Introduction
Glioma is the most common type of intracranial
primary malignant tumor, which is associated with a poor median
survival time of <15 months (1). Existing therapies include surgical
removal, chemotherapy and radiotherapy; however, they are often
unsuccessful (1–3). The difficulties in curing glioma are
due to uncontrollable proliferation and infiltrative growth
(1), which are considered to be
largely attributed to aberrant signaling (4).
Mitogen-activated protein kinase (MAPK) cascades
have been widely studied and are reported to be markedly altered in
glial tumors (4,5). Extracellular signal-regulated kinase
1/2 (ERK1/2) is a crucial member of the MAPK family, which contains
a conserved and dual-specificity motif (T-E-Y) that can be
phosphorylated on threonine (Thr)202 and tyrosine
(Tyr)204 residues. ERK1/2 is involved in the regulation
of cell cycle progression, proliferation, differentiation,
senescence and apoptosis (6). In
human glioma tissues, the expression levels of phosphorylated
(p)-ERK1/2 are significantly increased compared with in normal
brain tissues, and expression is correlated with glioma grade
(7,8), thus suggesting that aberrant
upregulation or activation of ERK1/2 may lead to malignant
progression of glioma. However, pharmacological inhibitors of
ERK1/2 are cytostatic at best, and only in a subset of patients
(4), thus indicating that other
unidentified factors or compensatory signals may affect the
survival and growth of tumor cells.
Nitric oxide (NO) is a short-lived free radical,
which serves critical roles in the regulation of cardiovascular,
immune and central nervous systems (9). S-nitrosylation refers to the
covalent addition of a NO group to a cysteine (Cys) thiol, and is
considered one of the important ways through which NO functions
(10). Protein S-nitrosylation
can alter spatial structure of proteins, and increase or decrease
protein activity and stability and subsequent signal transduction
and cellular processes (11).
Feng et al reported that ERK1 harbors six Cys residues and
that Cys183 is the key site for ERK1 nitrosylation
(12). The present study aimed to
investigate the association between ERK1/2 nitrosylation and ERK1/2
phosphorylation, and the effects of ERK1 S-nitrosylation at
Cys183 on glioma cell survival.
The results of the present study demonstrated that
treatment with the NO donors sodium nitroprusside (SNP) or
S-nitrosoglutathione (GSNO) induced an increase in ERK1/2
S-nitrosylation, and a reduction in ERK1/2 phosphorylation, which
were accompanied by growth inhibition of U251 glioma cells.
Mutational analysis [Cys183 to alanine
(Ala)183] uncovered that S-nitrosylation of ERK1
attenuated ERK1/2 phosphorylation, inhibited cell survival and
promoted apoptosis. In addition, the results detected an increase
in phosphorylation of ERK1/2 and a decrease in ERK1/2
S-nitrosylation in human glioma tissues. These findings identified
a novel mechanism of ERK1/2 underlying tumor cell development and
apoptotic resistance in glioma.
Materials and methods
Reagents and antibodies
Methyl methylthiomethyl sulfoxide (MMTS),
neocuproine, sodium ascorbate and GSNO were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany). SNP was obtained
from Beyotime Institute of Biotechnology (Haimen, China). PolyJet™
and Biotin-HPDP were purchased from Thermo fisher Scientific, Inc.
(Waltham, MA, USA). Antibodies against Flag (F1084; 1:1,000;
Sigma-Aldrich; Merck KGaA), ERK1/2 (ab17942; 1:1,000; Abcam,
Cambridge, UK), p-ERK1/2 (sc-81492; 1:1,000; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), and caspase-3 (GTX110543;
1:1,000; GeneTex, Inc., Irvine, CA, USA) were commercially
available.
Cell culture
The U251 glioma cell line was purchased from
Shanghai Cell Bank, Type Culture Collection Committee, Chinese
Academy of Sciences (Shanghai, China). The cells were cultured in
Dulbecco's modified Eagle's medium supplemented with 10% fetal
bovine serum (HyClone; GE Healthcare Life Sciences, Logan, UT, USA)
in a cell incubator containing 5% CO2 under saturated
humidity at 37°C. Cells were treated at 37°C with NO donors SNP
(0–2 µM) or GSNO (0–500 µM) for 36 h or with SNP (2
µM) or GSNO (500 µM) for different time (0–36 or 48
h) prior to further analysis.
Cell viability detection
Cell viability was measured using Cell Counting
kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto,
Japan). A single cell suspension (5×103/ml, 100
µl) was seeded into a 96-well plate. Subsequently, 10
µl CCK-8 reagent was added to each well and the plates were
incubated for 2 h at 37°C. Finally, the absorbance was measured at
450 nm using a scanning microplate reader. Cell viability at
individual time-points was normalized to the untreated group.
Flow cytometric apoptotic assay
Cells were harvested and washed twice with ice-cold
PBS, after which they were resuspended in 1X binding buffer [0.01 M
HEPES/NaOH (pH 7.4), 0.14 M NaCl, 2.5 mM CaCl2] at a
concentration of 106 cells/ml. Subsequently, 100
µl solution was transferred to a 5 ml cell culture tube and
was treated with fluorescein isothiocyanate-conjugated Annexin V
apoptosis detection kit I (BD Biosciences, Franklin Lakes, NJ, USA)
according to the manufacturer's protocol. The cells were analyzed
using flow cytometry (DiVa 8.0.1; BD Biosciences) and a total of
10,000 cells/sample were analyzed to determine the percentage of
apoptotic cells.
Western blot analysis
Total protein was extracted from the cells or
tissues, as described previously (13). Protein concentrations were
determined by a BCA protein assay kit (Beyotime Institute of
Biotechnology) according to the manufacturer's instructions. Equal
amounts of protein (30 µg) were mixed with SDS sample
buffer, separated by 10% SDS-PAGE and transferred to polyvinylidene
fluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were then incubated with 3% bovine serum albumin
(Sigma-Aldrich; Merck KGaA) in PBS at room temperature for 2 h, and
were treated with primary antibodies overnight at 4°C. β-actin
(sc-47778; 1:1,000; Santa Cruz Biotechnology, Inc.) was used as a
protein-loading control. The next day, membranes were incubated
with horseradish peroxidase-conjugated goat anti-rabbit
(31460)/mouse (31430) immunoglobulin G (1:4,000; Invitrogen; Thermo
fisher Scientific, Inc.) at room temperature for 2 h and were then
detected using a standard chemiluminescence detection system
(Pierce; Thermo fisher Scientific, Inc.). Band densities were
analyzed using ImageJ software (Image J 1.43u; National Institute
of Health, Bethesda, MD, USA).
NO detection
NO levels were determined by Griess assay using a
commercial kit (Nanjing Jiancheng Bioengineering Institute,
Nanjing, China). Cell supernatants (100 µl) were thoroughly
mixed with reagent I (200 µl). The supernatants were
obtained simply by a 200 µl pipette from the cell medium.
Subsequently, reagent II (100 µl) was added and mixed for 10
min at room temperature, followed by centrifugation at 1,700 × g
for 15 min at room temperature. Finally, 160 µl supernatants
were mixed with 80 µl chromogenic reagent for 15 min at room
temperature. The optical density of the samples was measured using
a spectrophotometer with absorbance set at 550 nm. Sodium nitrite
was used as a standard.
