Introduction
Cardiac hypertrophy refers to a complex cardiac
remodeling process that is induced by various stressors, including
hypertension, valve stenosis or regurgitation, and myocardial
infarction, thus resulting in an increase in myocardial cell size
and heart weight (HW), and ultimately cardiac dysfunction and heart
failure (1,2). During this process, the arrested
fetal genes that are associated with hypertrophy, including
natriuretic peptide type A (Nppa), natriuretic peptide type
B (Nppb) and myosin heavy polypeptide 7 (Myh7), are
reactivated (3). Previous studies
have reported that histone deacetylase-2 (HDAC2) serves an
important role in cardiac hypertrophy (4,5).
HDACs are post-translational modifying enzymes, which can modify
the structure of chromosomes and regulate gene expression (6). HDAC2 belongs to class I HDACs and it
has previously been demonstrated that inhibiting HDAC2 activity can
suppress the progression of cardiac hypertrophy (7,8).
Recent progress towards understanding the underlying mechanisms has
revealed that the transcriptional regulator Krüppel-like factor 4
(KLF4) mediates HDAC inhibitor-induced prevention of cardiac
hypertrophy (9).
Sphingosine-1-phosphate (S1P) is a lysophospholipid
mediator that circulates in the blood, which has been reported to
be able to inhibit HDAC2 activity, and has been proposed to protect
against numerous cardiovascular disorders, including coronary
artery disease, atherosclerosis, myocardial infarction and heart
failure (10,11). The majority of known S1P effects
are based on three specific G protein-coupled receptors (GPCRs),
termed S1P receptor (R)1-S1PR3, which are expressed in the
cardiovascular system (12).
Nevertheless, whether S1P can function through inhibiting HDAC2 in
the heart is unknown.
As aforementioned, S1P exerts cardioprotective
effects and can suppress HDAC2 activity, which is closely involved
in cardiac hypertrophy. However, whether S1P can alleviate cardiac
hypertrophy and whether S1PRs participate in it has not been
elucidated. Therefore, to further explore the role of S1P in the
heart, the present study investigated the effects of S1P in
vivo and in vitro. In vivo experiments were
performed on mice under transverse aortic constriction (TAC), which
were treated with or without S1P. In vitro experiments were
performed with H9c2 cells to explore the effects and mechanisms of
S1P.
Materials and methods
Reagents and antibodies
S1P was purchased from Cayman Chemical Company (Ann
Arbor, MI, USA). Phenylephrine (Pe) and fluorescein isothiocyanate
(FITC)-conjugated wheat germ agglutinin were obtained from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). Nuclear and
Cytoplasmic Protein Extraction kit and DAPI were purchased from
Wuhan Boster Biological Technology, Ltd. (Wuhan, China). Cell lysis
buffer for western blotting and immunoprecipitation, and
actin-Tracker Green were purchased from Beyotime Institute of
Biotechnology (Shanghai, China). HDAC2 Fluorimetric Drug Discovery
kit was purchased from Enzo Life Sciences, Inc. (Farmingdale, NY,
USA). Fetal bovine serum (FBS) and Dulbecco's modified Eagle's
medium (DMEM) for cell culture were obtained from Gibco (Thermo
Fisher Scientific, Inc., Waltham, MA, USA). Small interfering
(si)RNAs were purchased from Guangzhou RiboBio Co., Ltd.(Guangzhou,
China). Mega Tran1.0 was purchased from OriGene Technologies, Inc.
(Rockville, MD, USA). Antibodies against atrial natriuretic peptide
(ANP; sc-20158), brain natriuretic peptide (BNP; sc-271185), S1PR2
(sc-25491) and GAPDH (sc-32233) were from Santa Cruz Biotechnology,
Inc. (Dallas, TX, USA). Antibodies against β-myosin heavy chain
(β-MHC; 22280-1-AP), HDAC2 (12922-3-AP) and histone H3 (17168-1-AP)
were obtained from ProteinTech Group, Inc. (Chicago, IL, USA). An
antibody against KLF4 (BA3453) was purchased from Wuhan Boster
Biological Technology, Ltd. Antibodies against histone H3 with
lysine 9 acetylation (H3K9-Ac; ab32129), S1PR1 (ab125074) and S1PR3
(ab108370) were obtained from Abcam (Cambridge, MA, USA).
Animals and animal treatment
Male C57BL/6 mice (age, 8 weeks; weight, 23-26 g;
n=24) used in the present study were obtained from the Experimental
Animal Center of Changsha (Changsha, China). The present study was
approved by the Institutional Animal Research Committee of Tongji
Medical College (Wuhan, China). All animal experimental protocols
complied with the Guide for the Care and use of Laboratory Animals
published by the United States National Institutes of Health
(13). Mice were housed at the
Animal Experimental Center of Tongji Hospital (Wuhan, China) at
25°C, under a 12-h light/dark cycle, and were allowed free access
to normal mice chow and water throughout the study period.
Following 1 week acclimation, the mice were randomly
divided into various treatment groups. Pressure overload by TAC was
used to induce cardiac hypertrophy, as described previously
(14). Briefly, a medial skin
incision was made from the neck to the upper chest, and the
manubrium of the sternum was opened. The transverse aorta between
the right innominate artery and left carotid artery was constricted
using 7-0 silk suture tied around a 27-gauge needle. Sham surgery
was performed without constricting the aorta. A total of 1 week
after surgery, TAC-operated mice were randomized into various
cohorts, and were intraperitoneally injected with S1P (6
µg/g/day) or vehicle (saline) for a further 2 weeks
(15). The mice were grouped as
follows: Sham (n=8), TAC (n=5) and TAC + S1P (n=6) groups.
Originally, there were 8 mice in each group; however, following TAC
surgery, 3 mice died in the TAC group and 2 died in the TAC + S1P
group. This may be the result of the impact of TAC and the
specificity of different mice. Hypertrophic responses at the end of
the treatment were analyzed by echocardiography, and hemodynamic,
histological and biochemical analyses.
Sampling method
Following treatment with S1P for 2 weeks, mice were
sacrificed by exsanguination through the carotid artery under
pentobarbital (100 mg/kg) anesthesia; euthanasia was performed in
the same way for all of the groups. Subsequently, the whole heart
was collected, including the aorta, using ophthalmic scissors to
cut along the backbone. The root of the aorta was removed and the
heart was separated, weighed and washed with ice-cold saline; one
part was stored in liquid nitrogen at -80°C for western blotting
and HDAC2 activity assays. The other part of the heart was
maintained in 10% formalin solution for histochemical analysis.
Hemodynamic measurements
Ventricular hemodynamic measurements were performed
using a Millar Catheter system (Millar, Inc., Houston, TX, USA) via
the right carotid artery under intraperitoneal injection of 100
mg/kg pentobarbital, as described previously (16). Briefly, a pressure-volume catheter
(Millar 1.4F, SPR 835; Millar, Inc.) was inserted into the right
carotid artery and advanced into the left ventricle to measure
heart rate and instantaneous intraventricular pressure.
Analysis of cardiac function by
echocardiography
Cardiac function was assessed by echocardiography,
using a high-resolution imaging system equipped with a 30-MHz high
frequency scanhead (VisualSonics Vevo770; VisualSonics, Inc.,
Toronto, ON, Canada) applied to the chest wall. Ventricular
dimensions, ejection fraction (EF) and fractional shortening (FS)
were examined as described previously (17).
Histochemical analysis
Hearts were fixed with 10% formalin solution for 24
h at room temperature. They were then embedded in paraffin and
sectioned into slices (5 µm). To measure the area of
cardiomyocytes, heart sections were stained with hematoxylin and
eosin (H&E) and FITC-conjugated wheat germ agglutinin, as
previously described (18), and
were visualized by light microscopy. Image-Pro Plus Version 6.0
(Media Cybernetics, Inc., Bethesda, MD, USA) was used to measure
the area of each cell.
Cell culture and treatments
H9c2 cells, which are a subclone of the original
clonal cell line derived from the heart of embryonic BD1X rats,
were obtained from American Type Culture Collection (CRL-1446;
ATCC, Manassas, VA, USA). The cells were cultured in DMEM
supplemented with 10% FBS and penicillin-streptomycin (100 IU/ml)
in a humidified atmosphere containing 95% air and 5% CO2
at 37°C.
