Introduction
Diabetes severely affects human health, and
epidemiological studies have reported that the number of diabetic
patients is expected to reach 592 million worldwide by 2035
(1). Diabetes is tightly
associated with both microvascular (including neuropathy,
nephropathy and retinopathy) and macrovascular (including
cardiovascular diseases) complications (2–6).
As a common complication of diabetes, diabetic cardiomyopathy (DCM)
represents the main cause of morbidity and mortality among diabetic
patients (7). DCM is generally
considered to be manifested by a series of structural and
functional anomalies in the myocardium of diabetic patients,
including myocardial fibrosis, impaired diastolic and systolic
contractility, cardiomyocyte hypertrophy, cardiac autonomic
neuropathy and apoptosis (8–11).
Hyperglycaemia is the key element of diabetes, and plays a crucial
role in the evolution of DCM (11,12). Accumulating reports have revealed
that multifarious factors may contribute to hyperglycaemia-induced
myocardial damage, including reactive oxygen species (ROS)
generation (13–18), insufficiency of antioxidant
systems (16–21) and mitochondrial dysfunction
(13,21,22). Cardiac inflammatory reactions,
characterized by increased levels of pro-inflammatory cytokines,
may also play an important part in the manifestation of DCM
(23–25). However, the pathogenesis of
hyperglycaemia-induced cardiomyocyte injury has not been fully
elucidated.
The phosphoinositide 3-kinase and protein kinase B
(PI3K/Akt) signaling pathway plays a key role in the conditioning
of cell proliferation and survival (26). It has been reported that the
evolution of DCM is interlinked with Akt pathway deactivation
(27,28). In the myocardium of diabetic rats,
Akt phosphorylation may be inhibited by increased circulating free
fatty acids and inflammatory cytokines (29). However, in diabetic mice, cardiac
systolic function and cardiomyocyte proliferation may be improved
via benfotiamine-induced activation of the Akt pathway (27). Activation of the PI3K/Akt pathway
may protect cardiomyocytes against hyperglycaemia-triggered
oxidative stress as well as inflammation, along with an increase in
cell viability (29,30). Jadhav et al (31) also reported that increased
expression of the PI3K/Akt signaling pathway may lead to reduction
of pro-inflammatory cytokines and account for enhanced glucose
metabolism and amelioration of cardiac injury in DCM. Accordingly,
it is reasonable to hypothesize that the molecules that activate
PI3K/Akt signaling may exert cardioprotective effects against
hyperglycaemia-induced cardiomyocyte injury.
Angiotensin-(1-7) [Ang-(1-7)] is a heptapeptide,
mainly generated by cleavage of AngI and AngII by the
angiotensin-converting enzyme (ACE) 2 (32–34), that possesses cardioprotective
properties against myocardial hypertrophy, pathological cardiac
remodeling, fibrosis and inflammation (35–40). Ang-(1-7) has been found to
activate the PI3K/Akt pathway in cardiomyocytes (41–43); thus, it has been hypothesized that
Ang-(1-7) exerts protective effects on the myocardium against
diabetes, due to its range of therapeutic properties. The aim of
the present study was to investigate the cytoprotective effect of
Ang-(1-7) on H9c2 cardiomyoblasts against hyperglycaemia and its
effects on the PI3K/Akt signaling pathway, which is involved in
anti-inflammation and cell survival.
Materials and methods
Materials
Ang-(1-7) and D-Ala7-Ang-(1-7) (A-779) were
purchased from Sigma Chemicals Co. (St. Louis, MO, USA), and stored
at −20°C. 2′,7′-Dichlorodihydrofluorescein diacetate (DCFH-DA),
fetal bovine serum (FBS) and Dulbecco’s modified Eagle’s medium
(DMEM)-F12 were purchased from Gibco-BRL (Thermo Fisher Scientific;
Grand Island, NY, USA). Hoechst 332585, rhodamine 123 (Rh123) and
LY294002 (an inhibitor of PI3K/Akt) were obtained from
Sigma-Aldrich (Merck KGaA; St. Louis, MO, USA). The Cell Counting
Kit-8 (CCK-8) was supplied by Dojindo Laboratories (Kumamoto,
Japan). Anti-phospho-PI3K rabbit mAb (cat. no. 4228),
anti-total-PI3K rabbit mAb (cat. no. 4292), anti-phospho-Akt rabbit
mAb (cat. no. 12178), anti-total-Akt rabbit mAb (cat. no. 14702),
anti-cleaved caspase-1 rabbit mAb (cat. no. 2225), anti-cleaved
caspase-3 rabbit mAb (cat. no. 9662) and anti-cleaved caspase-12
rabbit mAb (cat. no. 2202) were supplied by Cell Signaling
Technology, Inc. (Boston, MA, USA), horseradish peroxidase
(HRP)-conjugated secondary antibody (cat. no. KC5G5) and
bicinchoninic acid (BCA) protein assay kit were obtained from
Kangchen Biotech, Inc. (Shanghai, China). Enhanced
chemiluminescence (ECL) solution was purchased from Nanjing KeyGen
Biotech Co., Ltd. (Nanjing, China). Interleukin (IL)-1β, -6 and
tumor necrosis factor (TNF)-α enzyme-linked immunosorbent assay
(ELISA) kits were provided by Abcam (Cambridge, UK).
Cell culture and treatments
H9c2 cells, a rat cardiac myoblast cell line, were
supplied by Sun Yat-Sen University Experimental Animal Center
(Guangzhou, China). H9c2 cardiomyoblasts were cultured in DMEM-F12
supplemented with 10% FBS under an atmosphere of 5% CO2
and at 37°C with 95% air. H9c2 cardiomyoblasts were treated with 35
mmol/l (mM) glucose (high glucose, HG) in the presence or absence
of 1 μmol/l (μM) Ang-(1-7) for 24 h. To further
ascertain whether the protective effect of Ang-(1-7) and the
activation of the PI3K/Akt pathway were induced by Ang-(1-7), H9c2
cardiomyoblasts were co-treated with 1 μM Ang-(1-7) and 35
mM glucose in the presence of 1 μM A-779 or 10 μM
LY294002 for 24 h.
Western blot analysis
After the indicated treatments, H9c2 cardiomyoblasts
were harvested and lysed with cell lysis solution at 4°C for 30 min
and total protein was quantified using the BCA protein assay kit.
Loading buffer was added to cytosolic extracts, followed by boiling
for 5 min; the same amount of supernatant from each sample was
fractionated by 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis, and the total proteins were transferred onto
polyvinylidene difluoride membranes. The membranes were blocked
with 5% fat-free milk for 60 min in fresh blocking buffer [0.1%
Tween-20 in Tris-buffered saline (TBS-T)] at room temperature, and
incubated with either anti-phospho-PI3K (1:1,000 dilution),
anti-total-PI3K (1:1,000 dilution), anti-phospho-Akt (1:1,000
dilution), anti-total-Akt (1:1,000 dilution), anti-cleaved
caspase-1 (1:1,000 dilution), anti-cleaved caspase-3 (1:1,000
dilution), or anti-cleaved caspase-12 (1:1,000 dilution) in freshly
prepared TBS-T with 3% free-fat milk overnight with gentle
agitation at 4°C. The membranes were washed for 15 min with TBS-T
and incubated with HRP-conjugated goat anti-rabbit secondary
antibody (1:3,000 dilution; Kangchen Biotech, Inc., Shanghai,
China) in TBS-T with 3% fat-free milk for 1.5 h at room
temperature. The membranes were then washed 3 times with TBS-T for
15 min. The immunoreactive signals were visualized using ECL
detection. In order to quantify protein expression, the X-ray films
were scanned and analyzed with ImageJ 1.47i software. The
experiment was performed 3 times.
Measurement of cell viability
H9c2 cardiomyoblasts were seeded in 96-well plates
at a density of 1×104/ml, incubated at 37°C, and the
CCK-8 assay was employed to assess cell viability. After the
indicated treatments, 10 μl CCK-8 solution (1/10 dilution)
was added to each well, and the plate was then incubated for 2 h in
the incubator. Absorbance at 450 nm was assayed using a microplate
reader (Molecular Devices, Sunnyvale, CA, USA) as previously
described (44). The means of the
optical density (OD) of 3 wells in the indicated groups were used
to calculate the percentage of cell viability as follows: Cell
viability (%) = (ODtreatment group/ODcontrol
group) × 100%. The experiment was performed 3 times.
Hoechst 33258 nuclear staining for
apoptosis assessment
Apoptotic cell death was tested using Hoechst 33258
staining followed by photofluorography. First, H9c2 cells were
plated in 35-mm dishes at a density of 1×106 cells/well.
After the above-mentioned indicated treatments, the H9c2 cells were
fixed with 4% paraformaldehyde in 0.1 mol/l phosphate-buffered
saline (PBS; pH 7.4) for 10 min at 4°C, and the slides were then
washed 5 times with PBS, followed by 5 mg/ml Hoechst 33258 for 10
min and washing 5 times with PBS. Finally, the cells were
visualized under a fluorescence microscope (BX50-FLA; Olympus,
Tokyo, Japan). Viable H9c2 cells displayed a uniform blue
fluorescence throughout the nucleus and normal nuclear size,
whereas apoptotic H9c2 cells exhibited condensed, distorted or
fractured nuclei. The experiment was performed 3 times.
Measurement of mitochondrial membrane
potential (MMP)
The MMP (ΔΨm) was tested using a fluorescent dye,
Rh123, a cell-permeable cationic dye that preferentially enters
mitochondria due to the highly negative MMP. Depolarization of the
membrane results in loss of MMP from the mitochondria and a
decrease in green and red fluorescence. H9c2 cells were cultured in
a slide with EMEM-F12. After the abovementioned treatments, the
slides were washed 3 times with PBS. The cells were incubated with
1 mg/l Rh123 at 37°C for 45 min in the incubator, washed briefly
with PBS 3 times and air-dried. Fluorescence was measured over the
entire field of vision using a fluorescence microscope connected to
an imaging system (BX50-FLA, Olympus). The mean fluorescence
intensity (MFI) of Rh123 from 5 random fields was analyzed using
the ImageJ 1.47i software; MFI was considered as an index of the
levels of MMP. The experiment was performed 3 times.