Biotin switch assay
Samples were lysed in non-denaturing lysis buffer
(25 mM HEPES, 50 mM NaCl, 0.1 mM EDTA, 1% NP-40 and 1X protease
inhibitor cocktail, pH 7.4). Protein concentration was determined
using the bicinchoninic acid protein assay kit (Beyotime Institute
of Biotechnology). Protein lysates (2 mg) were diluted to a final
volume of 1.8 ml using HEN buffer (100 mM HEPES, 1 mM EDTA and 0.1
mM neocuproine). Subsequently, 0.2 ml 25% SDS and 20 µl 10%
MMTS were added to block free thiols. After removing excess MMTS by
acetone precipitation, the S-nitrosothiol (SNO) groups in the
samples were reduced to thiols by 30 µl sodium ascorbate
(200 mM) and biotinylated by 30 µl Biotin-HPDP (2.5 mg/ml).
Finally, the biotinylated proteins were pulled down by
streptavidin-agarose beads, and the beads were eluted by SDS
loading buffer and subjected to western blot analysis.
Plasmids
Full-length wild-type ERK1 (ERK1WT) cDNA
clone was purchased from Sino Biological, Inc. (Beijing, China) and
was subcloned into the 3xFlag vector. The primer sequences used for
construction of Flag-ERK1WT were as follows: Forward,
5′-CCGGAATTCATGGCGGCGGCGGCGGCTCA-3′ and reverse,
5′-CGCGGATCCGGGGGCCTCCAGCACTCCGG-3′. C183A mutant ERK1
(ERK1C183A) cDNA was obtained by polymerase chain
reaction [PCR; Tiangen Biotech (Beijing) Co., Ltd., Beijing, China]
with the Flag-ERK1WT plasmid used as the template, and
was then subcloned into the 3xFlag vector. The primer sequences
used were as follows: forward, 5′-CCTTAAGATTGCTGATTTCGGCCTGGC-3′
and reverse, 5′-GCCGAAATCAGCAATCTTAAGGTCGCAG-3′. PCR thermocycling
was performed as follows: 94°C for 3 min; followed by 35 cycles at
90°C for 30 sec, 60°C for 45 sec and 72°C for 90 sec; 72°C for 10
min; hold at 4°C. The authenticity of the plasmids was confirmed by
DNA sequencing. Briefly, when U251 cells reached a confluency of
40–50%, the cell medium was replaced with 2 ml fresh DMEM medium at
30 min before transfection. Plasmid (1 µg) and PolyJet™
reagent (3 µl) were mixed in high glucose DMEM medium at
room temperature. The mixture was evenly added into the medium and
the cells were incubated at 37°C for 6–8 h before replacing the
medium with 2 ml fresh medium. After 48 h, the cells were used for
the experiments. Transient transfection was performed using
PolyJet™ according to the manufacturer's protocol. Expression of
proteins was verified by western blot analysis using ERK1/2 and
Flag antibodies.
Glioma and noncancerous human brain
tissue collection
Human glioma specimens, collected during surgical
resection, and noncancerous brain tissues, collected during
internal decompression after cerebral trauma, were obtained from
the Affiliated Hospital of Xuzhou Medical University (Xuzhou,
China). The clinicopathological characteristics of all of the
subjects are presented in Table
I. Surgically removed tissues were sampled for histological
diagnosis, and the remaining tissues were immediately frozen and
stored in liquid nitrogen for further analysis. All glioma
specimens had a confirmed pathological diagnosis and were
classified according to World Health Organization criteria
(14). Informed consent was
obtained from all subjects, or legal guardians, and the present
study was approved by the Medical Ethical Committee of Xuzhou
Medical University.
 | Table IClinicopathological characteristics
of the studied subjects. |
Table I
Clinicopathological characteristics
of the studied subjects.
| Case no. | Code no. | Gender | Age (years) | Location | WHO grade |
|---|
| 1 | 919616 | M | 57 | Right
cerebellum | Noncancerous |
| 2 | 912226 | F | 54 | Right temporal
lobe | Noncancerous |
| 3 | 972078 | F | 49 | Not available | Noncancerous |
| 4 | 968605 | F | 69 | Not available | Noncancerous |
| 5 | 981488 | M | 41 | Cerebellum | Noncancerous |
| 6 | 1095392 | M | 32 | Not available | Noncancerous |
| 7 | 1004728 | M | 63 | Right frontal
lobe | Noncancerous |
| 8 | 941814 | M | 20 | Cerebellum | Noncancerous |
| 9 | 928412 | M | 48 | Not available | Noncancerous |
| 10 | 970570 | M | 52 | Right frontal
lobe | Grade II |
| 11 | 1157139 | M | 42 | Right frontal
lobe | Grade II |
| 12 | 1145933 | F | 48 | Right frontal
lobe | Grade II |
| 13 | 1140811 | F | 49 | Right frontal
lobe | Grade II |
| 14 | 1164493 | M | 64 | Left insular
lobe | Grade II |
| 15 | 1190502 | M | 31 | Left frontal
lobe | Grade II |
| 16 | 1158620 | M | 43 | Right temporal
lobe | Grade II |
| 17 | 1196273 | M | 40 | Left frontal
lobe | Grade II |
| 18 | 1152968 | F | 63 | Left temporal
lobe | Grade II |
| 19 | 1110685 | F | 27 | Right frontal
lobe | Grade II |
| 20 | 1084447 | F | 52 | Not available | Grade II |
| 21 | 999737 | M | 52 | Left
frontal-temporal lobe | Grade III |
| 22 | 920498 | M | 50 | Right
temporal-parietal lobe | Grade III |
| 23 | 926714 | M | 56 | Bilateral temporal
lobe | Grade III |
| 24 | 1164248 | F | 66 | Left frontal
lobe | Grade III |
| 25 | 1191197 | M | 68 | Left
parietal-occipital lobe | Grade III |
| 26 | 922050 | F | 19 | Right frontal
lobe | Grade III |
| 27 | 1117547 | M | 47 | Right temporal
lobe | Grade III |
| 28 | 1081283 | F | 58 | Left temporal
lobe | Grade III |
| 29 | 947804 | F | 66 | Left
frontal-temporal-parietal lobe | Grade III |
| 30 | 1145935 | M | 23 | Cervical cord | Grade III |
| 31 | 1029589 | M | 31 | Right
frontal-temporal lobe | Grade III |
| 32 | 1147279 | F | 64 | Cerebellum | Grade IV |
| 33 | 1147166 | M | 58 | Left
frontal-temporal lobe | Grade IV |
| 34 | 1141904 | M | 47 | Right temporal
lobe | Grade IV |
| 35 | 1132842 | F | 62 | Left temporal
lobe | Grade IV |
| 36 | 1119597 | F | 50 | Right frontal
lobe | Grade IV |
| 37 | 1096129 | M | 61 | Left temporal
lobe | Grade IV |
| 38 | 1077922 | M | 43 | Left temporal
lobe | Grade IV |
| 39 | 1140776 | M | 26 | Right
parietal-occipital lobe | Grade IV |
| 40 | 1088070 | F | 58 | Right temporal
lobe | Grade IV |
| 41 | 1164493 | M | 66 | Left insular
lobe | Grade IV |
| 42 | 1184604 | F | 34 | Right temporal
lobe | Grade IV |
Statistical analysis
All quantitative data were obtained from at least
three independent experiments and are presented as the means ±
standard error of the mean [SPSS version 13.0 (SPSS, Inc., Chicago,
IL, USA)]. Data between two groups were assessed by Student's
t-test, whereas one-way analysis of variance followed by Dunnett
post hoc comparison was used to analyze data among three groups or
more. P<0.05 was considered to indicate a statistically
significant difference.