Cells were plated in 6-well or 12-well plates at
37°C, and were treated with 1 µM S1P for 1 h, followed by
100 µM PE for 24 h, after which cells were collected. For
some experiments, 2×105 cells per well were transfected
for 24 h at room temperature with small interfering (si)RNA
negative control (siNC) and siRNAs targeting rat S1PR1, S1PR2 and
S1PR3 (siR1, siR2 and siR3) using MegaTran 1.0, according to the
manufacturer's protocol. The concentration of siRNAs used was 50
nM. A total of 24 hours post-transfection, cells were treated with
S1P for 1 h and PE for 24 h.
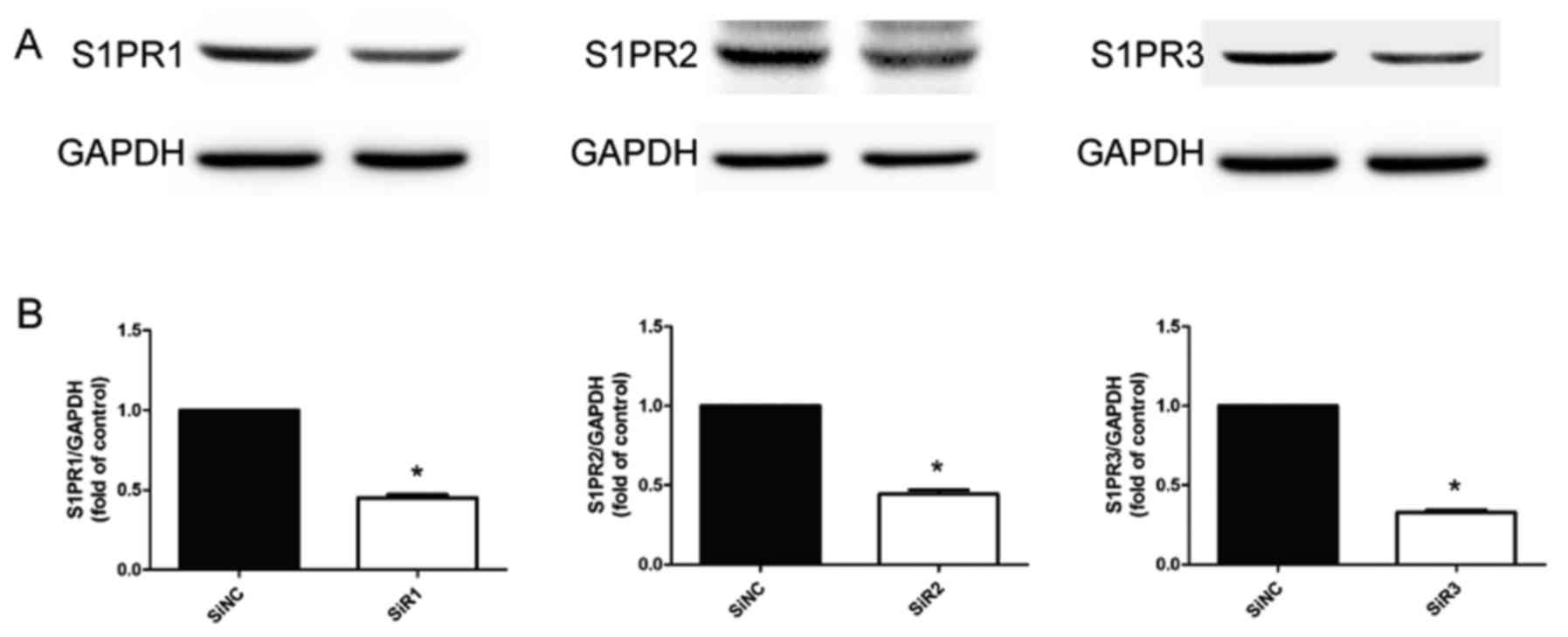
All in vitro experiments were repeated three
times with the same interventions. The siRNA sequences were as
follows: siRNA-S1PR1, 5′-CCUCCUUGCUAUCGCCAUUdTdT-3′ (sense) and
3′-dTdTGGAGGAACGAUAGCGGUAA-5′ (anti-sense); siRNA-S1PR2,
5′-CCUUGUACGUCCGAAUCUAdTdT-3′ (sense) and
3′-dTdTGGAACAUGCAGGCUUAGAU-5′ (antisense); siRNA-S1PR3,
5′-GCCACUCUCCAAAGGUCAAdTdT-3′ (sense) and
3′-dTdTCGGUGAGAGGUUUCCAGUU-5′ (antisense). Successful transfection
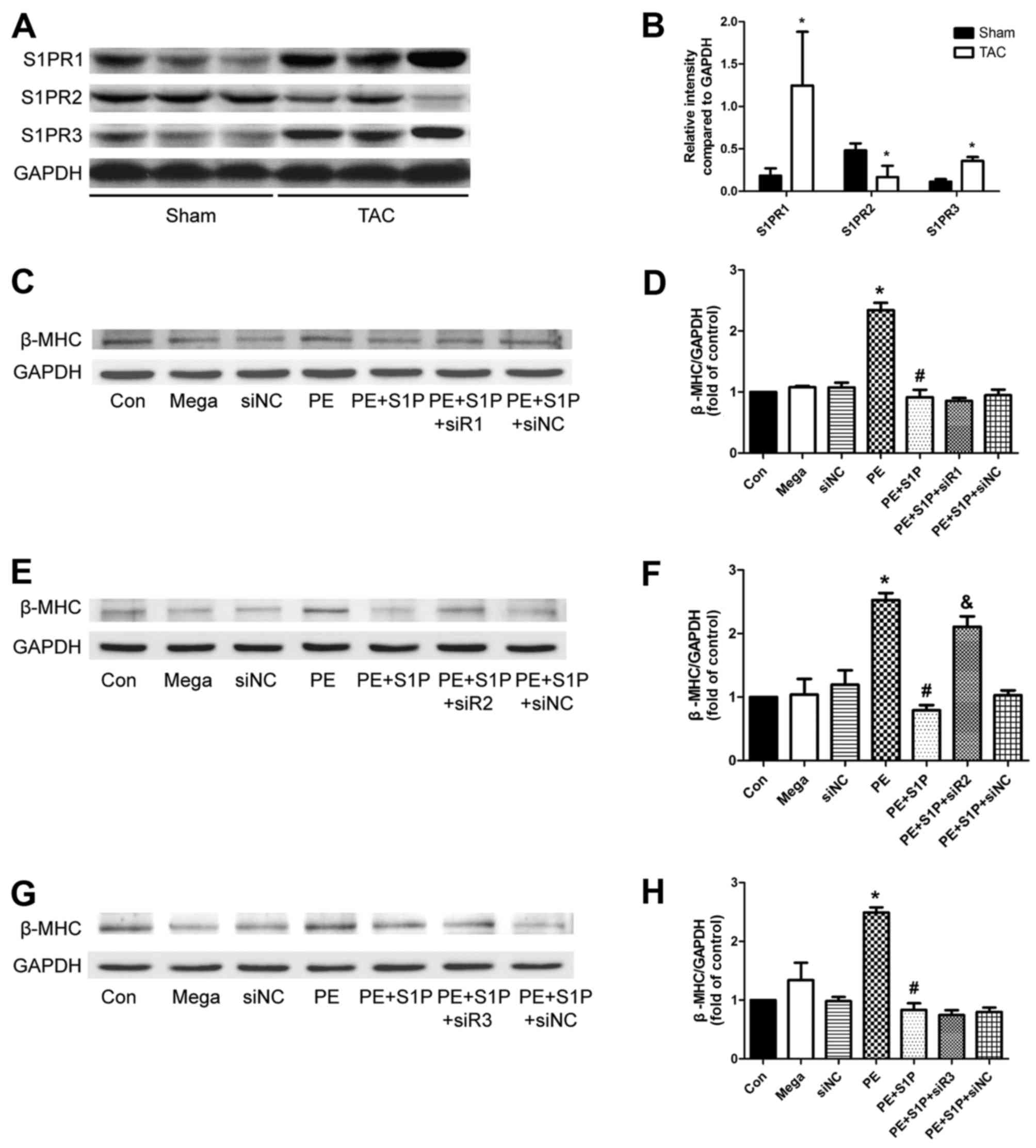
was confirmed by western blotting (Fig. 1).
Immunocytofluorescence
Cells were initially plated in 12-well plates and
were treated as aforementioned. Subsequently, cells were washed
with ice-cold PBS, fixed in 4% paraformaldehyde for 10 min and
treated with 0.3% Triton X-100 for 20 min at room temperature.
