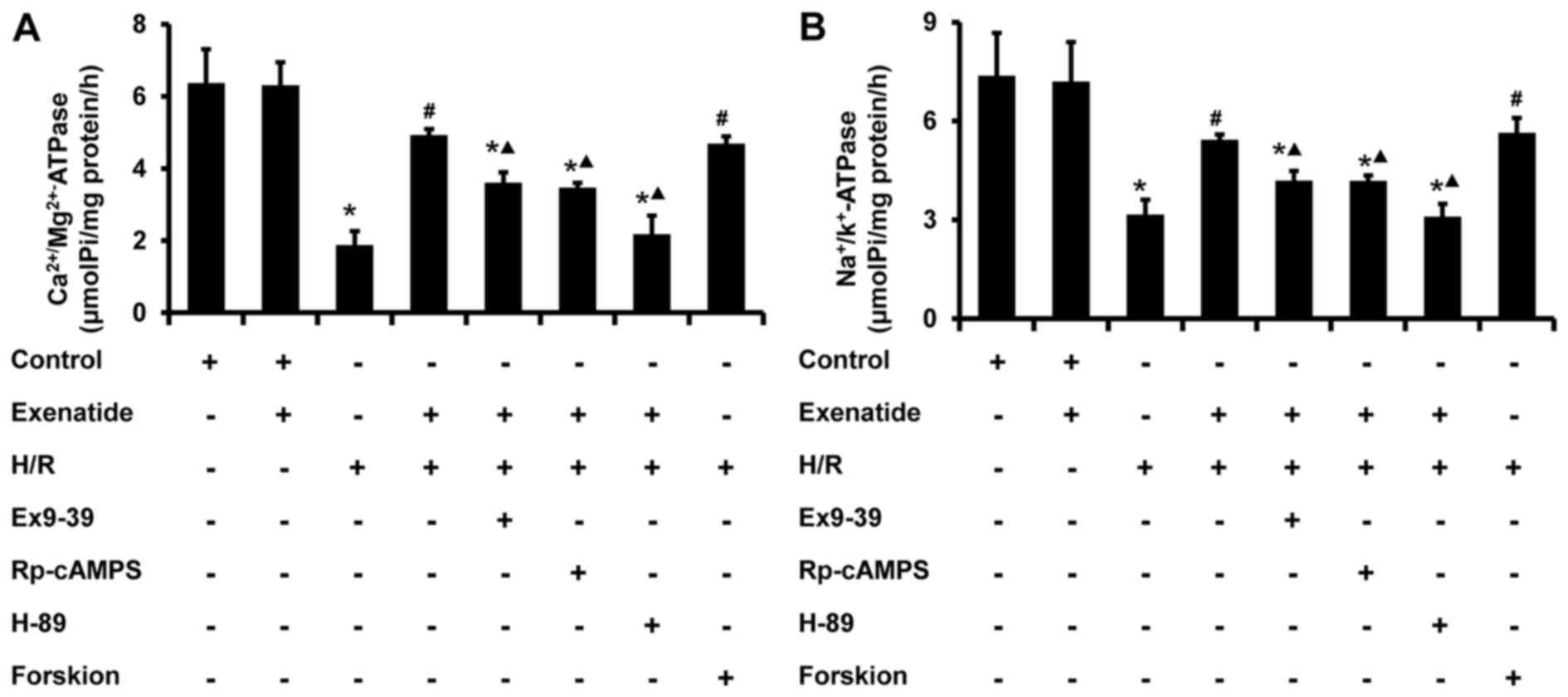
Exenatide inhibits the H/R-induced
decrease of mitochondrial ATPase activity in H9c2 cells via the
GLP-1 receptor/cAMP/PKA pathway
The activity of mitochondrial ATPase was further
examined. As shown in Fig. 8,
H/R-treated cells exhibited a significant decrease in the activity
of Na+/K+ ATPase (P<0.05) and
Ca2+/Mg2+ ATPase (P<0.05), whereas
exenatide pretreat ment significantly increased the activity of
Na+/K+ ATPase (P<0.05) and
Ca2+/Mg2+ ATPase (P<0.05) in H/R-treated
cells; these results were similar to those in the H/R + forskolin
group. The effects of exenatide on ATPase in mitochondria were also
inhibited by exendin-(9-39), Rp-cAMPS and H-89 (P<0.05). These
findings provide evidence that exenatide maintains mitochondrial
ATPase activity during H/R via activating the GLP-1
receptor/cAMP/PKA pathway.