Examination of intracellular ROS
generation
Intracellular ROS generation was determined based on
the oxidative conversion of cell-permeable oxidation of DCFH-DA to
fluorescent DCF. H9c2 cardiomyoblasts were cultured in a slide with
EMEM-F12 medium. After the abovementioned treatments, the slides
were washed twice with PBS. DCFH-DA (10 μmol/l) solution in
serum-free medium was added to the slides, and the cells were then
incubated at 37°C for a further 30 min in the incubator. The slides
were washed 5 times with PBS, and DCF fluorescence was measured
over the entire field of vision using a fluorescence microscope
connected to an imaging system (BX50-FLA, Olympus). The MFI from 5
random fields was measured using ImageJ 1.47i software and the MFI
was used as an index of the amount of ROS. The experiment was
performed 3 times.
ELISA
H9c2 cells were cultured in 96-well plates. After
the indicated treatments, the medium was collected and used for
ELISA. IL-1β, -6 and TNF-α assays were performed according to the
manufacturer’s instructions with the respective ELISA kits. The
experiment was performed 3 times.
Statistical analysis
All data are presented as the mean ± standard error
of the mean. Differences between groups were analyzed with one-way
analysis of variance using SPSS 13.0 software (SPSS, Inc., Chicago,
IL, USA), followed by the LSD post-hoc comparison test. Statistical
significance was set at P<0.05.
Results
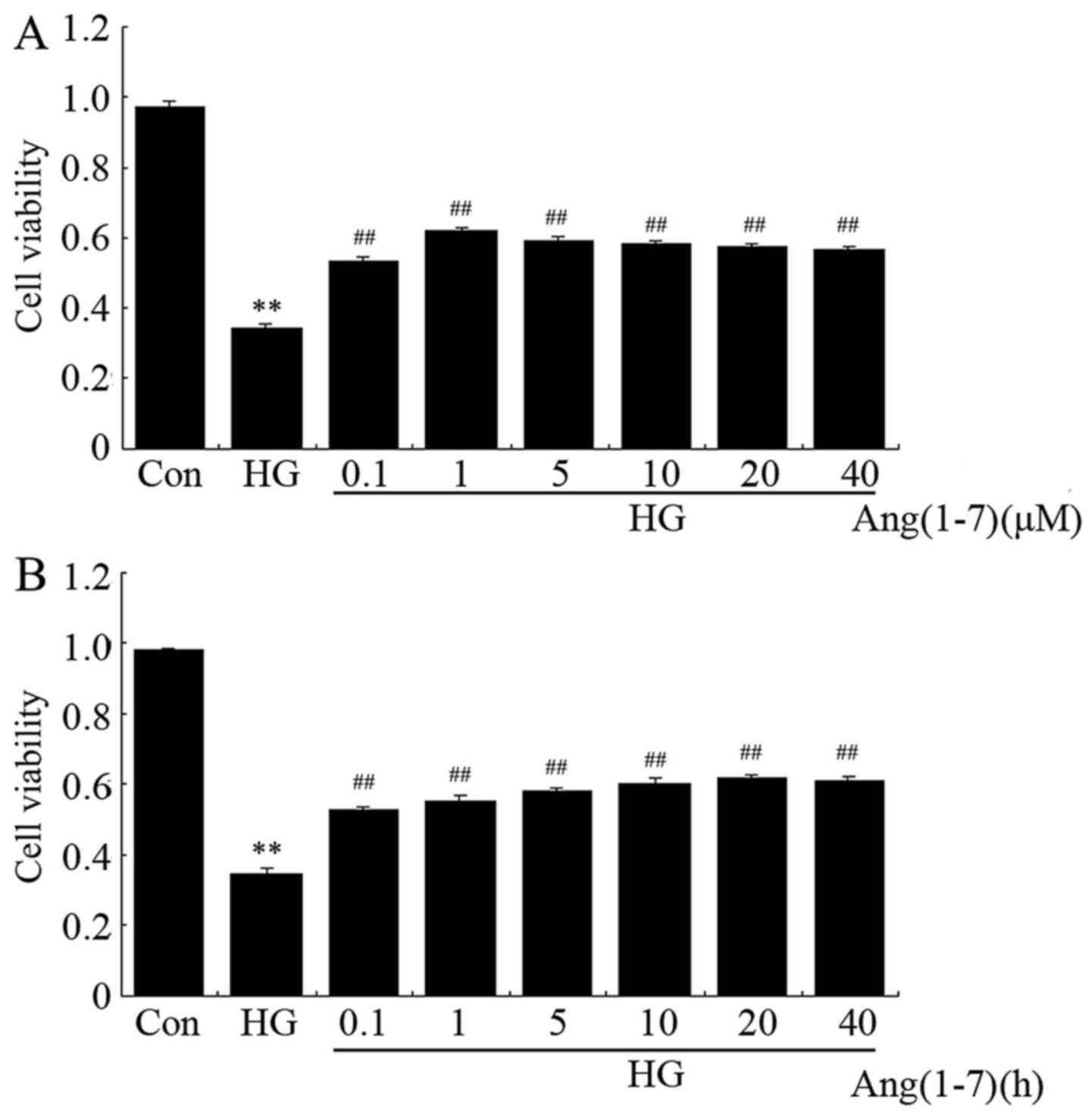
Ang-(1-7) attenuates HG-induced decrease
in cell viability in H9c2 cardiomyoblast cells
To evaluate whether Ang-(1-7) protects H9c2
cardiomyoblasts against HG (35 mM), a dose-response study with
varying doses of Ang-(1-7) (0.1, 1, 5, 10, 20 and 40 μM) was
performed to calculate the effective cytoprotective dose of
Ang-(1-7). The data shown in Fig.
1A indicate that exposure of H9c2 cells to 35 mM glucose for 24
h was markedly cytotoxic, decreasing cell viability to 34.7%
(P<0.01) compared with the non-treated group. However, the
cytotoxic effect of HG on H9c2 cells was notably inhibited by
treatment with Ang-(1-7) at the indicated concentrations for 24 h.
The maximum inhibitory effect was observed with 1 μM
Ang-(1-7). Ang-(1-7) (1 μM) alone did not obviously alter
the viability of H9c2 cells. Therefore, 1 μM Ang-(1-7) was
used in the subsequent time-response study with different
pretreatment times (1, 3, 6, 12, 24 and 48 h). As shown in Fig. 1B, co-treatment of H9c2 cells with
1 μM Ang-(1-7) and 35 mM glucose for the indicated times all
markedly reduced HG-induced cytotoxicity, achieving the maximal
inhibitory ability at 24 h. Based on the abovementioned results,
H9c2 cardiomyoblasts were co-treated with 1 μM Ang-(1-7) and
35 mM glucose for 24 h in all the subsequent experiments.
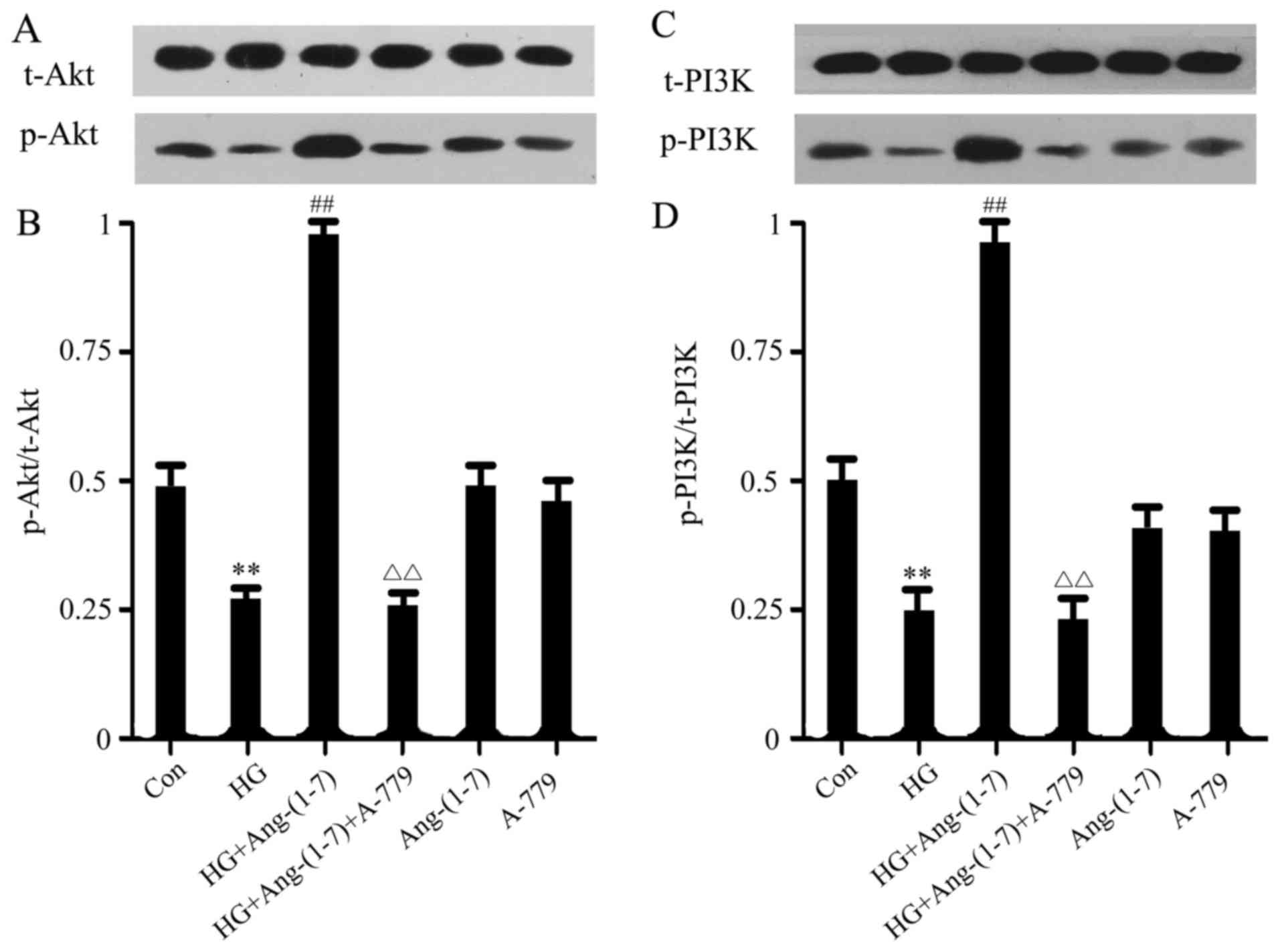
Ang-(1-7) alleviates HG-induced
dephosphorylation of PI3K/Akt in H9c2 cardiomyoblasts
To investigate the potential mechanism underlying
the cytoprotective effect of Ang-(1-7) on H9c2 cells, PI3K/Akt
activation was subsequently examined. As shown in Fig. 2, PI3K/Akt phosphorylation was
suppressed by HG treatment compared with the control group, but
this effect was abolished when the H9c2 cells were co-treated with
Ang-(1-7) and HG. Moreover, the function of Ang-(1-7) in restoring
PI3K/Akt phosphorylation may be abolished by the presence of 1
μM A-779 (an antagonist of the Mas receptor). Treatment with
either Ang-(1-7) or A-779 alone did not affect PI3K/Akt
phosphorylation.