Results
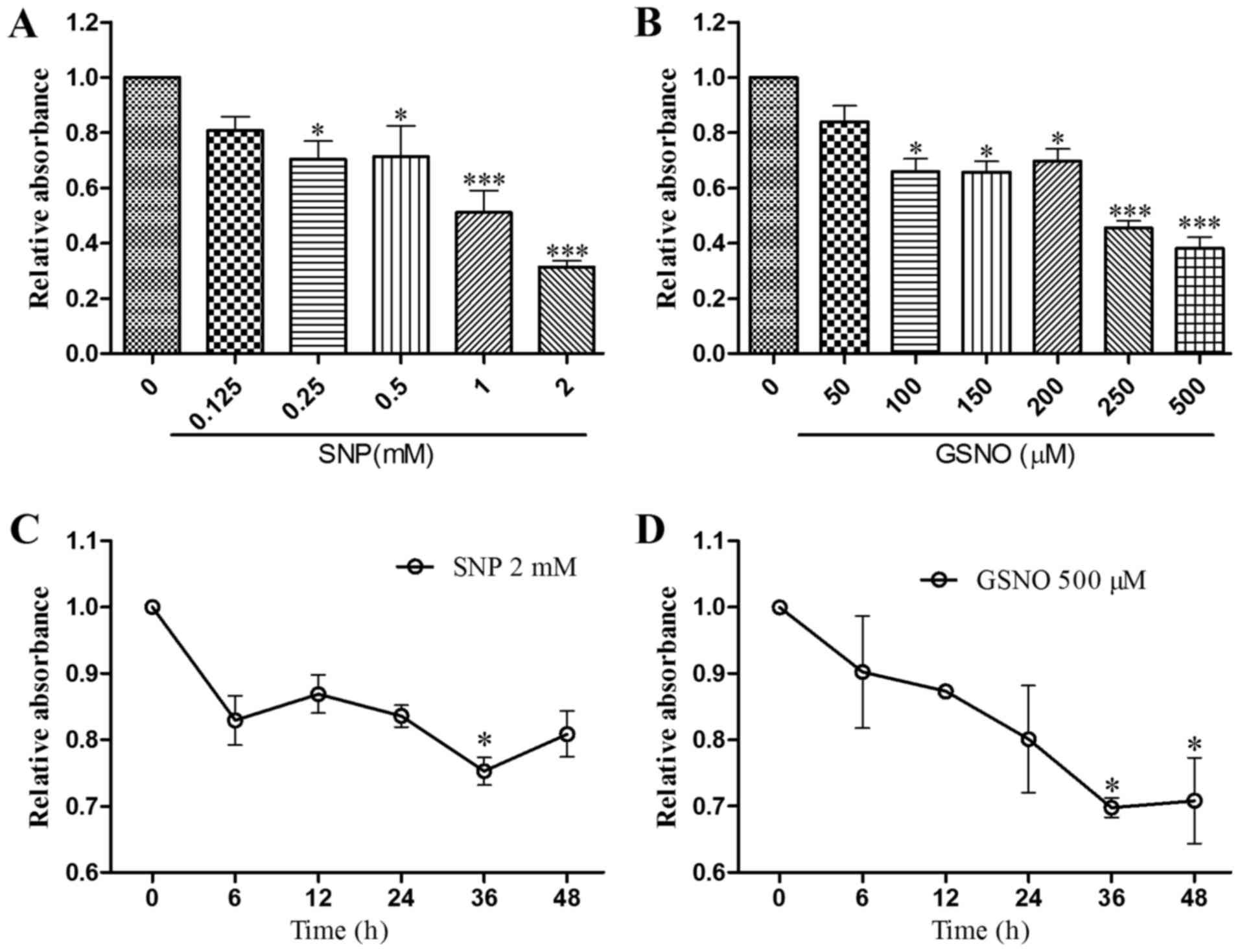
NO donor treatment inhibits growth of
glioma cells
According to the literature, treatment with the NO
donor SNP for 30–40 h significantly inhibits the growth of human
glioma cells (15); therefore,
the present study conducted a CCK-8 assay to evaluate the
concentration-dependent effects of SNP and GSNO on cell survival
after 36 h in U251 cells. Cell viability was significantly reduced
following treatment with 0.25 and 0.5 mM SNP or 100–200 µM
GSNO, and was reduced to a greater extent following treatment with
1 and 2 mM SNP or 250 and 500 µM GSNO (Fig. 1A and B). To determine the
appropriate duration of high-dose SNP or GSNO treatment, U251
glioma cells were treated with 2 mM SNP or 500 µM GSNO for
0, 6, 12, 24, 36 and 48 h. Significant inhibition of cell survival
was observed when cells were exposed to 2 mM SNP for 36 h or 500
µM GSNO for 36 and 48 h (Fig.
1C and D). These results indicated that high doses of NO donors
exert significant inhibitory effects on the growth of glioma
cells.
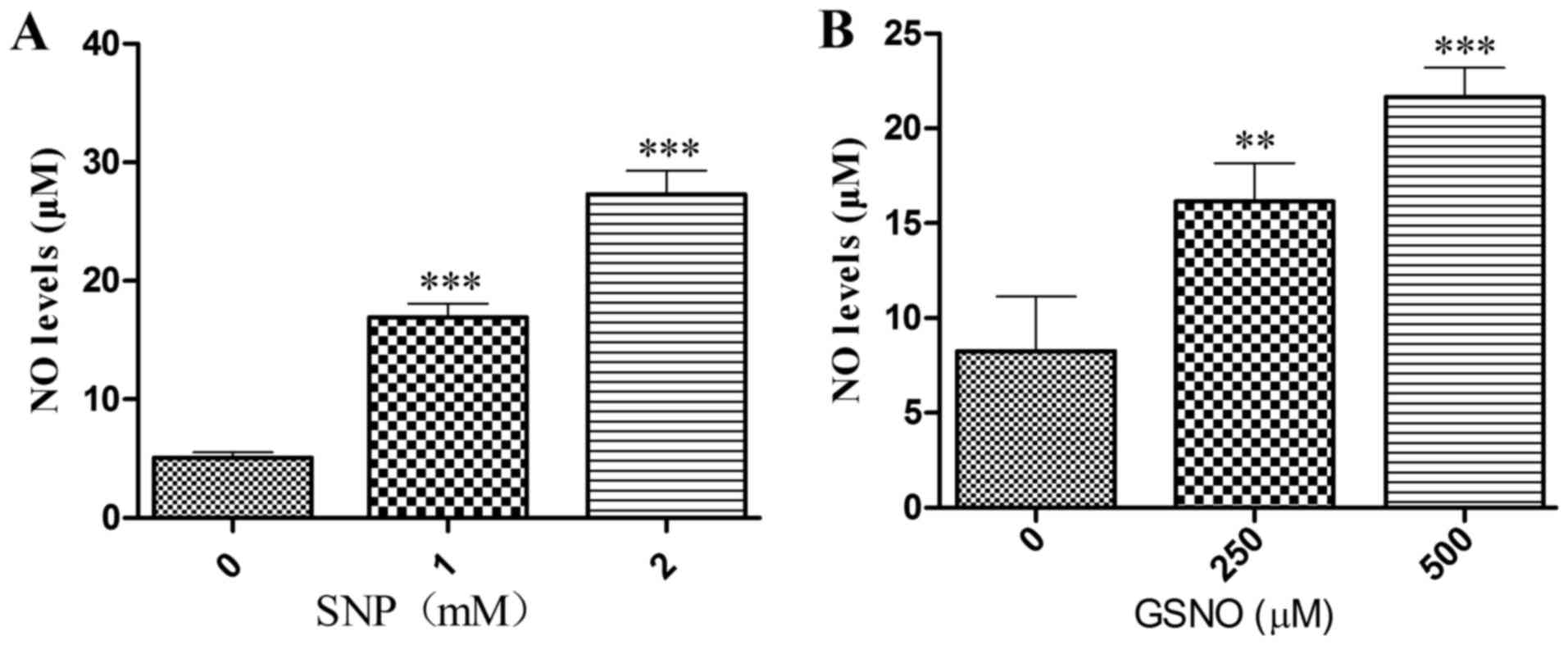
NO donor treatment increases NO release
into the cell supernatant
To ascertain whether the NO donors SNP and GSNO
could release NO, the Griess method was used to measure NO levels
in the supernatant of cultured U251 cells. Significant increases in
NO release were detected following treatment with 1 or 2 mM SNP
(Fig. 2A) and 250 or 500
µM GSNO (Fig. 2B). These
data suggested that SNP and GSNO may breakdown to release NO into
the culture supernatant of glioma cells.