After blocking with 5% bovine serum albumin (BSA; Abcam) for 30 min
at room temperature, cells were incubated in actin-tracker Green at
4°C overnight. Subsequently, the cells were stained with DAPI for
nuclear detection and were visualized under a Nikon DXM1200
fluorescence microscope (Nikon Corporation, Tokyo, Japan).
Image-Pro Plus 6.0 (Media Cybernetics, Inc., Bethesda, MD, USA) was
applied to merge images and measure the area of cells.
Immunoprecipitation
Nuclear proteins were extracted using Nuclear and
Cytoplasmic Protein Extraction kit according to the manufacturer's
protocol, after which they were precleared with protein A/G-agarose
beads and incubated overnight with HDAC2 antibody at 4°C.
Subsequently, protein A/G-agarose beads were added and incubated
for 2-3 h at 4°C on a rotator. Beads were collected, washed with
ice-cold PBS and used for HDAC2 activity assays. Aliquots of
agarose-bound immunocomplexes were boiled in SDS-PAGE sample buffer
and the released HDAC2 proteins were analyzed by western blot
analysis using HDAC2 antibody.
HDAC2 activity assays
HDAC2 activity was tested using a HDAC2 Fluorimetric
Drug Discovery kit according to the manufacturer's protocol.
Briefly, HDAC2 was isolated by immunoprecipitation and was then
subjected to an activity assay. Briefly, samples were incubated
with Fluor de Lys®-Green Substrate at 37°C for 30 min.
Fluor de Lys® Developer was then added and the samples
were incubated for 15 min at room temperature. HDAC2 activity
levels were expressed as arbitrary fluorescence units and were
determined using a Synergy 2 reader (Bio-Tek Instruments, Inc.,
Winooski, VT, USA) by measuring fluorescence with excitation at 485
nm and emission at 528 nm.
Western blot analysis
For protein extraction, heart samples and cells were
lysed with cell lysis buffer for western blotting and
immunoprecipitation. Subsequently, lysates were centrifuged at
12,000 × g for 20 min at 4°C and the supernatants were used for
western blot analysis. The protein concentration was quantified
using BCA Protein Assay kit. About 8.5 mg/ml protein was loaded
onto gels. Proteins were subjected to 10% SDS-PAGE and were
transferred onto polyvinylidene fluoride membranes, after which
they were incubated at room temperature for 2 h with blocking
solution (5% BSA), and were then incubated with the indicated
antibodies overnight at 4°C. Rabbit anti-ANP (1:1,000 dilution),
rabbit anti-BNP (1:1,000 dilution), rabbit anti-β-MHC (1:2,000
dilution), mouse anti-GAPDH (1:2,000 dilution), rabbit anti-HDAC2
(1:1,000 dilution), rabbit anti-H3K9-Ac (1:500 dilution), rabbit
anti-H3-total (1:2,000 dilution), rabbit anti-KLF4 (1:200
dilution), rabbit anti-S1PR1 (1:5,000 dilution), rabbit anti-S1PR2
(1:1,000 dilution) and rabbit anti-S1PR3 (1:5,000 dilution) were
used. Subsequently, these membranes were incubated with a secondary
antibody conjugated to horseradish peroxidase (1:5,000 dilution;
cat. nos. ab6734 and ab131368; Abcam) at room temperature for ~2 h.
Following incubation with each antibody, the membranes were washed
five times with Tris-buffered saline-Tween containing 10 mM Tris-Cl
(pH 7.5), 100 mM NaCl, and 0.1% Tween-20 at room temperature.
Immunoreactive bands were examined with enhanced chemiluminescence
solution (Thermo Fisher Scientific, Inc.), and were semi-quantified
by densitometry and normalized to GAPDH or histone H3 expression.
All groups were then normalized to their respective controls.
Statistical analysis
All continuous data were expressed as the means ±
standard error of the mean. The densitometry was realized using
Gel-Pro 32 analyzer and the statistical software used was GraphPad
Prism5. Differences between groups were evaluated using unpaired
Student's t-test or one-way analysis of variance (ANOVA) and
Bonferroni post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
S1P attenuates TAC-induced cardiac
dysfunction in mice
To determine the effects of S1P on cardiac
performance, heart tissues obtained from the various groups
underwent hemodynamic and echocardiographic assessments. The
results demonstrated that, compared with in the TAC group, maximal
slope of systolic pressure increment and minimal slope of diastolic
pressure decrement were consistently increased in the TAC + S1P
group; there were no significant alterations in heart rate between
the groups (Table I). Enlargement
of ventricular chambers at end-systole and end-diastole, and
decreased EF and FS were observed following TAC, thus indicating
impaired cardiac function. Conversely, compromised cardiac function
was significantly improved following S1P treatment (Table I). These data indicated that S1P
may ameliorate TAC-induced cardiac function deterioration.
 | Table IHemodynamic and echocardiographic
results. |
Table I
Hemodynamic and echocardiographic
results.
| Variable | Sham | TAC | TAC + S1P |
|---|
| Hemodynamics | | | |
| HR (bpm) | 409±33 | 375±9 | 432±20 |
|
dp/dtmax (mmHg/sec) | 5,489±599 | 2,356±390a | 6,330±1241b |
|
dp/dtmin (mmHg/sec) | −4,427±562 | −1,620±369a | −4,997±782b |
|
Echocardiography | | | |
| EF (%) | 51.5±0.9 | 38.4±2.5a | 55.0±2.9b |
| FS (%) | 26.1±0.5 | 18.7±1.4a | 28.2±1.8b |
| LVID (d) (mm) | 4.30±0.13 | 4.77±0.10a | 3.81±0.14b |
| LVID (s) (mm) | 3.22±0.13 | 3.85±0.11a | 2.84±0.11b |
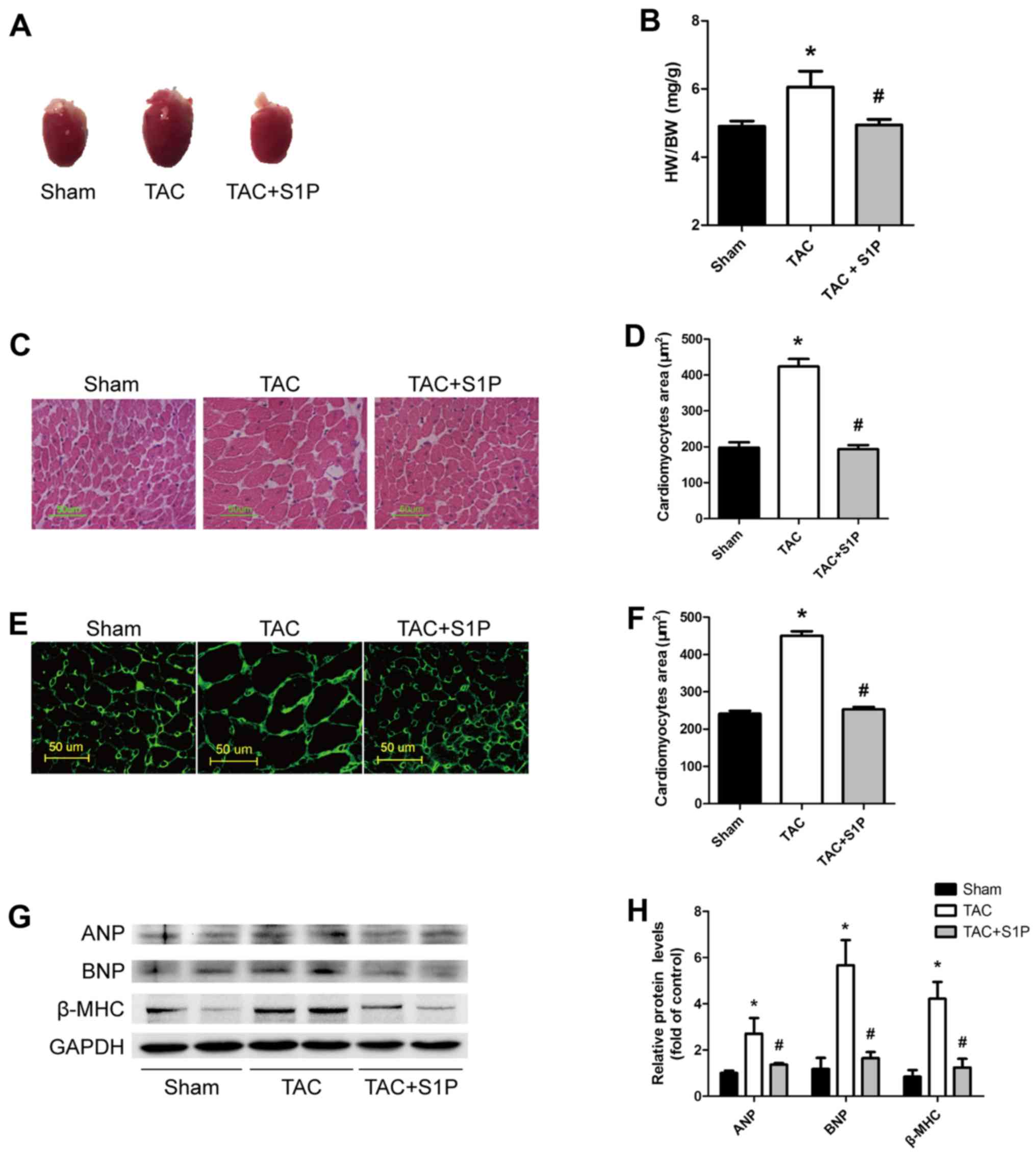
S1P treatment prevents the development of
TAC-induced cardiac hypertrophy and reduces the expression of
cardiac hypertrophic proteins in mice
To determine the effects of S1P on cardiac
hypertrophy, heart weight (HW)/body weight (BW) ratios and cardiac
morphology were compared in mice under various treatment
conditions. Heart size and HW/BW ratios were markedly increased in
the TAC group; however, in TAC mice treated with S1P, these
parameters were reduced to normal levels (Fig. 2A and B). Consistent findings were
observed when paraffin-embedded heart sections were stained with
H&E and FITC-conjugated wheat germ agglutinin to evaluate
cardiac hypertrophy. The TAC group exhibited larger sizes of
cardiomyocytes compared with in the sham group, whereas the TAC +
S1P group exhibited similar sizes as the sham group (Fig. 2C–F).