Activation of the PI3K/Akt pathway
contributes to the cytoprotective effect of Ang-(1-7) against the
HG-induced decline in H9c2 cell viability
To determine whether the increase in PI3K/Akt
phosphorylation by Ang-(1-7) contributes to the cardioprotective
effect of Ang-(1-7) against HG-induced cytotoxicity, H9c2
cardiomyoblasts were co-conditioned with 1 μM Ang-(1-7) and
35 mM glucose in the presence of 10 μM LY294002 (a selective
inhibitor of PI3K/Akt). As shown in Fig. 3, co-treatment with HG and
Ang-(1-7) blunted the cytotoxic effect and increased cell
viability, but the presence of A-779 eliminated the cytoprotective
effect of Ang-(1-7). Of note, treatment with LY294002 eliminated
the protective effect of Ang-(1-7) in H9c2 cardiomyoblasts against
HG-induced decreased cell viability. However, treatment with
Ang-(1-7) or A-779 or LY294002 alone did not decrease H9c2 cell
viability. These findings indicate that Ang-(1-7) protects H9c2
cardiomyoblasts against HG-induced cytotoxicity, at least partially
via PI3K/Akt pathway activation.
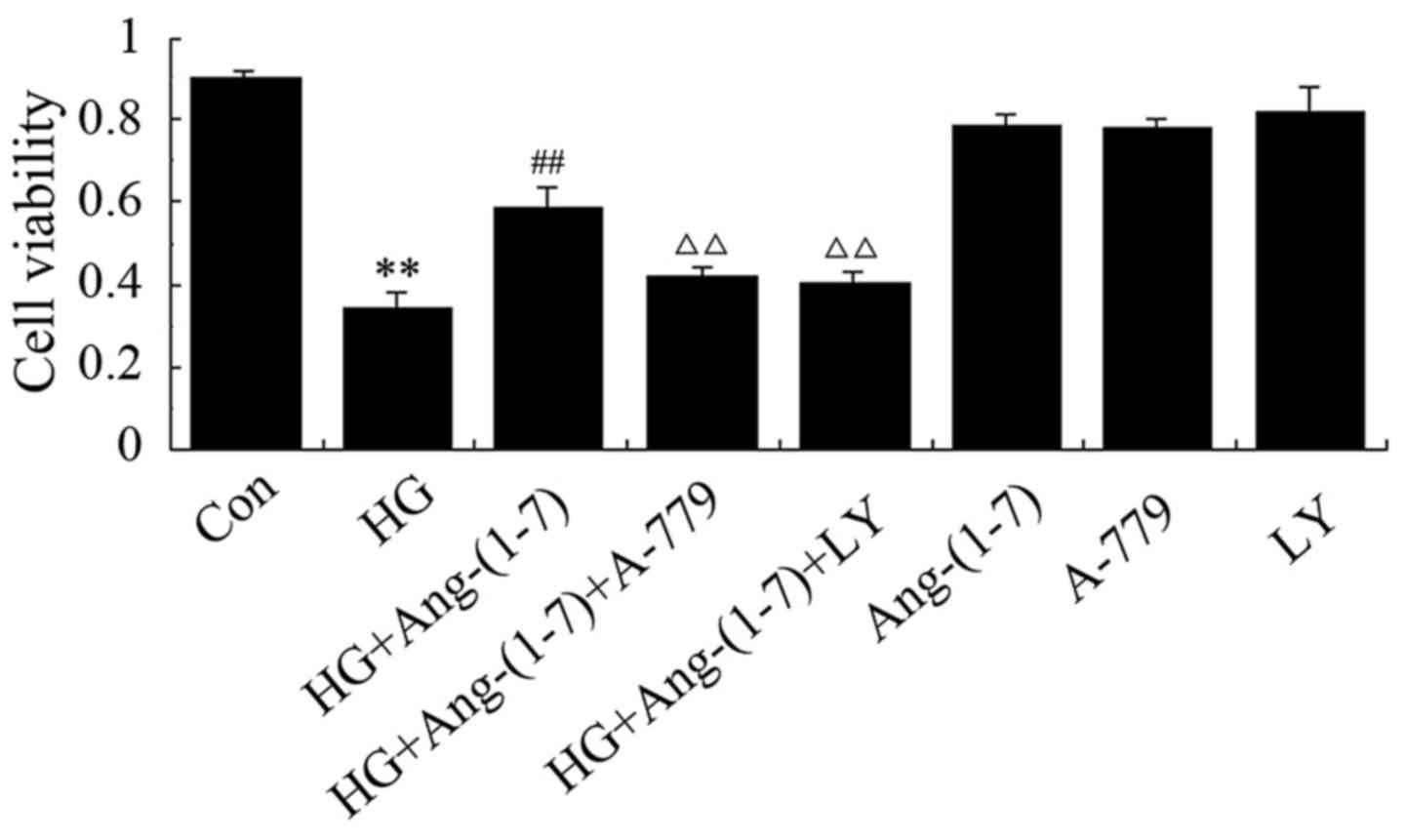 | Figure 3Activation of phosphoinositide
3-kinase and protein kinase B (PI3K/Akt) participates in the
protective effect of Ang-(1-7) against HG-induced decreased
viability of H9c2 cells. H9c2 cells were treated for 24 h with 35
mM glucose alone, or co-treated with 1 μM Ang-(1-7) and 35
mM glucose, or co-treated with 35 mM glucose, 1 μM Ang-(1-7)
and 1 μM A-779, or co-treated with 35 mM glucose, 1
μM Ang-(1-7) and 10 μM LY294002, or treated with 1
μM Ang-(1-7) alone, or 1 μM A-779 alone, or 10
μM LY294002 alone. Cell viability was detected using the
Cell Counting Kit-8. Data are presented as mean ± standard error of
the mean (n=3). **P<0.01 compared with the control
group; ##P<0.01 compared with HG-treated group;
ΔΔP<0.01 vs. the group co-treated with Ang-(1-7) and
HG. Ang-(1-7), angiotensin-(1-7); HG, high glucose (35 mM); LY,
LY294002. |
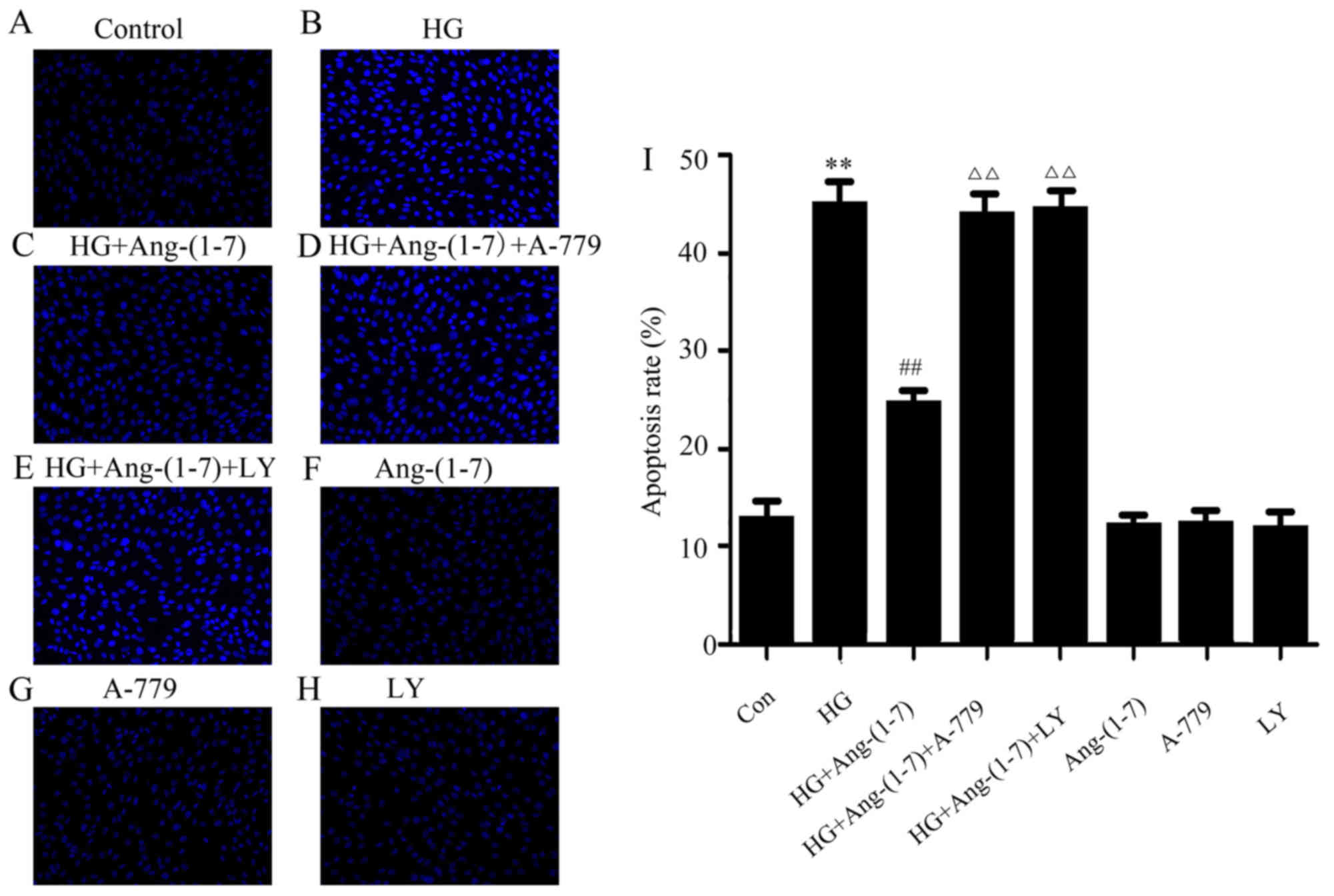
Activation of the PI3K/Akt pathway
promotes the cytoprotective effect of Ang-(1-7) against HG-induced
apoptosis in H9c2 cardiomyoblasts
An increasing number of studies have proposed that
HG leads to inceased apoptosis in myocardial injury. Thus, the
effect of Ang-(1-7) on HG-induced cell apoptosis was observed in
H9c2 cardiomyoblasts. It was demonstrated that treatment of H9c2
cells with 35 mM glucose for 24 h significantly increased apoptosis
(Fig. 4B). However, the
abovementioned phenomenon may be clearly reversed by co-treatment
with Ang-(1-7) and HG for 24 h (Fig.