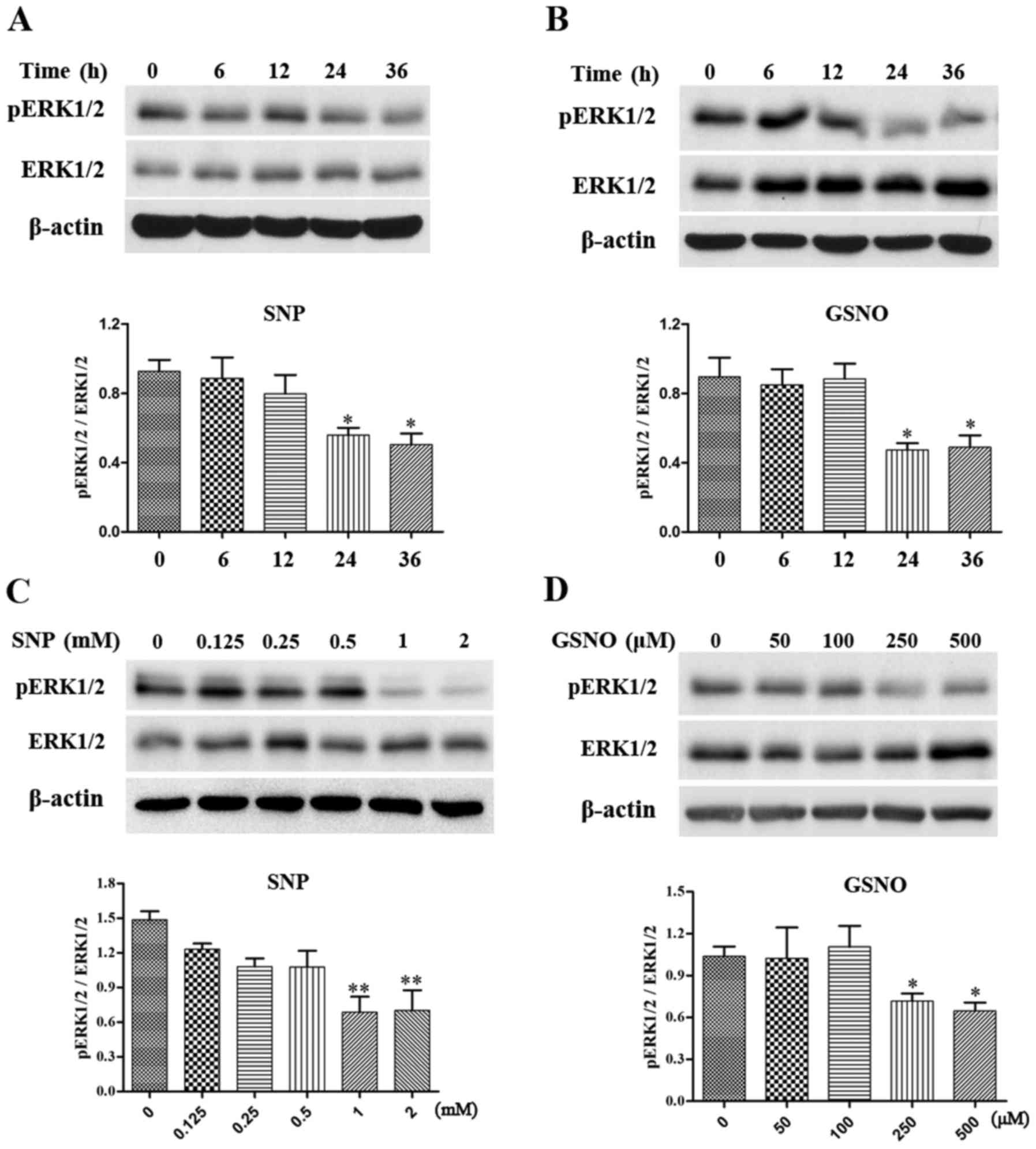
NO donor treatment attenuates
phosphorylation of ERK1/2 in glioma cells
The MAPK pathway has been reported to serve a
critical role in cell survival (6), and increased phosphorylation of
ERK1/2 has been detected in various grades of glioma (7,8).
The present study investigated the effects of NO donor treatment on
the expression levels of p-ERK1/2 in glioma cells. Initially, a
time-dependent assay was performed in U251 glioma cells. The
expression levels of p-ERK1/2 were significantly reduced following
treatment with 2 mM SNP or 500 µM GSNO treatment for 24 and
36 h (Fig. 3A and B).
Subsequently, U251 cells were exposed to various concentrations of
SNP or GSNO for 36 h. A concentration-dependent decrease in
p-ERK1/2 expression was evident in response to SNP and GSNO
treatment. Significant decreases in the expression levels of
p-ERK1/2 were observed following treatment with 1 and 2 mM SNP
(Fig. 3C). Similarly, p-ERK1/2
expression was significantly reduced following 250 and 500
µM GSNO treatment (Fig.
3D). These data indicated that NO donor treatment, particularly
in high concentrations, may attenuate the phosphorylation of ERK1/2
in glioma cells.
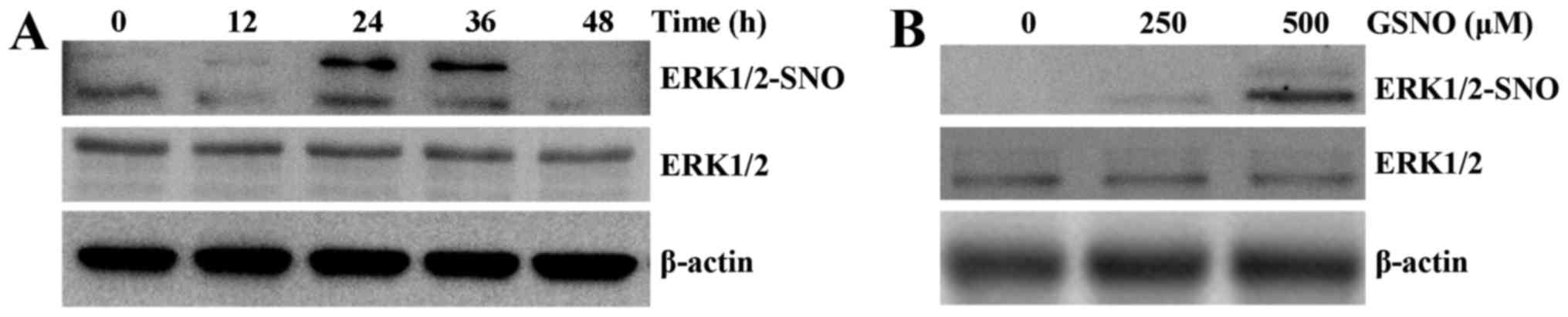
NO donor treatment promotes
S-nitrosylation of ERK1/2 in glioma cells
To determine whether ERK could be nitrosylated by
NO, S-nitrosylation of ERK1/2 (SNO-ERK1/2) was analyzed by a biotin
switch assay, followed by western blot analysis. Time- and
dose-dependent increases in the levels of SNO-ERK1/2 were detected
in response to GSNO treatment of U251 cells. The levels of
ERK1/2-SNO were markedly increased at 24 and 36 h, and then
returned to control levels at 48 h (Fig. 4A). ERK1/2-SNO was initially
detected following treatment with 250 µM GSNO and was
amplified with 500 µM GSNO treatment (Fig. 4B). These results suggested that
ERK1/2 may be nitrosylated in a time- and dose-dependent
manner.
