Introduction
Ghrelin was first identified in 1999 by Kojima et
al as the endogenous ligand of the long-known growth hormone
secretagogue receptor 1a (GHS-R1a) isoform (1). Ghrelin-positive X/A-like cells
distributed throughout the gastric oxyntic mucosa (2,3)
are the main source of circulating ghrelin (4), as demonstrated by the sharp decline
in ghrelin levels following gastrectomy (5). Ghrelin has been also been detected
in the central nervous system in the arcuate nucleus (ARC) of the
hypothalamus (6), as well as in
neurons adjacent to the third ventricle (7). The ARC is strongly implicated in the
regulation of food intake. Ghrelin-containing neurons in the ARC
send projections to neuropeptide Y (NPY) and agouti-related peptide
(AgRP)-positive neurons (8). NPY
and AgRP are orexigenic neuropeptides and are regulated by ghrelin
(9). The peripheral injection of
ghrelin was found to selectively activate NPY-containing neurons in
the ARC in mice (10). Similarly,
the intracerebroventricular (ICV) administration of ghrelin
activates NPY/AgRP-expressing neurons and stimulates the expression
of NPY and AgRP mRNA in the ARC (11). Total ghrelin levels are inversely
correlated with the body mass index, as they increase in anorexic
and cachectic patients and decrease under conditions of obesity
(12). In humans and other
mammals, ghrelin levels increase before meals, and decline rapidly
postprandially (13,14). The postprandial suppression of
plasma ghrelin has been considerably more extensively investigated
compared with the preprandial peak. Although the physiological
importance of this event remains unclear, the suppression of this
orexigenic hormone may play a role in the satiating effect of
ingested nutrients (15).
Furthermore, the brain mechanism underlying the preprandial peak of
plasma ghrelin remains unknown. ARC neurons are destroyed by
neonatal administration of monosodium glutamate (MSG). The
destruction of the ARC by neonatal administration of MSG leads to a
significant decrease in the number of ARC neurons (16). This is an effect attributed to the
underdeveloped blood-brain barrier (BBB) in this area, allowing MSG
to penetrate the brain (16,17). Other areas with a weak BBB were
destroyed by neonatal MSG administration, including the area
postrema (AP) (16,18). One of the most notable effects of
neonatal MSG treatment is obesity in adult mice (16) and rats (19). The aim of the present study was to
test whether ARC lesions affect the ghrelin level in the plasma and
stomach in MSG-treated mice.
Materials and methods
Animals
All animal experiments (total number of animals, 58)
were conducted in accordance with the guidelines for animal care of
Qingdao University. A total of 33 neonatal Kunming mice (obtained
from the Laboratory Animal Center of Shandong University of
Traditional Chinese Medicine; license: SCXK Lu 20050015) were
subcutaneously injected into the dorsal dermis area, just below the
interscapular region on days 1, 3, 5, 7 and 9 after birth with 10
μl MSG (Sigma-Aldrich; Merck KGaA, St. Louis, MO, USA) to
deliver 4 mg/g (body mass), or with equivalent volumes of 0.9%
saline. A 24.2% death rate occurred in MSG-treated pups, while
there were no deaths among the 25 pups injected with saline.
However, 6 animals (3 treated with MSG and 3 with saline) were
excluded from the analysis, as their body mass index was low and
>2 standard deviations from the mean. Finally, 22 animals were
treated with MSG (10 male and 12 female) and 22 with saline (10
male and 12 female). At 4 weeks of age, the pups were weaned, bred
and housed in groups according to treatment. The mice were housed
in air-conditioned animal quarters, with lights on from 8:00 a.m.
to 9:00 p.m., and were provided with food and water ad
libitum.
Food deprivation, serum and tissue
harvesting
Food intake was measured weekly after weaning. At 12
weeks of age, food was discontinued; in addition, the beddings were
removed and replaced with new beddings. In order to measure the
response to fasting, mice (n=8 per group) were provided with water
(but no food) for 48 h. After fasting, food was provided. The body
mass index was measured before and after food deprivation. Before
fasting, at 48 h of fasting and after re-feeding, the animals were
lightly anesthetized with diethyl ether and an orbital blood sample
was collected for the measurement of serum ghrelin. Blood was
collected into EDTA tubes containing 500 KIU of aprotinin,
centrifuged at 4°C for 15 min at 1,500 × g, and the separated serum
was stored at −80°C until use. All samples obtained from each
subject were run in duplicate in the same assay. A commercially
available mouse ghrelin EIA kit (Phoenix Pharmaceuticals, Belmont,
CA, USA) was used. The sensitivity of the assay was 0.07 ng/ml. The
intra- and inter-assay error was 5-10% and <15%, respectively.