4C). It was observed that treating the H9c2 cardiomyoblasts
with 35 mM glucose and 1 μM Ang-(1-7) in the presence of
A-779 for 24 h did not significantly reduce apoptosis (Fig. 4D). Of note, apoptosis was
increased in H9c2 cardiomyoblasts by co-treatment with 1 μM
Ang-(1-7) and 35 mM glucose in the presence of 10 μM
LY294002 (Fig. 4E). Ang-(1-7),
A-779 or LY294002 alone did not exert any effect on myocardial
apoptosis (Fig. 4F–H). These
findings indicated that the PI3K/Akt pathway may participate in the
anti-apoptotic function of Ang-(1-7) in HG-exposed H9c2
cardiomyoblast cells.
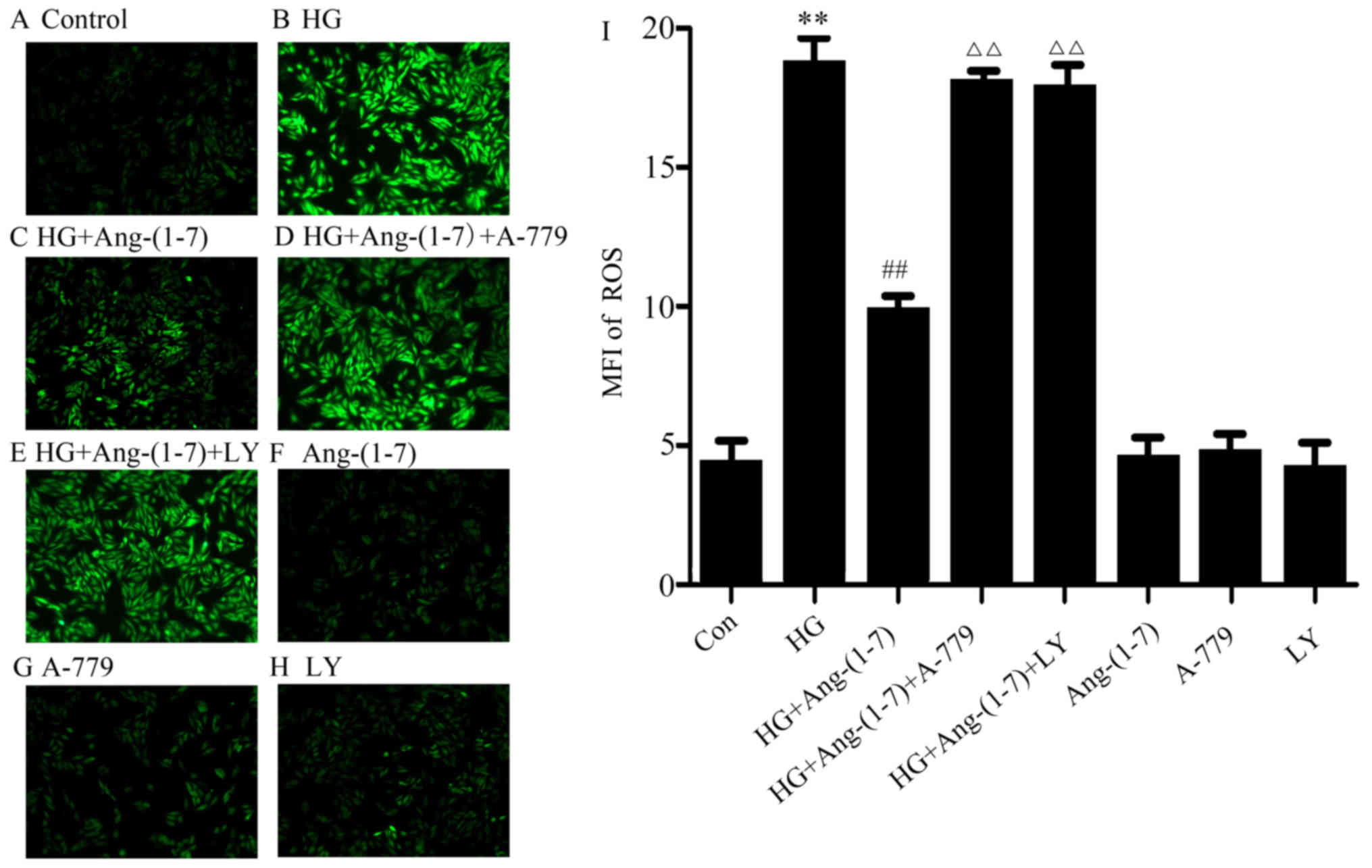
Activation of the PI3K/Akt pathway is
associated with the cytoprotection of Ang-(1-7) against HG-induced
ROS production in H9c2 cardiomyoblast cells
As shown in Fig.
5B, exposure to 35 mM glucose for 24 h induced an increase in
the generation of ROS in H9c2 cardiomyoblast cells, and the
increased ROS production was suppressed by the presence of
Ang-(1-7) (Fig. 5C). Furthermore,
co-treatment with A-779, Ang-(1-7) and HG diminished the
aforementioned effect of Ang-(1-7), further indicating the
cardioprotective function of Ang-(1-7) against HG-induced ROS
production (Fig. 5D). However,
ROS production increased when H9c2 cardiomyoblasts were co-treated
with 1 μM Ang-(1-7) and 35 mM glucose in the presence of 10
μM LY294002 (Fig. 5E). Our
study demonstrated that Ang-(1-7), A-779 or LY294002 alone did not
affect ROS production in H9c2 cells (Fig. 5F–H). These results revealed that
PI3K/Akt pathway activation is involved in the cytoprotective
function of Ang-(1-7) against HG-triggered ROS overproduction in
H9c2 cardiomyoblasts.