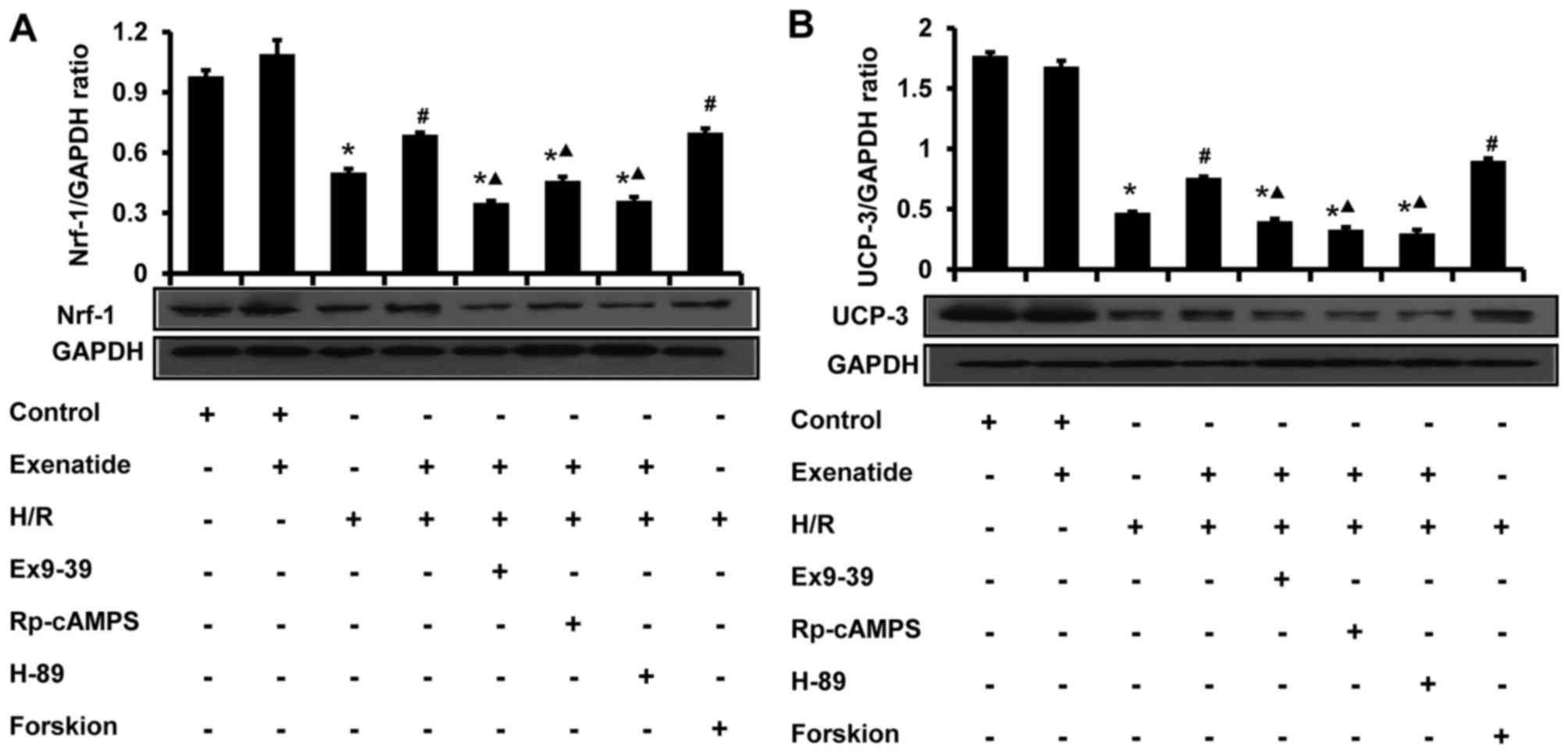
Exenatide inhibits the H/R-induced
reduction of UCP-3 and Nrf-1 protein expression in H9c2 cells via
the GLP-1 receptor/cAMP/PKA pathway
The effects of exenatide on UCP-3 and Nrf-1 protein
expression in H9c2 cells were analyzed by western blotting
(Fig. 9). Compared with the
control group, H/R treatment significantly decreased the levels of
UCP-3 and Nrf-1 (P<0.05). Compared with the H/R group, exenatide
significantly increased the UCP-3 and Nrf-1 levels (P<0.05),
whereas UCP-3 and Nrf-1 levels did not differ significantly between
the H/R + exenatide and the H/R + forskolin groups (P>0.05).