To determine whether TAC induced upregulation of
cardiac hypertrophic proteins, western blot analysis was performed
to examine the expression levels of ANP, BNP and β-MHC. The results
revealed that TAC increased the expression of these proteins;
however, S1P treatment inhibited the expression of these proteins
(Fig. 2G and H).
These findings suggested that S1P treatment may
prevent the development of TAC-induced cardiac hypertrophy in
mice.
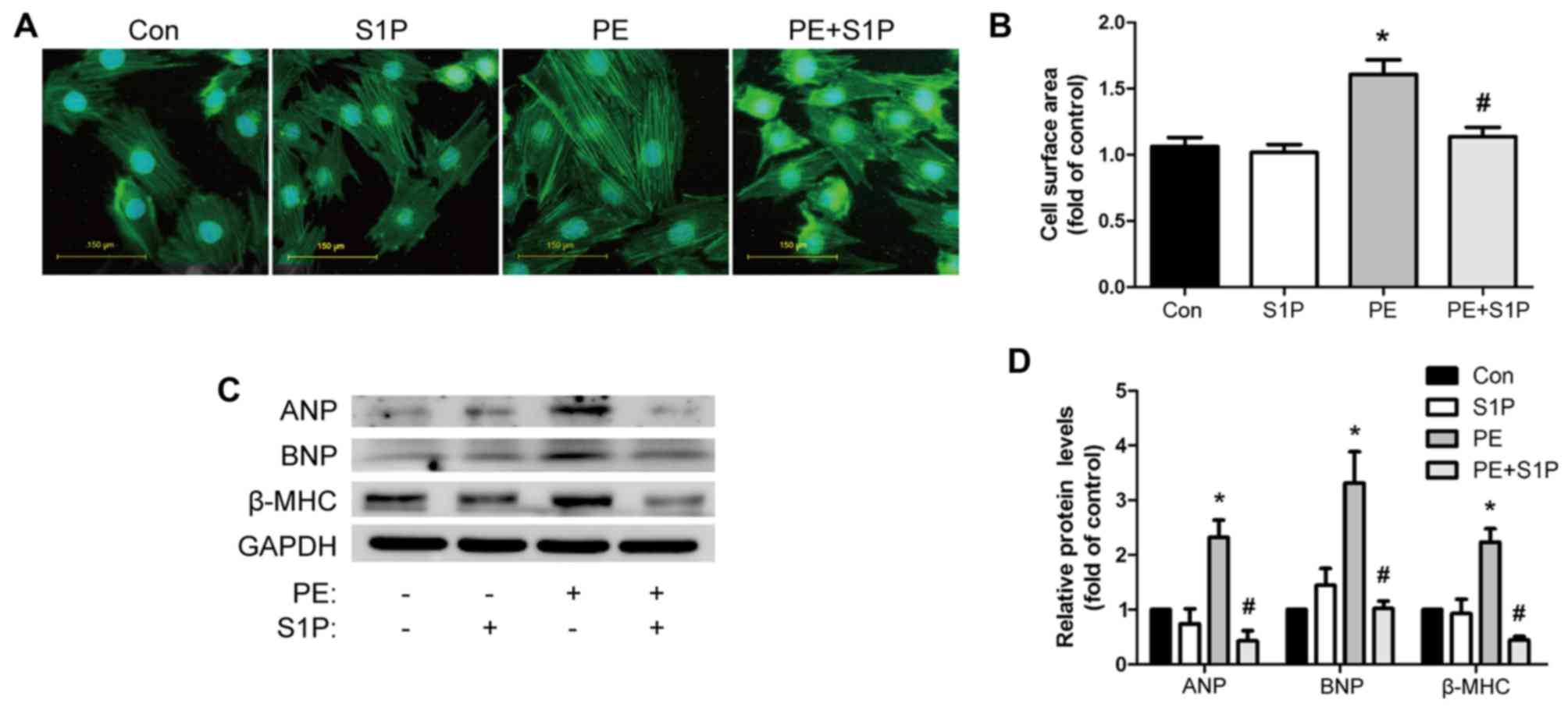
S1P administration inhibits the
PE-induced cardiac hypertrophic response in cultured H9c2
cells
The present study further investigated the effects
of S1P on the PE-induced hypertrophic response in H9c2 cells.
Consistent with the in vivo findings, PE (100 µM)
increased cardiomyocyte size, as detected by cell surface area
measurement. Notably, pretreatment with S1P (1 µM)
restricted this effect (Fig. 3A and
B). Furthermore, western blot analysis demonstrated that S1P
suppressed the expression of PE-induced markers of cardiac
hypertrophy, including ANP, BNP and β-MHC (Fig. 3C and D). S1P treatment alone had
no effect on cardiac hypertrophy compared with in the control group
(Fig. 3). These data indicated
that S1P could attenuate the cardiac hypertrophic response in
vitro.
S1P regulates the cardiac hypertrophic
response through inhibiting HDAC2 activity, but not by influencing
HDAC2 expression
To test the hypothesis that S1P may regulate the
cardiac hypertrophic response by inhibiting HDAC2, the expression
levels of HDAC2 were detected in heart samples and H9c2 cells; S1P
was revealed to have no effect on HDAC2 expression levels (Fig 4A–D). Therefore, the present study
investigated whether S1P could affect HDAC2 activity. HDAC2 was
initially extracted from nuclear proteins by immunoprecipitation
(Fig. 4Ex and F) and underwent
HDAC2 activity assays. The results indicated that in vivo
and in vitro, HDAC2 activity was upregulated under
hypertrophic conditions, whereas it was significantly suppressed in
groups treated with S1P (Fig. 4G and
H). To further verify these findings, the expression of H3K9-Ac
was detected; treatment with S1P was shown to increase acetylation
of H3K9 (Fig. 4I–L).