S-nitrosylation of ERK1 is prevented by
mutation at Cys183
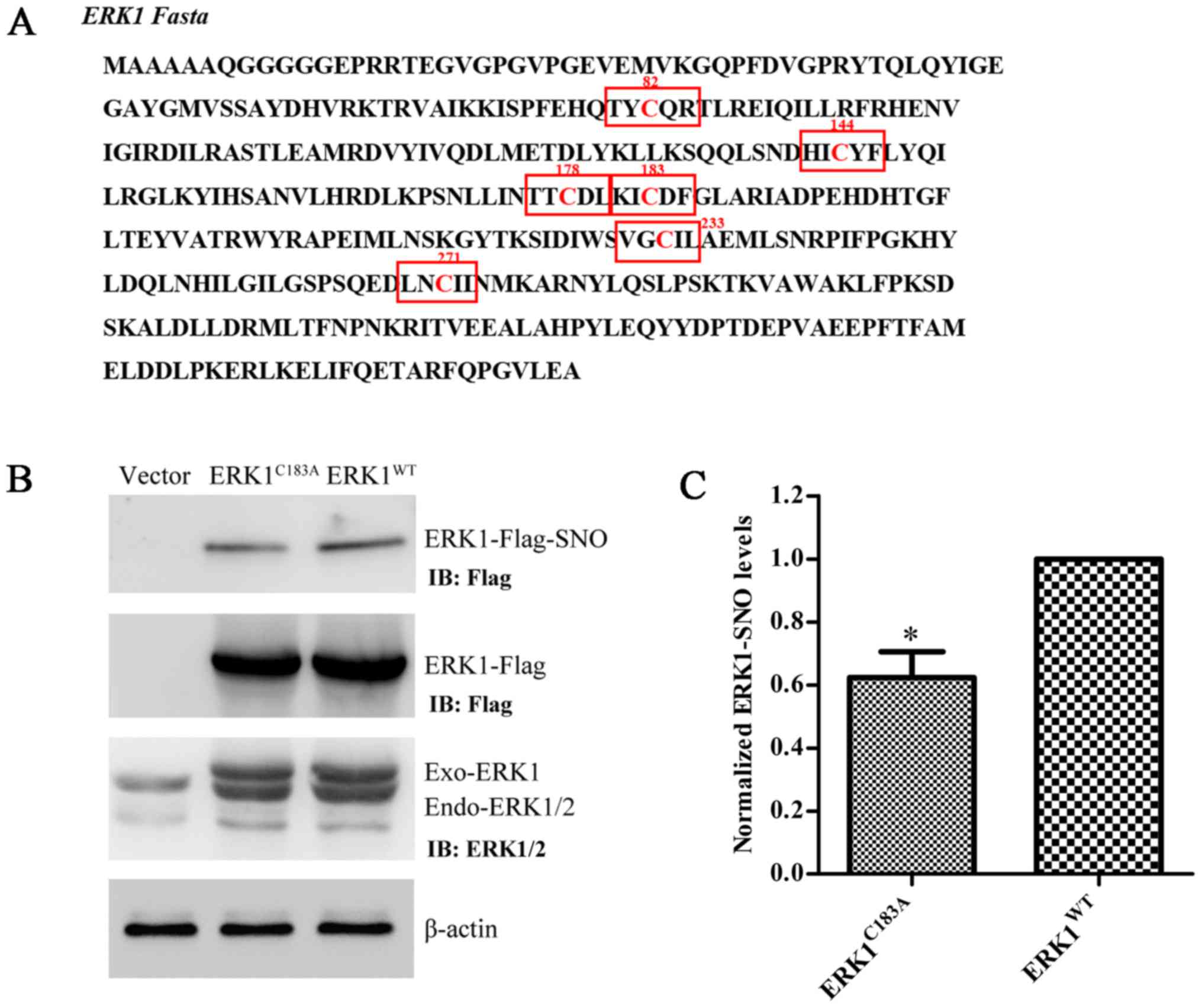
ERK1 has six Cys residues in its FASTA sequence
(Fig. 5A). A preliminary
computational prediction indicated that Cys183 is the
most probable site for nitrosylation, according to the previously
reported nitrosylation motif (K/R/H/D/E+C+D/E) (16). Therefore, Cys183 was
replaced with Ala, and the ERK1 mutant plasmid
(ERK1C183A) was constructed. Transfection efficiency of
ERK1WT and ERK1C183A was verified by western
blot analysis using anti-Flag and anti-ERK1/2 antibodies (Fig. 5B). The expression levels of
ERK1-SNO were significantly attenuated following transfection of
U251 cells with the ERK1C183A plasmid (Fig. 5B and C). These results suggested
that mutation at Cys183 may partially prevent
S-nitrosylation of ERK1/2 in glioma cells.
Preventing S-nitrosylation of ERK1
promotes ERK phosphorylation and cell survival
To determine the relationship between ERK1/2
nitrosylation and phosphorylation, U251 glioma cells were
transfected with either ERK1WT or ERK1C183A
plasmids, and were then treated with GSNO (500 µM). The
results indicated that the expression levels of p-ERK were
significantly decreased in the ERK1WT-transfected cells
when GSNO was added (Fig. 6A and
B), which was consistent with the previous findings presented
in Fig. 3D. However, GSNO failed
to reduce p-ERK1/2 levels when U251 cells were transfected with the
ERK1C183A mutant (Fig. 6A
and B). These findings indicated that preventing ERK1
S-nitrosylation may increase the phosphorylation of ERK1/2 in
glioma cells.