However, the GLP-1 receptor antagonist exendin-(9-39), the cAMP
inhibitor Rp-cAMPS and the PKA inhibitor H-89 attenuated the
effects of exenatide on UCP-3 and Nrf-1 (P<0.05). These results
suggest that exenatide prevents the reduction of UCP-3 and Nrf-1
protein expression induced by H/R via activating the GLP-1
receptor/cAMP/PKA pathway in H9c2 cells.
Discussion
The main findings of this study revealed that
exenatide exerted cardioprotective effects in an in vitro
model of H/R, which resembles ischemia-reperfusion in vivo,
by improving mitochondrial function, namely inhibiting the
development of morphological abnormalities, opening of mPTP and
depolarization of ΔΨm, decreasing mitochondrial oxidative stress
and (Ca2+)m overload, enhancing ATP synthesis
and the activity of Na+/K+ ATPase and
Ca2+/Mg2+ ATPase. Importantly, these
beneficial effects were abolished by treatment with exendin-(9-39),
Rp-cAMPS and H-89, demonstrating that exenatide protects against
ischemia-reperfusion injury via mitochondrial function improvement
involving the GLP-1 receptor/cAMP/PKA signaling pathway.
GLP-1 must bind to the GLP-1 receptor, a specific G
protein-coupled receptor, in order to perform its cellular
functions (13). The presence of
the GLP-1 receptor has been demonstrated in a number of human
organs and tissues, including the pancreas, heart, lung, kidney and
brain (13,14). However, there was no report on
whether the GLP-1 receptor is expressed in the H9c2 cell line. In
the present study, it was first proven that H9c2 cells express the
GLP-1 receptor using confocal laser scanning microscopy and western
blot analysis.
Mitochondrial dysfunction plays a key role in
myocardial injury during ischemia-reperfusion (3–5).
In the present study, mitochondrial function was found to be
severely impaired in H9c2 cells subjected to H/R, as evidenced by
reduced ATP synthesis, decreased activity of mitochondrial ATPases,
opening of mPTP and depolarization of ΔΨm. Previous studies
reported that changes in mitochondrial morphology may affect their
biological processes and function (27). It was observed that H/R treatment
caused mitochondrial abnormalities, including swelling and
disarrayed cristae, and these changes in shape are associated with
the decrease in ATP synthesis and activity of mitochondrial
ATPases. It is well-known that mitochondria, being a store of
intracellular calcium, a source of ROS and a sensor of oxidative
stress, play a key role in triggering necrotic and apoptotic cell
death under a variety of pathological conditions, including
ischemia-reperfusion injury (28–30). In the present study, exposure to
H/R was found to reduce cell viability, increase the cell apoptotic
rate, and increase the LDH and CK-MB levels in the cultured
supernatant. Furthermore, H/R injury also increased mitochondrial
oxidative stress, as evidenced by increased ROS and RNS generation
and (Ca2+)m overload. Taken together, these
findings indicate that H/R treatment compromised the mitochondrial
function, further contributing to cellular injury in the H/R
model.