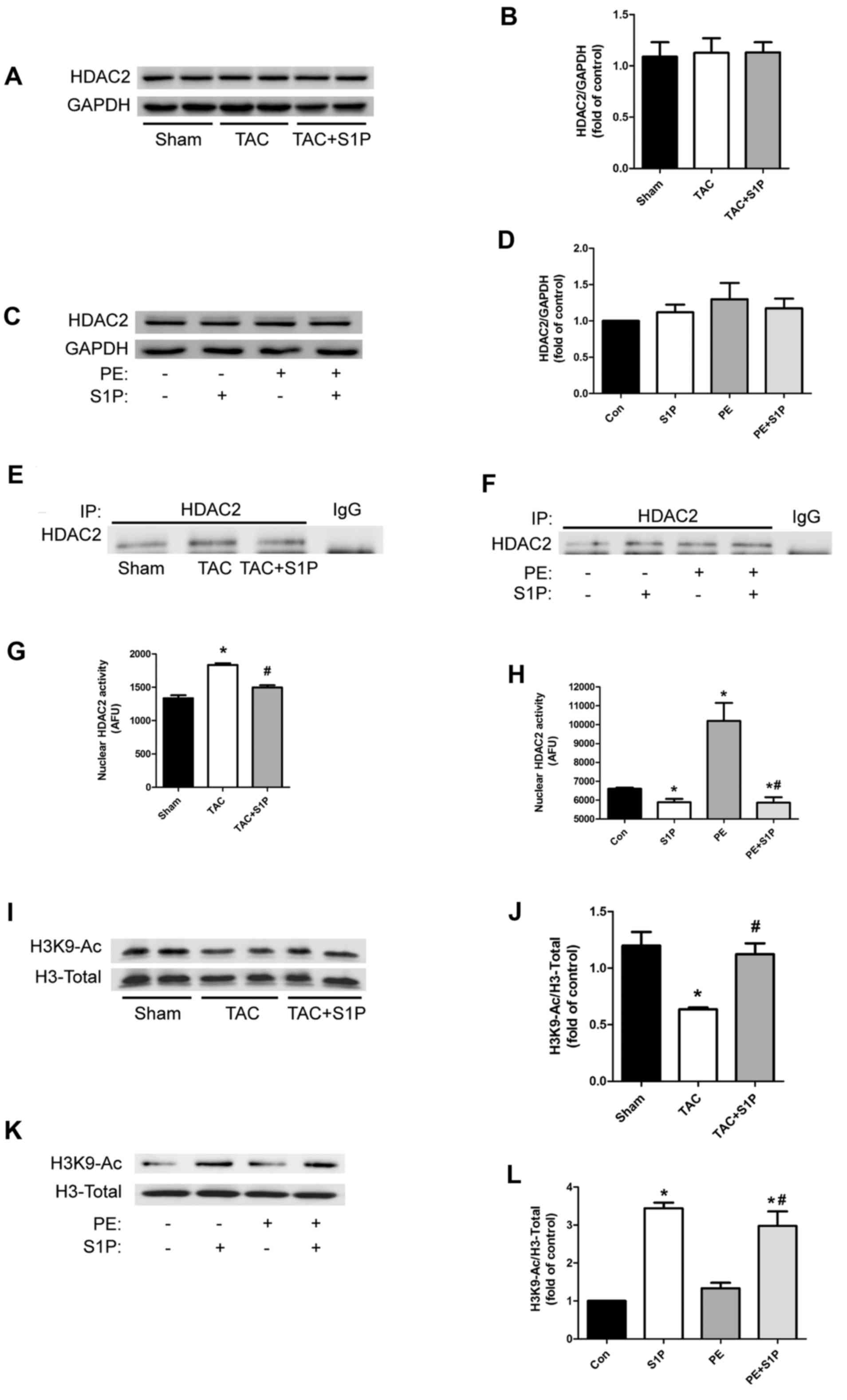 | Figure 4S1P regulates cardiac hypertrophic
response through inhibiting HDAC2 activity, but not by affecting
HDAC2 expression. (A–D) Representative immunoblots and
semi-quantification of HDAC2 expression in heart samples and H9c2
cells. (e and F) IP of HDAC2 in nuclear proteins from various
groups. (G and H) nuclear HDAC2 activity was determined in
duplicate cardiac samples and H9c2 cells. (I–L) H3K9-Ac expression
was determined by western blotting with the indicated antibodies.
Data are presented as the means ± standard error of the mean. In
vivo experiments, n≥5 for each group; *P<0.05 vs.
Sham group; #P<0.05 vs. TAC group; in vitro
experiments, n≥3 for each experiment; *P<0.05 vs. Con
group; #P<0.05 vs. PE group. Con, control; H3K9-Ac,
histone H3 with lysine 9 acetylation; HDAC2, histone deacetylase-2;
IP, immunoprecipitation; PE, phenylephrine; S1P,
sphingosine-1-phosphate; TAC, transverse aortic constriction. |
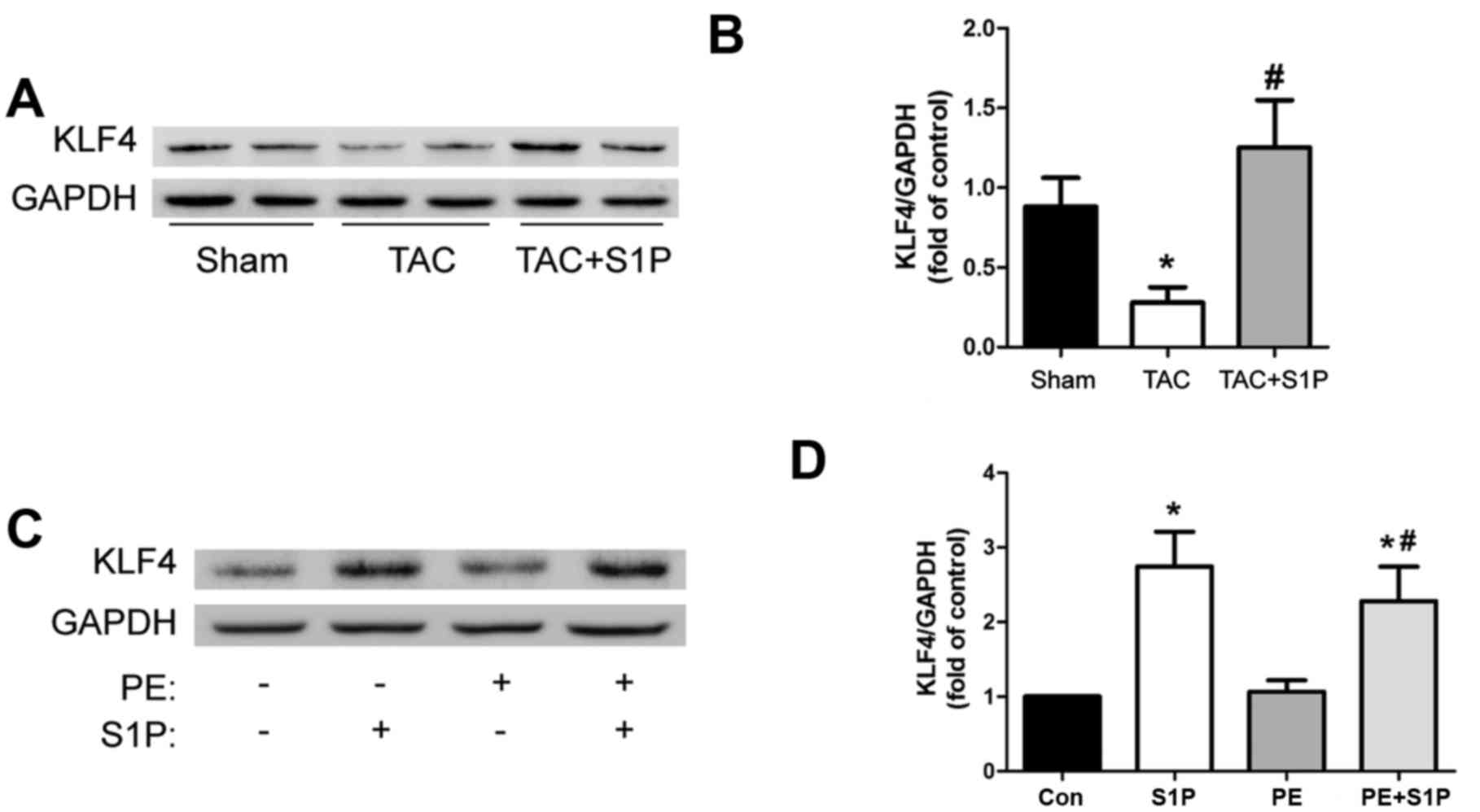
KLF4, an antihypertrophic factor, is
upregulated by S1P in vivo and in vitro
To determine whether S1P, like other HDAC
inhibitors, such as trichostatin A, may upregulate KLF4 to prevent
the progression of cardiac hypertrophy, the present study measured
the expression levels of KLF4 in heart samples. The results
demonstrated that KLF4 was significantly upregulated by treatment
with S1P compared with in the TAC group (Fig. 5A and B). This increase in KLF4
expression was confirmed in H9c2 cells (Fig. 5C and D).