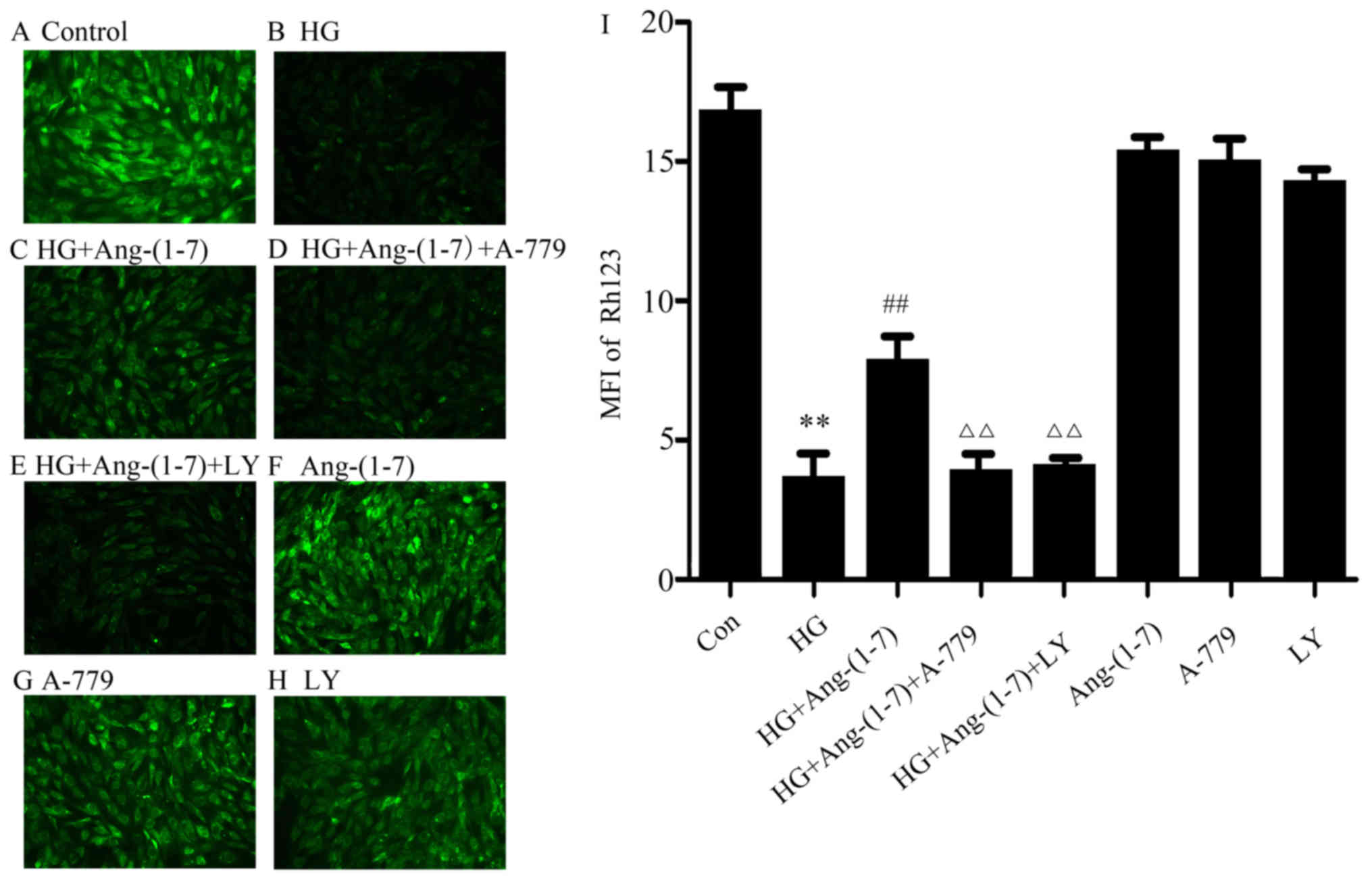
Activation of the PI3K/Akt pathway
facilitates the cytoprotective function of Ang-(1-7) against
HG-induced loss of MMP in H9c2 cardiomyoblasts
The cardioprotective effect of Ang-(1-7) on
HG-induced loss of MMP was further examined in H9c2 cardiomyoblast
cells. As shown in Fig. 6B,
treatment of H9c2 cells with 35 mM glucose for 24 h diminished MMP,
while MMP was elevated by co-treatment with 1 μM Ang-(1-7)
and 35 mM glucose (Fig. 6C). Of
note, exposure to 35 mM glucose in the presence of Ang-(1-7) and
A-779 still resulted in loss of MMP (Fig. 6D). Importantly, co-treatment of
H9c2 cells with 10 μM LY294002, 1 μM Ang-(1-7) and 35
mM glucose did not attenuate the loss of MMP caused by HG (Fig. 6E). These findings demonstrated
that the cytoprotective effect of Ang-(1-7) on the HG-induced loss
of MMP in H9c2 cardiomyoblasts was mediated in part by PI3K/Akt
pathway activation. Ang-(1-7), A-779 or LY294002 alone did not have
any effect on MMP in H9c2 cardiomyoblasts (Fig. 6F–H).
Ang-(1-7) decreases HG-induced
inflammation in H9c2 cells, while inhibitors of PI3K/Akt reverse
the effect of Ang-(1-7)
In the present study, it was examined by ELISA
whether exposure to HG in H9c2 cardiomyoblasts triggers
inflammatory responses and the role of Ang-(1-7) in this process.
It was demonstrated that cardiac expression of IL-1β, -6 and TNF-α
increased following exposure to 35 mM glucose for 24 h, while
co-treatment with 1 μM Ang-(1-7) and 35 mM glucose
significantly lowered the level of these inflammatory cytokines. By
contrast, exposure of H9c2 cardiomyoblasts to 35 mM glucose in the
presence of both Ang-(1-7) and A-779 increased the expression of
these inflammatory mediators. Of note, inflammatory reactions were
suppressed by co-treatment of H9c2 cells with 10 μM
LY294002, 1 μM Ang-(1-7) and 35 mM glucose, whereas
treatment with Ang-(1-7), A-779 or LY294002 alone did not affect
the inflammatory responses in H9c2 cardiomyoblasts (Fig. 7).
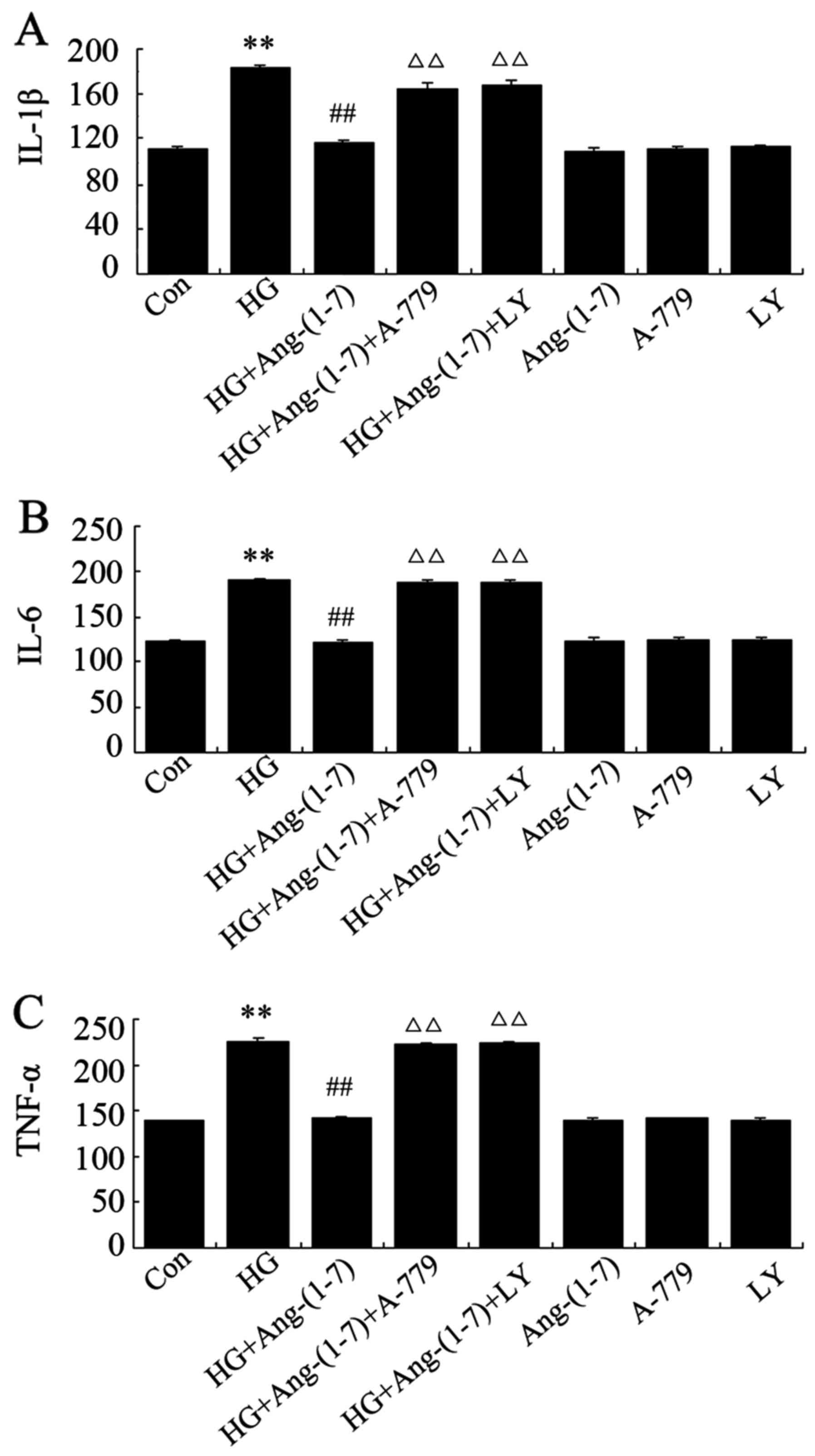 | Figure 7Ang-(1-7) protects against
HG-triggered inflammation in H9c2 cells. H9c2 cells were treated
for 24 h with 35 mM glucose alone, or co-treated with 1 μM
Ang-(1-7) and 35 mM glucose, or co-treated with 35 mM glucose, 1
μM Ang-(1-7) and 1 μM A-779, or co-treated with 35 mM
glucose, 1 μM Ang-(1-7) and 10 μM LY294002, or
treated with 1 μM Ang-(1-7) alone, 1 μM A-779 alone,
or 10 μM LY294002 alone. After the indicated treatments, the
release of (A) IL-1β, (B) IL-6 and (C) TNF-α was assessed via
ELISA. Data are presented as mean ± standard error of the mean
(n=3). **P<0.01 compared with the control group;
##P<0.01 compared with the HG-treated group;
ΔΔP<0.01 vs. the group co-treated with Ang-(1-7) and
HG. Ang-(1-7), angiotensin-(1-7); HG, high glucose (35 mM); LY,
LY294002; IL, interleukin; TNF, tumor necrocis factor. |
Ang-(1-7) diminishes the HG-induced
increased expression of cleaved caspase-1, -3 and -12 in H9c2
cells, while PI3K/Akt inhibitors block the action of Ang-(1-7)
In order to further verify the protective effect of
Ang-(1-7) against HG-induced cardiomyoblast apoptosis and
inflammation, the expression level of cleaved caspase-1, -3 and -12
in H9c2 cardiomyoblasts was evaluated by western blot analysis. As
shown in Fig. 8, exposure to 35
mM glucose for 24 h induced a significant increase of cleaved
caspase-1, -3 and -12 expression level. As we hypothesized,
co-treatment with 1 μM Ang-(1-7) and 35 mM glucose markedly
reduced the HG-induced increased expression level of these
proteins. Incubating H9c2 cardio-myoblasts with 35 mM glucose and 1
μM Ang (1-7) in the presence of 1 μM A-779 enhanced
the expression of these proteins; similarly, co-treatment of H9c2
cells with 10 μM LY294002, 1 μM Ang-(1-7) and 35 mM
glucose increased the expression level of these proteins. Finally,
treatment with Ang-(1-7), A-779 or LY294002 alone did not affect
the expression level of these proteins.