It is noteworthy that improved mitochondrial
function with endogenous adjustment or artificial intervention
accelerates recovery of cardiac and cellular functions subsequent
to ischemia-reperfusion injury (6,7,31,32). Therefore, treatments focused on
preserving mitochondrial integrity and function hoping to minimize
the impact of ischemia-reperfusion injury have become an area of
intensive research. Various cardioprotective effects of GLP-1 and
its analogues have been reported (15–19). Treatment with GLP-1 and its
analogues may improve myocardial glucose uptake (33,34) and metabolism (35,36), as well as cardiac function
(34,37) in both animal models and clinical
studies (15,16). Several mechanisms underlying this
cardioprotection have been proposed, such as activating the
pro-survival kinase associated with reperfusion injury signaling
kinase pathway (38), reducing
oxidative stress and increasing antioxidants (37). Recently, Brown et al
reported that the GLP-1 analogue exendin-4 exerted a persistent
beneficial effect via altering the mitochondrial phenotype, which
decreased the cardiac (Ca2+)m uptake and
reduced oxidative phosphorylation (20). To the best of our knowledge, the
present study is the first to provide evidence that exenatide
improves several characteristics of mitochondrial function (ATP
synthesis, ΔΨm, mPTP and mitochondrial ATPase activity) following
H/R injury. We also demonstrated that exenatide treatment decreases
the mitochondrial oxidative stress (decreased ROS and RNS
generation and (Ca2+)m overload) in the H/R
model. Moreover, in line with previous results (15,16,18), exenatide was found to reduce cell
apoptosis and cell injury, resulting in increased viability of H9c2
cells subjected to H/R. Based on the abovementioned results,
exenatide was proven to exert cardioprotective effects in this
cellular model of H/R via improving mitochondrial function.
The cAMP/cAMP-dependent PKA signaling pathway
(cAMP/PKA) is well-known to regulate cellular energy metabolism,
critically affecting glucose transport and utilization (39), mitochondrial respiration and
dynamics (40–42). It was recently revealed that
activation of the cAMP/PKA pathway may be involved in
GLP-1-mediated protective effects. Wang et al demonstrated
that GLP-1 and its analogue exenatide protected against cardiac
microvascular injury in diabetes via a cAMP/PKA/Rho-dependent
mechanism (43). Xiao et
al observed that GLP-1 enhanced cardiac L-type Ca2+
currents through the cAMP/PKA pathway (44). Bose et al reported that
GLP-1 treatment may attenuate ischemia-reperfusion injury, at least
in part via activation of PKA (45–47). Based on the abovementioned
findings, we hypothesized that the protective effects of exenatide
on mitochondrial function in H/R injury may be associated with
activation of the cAMP/PKA pathway via binding to the GLP-1
receptor. In the present study, it was observed that the
exenatide-induced improvement of mitochondrial function (ATP
synthesis, ΔΨm, mPTP and mitochondrial ATPase activity) was
abolished by the GLP-1 receptor inhibitor exendin-(9-39), the cAMP
inhibitor Rp-cAMPS, and the PKA inhibitor H-89. To further examine
the mechanism underlying the effect of exenatide on mitochondrial
function, forskolin, a potent activator of cAMP, was employed to
activate the cAMP/PKA pathway. We observed that both exenatide and
forskolin exerted a similar protective effect on mitochondrial
function in H/R injury. These results strongly suggest that
exenatide may improve mitochondrial function in H/R injury, at
least in part though activation of the GLP-1 receptor/cAMP/PKA
signaling pathway.
Mitochondrial UCPs and Nrf-1 are both known
downstream effectors of the cAMP/PKA pathway. UCPs belong to the
superfamily of anion carrier proteins and are located in the inner
mitochondrial membrane (48).