S1PR expression is altered following TAC,
and S1PR2 may be involved in the antihypertrophic effects of
S1P
Since S1PRs have been reported to serve a role in
mediating cardioprotection, the present study aimed to determine
whether they participate in the antihypertrophic effects of S1P.
Initially, the expression levels of S1PR1, 2 and 3 were analyzed,
which have been suggested to be expressed in the cardiovascular
system of mice (12,26). The results indicated that the
expression of all three S1PRs was altered following TAC; S1PR1 and
S1PR3 were upregulated, whereas S1PR2 was downregulated, thus
suggesting that S1PRs may be involved in cardiac hypertrophy
(Fig. 6A and B). Subsequently,
S1PR-specific siRNAs were used to investigate whether S1P
attenuated cardiac hypertrophic responses through S1PR in H9c2
cells. The results demonstrated that the representative
hypertrophic marker, β-MHC, was upregulated by PE; however,
expression was decreased by S1P and this protective effect was
partially suppressed by siR2, but not by siR1 or siR3 (Fig. 6C–H). These findings suggested that
S1PR2 may be involved in the antihypertrophic effects of S1P.
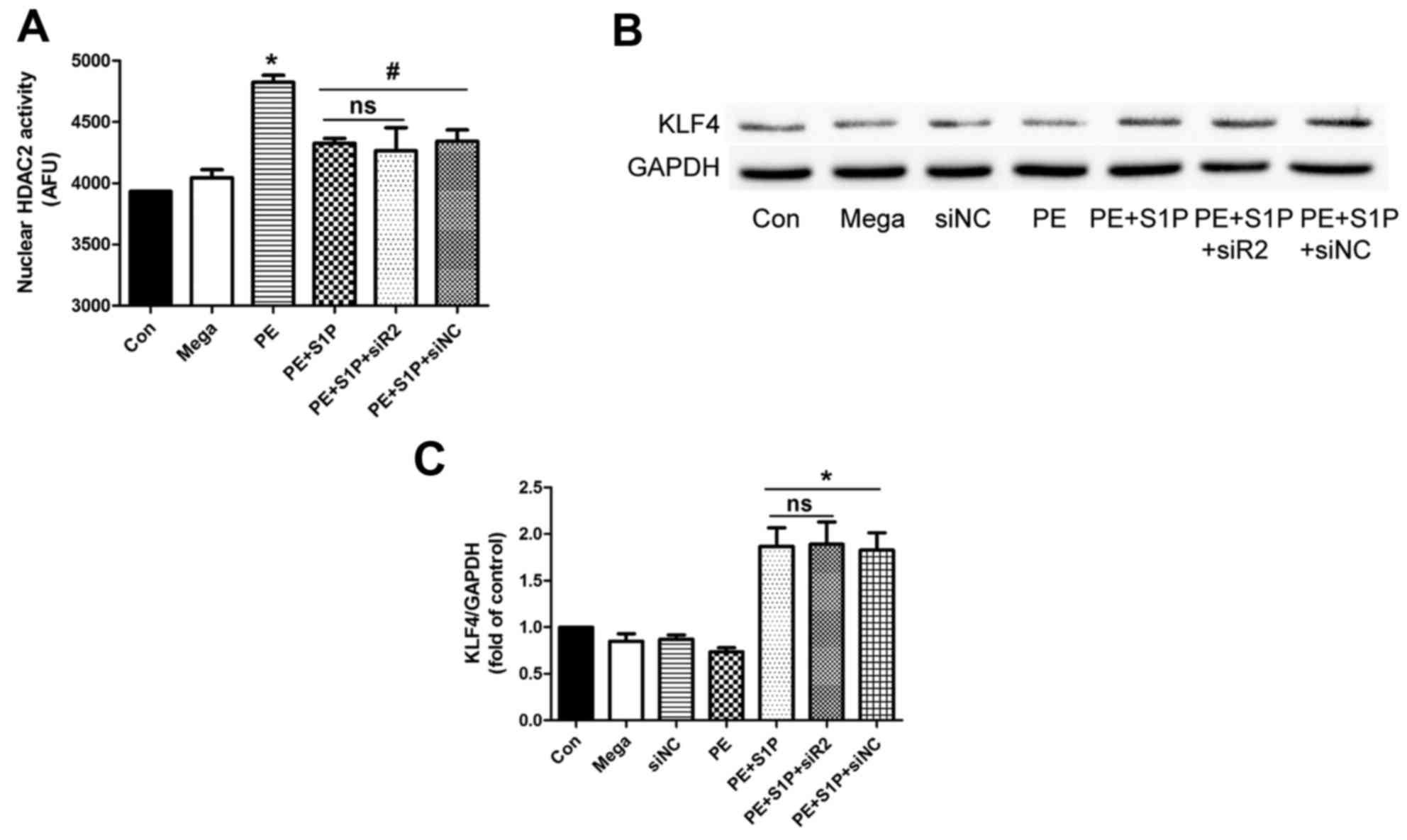
Suppressive effects of S1P on HDAC2
activity are independent of S1PR2
Since S1PR2 may be involved in the antihypertrophic
effects of S1P, the present study aimed to determine whether
inhibition of the effects of S1P on HDAC2 activity was
S1PR2-dependent. HDAC2 was extracted from nuclear proteins by
immunoprecipitation and its activity was detected following siR2
transfection. The results indicated that S1P significantly
inhibited PE-induced upregulation of HDAC2 activity; however, siR2
did not suppress this effect (Fig.
7A). Furthermore, the downstream factor of HDAC2, KLF4, was
investigated. The results demonstrated that siR2 did not affect the
S1P-induced upregulation of KLF4 (Fig. 7B and C). These findings suggested
that the suppressive effects of S1P on HDAC2 activity were
independent of S1PR2.
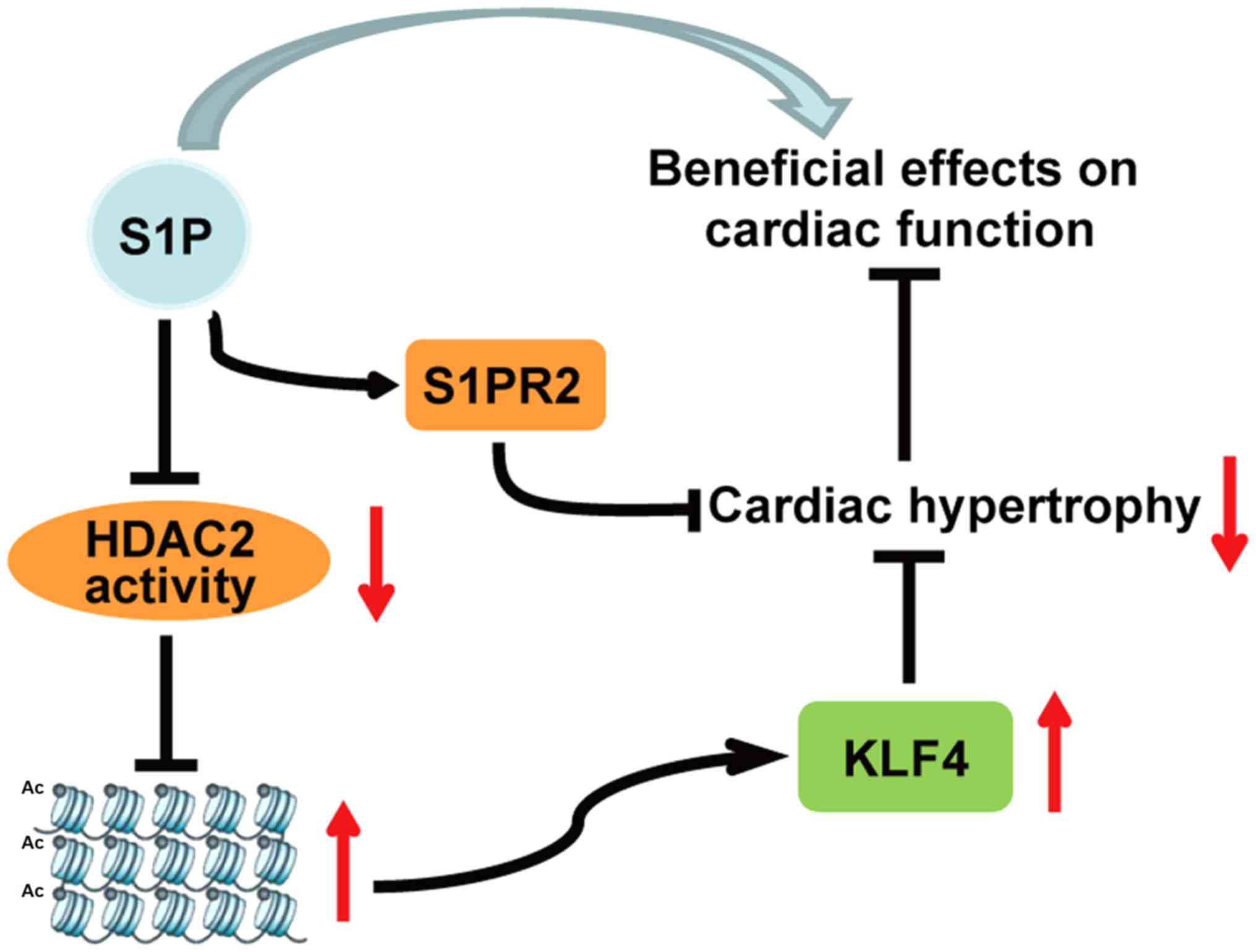
Discussion
In the present study, a mouse model of TAC-induced
cardiac hypertrophy was generated, as well as a PE-induced H9c2
cardiac hypertrophy cell model. Mice and cells were administered
S1P in order to investigate its effects on the cardiac hypertrophic
response and the underlying mechanisms. The results indicated that
S1P could attenuate the progression of cardiac hypertrophy, and
this may be mediated by inhibiting HDAC2 activity, and upregulating
KLF4 independently of S1PRs. The results demonstrated that
administration of S1P ameliorated TAC-induced cardiac function
deterioration, and reduced the hypertrophic response in TAC-treated
mice and PE-treated H9c2 cells. Furthermore, the expression levels
and activity of HDAC2 were detected; the results suggested that S1P
treatment did not affect HDAC2 expression but it did decrease its
activity. In addition, the downstream antihypertrophic factor of
HDAC2, KLF4, was examined and the results indicated that S1P
upregulated KLF4, which may be associated with the suppression of
HDAC2 activity. S1PR2 was revealed to be potentially involved in
the anti-hypertrophic effects of S1P, whereas S1P functioned
independently of it. These findings are summarized in the schematic
diagram presented in Fig. 8.