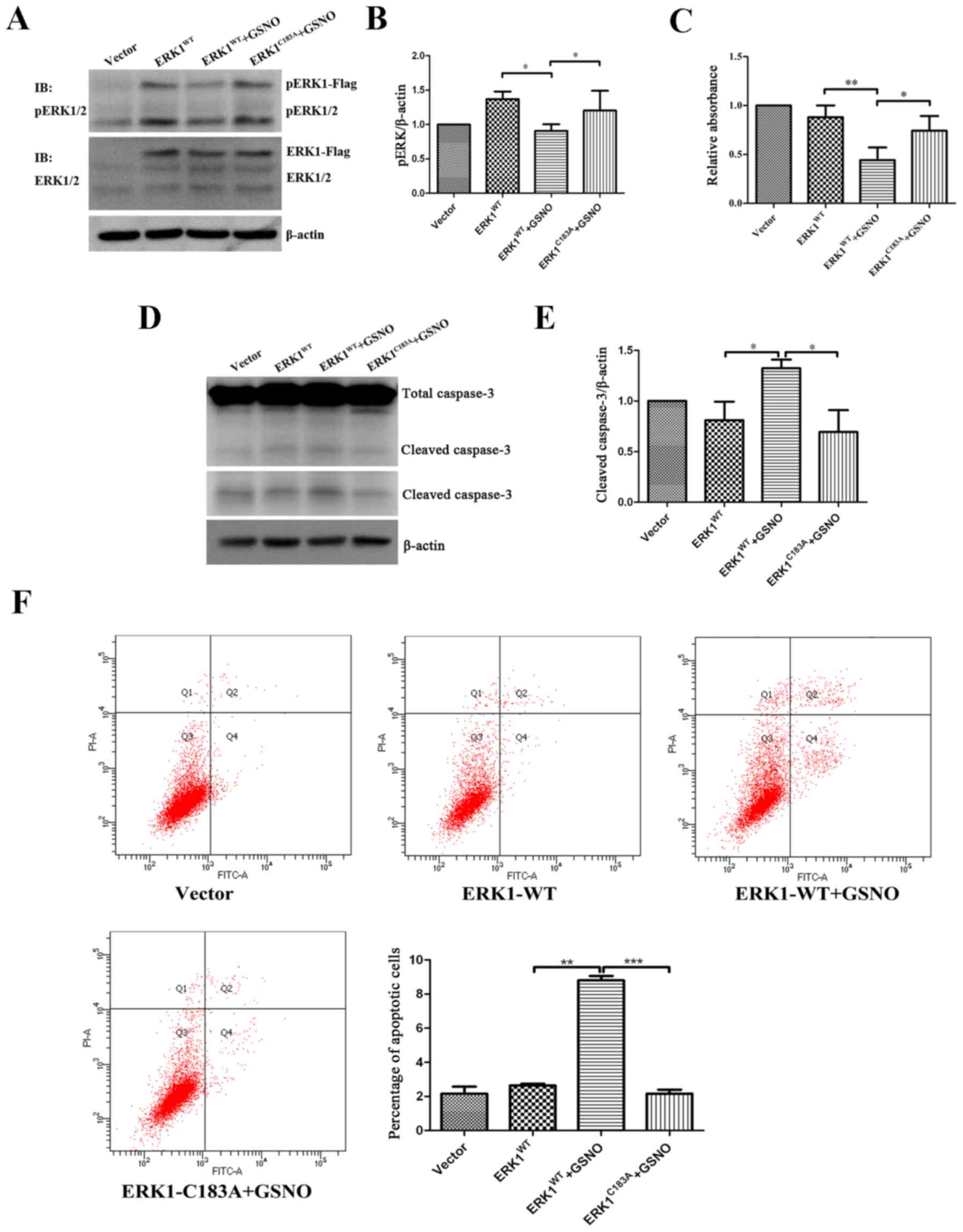 | Figure 6Preventing S-nitrosylation of ERK1
promotes ERK phosphorylation and cell survival, and suppresses
apoptosis. Following transfection of U251 glioma cells with
ERK1-Flag or ERK1 mutant form (ERKC183A), cells were
treated with 500 µM GSNO for 24 h. (A and B) p-ERK1/2 was
detected by western blotting and was semi-quantified. p-ERK1/2
levels were compared with β-actin levels, and results were
normalized to vector group. (C) Cell Counting kit-8 assay was
performed to examine the viability of U251 glioma cells. Cell
survival percentage was normalized to the vector group. (D and E)
Caspase-3 protein expression was detected by western blotting and
semi-quantified. Cleaved caspase-3 levels were compared with
β-actin levels, and results were normalized to the vector group.
The lower panel of cleaved caspase-3 blot in part D was obtained
after a longer exposure time compared with the upper panel. (F)
Flow cytometric detection of apoptosis of U251 glioma cells. The
percentage of apoptotic cells was quantified and compared.
*P<0.05, **P<0.01 and
***P<0.001. IB, immunoblotting; ERK, extracellular
signal-regulated kinase; fITC, fluorescein isothiocyanate; GSNO,
S-nitrosoglutathione; p-ERK1/2, phosphorylated ERK1/2; PI,
propidium iodide; WT, wild-type. |
The present study also investigated the effects of
ERK1 S-nitrosylation prevention on the survival of glioma cells. In
line with the data presented in Fig.
1, cell viability was significantly reduced following treatment
with GSNO; however, the reduction in cell viability was reversed by
a point mutation at Cys183 of ERK1 (Fig. 6C). Furthermore, western blot
analysis demonstrated that GSNO treatment induced an increase in
cleaved caspase-3 expression; however, this was reversed following
transfection with the ERK1C183A mutant (Fig. 6D and E). In addition, flow
cytometric apoptotic assay indicated that the percentage of U251
apoptotic cells transfected with ERK1C183A mutant was
significantly reduced following GSNO treatment compared with in the
cells transfected with the ERK1WT plasmid (Fig. 6F). Taken together, these results
suggested that preventing S-nitrosylation of ERK1, via transfection
with a ERK1C183A mutant, partially reversed GSNO-induced
decreases in ERK phosphorylation and cell survival in U251 glioma
cells.
Alterations in ERK1/2 phosphorylation and
S-nitrosylation levels in human glioma tissues
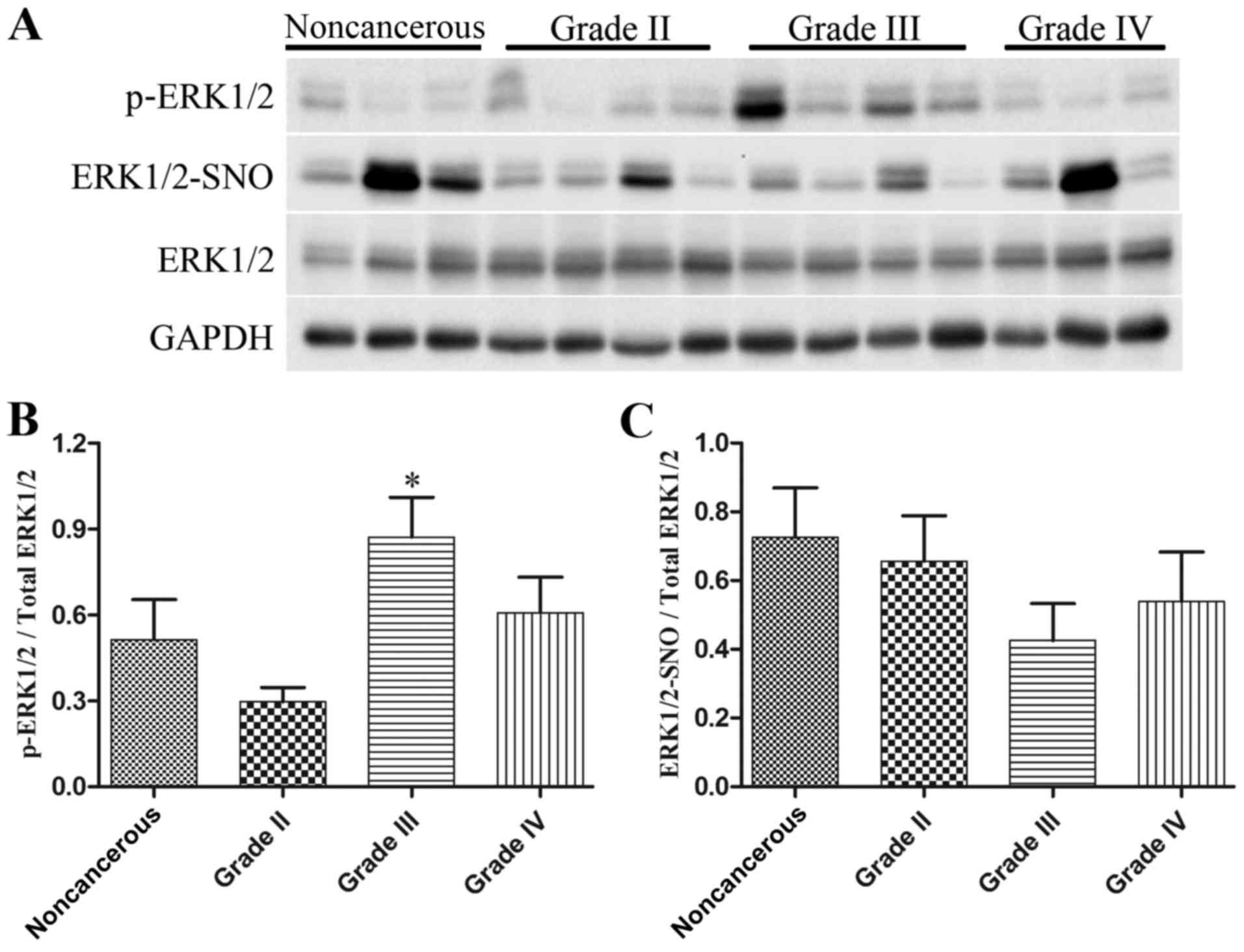
Western blot analysis was employed to detect
p-ERK1/2 and total ERK1/2 levels, and a biotin switch assay
followed by western blot analysis was used to measure ERK1/2-SNO
levels, in 9 noncancerous brain tissues and 33 glioma specimens
(n=11/grade). As presented in Fig.