Previous studies demonstrated that overexpression of UCPs in
cardiomyocytes may prevent cell death by preserving mitochondrial
function and structure (49,50). The present study demonstrated that
exenatide treatment enhanced the expression of UCP-3, which was
associated with improvement of mitochondrial function in H9c2
cardiomyocytes subjected to H/R, while these effects were abolished
by the GLP-1 receptor inhibitor exendin-(9-39), the cAMP inhibitor
Rp-cAMPS and the PKA inhibitor H-89. Nrf-1 is a key nuclear
transcription factor that regulates the expression of nuclear
mitochondrial genes encoding proteins of the mitochondrial
respiratory chain and oxidative phosphorylation (51,52). Therefore, Nrf-1 plays an important
role in regulating mitochondrial biogenesis and respiratory
function (53). The present study
demonstrated that exenatide upregulated the expression of Nrf-1 in
H/R-treated H9c2 cardiomyocytes. Furthermore, the results revealed
that upregulation of Nrf-1 was associated with improvement of
mitochondrial function. These beneficial effects were also
abolished by exendin-(9-39), Rp-cAMPS and H-89. Although the
present study did not evaluate any other proteins responsible for
mitochondrial function, the cAMP/PKA pathway was correlated with
its downstream factors (UCP-3 and Nrf-1) using the inhibitors of
cAMP/PKA. Taken together, our data further indicated that exenatide
prevented H/R-induced mitochondrial dysfunction, possibly through
upregulation of UCP-3 and Nrf-1 via activation of the GLP-1
receptor/cAMP/PKA signaling pathway.
In conclusion, the data of the present study
demonstrated that the GLP-1 analogue exenatide exerted
cardioprotective effects in an in vitro model of H/R, which
resembles ischemia-reperfusion in vivo, and that this
cardioprotection may be attributed to the improvement of
mitochondrial function. These effects are most likely associated
with activation of the GLP-1 receptor and the cAMP/PKA signaling
pathway. These findings highlight a novel mechanism underlying the
cardio-protective effects of GLP-1 analogues and the improvement of
myocardial ischemia-reperfusion injury.
Acknowledgments
The present study was supported by the National
Natural Science Fund (grant no. 85170212), the Natural Science
Foundation Project of CQ CSTC (grant no. cstc2011jjA10008) and the
National key Clinical Specialties Construction Program of China
(grant no. 2011-170). The authors greatly appreciate the excellent
technical support of Mr. Jianyong Wu and Mr. Dezhang Zhao
(Institute of Life Sciences, Chongqing Medical University) for the
flow cytometry analysis.
References
|
1
|
Braunwald E and Kloner RA: Myocardial
reperfusion: A double-edged sword? J Clin Invest. 76:1713–1719.
1985. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Acar E, Ural D, Bildirici U, Sahin T and
Yılmaz I: Diabetic cardiomyopathy. Anadolu Kardiyol Derg.
11:732–737. 2011.PubMed/NCBI
|
|
3
|
Ong SB and Gustafsson AB: New roles for
mitochondria in cell death in the reperfused myocardium. Cardiovasc
Res. 94:190–196. 2012. View Article : Google Scholar :
|
|
4
|
Honda HM, Korge P and Weiss JN:
Mitochondria and ischemia/reperfusion injury. Ann NY Acad Sci.
1047:248–258. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lesnefsky EJ and Hoppel CL:
Ischemia-reperfusion injury in the aged heart: Role of
mitochondria. Arch Biochem Biophys. 420:287–297. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sun L, Zhao M, Yu XJ, Wang H, He X, Liu JK
and Zang WJ: Cardioprotection by acetylcholine: A novel mechanism
via mitochondrial biogenesis and function involving the PGC-1α
pathway. J Cell Physiol. 228:1238–1248. 2013. View Article : Google Scholar
|
|
7
|
Yue R, Hu H, Yiu KH, Luo T, Zhou Z, Xu L,
Zhang S, Li K and Yu Z: Lycopene protects against
hypoxia/reoxygenation-induced apoptosis by preventing mitochondrial
dysfunction in primary neonatal mouse cardiomyocytes. PLoS One.