Cardiac hypertrophy is a common pathological
characteristic of numerous heart diseases and usually occurs in
response to pressure overload, volume overload or myocardial
infarction (19). S1P is a lipid
mediator formed by metabolism of sphingomyelin, which regulates
important functions in cardiac and vascular homeostasis (20). It can increase viability of
cardiomyocytes incubated under hypoxic conditions and reduce
infarct size in isolated, perfused rat hearts following
ischemia/reperfusion (21,22).
Furthermore, it participates in the regulation of vascular tone and
permeability of vessels (12,23–25). The majority of these effects are
mediated by the activation of S1PRs. It has previously been
reported that S1P has five specific GPCRs, S1PR1-S1PR5; however,
only S1PR1, S1PR2 and S1PR3 are mainly expressed in the heart
(12,26). Binding of S1P to each of these
receptors activates distinct intracellular signals. A previous
study indicated that S1PR signaling is important in heart
development, and influences the migration, differentiation and
survival of embryonic cardiomyocytes (27). In the present study, the
expression levels of S1PRs were markedly altered in hypertrophic
hearts; S1PR1 and S1PR3 were upregulated, whereas S1PR2 was
downregulated, thus suggesting an association between S1PRs and
cardiac hypertrophy.
Hait et al previously reported that S1P could
function independently of S1PRs, through binding HDAC2 and
inhibiting its enzymatic activity (11). HDACs are post-translational
modifying enzymes that can remove acetyl functional groups from
lysine residues of histone and nonhistone proteins (28). Previous studies regarding HDAC
inhibitors have provided evidence to suggest that class I HDACs are
prohypertrophic, among which HDAC2 is predominantly activated by
hypertrophic stress; activated HDAC2 triggers hypertrophy through
inhibiting the signaling cascades of KLF4 (6,9).
Therefore, a selective inhibitor of HDAC2 is considered an
effective treatment for cardiac hypertrophy (7,8).
In the present study, the effects of S1P on cardiac
hypertrophy were investigated. There are conflicting data regarding
the role of S1P in cardiac hypertrophy. A previous study performed
in neonatal rat cardiomyocytes demonstrated that S1P did not induce
hypertrophy, as determined by measuring ANP expression and
phenylalanine incorporation, whereas the related sphingolipid,
sphingosylphosphorylcholine, was able to induce hypertrophy
(29). However, the underlying
mechanism remained unclear. Data from another study indicated that
S1P induced hypertrophy in neonatal rat cardiomyocytes, as assessed
by cell size, cytoskeletal organization, phenylanine incorporation
and BNP expression; this hypertrophic response appeared to be
mediated by S1PR1 (30).
Nevertheless, S1P treatment has not previously been performed under
hypertrophic conditions, and it should be recognized that
S1P-induced cardiac hypertrophy is less robust and occurs more
slowly than the canonical hypertrophic responses elicited by
phenylyephrine and endothelin (31).
Since Hait et al reported that S1P could
inhibit HDAC2 activity, the effects of S1P have been studied on
diseases associated with HDAC2. For example, a recent study
demonstrated that S1P increased the ability of muscle cells to use
fatty acids as an energy source in mice with Duchenne muscular
dystrophy through inhibiting HDAC2 activity and increasing the
expression of beneficial muscle genes (32). S1P has also been reported to
protect the liver from lipid metabolism dysfunction in mice fed a
high fat diet by decreasing HDAC2 activity and upregulating key
genes encoding nuclear receptors/enzymes involved in nutrient
metabolism (33). The present
study demonstrated that S1P was able to prevent cardiac hypertrophy
via the suppression of HDAC2 activity; this finding was in
agreement with previous reports, which suggested that inhibition of
HDAC2 may suppress cardiac hypertrophy (7,8).
KLF4 belongs to a large family of transcription
factors named KLFs, which have common structures, including a
transcriptional activation/repression domain and three Krüppel-like
zinc fingers (34). Previous
studies have indicated that KLF4 is a novel regulator of cardiac
hypertrophy that is responsible for HDAC inhibitor-induced
prevention of cardiac hypertrophy (9,35,36). It was previously revealed that
KLF4 bound to the Nppa promoter region from −130 to ~−105 bp
downregulates its expression and suppresses cardiac hypertrophy
(9). The present study expanded
on this established mechanism to determine how S1P regulates
cardiac hypertrophy; the results demonstrated that the protective
effects of S1P may be mediated by upregulating KLF4.
Notably, cardiac function in the TAC + S1P group was
a little better than in the Sham group. We considered that cardiac
function would be improved after one week of TAC when cardiac
hypertrophy was adapted and compensatory and at that time we
treated mice with S1P. Furthermore, the known beneficial effects of
S1P on the cardiovascular system may be responsible for this
phenomenon. H3K9-Ac and KLF4 were downregulated in mice in the TAC
group compared with in the Sham group, whereas PE had no effects on
H3K9-Ac and KLF4 expression in H9c2 cells. This may be due to the
different in vivo and in vitro environments. In
addition, their expression levels were originally low in control
H9c2 cells. In cultured H9c2 cells, S1P upregulated H3K9-Ac and
KLF4 expression; however, it had no effect on the expression of
hypertrophic markers (ANP, BNP and β-MHC) compared with in the
control group. The reason for this may be that the expression
levels of hypertrophic markers were originally low under control
condition.
In conclusion, the present study indicated that S1P
attenuates cardiac hypertrophy by inhibiting HDAC2 activity, thus
resulting in the upregulation of KLF4 in a S1PR2-independent
manner. These findings may provide important information regarding
the potential clinical applications to prevent cardiac hypertrophy.
However, whether S1P directly binds to HDAC2 in cardiomyocytes and
in mouse models, or whether there are other possible mechanisms
underlying S1P-induced inactivation of HDAC2, requires further
exploration in future studies, and the role of S1PR2 in hypertrophy
should be confirmed in S1PR2-knockout mice.
Glossary
Abbreviations
Abbreviations:
|
H3K9-Ac
|
histone H3 with lysine 9
acetylation
|
|
S1PR
|
sphingosine-1-phosphate receptor
|
|
siR
|
small interfering RNA-S1PR
|
Acknowledgments
The authors would like to thank Dr Xingxu Wang for
assistance in echocardiography and Xingwei He for assistance in
language editing.