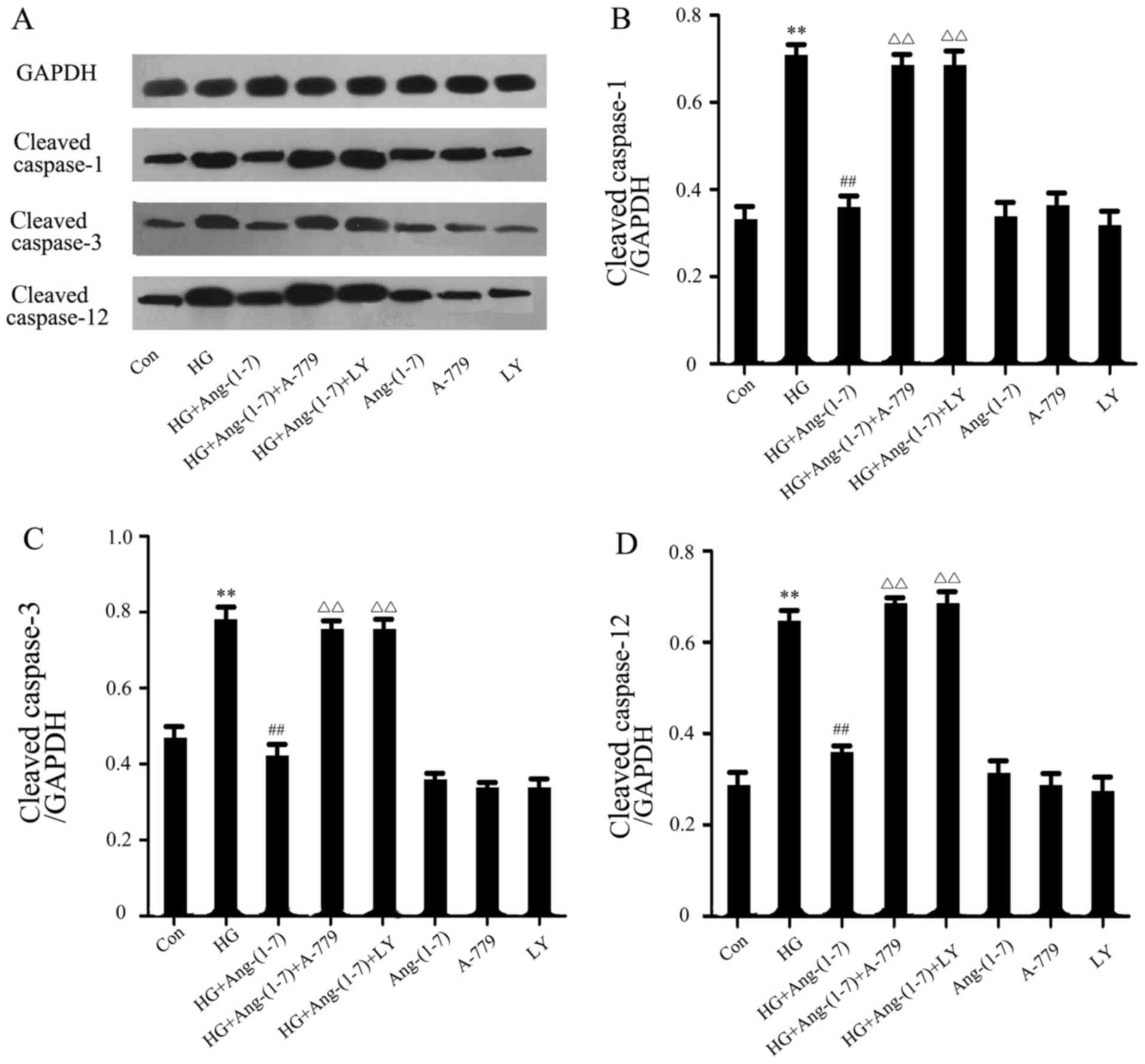 | Figure 8Ang-(1-7) reduced HG-induced
increased expression levels of cleaved caspase-1, -3 and -12 in
H9c2 cells. H9c2 cells were treated for 24 h with 35 mM glucose
alone, or co-treated with 1 μM Ang-(1-7) and 35 mM glucose,
or co-treated with 35 mM glucose, 1 μM Ang-(1-7) and 1
μM A-779, or co-treated with 35 mM glucose, 1 μM
Ang-(1-7) and 10 μM LY294002, or treated with 1 μM
Ang-(1-7) alone, or 1 μM A-779 alone, or 10 μM
LY294002 alone. The expression of cleaved caspase-1, -3 and -12 was
detected by western blot analysis. (A) Changes in the expression
levels of cleaved caspase-1, -3 and -12 in the indicated groups.
(B-D) Densitometric analysis of the results in (A). Data are
presented as mean ± standard error of the mean (n=3).
**P<0.01 compared with the control group;
##P<0.01 compared with the HG-treated group;
ΔΔP<0.01 vs. the group co-treated with Ang-(1-7) and
HG. Ang-(1-7), angiotensin-(1-7); HG, high glucose (35 mM); LY,
LY294002. |
Discussion
Growing evidence indicates that hyperglycaemia plays
a pivotal role in the development of DCM, but the
pathophysiological and molecular mechanisms of
hyperglycaemia-induced cardiomyocyte injury remain unclear. In the
present study, the HG (35 mM glucose)-induced H9c2 cardiomyoblast
injury model was used to investigate the cardioprotective effects
of Ang-(1-7) against HG-induced cardiomyocyte injury and the
underlying mechanisms.
Consistent with previous studies (13–22), our findings verified several
detrimental events induced by HG on H9c2 cardiomyoblasts, such as
cytotoxicity, apoptosis, oxidative damage, mitochondrial
dysfunction and inflammation, as demonstrated by an increase in the
apoptotic cell percentage, ROS generation and inflammatory cytokine
level, as well as a decline in cell viability and mitochondrial
luminosity. In addition, caspase-1, -3 and -12 are known to be
involved in cell apoptosis and inflammatory response (29,30,45–47); thus we investigated the expression
of these proteins and found that HG treatment significantly
increased their levels, further confirming that HG treatment may
trigger apoptosis and inflammation in H9c2 cardiomyoblasts.
An important finding in the present study was the
protective effects of Ang-(1-7) against HG-induced injury of H9c2
cardiomyoblasts. Ang-(1-7), which is formed from AngI and AngII by
the action of ACE2 (32–34), exhibits physiological functions
that are different from those of AngII, including prevention of
myocardial hypertrophy, mitigation of cardiac remodeling,
antifibrotic effect and vasodilatory function (35–39,48–50). It has been reported that Ang-(1-7)
may enable glucose uptake in neonatal cardiomyocytes (51). Additionally, in
streptozotocin-induced diabetic rats, Ang-(1-7) treatment may
suppress right ventricular (RV) fibrosis and ameliorate RV
oxidative stress (52). Taking
into consideration these reports, we further investigated the
protective role of Ang-(1-7) against hyperglycaemia in H9c2
cardiomyoblasts. First, it was observed that Ang-(1-7) clearly
restrained HG-induced cytotoxicity, since co-treatment with
Ang-(1-7) and HG increased cell survival rate compared with the HG
treatment group. These results are consistent with those of
previous studies (36,38,39,52). Second, we investigated the
anti-apoptotic function of Ang-(1-7) in HG-treated H9c2 cells,
which is supported by recent reports that
ischemia̸reperfusion-induced cardiomyocyte apoptosis may be
significantly inhibited by Ang-(1-7) (53). Third, in line with previous
reports (52,53), we observed that Ang-(1-7)
suppresses HG-induced oxidative stress in H9c2 cardiomyoblasts, as
shown by a marked decrease in the generation of ROS. Fourth, the
results of the present study demonstrated that Ang-(1-7) protected
mitochondria against HG-triggered loss of MMP, which was consistent
with the findings of a previous study (54) demonstrating that the Ang-(1-7)
peptidomimetic AVE 0991 exerted protective effects in the kidneys
in ApoE-knockout mice by partially reversing
atherosclerosis-related changes in the mitochondrial proteome.
Fifth, the HG-induced cardiac inflammatory reaction may be blocked
by Ang-(1-7), with lower levels of IL-1β, -6 and TNF-α compared
with the HG group. Similarly, Papinska et al (55) observed that Ang-(1-7) treatment
reduced inflammatory cell infiltration of the heart tissue in a
mouse model of type 2 diabetes. Finally, Ang-(1-7) treatment in
H9c2 cardiomyoblasts decreased cleaved caspase-1, -3 and -12
expression under HG conditions, further verifying the protective
effect of Ang-(1-7) against HG-induced apoptosis and inflammation
in H9c2 cardiomyoblasts. Of note, co-administration of Ang-(1-7)
and A-779 reversed the abovementioned protective effects of
Ang-(1-7), suggesting that HG-related injuries may reappear with
inhibition of Ang-(1-7). The findings of the present study offer
convincing evidence regarding the cardioprotective effects of
Ang-(1-7) against HG-induced injury.