7:e507782012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rehman H, Shi Y and Zhong Z:
Ischemia/reperfusion inhibits mitochondrial biogenesis after
partial hepatectomy in mice: 1738. Transplantation. 90:8392010.
View Article : Google Scholar
|
|
9
|
Ren J, Pulakat L, Whaley-Connell A and
Sowers JR: Mitochondrial biogenesis in the metabolic syndrome and
cardiovascular disease. J Mol Med (Berl). 88:993–1001. 2010.
View Article : Google Scholar
|
|
10
|
Rimbaud S, Garnier A and Ventura-Clapier
R: Mitochondrial biogenesis in cardiac pathophysiology. Pharmacol
Rep. 61:131–138. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Garber AJ: Novel GLP-1 receptor agonists
for diabetes. Expert Opin Investig Drugs. 21:45–57. 2012.
View Article : Google Scholar
|
|
12
|
Mundil D, Cameron-Vendrig A and Husain M:
GLP-1 receptor agonists: A clinical perspective on cardiovascular
effects. Diab Vasc Dis Res. 9:95–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Baggio LL and Drucker DJ: Biology of
incretins: GLP-1 and GIP. Gastroenterology. 132:2131–2157. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei Y and Mojsov S: Tissue-specific
expression of the human receptor for glucagon-like peptide-I:
Brain, heart and pancreatic forms have the same deduced amino acid
sequences. FEBS Lett. 358:219–224. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chinda K, Chattipakorn S and Chattipakorn
N: Cardioprotective effects of incretin during
ischaemia-reperfusion. Diab Vasc Dis Res. 9:256–269. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ravassa S, Zudaire A and Díez J: GLP-1 and
cardioprotection: From bench to bedside. Cardiovasc Res.
94:316–323. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bao W, Holt LJ, Prince RD, Jones GX,
Aravindhan K, Szapacs M, Barbour AM, Jolivette LJ, Lepore JJ,
Willette RN, et al: Novel fusion of GLP-1 with a domain antibody to
serum albumin prolongs protection against myocardial
ischemia/reperfusion injury in the rat. Cardiovasc Diabetol.
12:1482013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao TC: Glucagon-like peptide-1 (GLP-1)
and protective effects in cardiovascular disease: A new therapeutic
approach for myocardial protection. Cardiovasc Diabetol. 12:902013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu Q, Anderson C, Broyde A, Polizzi C,
Fernandez R, Baron A and Parkes DG: Glucagon-like peptide-1 and the
exenatide analogue AC3174 improve cardiac function, cardiac
remodeling, and survival in rats with chronic heart failure.
Cardiovasc Diabetol. 9:762010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Brown SB, Libonati JR, Selak MA, Shannon
RP and Simmons RA: Neonatal exendin-4 leads to protection from
reperfusion injury and reduced rates of oxidative phosphorylation
in the adult rat heart. Cardiovasc Drugs Ther. 24:197–205. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tomas E, Stanojevic V and Habener JF:
GLP-1-derived nonapeptide GLP-1(28-36)amide targets to mitochondria
and suppresses glucose production and oxidative stress in isolated
mouse hepatocytes. Regul Pept. 167:177–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Park M, Youn B, Zheng XL, Wu D, Xu A and
Sweeney G: Globular adiponectin, acting via AdipoR1/APPL1, protects
H9c2 cells from hypoxia/reoxygenation-induced apoptosis. PLoS One.