References
|
1
|
Eom GH and Kook H: Posttranslational
modifications of histone deacetylases: Implications for
cardiovascular diseases. Pharmacol Ther. 143:168–180. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Frey N and Olson EN: Cardiac hypertrophy:
The good, the bad, and the ugly. Annu Rev Physiol. 65:45–79. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Chang L, Kiriazis H, Gao XM, Du XJ and
El-Osta A: Cardiac genes show contextual SWI/SNF interactions with
distinguishable gene activities. Epigenetics. 6:760–768. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Eom GH and Kook H: Role of histone
deacetylase 2 and its post-translational modifications in cardiac
hypertrophy. BMB Rep. 48:131–138. 2015. View Article : Google Scholar :
|
|
5
|
Trivedi CM, Luo Y, Yin Z, Zhang M, Zhu W,
Wang T, Floss T, Goettlicher M, Noppinger PR, Wurst W, et al: Hdac2
regulates the cardiac hypertrophic response by modulating Gsk3 beta
activity. Nat Med. 13:324–331. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kee HJ and Kook H: Roles and targets of
class I and IIa histone deacetylases in cardiac hypertrophy. J
Biomed Biotechnol. 2011:9283262011. View Article : Google Scholar
|
|
7
|
Kee HJ, Sohn IS, Nam KI, Park JE, Qian YR,
Yin Z, Ahn Y, Jeong MH, Bang YJ, Kim N, et al: Inhibition of
histone deacetylation blocks cardiac hypertrophy induced by
angiotensin II infusion and aortic banding. Circulation. 113:51–59.
2006. View Article : Google Scholar
|
|
8
|
Kong Y, Tannous P, Lu G, Berenji K,
Rothermel BA, Olson EN and Hill JA: Suppression of class I and II
histone deacetylases blunts pressure-overload cardiac hypertrophy.
Circulation. 113:2579–2588. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kee HJ and Kook H: Krüppel-like factor 4
mediates histone deacetylase inhibitor-induced prevention of
cardiac hypertrophy. J Mol Cell Cardiol. 47:770–780. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Takuwa Y, Okamoto Y, Yoshioka K and Takuwa
N: Sphingosine-1-phosphate signaling and biological activities in
the cardiovascular system. Biochim Biophys Acta. 1781:483–488.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hait NC, Allegood J, Maceyka M, Strub GM,
Harikumar KB, Singh SK, Luo C, Marmorstein R, Kordula T, Milstien
S, et al: Regulation of histone acetylation in the nucleus by
sphingosine-1-phosphate. Science. 325:1254–1257. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Means CK and Brown JH:
Sphingosine-1-phosphate receptor signalling in the heart.
Cardiovasc Res. 82:193–200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
National Research Council (US) Committee
for the Update of the Guide for the Care and use of Laboratory
Animals. Washington (DC): National Academies Press (US); 2011
|
|
14
|
Takimoto E, Champion HC, Li M, Belardi D,
Ren S, Rodriguez ER, Bedja D, Gabrielson KL, Wang Y and Kass DA:
Chronic inhibition of cyclic GMP phosphodiesterase 5A prevents and
reverses cardiac hypertrophy. Nat Med. 11:214–222. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Liu W, Zi M, Tsui H, Chowdhury SK, Zeef L,
Meng QJ, Travis M, Prehar S, Berry A, Hanley NA, et al: A novel
immunomodulator, FTY-720 reverses existing cardiac hypertrophy and
fibrosis from pressure overload by targeting NFAt (nuclear factor
of activated T-cells) signaling and periostin. Circ Heart Fail.
6:833–844. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cingolani OH, Yang XP, Cavasin MA and
Carretero OA: Increased systolic performance with diastolic
dysfunction in adult spontaneously hypertensive rats. Hypertension.
41:249–254. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ma H, Gong H, Chen Z, Liang Y, Yuan J,
Zhang G, Wu J, Ye Y, Yang C, Nakai A, et al: Association of Stat3
with HSF1 plays a critical role in G-CSF-induced cardio-protection
against ischemia/reperfusion injury. J Mol Cell Cardiol.
52:1282–1290. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dolber PC, Bauman RP, Rembert JC and
Greenfield JC Jr: Regional changes in myocyte structure in model of
canine right atrial hypertrophy. Am J Physiol. 267:H1279–H1287.
1994.PubMed/NCBI
|
|
19
|
Sag CM, Santos CX and Shah AM: Redox
regulation of cardiac hypertrophy. J Mol Cell Cardiol. 73:103–111.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Knapp M: Cardioprotective role of
sphingosine-1-phosphate. J Physiol Pharmacol. 62:601–607. 2011.
|
|
21
|
Karliner JS, Honbo N, Summers K, Gray MO
and Goetzl EJ: The lysophospholipids sphingosine-1-phosphate and
lysophosphatidic acid enhance survival during hypoxia in neonatal
rat cardiac myocytes. J Mol Cell Cardiol. 33:1713–1717. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lecour S, Smith RM, Woodward B, Opie LH,
Rochette L and Sack MN: Identification of a novel role for
sphingolipid signaling in TNF alpha and ischemic preconditioning
mediated cardioprotection. J Mol Cell Cardiol. 34:509–518. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Alvarez SE, Milstien S and Spiegel S:
Autocrine and paracrine roles of sphingosine-1-phosphate. Trends
Endocrinol Metab. 18:300–307. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fyrst H and Saba JD: An update on
sphingosine-1-phosphate and other sphingolipid mediators. Nat Chem
Biol. 6:489–497. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Strub GM, Maceyka M, Hait NC, Milstien S
and Spiegel S: Extracellular and intracellular actions of
sphingosine-1-phosphate. Adv Exp Med Biol. 688:141–155. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Spiegel S and Milstien S: Functions of a
new family of sphin-gosine-1-phosphate receptors. Biochim Biophys
Acta. 1484:107–116. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wendler CC and Rivkees SA:
Sphingosine-1-phosphate inhibits cell migration and endothelial to
mesenchymal cell transformation during cardiac development. Dev
Biol. 291:264–277. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Seto E and Yoshida M: Erasers of histone
acetylation: The histone deacetylase enzymes. Cold Spring Harb
Perspect Biol. 6:a0187132014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sekiguchi K, Yokoyama T, Kurabayashi M,
Okajima F and Nagai R: Sphingosylphosphorylcholine induces a
hypertrophic growth response through the mitogen-activated protein
kinase signaling cascade in rat neonatal cardiac myocytes. Circ
Res. 85:1000–1008. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Robert P, Tsui P, Laville MP, Livi GP,
Sarau HM, Bril A and Berrebi-Bertrand I: EDG1 receptor stimulation
leads to cardiac hypertrophy in rat neonatal myocytes. J Mol Cell
Cardiol. 33:1589–1606. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wei L: Lysophospholipid signaling in
cardiac myocyte hypertrophy. J Mol Cell Cardiol. 36:465–468. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Nguyen-Tran DH, Hait NC, Sperber H, Qi J,
Fischer K, Ieronimakis N, Pantoja M, Hays A, Allegood J, Reyes M,
et al: Molecular mechanism of sphingosine-1-phosphate action in
Duchenne muscular dystrophy. Dis Model Mech. 7:41–54. 2014.
View Article : Google Scholar :
|
|
33
|
Nagahashi M, Takabe K, Liu R, Peng K, Wang
X, Wang Y, Hait NC, Wang X, Allegood JC, Yamada A, et al:
Conjugated bile acid-activated S1P receptor 2 is a key regulator of
sphingosine kinase 2 and hepatic gene expression. Hepatology.
61:1216–1226. 2015. View Article : Google Scholar :
|
|
34
|
Pearson R, Fleetwood J, Eaton S, Crossley
M and Bao S: Krüppel-like transcription factors: A functional
family. Int J Biochem Cell Biol. 40:1996–2001. 2008. View Article : Google Scholar
|
|
35
|
Yoshida T, Yamashita M, Horimai C and
Hayashi M: Kruppel-like factor 4 protein regulates
isoproterenol-induced cardiac hypertrophy by modulating myocardin
expression and activity. J Biol Chem. 289:26107–26118. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Liao X, Haldar SM, Lu Y, Jeyaraj D,
Paruchuri K, Nahori M, Cui Y, Kaestner KH and Jain MK: Krüppel-like
factor 4 regulates pressure-induced cardiac hypertrophy. J Mol Cell
Cardiol. 49:334–338. 2010. View Article : Google Scholar : PubMed/NCBI
|