The potential mechanism underlying the
cardioprotective effect of Ang-(1-7) against HG-induced injury was
then investigated. As is known, a group of survival protein
kinases, including PI3K/Akt, constitute a target for
cardioprotection against ischemia/reperfusion injury (56,57). Therefore, in this study the
function of the PI3K/Akt signaling pathway was examined under HG
conditions. Another novel finding of our study was that the
activation of the PI3K/Akt signaling pathway is involved in the
cardioprotective effect of Ang-(1-7) against HG. First, we observed
that HG treatment triggered the dephosphorylation of the PI3K and
Akt proteins in H9c2 cardiomyoblasts, in accordance with previous
findings (27–29). Furthermore, co-treatment with
Ang-(1-7) and HG not only protects H9c2 cells against HG, but also
considerably reverses the HG-induced dephosphorylation of PI3K/Akt
in these cells. Of note, the presence of LY294002, an inhibitor of
PI3K/Akt, markedly inhibited the cardioprotective effect of
Ang-(1-7) against HG-triggered cytotoxicity, cell apoptosis,
oxidative stress, mitochondrial damage and inflammatory reaction.
Therefore, activation of PI3K/Akt signaling by Ang-(1-7) may, at
least in part, be involved in its cardioprotective effect against
HG. Several recent studies reported that PI3K/Akt pathway
activation participates in cardiac cell resistance to apoptosis,
oxidative stress and inflammation, and improves myocardial systolic
function (27–32); those findings were supported by
our results.
Interestingly, the mechanisms of the
cardioprotection of Ang-(1-7) against HG may be multifarious.
Endoplasmic reticulum stress (ERS) is known to play a key role in
the progression of DCM. HG-activated ERS may reduce the myocardial
protein expression of p-PI3K and p-Akt (58), whereas overexpression of p-Akt may
successfully withstand ERS-induced apoptosis and protect the
myocardium against hyperglycaemia-induced dysfunction (59). In addition, ROS-stimulated
mitogen-activated protein kinase (MAPK) pathways, including the p38
MAPK, ERK1/2 and JNK signaling pathways, are involved in HG-induced
injuries, and suppression of these signaling pathways may also
significantly mitigate HG-induced cytotoxicity, apoptosis,
overproduction of ROS and dissipation of MMP (13). Based on these reports, the
cardioprotective function of Ang-(1-7) may be associated with the
regulation of ERS and MAPK pathways. To confirm this hypothesis,
further investigation is required.
In summary, Ang-(1-7) protects H9c2 cardiomyoblasts
against HG-induced cytotoxicity, cell apoptosis, oxidative stress,
mitochondrial damage and inflammation, and PI3K/Akt signaling
pathway activation may play a key role in the protective function
of Ang-(1-7). These conclusions offer a basis for further studies
on the cytoprotective effect of Ang-(1-7) against diabetic
cardiovascular complications, in order to identify novel methods
for the prevention of hyperglycaemia-induced cardiomyocyte
injury.
References
|
1
|
Hu FB, Satija A and Manson JE: Curbing the
diabetes pandemic: the need for global policy solutions. JAMA.
313:2319–2320. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rahman S, Rahman T, Ismail AA and Rashid
AR: Diabetes-associated macrovasculopathy: pathophysiology and
pathogenesis. Diabetes Obes Metab. 9:767–780. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Campos C: Chronic hyperglycemia and
glucose toxicity: pathology and clinical sequelae. Postgrad Med.
124:90–97. 2012. View Article : Google Scholar
|
|
4
|
Tabák AG, Herder C, Rathmann W, Brunner EJ
and Kivimäki M: Prediabetes: a high-risk state for diabetes
development. Lancet. 379:2279–2290. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arora MK and Singh UK: Molecular
mechanisms in the pathogenesis of diabetic nephropathy: an update.
Vascul Pharmacol. 58:259–271. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nguyen DV, Shaw LC and Grant MB:
Inflammation in the pathogenesis of microvascular complications in
diabetes. Front Endocrinol (Lausanne). 3:1702012.
|
|
7
|
Chavali V, Tyagi SC and Mishra PK:
Predictors and prevention of diabetic cardiomyopathy. Diabetes
Metab Syndr Obes. 6:151–160. 2013.PubMed/NCBI
|
|
8
|
Gao X, Xu Y, Xu B, Liu Y, Cai J, Liu HM,
Lei S, Zhong YQ, Irwin MG and Xia Z: Allopurinol attenuates left
ventricular dysfunction in rats with early stages of
streptozotocin-induced diabetes. Diabetes Metab Res Rev.
28:409–417. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Huynh K, Bernardo BC, McMullen JR and
Ritchie RH: Diabetic cardiomyopathy: mechanisms and new treatment
strategies targeting antioxidant signaling pathways. Pharmacol
Ther. 142:375–415. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Goyal BR and Mehta AA: Diabetic
cardiomyopathy: pathophysiological mechanisms and cardiac
dysfuntion. Hum Exp Toxicol. 32:571–590. 2013. View Article : Google Scholar
|
|
11
|
Ren J and Davidoff AJ: Diabetes rapidly
induces contractile dysfunctions in isolated ventricular myocytes.
Am J Physiol. 272:H148–H158. 1997.PubMed/NCBI
|
|
12
|
Tarquini R, Lazzeri C, Pala L, Rotella CM
and Gensini GF: The diabetic cardiomyopathy. Acta Diabetol.
48:173–181. 2011. View Article : Google Scholar
|
|
13
|
Chen J, Guo R, Yan H, Tian L, You Q, Li S,
Huang R and Wu K: Naringin inhibits ROS-activated MAPK pathway in
high glucose-induced injuries in H9c2 cardiac cells. Basic Clin
Pharmacol Toxicol. 114:293–304. 2014. View Article : Google Scholar
|
|
14
|
Kamalakkannan N and Prince PS:
Antihyperglycaemic and antioxidant effect of rutin, a polyphenolic
flavonoid, in streptozotocin-induced diabetic wistar rats. Basic
Clin Pharmacol Toxicol. 98:97–103. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Privratsky JR, Wold LE, Sowers JR, Quinn
MT and Ren J: AT1 blockade prevents glucose-induced cardiac
dysfunction in ventricular myocytes: role of the AT1 receptor and
NADPH oxidase. Hypertension. 42:206–212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Peake BF, Nicholson CK, Lambert JP, Hood
RL, Amin H, Amin S and Calvert JW: Hydrogen sulfide preconditions
the db/db diabetic mouse heart against ischemia-reperfusion injury
by activating Nrf2 signaling in an Erk-dependent manner. Am J
Physiol Heart Circ Physiol. 304:H1215–H1224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murali R, Karthikeyan A and Saravanan R:
Protective effects of D-limonene on lipid peroxidation and
antioxidant enzymes in streptozotocin-induced diabetic rats. Basic
Clin Pharmacol Toxicol. 112:175–181. 2013. View Article : Google Scholar
|
|
18
|
Saandeep K, Vikram A, Tripathi DN, Ramarao
P and Jena G: Influence of hyperglycaemia on chemical-induced
toxicity: study with cyclophosphamide in rat. Basic Clin Pharmacol
Toxicol. 105:236–242. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Maritim AC, Sanders RA and Watkins JB III:
Diabetes, oxidative stress, and antioxidants: a review. J Biochem
Mol Toxicol. 17:24–38. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ashour M, Al-Kattan K, Rafay MA, Saja KF,
Hajjar W and Al-Fraye AR: Current surgical therapy for
bronchiectasis. World J Surg. 23:1096–1104. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ceriello A: Cardiovascular effects of
acute hyperglycaemia: pathophysiological underpinnings. Diab Vasc
Dis Res. 5:260–268. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Boudina S, Sena S, Theobald H, Sheng X,
Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, et al:
Mitochondrial energetics in the heart in obesity-related diabetes:
direct evidence for increased uncoupled respiration and activation
of uncoupling proteins. Diabetes. 56:2457–2466. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Westermann D, Van Linthout S, Dhayat S,
Dhayat N, Schmidt A, Noutsias M, Song XY, Spillmann F, Riad A,
Schultheiss HP, et al: Tumor necrosis factor-alpha antagonism
protects from myocardial inflammation and fibrosis in experimental
diabetic cardiomyopathy. Basic Res Cardiol. 102:500–507. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Di Filippo C, Marfella R, Cuzzocrea S,
Piegari E, Petronella P, Giugliano D, Rossi F and D’Amico M:
Hyperglycemia in strep-tozotocin-induced diabetic rat increases
infarct size associated with low levels of myocardial HO-1 during
ischemia/reperfusion. Diabetes. 54:803–810. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Venkatachalam K, Mummidi S, Cortez DM,
Prabhu SD, Valente AJ and Chandrasekar B: Resveratrol inhibits high
glucose-induced PI3K/Akt/ERK-dependent interleukin-17 expression in
primary mouse cardiac fibroblasts. Am J Physiol Heart Circ Physiol.
294:H2078–H2087. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Manukyan MC, Weil BR, Wang Y, Abarbanell
AM, Herrmann JL, Poynter JA and Meldrum DR: The phosphoinositide-3
kinase survival signaling mechanism in sepsis. Shock. 34:442–449.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Katare RG, Caporali A, Oikawa A, Meloni M,
Emanueli C and Madeddu P: Vitamin B1 analog benfotiamine prevents
diabetes-induced diastolic dysfunction and heart failure through
Akt/Pim-1-mediated survival pathway. Circ Heart Fail. 3:294–305.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sun D, Shen M, Li J, Li W, Zhang Y, Zhao
L, Zhang Z, Yuan Y, Wang H and Cao F: Cardioprotective effects of
tanshinone IIA pretreatment via kinin B2 receptor-Akt-GSK-3β
dependent pathway in experimental diabetic cardiomyopathy.
Cardiovasc Diabetol. 10:42011. View Article : Google Scholar
|
|
29
|
Yu W, Wu J, Cai F, Xiang J, Zha W, Fan D,
Guo S, Ming Z and Liu C: Curcumin alleviates diabetic
cardiomyopathy in experimental diabetic rats. PLoS One.