6:e191432011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wang L, Wang ZH, Shen CY, You ML, Xiao JF
and Chen GQ: Differentiation of human bone marrow mesenchymal stem
cells grown in terpolyesters of 3-hydroxyalkanoates scaffolds into
nerve cells. Biomaterials. 31:1691–1698. 2010. View Article : Google Scholar
|
|
24
|
Kumar S, Kain V and Sitasawad SL: High
glucose-induced Ca2þ overload and oxidative stress contribute to
apoptosis of cardiac cells through mitochondrial dependent and
independent pathways. Biochim Biophys Acta. 1820:907–920. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Odagiri K, Katoh H, Kawashima H, Tanaka T,
Ohtani H, Saotome M, Urushida T, Satoh H and Hayashi H: Local
control of mitochondrial membrane potential, permeability
transition pore and reactive oxygen species by calcium and
calmodulin in rat ventricular myocytes. J Mol Cell Cardiol.
46:989–997. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tominaga H, Katoh H, Odagiri K, Takeuchi
Y, Kawashima H, Saotome M, Urushida T, Satoh H and Hayashi H:
Different effects of palmitoyl-L-carnitine and palmitoyl-CoA on
mitochondrial function in rat ventricular myocytes. Am J Physiol
Heart Circ Physiol. 295:H105–H112. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Ong SB, Subrayan S, Lim SY, Yellon DM,
Davidson SM and Hausenloy DJ: Inhibiting mitochondrial fission
protects the heart against ischemia/reperfusion injury.
Circulation. 121:2012–2022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Halestrap AP, Clarke SJ and Khaliulin I:
The role of mitochondria in protection of the heart by
preconditioning. Biochim Biophys Acta. 1767:1007–1031. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li Q, Zhou LY, Gao GF, Jiao JQ and Li PF:
Mitochondrial network in the heart. Protein Cell. 3:410–418. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Crow MT, Mani K, Nam YJ and Kitsis RN: The
mitochondrial death pathway and cardiac myocyte apoptosis. Circ
Res. 95:957–970. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Garlid KD, Costa AD, Quinlan CL, Pierre SV
and Dos Santos P: Cardioprotective signaling to mitochondria. J Mol
Cell Cardiol. 46:858–866. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Perrelli MG, Pagliaro P and Penna C:
Ischemia/reperfusion injury and cardioprotective mechanisms: Role
of mitochondria and reactive oxygen species. World J Cardiol.
3:186–200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhao T, Parikh P, Bhashyam S, Bolukoglu H,
Poornima I, Shen YT and Shannon RP: Direct effects of glucagon-like
peptide-1 on myocardial contractility and glucose uptake in normal
and postischemic isolated rat hearts. J Pharmacol Exp Ther.
317:1106–1113. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nikolaidis LA, Elahi D, Hentosz T,
Doverspike A, Huerbin R, Zourelias L, Stolarski C, Shen YT and
Shannon RP: Recombinant glucagon-like peptide-1 increases
myocardial glucose uptake and improves left ventricular performance
in conscious dogs with pacing-induced dilated cardiomyopathy.
Circulation. 110:955–961. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Luque MA, González N, Márquez L, Acitores
A, Redondo A, Morales M, Valverde I and Villanueva-Peñacarrillo ML:
Glucagon-like peptide-1 (GLP-1) and glucose metabolism in human
myocytes. J Endocrinol. 173:465–473. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bao W, Aravindhan K, Alsaid H, Chendrimada
T, Szapacs M, Citerone DR, Harpel MR, Willette RN, Lepore JJ and
Jucker BM: Albiglutide, a long lasting glucagon-like peptide-1
analog, protects the rat heart against ischemia/reperfusion injury:
Evidence for improving cardiac metabolic efficiency. PLoS One.
6:e235702011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Timmers L, Henriques JP, de Kleijn DP,
Devries JH, Kemperman H, Steendijk P, Verlaan CW, Kerver M, Piek
JJ, Doevendans PA, et al: Exenatide reduces infarct size and
improves cardiac function in a porcine model of ischemia and
reperfusion injury. J Am Coll Cardiol. 53:501–510. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ban K, Noyan-Ashraf MH, Hoefer J, Bolz SS,
Drucker DJ and Husain M: Cardioprotective and vasodilatory actions
of glucagon-like peptide 1 receptor are mediated through both
glucagon-like peptide 1 receptor-dependent and -independent
pathways. Circulation. 117:2340–2350. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Depre C, Ponchaut S, Deprez J, Maisin L
and Hue L: Cyclic AMP suppresses the inhibition of glycolysis by
alternative oxidizable substrates in the heart. J Clin Invest.