7, the expression levels of p-ERK1/2 were increased in
high-grade glioma tissues, particularly in glioma grade III
(Fig. 7A and B), whereas there
was a marked reduction in ERK1/2-SNO levels in glioma tissues,
which was also evident in glioma grade III (Fig. 7A and C). These data provided in
vivo evidence for the possible influence of ERK1/2
S-nitrosylation on ERK1/2 phosphorylation during glioma
proliferation.
Discussion
NO donors, SNP and GSNO, breakdown to release NO and
exert an inhibitory effect on cell survival in glioma cells. In the
present study, NO donor treatment induced a significant decrease in
p-ERK1/2 expression (Fig. 3) and
a marked increase in ERK1/2-SNO levels (Fig. 4) in U251 cells, thus suggesting a
link between ERK1/2-SNO and p-ERK/2. Further mutational analysis
demonstrated that Cys183 was vital for S-nitrosylation
of ERK1 (Fig. 5) and that
preventing the formation of ERK-SNO by ERK1C183A
mutation reversed NO-induced suppression of cell viability and
p-ERK1/2 expression, and increased cell apoptosis of glioma cells
(Fig. 6). In addition, increased
p-ERK1/2 levels were observed in human glioma tissues, which were
accompanied by a marked decrease in ERK1/2-SNO levels (Fig. 7). These findings indicated a novel
mechanism underlying the antitumor role of NO-associated chemicals
and provided insights into gene therapy targeting the ERK1/2
pathway in glioma.
NO is a free radical, which predominantly functions
as a messenger or effector molecule. Previous studies have reported
that the viability of U87 and C6 cells may be significantly
inhibited following exposure to high concentrations of NO donors
(15,17). The present study demonstrated that
treatment with the NO donors SNP or GSNO resulted in a significant
reduction in the viability of U251 cells. These data suggested that
the inhibitory effects of NO on cell survival could be generalized
in various glioma cell lines. However, previous evidence also
suggests that NO displays stimulatory and inhibitory effects in the
context of cell survival and apoptosis. Maejima et al
reported that low doses of the NO donor
S-mitroso-N-acetyl-D,L-penicillamine favor cell survival, whereas
high doses may reduce cell viability of cardiomyocytes (18). Lechner et al demonstrated
that low levels of NO produced by the tumor microenvironment favor
tumor cell survival, whereas tumor cells with high NO levels
undergo cell death (19). The
dual effects of NO may be ascribed to the availability of enzymes,
timing of apoptotic stimuli, redox state, donor doses, spatial
location of key reactants and interactions with other molecules
(20).
S-nitrosylation is involved in the regulation of
numerous biological processes, including cell proliferation and
survival, and particularly apoptosis (11,21). S-nitrosylation of B-cell lymphoma
2 enhances its stability, inhibits ubiquitous degradation in
numerous tumor types and induces resistance to cis-platinum
chemotherapy in breast cancer (22,23). Furthermore, S-nitrosylation of the
death receptor Fas initiates its redistribution on lipid rafts and
promotes Fas ligand-mediated apoptosis in cancer (24). ERK1 harbors six Cys residues, as
indicated in Fig. 5A.
Cys183 is the most probable site for S-nitrosylation,
according to the S-nitrosylation motif (K/R/H/D/E+C+D/E) reported
previously (16). The present
study indicated that ERK1/2 may be nitrosylated by the NO donor
GSNO, and that replacing Cys183 with alanine may prevent
the S-nitrosylation of ERK1/2 in glioma cells. The small decrease
in ERK1-SNO levels in response to Cys183 mutation
indicates that other ERK1 Cys residues may also contribute to
S-nitrosylation. Nevertheless, the ERK1C183A mutation
significantly reversed GSNO-induced suppression of cell viability
and enhancement of apoptosis of glioma cells. Together with a
previous study in breast cancer cells (12), these findings suggested that
ERK-SNO may promote tumor cell apoptosis.
Within the MAPK cascades, the ERK1/2 signaling
pathway is a principle pathway that regulates cell proliferation
and survival when activated by phosphorylation at Thr202
and Tyr204 residues of ERK1 (5,25).
Activation of ERK1/2 signaling in glioma tissues, as determined by
increased p-ERK1/2 levels, has been detected in the present study,
as well as in previous studies (7,8).
The S-nitrosylation site Cys183 is spatially close to
Thr202 and Tyr204, thus suggesting the
possibility of mutual regulation between S-nitrosylation and
phosphorylation of ERK1/2. The present in vitro results
indicated that treatment with GSNO induced a reduction in p-ERK1/2
expression, an increase in ERK1/2-SNO levels, and cell growth
inhibition in glioma cells. In vivo, the results
demonstrated that p-ERK1/2 levels were increased, whereas
ERK1/2-SNO levels were decreased in glioma tissues, particularly in
glioma grade III. Furthermore, a point mutation at
Cys183 confirmed that preventing formation of ERK1-SNO
significantly increased p-ERK1/2 expression and reversed
GSNO-induced cell apoptosis in U251 glioma cells. These findings
suggested a regulatory role of ERK1/2 S-nitrosylation on ERK1/2
phosphorylation, which may provide novel information regarding
ERK1/2 targeting in glioma therapy.
In addition to ERK1/2, other important signaling
proteins are also modified by S-nitrosylation. Murillo-Carretero
et al reported that S-nitrosylation of epidermal growth
factor receptor (EGFR) inhibited EGFR phosphorylation and cell
proliferation in neuroblastoma cells (26). In head and neck squamous cell
carcinoma, S-nitrosylation of signal transducer and activator of
transcription 3 (STAT3) and nuclear factor (NF)-κB inhibited
phosphorylation of STAT3 and activation of NF-κB, and decreased
cell proliferation and increased apoptosis (27). These studies indicated a critical
role of S-nitrosylation in the regulation of protein
phosphorylation and cellular biological functions. Notably, several
NO-hybridized drugs have been developed to inhibit cancer cell
growth in vitro and in vivo (28,29), thus suggesting the potential
translational relevance of NO-mediated S-nitrosylation in the
future.