7:e520132012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Tsai CY, Wang CC, Lai TY, Tsu HN, Wang CH,
Liang HY and Kuo WW: Antioxidant effects of diallyl trisulfide on
high glucose-induced apoptosis are mediated by the
PI3K/Akt-dependent activation of Nrf2 in cardiomyocytes. Int J
Cardiol. 168:1286–1297. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Jadhav A, Tiwari S, Lee P and Ndisang JF:
The heme oxygenase system selectively enhances the
anti-inflammatory macrophage-M2 phenotype, reduces pericardial
adiposity, and ameliorated cardiac injury in diabetic
cardiomyopathy in Zucker diabetic fatty rats. J Pharmacol Exp Ther.
345:239–249. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Tipnis SR, Hooper NM, Hyde R, Karran E,
Christie G and Turner AJ: A human homolog of angiotensin-converting
enzyme. Cloning and functional expression as a
captopril-insensitive carboxypeptidase. J Biol Chem.
275:33238–33243. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Reudelhuber TL: The renin-angiotensin
system: peptides and enzymes beyond angiotensin II. Curr Opin
Nephrol Hypertens. 14:155–159. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Santos RA, Ferreira AJ, Pinheiro SV,
Sampaio WO, Touyz R and Campagnole-Santos MJ: Angiotensin-(1-7) and
its receptor as a potential targets for new cardiovascular drugs.
Expert Opin Investig Drugs. 14:1019–1031. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Gomes ER, Lara AA, Almeida PW, Guimarães
D, Resende RR, Campagnole-Santos MJ, Bader M, Santos RA and
Guatimosim S: Angiotensin-(1-7) prevents cardiomyocyte pathological
remodeling through a nitric oxide/guanosine 3′,5′-cyclic
mono-phosphate-dependent pathway. Hypertension. 55:153–160. 2010.
View Article : Google Scholar
|
|
36
|
Sukumaran V, Veeraveedu PT, Gurusamy N,
Lakshmanan AP, Yamaguchi K, Ma M, Suzuki K, Kodama M and Watanabe
K: Telmisartan acts through the modulation of ACE-2/ANG 1-7/mas
receptor in rats with dilated cardiomyopathy induced by
experimental autoimmune myocarditis. Life Sci. 90:289–300. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Giani JF, Muñoz MC, Mayer MA, Veiras LC,
Arranz C, Taira CA, Turyn D, Toblli JE and Dominici FP:
Angiotensin-(1-7) improves cardiac remodeling and inhibits
growth-promoting pathways in the heart of fructose-fed rats. Am J
Physiol Heart Circ Physiol. 298:H1003–H1013. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Iwata M, Cowling RT, Gurantz D, Moore C,
Zhang S, Yuan JX and Greenberg BH: Angiotensin-(1-7) binds to
specific receptors on cardiac fibroblasts to initiate antifibrotic
and antitrophic effects. Am J Physiol Heart Circ Physiol.
289:H2356–H2363. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Grobe JL, Mecca AP, Mao H and Katovich MJ:
Chronic angiotensin-(1-7) prevents cardiac fibrosis in DOCA-salt
model of hypertension. Am J Physiol Heart Circ Physiol.
290:H2417–H2423. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Patel VB, Mori J, McLean BA, Basu R, Das
SK, Ramprasath T, Parajuli N, Penninger JM, Grant MB, Lopaschuk GD,
et al: ACE2 deficiency worsens epicardial adipose tissue
inflammation and cardiac dysfunction in response to diet-induced
obesity. Diabetes. 65:85–95. 2016.
|
|
41
|
Dias-Peixoto MF, Santos RA, Gomes ER,
Alves MN, Almeida PW, Greco L, Rosa M, Fauler B, Bader M, Alenina
N, et al: Molecular mechanisms involved in the
angiotensin-(1-7)/Mas signaling pathway in cardiomyocytes.
Hypertension. 52:542–548. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Giani JF, Gironacci MM, Muñoz MC, Peña C,
Turyn D and Dominici FP: Angiotensin-(1-7) stimulates the
phosphorylation of JAK2, IRS-1 and Akt in rat heart in vivo: role
of the AT1 and Mas receptors. Am J Physiol Heart Circ Physiol.
293:H1154–H1163. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shah A, Gul R, Yuan K, Gao S, Oh YB, Kim
UH and Kim SH: Angiotensin-(1-7) stimulates high atrial
pacing-induced ANP secretion via Mas/PI3-kinase/Akt axis and
Na+/H+ exchanger. Am J Physiol Heart Circ
Physiol. 298:H1365–H1374. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liang W, Chen J, Mo L, Ke X, Zhang W,
Zheng D, Pan W, Wu S, Feng J, Song M and Liao X: ATP-sensitive
K+ channels contribute to the protective effects of
exogenous hydrogen sulfide against high glucose-induced injury in
H9c2 cardiac cells. Int J Mol Med. 37:763–772. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Latz E, Xiao TS and Stutz A: Activation
and regulation of the inflammasomes. Nat Rev Immunol. 13:397–411.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mezzaroma E, Toldo S, Farkas D, Seropian
IM, Van Tassell BW, Salloum FN, Kannan HR, Menna AC, Voelkel NF and
Abbate A: The inflammasome promotes adverse cardiac remodeling
following acute myocardial infarction in the mouse. Proc Natl Acad
Sci USA. 108:19725–19730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Kerr LE, McGregor AL, Amet LE, Asada T,
Spratt C, Allsopp TE, Harmar AJ, Shen S, Carlson G, Logan N, et al:
Mice overexpressing human caspase 3 appear phenotypically normal
but exhibit increased apoptosis and larger lesion volumes in
response to transient focal cerebral ischaemia. Cell Death Differ.
11:1102–1111. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Campagnole-Santos MJ, Diz DI, Santos RA,
Khosla MC, Brosnihan KB and Ferrario CM: Cardiovascular effects of
angiotensin-(1-7) injected into the dorsal medulla of rats. Am J
Physiol. 257:H324–H329. 1989.PubMed/NCBI
|
|
49
|
Olivon VC, Aires RD, Santiago LB, Ramalho
LZ, Cortes SF and Lemos VS: Mas receptor overexpression increased
Ang-(1-7) relaxation response in renovascular hypertensive rat
carotid. Peptides. 71:250–258. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Souza AP, Sobrinho DB, Almeida JF, Alves
GM, Macedo LM, Porto JE, Vêncio EF, Colugnati DB, Santos RA,
Ferreira AJ, et al: Angiotensin II type 1 receptor blockade
restores angiotensin-(1-7)-induced coronary vasodilation in
hypertrophic rat hearts. Clin Sci (Lond). 125:449–459. 2013.
View Article : Google Scholar
|
|
51
|
Santos SH, Giani JF, Burghi V, Miquet JG,
Qadri F, Braga JF, Todiras M, Kotnik K, Alenina N, Dominici FP, et
al: Oral administration of angiotensin-(1-7) ameliorates type 2
diabetes in rats. J Mol Med Berl. 92:255–265. 2014. View Article : Google Scholar
|
|
52
|
Hao PP, Yang JM, Zhang MX, Zhang K, Chen
YG, Zhang C and Zhang Y: Angiotensin-(1-7) treatment mitigates
right ventricular fibrosis as a distinctive feature of diabetic
cardiomyopathy. Am J Physiol Heart Circ Physiol. 308:H1007–H1019.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Liao X, Wang L, Yang C, He J, Wang X, Guo
R, Lan A, Dong X, Yang Z, Wang H, et al: Cyclooxygenase mediates
cardioprotection of angiotensin-(1-7) against
ischemia/reperfusion-induced injury through the inhibition of
oxidative stress. Mol Med Rep. 4:1145–1150. 2011.PubMed/NCBI
|
|
54
|
Suski M, Olszanecki R, Stachowicz A, Madej
J, Bujak-Giżycka B, Okoń K and Korbut R: The influence of
angiotensin-(1-7) Mas receptor agonist (AVE 0991) on mitochondrial
proteome in kidneys of apoE knockout mice. Biochim Biophys Acta.
1834:2463–2469. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Papinska AM, Mordwinkin NM, Meeks CJ,
Jadhav SS and Rodgers KE: Angiotensin-(1-7) administration benefits
cardiac, renal and progenitor cell function in db/db mice. Br J
Pharmacol. 172:4443–4453. 2015. View Article : Google Scholar :
|
|
56
|
Hausenloy DJ and Yellon DM: New directions
for protecting the heart against ischaemia-reperfusion injury:
targeting the reperfusion injury salvage kinase (RISK)-pathway.
Cardiovasc Res. 61:448–460. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Wang J, Ji SY, Liu SZ, Jing R and Lou WJ:
Cardioprotective effect of breviscapine: inhibition of apoptosis in
H9c2 cardiomyocytes via the PI3K/Akt/eNOS pathway following
simulated ischemia/reperfusion injury. Pharmazie. 70:593–597.
2015.PubMed/NCBI
|
|
58
|
Lakshmanan AP, Harima M, Suzuki K,
Soetikno V, Nagata M, Nakamura T, Takahashi T, Sone H, Kawachi H
and Watanabe K: The hyperglycemia stimulated myocardial endoplasmic
reticulum (ER) stress contributes to diabetic cardiomyopathy in the
transgenic non-obese type 2 diabetic rats: a differential role of
unfolded protein response (UPR) signaling proteins. Int J Biochem
Cell Biol. 45:438–447. 2013. View Article : Google Scholar
|
|
59
|
Cicek FA, Toy A, Tuncay E, Can B and Turan
B: Beta-blocker timolol alleviates hyperglycemia-induced cardiac
damage via inhibition of endoplasmic reticulum stress. J Bioenerg
Biomembr. 46:377–387. 2014. View Article : Google Scholar : PubMed/NCBI
|