101:390–397. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
De Rasmo D, Gattoni G, Papa F, Santeramo
A, Pacelli C, Cocco T, Micelli L, Sardaro N, Larizza M, Scivetti M,
et al: The β-adrenoceptor agonist isoproterenol promotes the
activity of respiratory chain complex I and lowers cellular
reactive oxygen species in fibroblasts and heart myoblasts. Eur J
Pharmacol. 652:15–22. 2011. View Article : Google Scholar
|
|
41
|
Acin-Perez R, Salazar E, Kamenetsky M,
Buck J, Levin LR and Manfredi G: Cyclic AMP produced inside
mitochondria regulates oxidative phosphorylation. Cell Metab.
9:265–276. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Valsecchi F, Ramos-Espiritu LS, Buck J,
Levin LR and Manfredi G: cAMP and mitochondria. Physiology
(Bethesda). 28:199–209. 2013.
|
|
43
|
Wang D, Luo P, Wang Y, Li W, Wang C, Sun
D, Zhang R, Su T, Ma X, Zeng C, et al: Glucagon-like peptide-1
protects against cardiac microvascular injury in diabetes via a
cAMP/PKA/Rho-dependent mechanism. Diabetes. 62:1697–1708. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Xiao YF, Nikolskaya A, Jaye DA and Sigg
DC: Glucagon-like peptide-1 enhances cardiac L-type Ca2+
currents via activation of the cAMP-dependent protein kinase A
pathway. Cardiovasc Diabetol. 10:62011. View Article : Google Scholar
|
|
45
|
Bose AK, Mocanu MM, Carr RD and Yellon DM:
Glucagon like peptide-1 is protective against myocardial
ischemia/reperfusion injury when given either as a preconditioning
mimetic or at reperfusion in an isolated rat heart model.
Cardiovasc Drugs Ther. 19:9–11. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Bose AK, Mocanu MM, Carr RD, Brand CL and
Yellon DM: Glucagon-like peptide 1 can directly protect the heart
against ischemia/reperfusion injury. Diabetes. 54:146–151. 2005.
View Article : Google Scholar
|
|
47
|
Bose AK, Mocanu MM, Carr RD and Yellon DM:
Myocardial ischaemia-reperfusion injury is attenuated by intact
glucagon like peptide-1 (GLP-1) in the in vitro rat heart and may
involve the p70s6K pathway. Cardiovasc Drugs Ther. 21:253–256.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Krauss S, Zhang CY and Lowell BB: The
mitochondrial uncoupling-protein homologues. Nat Rev Mol Cell Biol.
6:248–261. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Teshima Y, Akao M, Jones SP and Marbán E:
Uncoupling protein-2 overexpression inhibits mitochondrial death
pathway in cardiomyocytes. Circ Res. 93:192–200. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Bienengraeber M, Ozcan C and Terzic A:
Stable transfection of UCP1 confers resistance to
hypoxia/reoxygenation in a heart-derived cell line. J Mol Cell
Cardiol. 35:861–865. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Scarpulla RC: Nuclear control of
respiratory gene expression in mammalian cells. J Cell Biochem.
97:673–683. 2006. View Article : Google Scholar
|
|
52
|
Scarpulla RC: Nuclear control of
respiratory chain expression by nuclear respiratory factors and
PGC-1-related coactivator. Ann NY Acad Sci. 1147:321–334. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Javadov S, Purdham DM, Zeidan A and
Karmazyn M: NHE-1 inhibition improves cardiac mitochondrial
function through regulation of mitochondrial biogenesis during
postinfarction remodeling. Am J Physiol Heart Circ Physiol.
291:H1722–H1730. 2006. View Article : Google Scholar : PubMed/NCBI
|