A few limitations should be mentioned with regards
to the present study. All in vitro work presented in this
study was performed in U251 glioma cells. In this respect,
duplication of efforts in other glioma cell lines would be
beneficial. In addition, it is necessary to perform brain xenograft
experiments to confirm the inhibitory role of ERK1 S-nitrosylation
on ERK1/2 phosphorylation and glioma growth.
In conclusion, NO donor treatment inhibited cell
survival and induced apoptosis of U251 glioma cells.
S-nitrosylation of ERK1/2 and ERK1/2 phosphorylation exhibited
inverse alterations in GSNO-treated glioma cells and in human
glioma tissues. Preventing ERK1 nitrosylation via a mutation at
Cys183 partially reversed NO-induced decreases in ERK
phosphorylation and cell survival. These findings revealed a novel
mechanism of ERK1/2 underlying tumor cell development and apoptotic
resistance of glioma.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 31400930, 81472345
and 81302175), the Natural Science Foundation of Jiangsu province
(grant no. BK20140217), the China Postdoctoral Science Foundation
(grant nos. 2015M570480 and 2016T90505) and the Key Research and
Development Plan of Jiangsu Province (grant no. BE2016646).
References
|
1
|
Stupp R, Hegi ME, Gilbert MR and
Chakravarti A: Chemoradiotherapy in malignant glioma: Standard of
care and future directions. J Clin Oncol. 25:4127–4136. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sanai N, Alvarez-Buylla A and Berger MS:
Neural stem cells and the origin of gliomas. N Engl J Med.
353:811–822. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Roberts PJ and Der CJ: Targeting the
Raf-MEK-ERK mitogen-activated protein kinase cascade for the
treatment of cancer. Oncogene. 26:3291–3310. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pandey V, Bhaskara VK and Babu PP:
Implications of mitogen-activated protein kinase signaling in
glioma. J Neurosci Res. 94:114–127. 2016. View Article : Google Scholar
|
|
6
|
Dong Chen, Waters SB, Holt KH and Pessin
JE: SOS phosphorylation and disassociation of the Grb2-SOS complex
by the ERK and JNK signaling pathways. J Biol Chem. 271:6328–6332.
1996. View Article : Google Scholar
|
|
7
|
Bhaskara VK, Panigrahi M, Challa S and
Babu PP: Comparative status of activated ERK1/2 and PARP cleavage
in human gliomas. Neuropathology. 25:48–53. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Xie H, Xue YX, Liu LB, Wang P, liu YH and
Ying HQ: Expressions of matrix metalloproteinase-7 and matrix
metalloproteinase-14 associated with the activation of
extracellular signal-regulated kinase1/2 in human brain gliomas of
different pathological grades. Med Oncol. 28(Suppl 1): S433–S438.
2011. View Article : Google Scholar
|
|
9
|
Lundberg JO, Gladwin MT and Weitzberg E:
Strategies to increase nitric oxide signalling in cardiovascular
disease. Nat Rev Drug Discov. 14:623–641. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nakamura T and Lipton SA: Protein
S-nitrosylation as a therapeutic target for neurodegenerative
diseases. Trends Pharmacol Sci. 37:73–84. 2016. View Article : Google Scholar :
|
|
11
|
Wang Z: Protein S-nitrosylation and
cancer. Cancer Lett. 320:123–129. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Feng X, Sun T, Bei Y, Ding S, Zheng W, lu
Y and Shen P: S-nitrosylation of ERK inhibits ERK phosphorylation
and induces apoptosis. Sci Rep. 3:18142013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shen A, Gao S, Ben Z, Wang H, Jia J, Tao
T, Niu S, Li X and Cheng C: Identification and potential role of
PSD-95 in Schwann cells. Neurol Sci. 29:321–330. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Louis DN, Ohgaki H, Wiestler OD and
Cavenee WK: WHO Classification of Tumours of the Central Nervous
System. IARC WHO Classification of Tumours; Lyon: 2016
|
|
15
|
Kurimoto M, Endo S, Hirashima Y, Hamada H,
Ogiichi T and Takaku A: Growth inhibition and radiosensitization of
cultured glioma cells by nitric oxide generating agents. J
Neurooncol. 42:35–44. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Stamler JS, Toone EJ, Lipton SA and Sucher
NJ: (S)NO signals: Translocation, regulation, and a consensus
motif. Neuron. 18:691–696. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Weyerbrock A, Baumer B and Papazoglou A:
Growth inhibition and chemosensitization of exogenous nitric oxide
released from NONOates in glioma cells in vitro. J Neurosurg.
110:128–136. 2009. View Article : Google Scholar
|
|
18
|
Maejima Y, Adachi S, Morikawa K, Ito H and
Isobe M: Nitric oxide inhibits myocardial apoptosis by preventing
caspase-3 activity via S-nitrosylation. J Mol Cell Cardiol.
38:163–174. 2005. View Article : Google Scholar
|
|
19
|
Lechner M, Lirk P and Rieder J: Inducible
nitric oxide synthase (iNOS) in tumor biology: The two sides of the
same coin. Semin Cancer Biol. 15:277–289. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lancaster JR Jr and Xie K: Tumors face NO
problems. Cancer Res. 66:6459–6462. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang Y, Chen C, Loake GJ and Chu C: Nitric
oxide: Promoter or suppressor of programmed cell death. Protein
Cell. 1:133–142. 2010. View Article : Google Scholar
|
|
22
|
Azad N, Vallyathan V, Wang L,
Tantishaiyakul V, Stehlik C, leonard SS and Rojanasakul Y:
S-nitrosylation of Bcl-2 inhibits its ubiquitin-proteasomal
degradation. A novel antiapoptotic mechanism that suppresses
apoptosis. J Biol Chem. 281:34124–34134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chanvorachote P, Nimmannit U, Stehlik C,
Wang L, Jiang BH, Ongpipatanakul B and Rojanasakul Y: Nitric oxide
regulates cell sensitivity to cisplatin-induced apoptosis through
S-nitrosylation and inhibition of Bcl-2 ubiquitination. Cancer Res.
66:6353–6360. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Leon-Bollotte L, Subramaniam S, Cauvard O,
Plenchette-Colas S, Paul C, Godard C, Martinez-Ruiz A, Legembre P,
Jeannin JF and Bettaieb A: S-nitrosylation of the death receptor
fas promotes fas ligand-mediated apoptosis in cancer cells.
Gastroenterology. 140:2009–2018. 2018.e2001–2004. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sebolt-Leopold JS and Herrera R: Targeting
the mitogen-activated protein kinase cascade to treat cancer. Nat
Rev Cancer. 4:937–947. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Murillo-Carretero M, Torroglosa A, Castro
C, Villalobo A and Estrada C: S-nitrosylation of the epidermal
growth factor receptor: A regulatory mechanism of receptor tyrosine
kinase activity. Free Radic Biol Med. 46:471–479. 2009. View Article : Google Scholar
|
|
27
|
Kaliyaperumal K, Sharma A K, McDonald DG,
Dhindsa JS, Yount C, Singh AK, Won JS and Singh I:
S-nitrosoglutathione-mediated STAT3 regulation in efficacy of
radiotherapy and cisplatin therapy in head and neck squamous cell
carcinoma. Redox Biol. 6:41–50. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chattopadhyay M, Goswami S, Rodes DB,
Kodela R, Velazquez CA, Boring D, Crowell JA and Kashfi K:
NO-releasing NSAIDs suppress NF-κB signaling in vitro and in vivo
through S-nitrosylation. Cancer Lett. 298:204–211. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Szabo C: Gasotransmitters in cancer: From
pathophysiology to experimental therapy. Nat Rev Drug Discov.
15:185–203. 2016. View Article : Google Scholar
|