The remaining animals were divided into the before fasting (MSG,
n=7; saline, n=7) and fasting for 48 h (MSG, n=7; saline, n=7)
groups. The mice were deeply anesthetized with a lethal dose of
pentobarbital (30 mg/kg, i.p.) before fasting or after fasting for
48 h. Then, the stomachs and brains were quickly removed and rinsed
with double-distilled water. The stomach was deep-frozen in liquid
nitrogen for reverse transcription (RT)-polymerase chain reaction
or western blot analysis. The bilateral inguinal white adipose
tissue (IWAT), bilateral retroperitoneal WAT (RWAT), bilateral
gonadal WAT (GWAT) and interscapular brown adipose tissue depots
were quickly removed and weighed.
Perfusions and immunohistochemistry
The animals were perfused with 25 ml isotonic
saline, followed by 25 ml 4% paraformaldehyde in 0.01 M
phosphate-buffered saline (PBS) solution (pH 7.2). The brains were
quickly dissected out and stored in 4% paraformaldehyde, then
transferred to 20% sucrose (0.1% sodium azide) for 24 h and 30%
sucrose (0.1% sodium azide) for 48 h. The brains were embedded in
optimal cutting temperature compound (Sakura Finetek USA, Inc.,
Torrance, CA, USA) and sectioned in the coronal plane on a freezing
microtome (Kryostat 1720; Leica, Mannheim, Germany) at a thickness
of 20 μm. Brain sections were stored in an ultra-cold
freezer at −40°C for immunohistochemistry staining.
For immunohistochemistry staining, the sections were
first rinsed with distilled water and immersed in PBS for 5 min.
Next, the sections were treated with a solution containing 3%
H2O2 (V/V) and 0.5% Triton X-100 in 0.01 M
PBS for 30 min to block endogenous peroxidase activity.
Subsequently, the sections were treated with PBS containing 10%
normal goat serum for 30 min to prevent non-specific binding of
secondary antibodies, followed by incubation overnight at 4°C with
primary antibodies: Rabbit anti-NPY (dilution, 1:6,000; N9528), and
mouse anti-tyrosine hydroxylase (TH; dilution, 1:4,000; T1299)
(both from Sigma-Aldrich; Merck KGaA). The sections were next
rinsed with PBS three times and incubated for 30 min at room
temperature with biotin-conjugated secondary antibodies: Goat
anti-mouse (TH; dilution, 1:500; sc-2039) and goat anti-rabbit
(NPY; dilution, 1:500; sc-2040) (both from Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA). Subsequently, the
sections were rinsed with PBS three times and immersed in a
horseradish peroxidase-conjugated streptavidin complex for 30 min
at room temperature, then rinsed again with PBS three times. The
immunoreaction was visualized by DAB staining (DAB substrate kit;
Zhongshan Golden Bridge Biotechnology Co., Ltd., Beijing, China)
for 5 min and observed under an Olympus BX50 microscope (Olympus
Corporation, Tokyo, Japan). The reaction was terminated by rinsing
the sections with distilled water. The sections were covered by
neutral balata after counterstaining and dehydration. Negative
controls were determined by omission of the primary antibody. Three
representative sections of ARC or AP were selected from each mouse.
In each section, an area within the ARC or AP was selected for
counting TH- or NPY-positive neurons and optical density analysis.
The amount and mean optical density of TH- or NPY-IR fiber
staining, as well as Nissl staining, were obtained using an Olympus
BX50 microscope (Olympus Corporation); these were analyzed using
image analysis software (Compix, Inc., Arizona, USA).
RT of extracted tissue RNA
Total RNA was extracted from the stomach tissues
(~200 mg) of each mouse using TRIzol reagent, according to the
manufacturer's instructions. RT was performed using the AMV Reverse
Transcriptase system (Promega Corp., Madison, WI, USA). The ghrelin
cDNA fragment (108 bp) was amplified with the following primers:
Forward, 5′-TCAGGAGCTCAGTATCAGCAGCA-3′ and reverse,
5′-GCCTGTCCGTGGTTACTTGTCA-3′; β-actin (171 bp) was amplified with
the following primers: Forward, 5′-CATCCGTAAAGACCTCTATGCCAAC-3′ and
reverse, 5′-ATGGAGCCACCGATCCACA-3′. The DNA was immediately
amplified with a single cycle at 94°C for 5 min, followed by 30
cycles at 94°C for 30 sec, 58°C for 30 sec and 72°C for 30 sec; and
a final extension step was performed at 72°C for 10 min. Ethidium
bromide stained gels were scanned and qualified using Tanon Image
Software (Tanon 1600R; Tanon, Shanghai, China). Ghrelin mRNA levels
were expressed as ratios to β-actin mRNA.
Western blot analysis
Western blot analysis was performed to detect the
expression of proghrelin peptide in mouse gastric tissues. Tissue
protein was extracted in lysis buffer (50 mmol/l Tris-HCl, 150
mmol/l NaCl, 1% Nonidet P-40, 0.5% sodium deoxycholate, 1 mmol/l
EDTA and 1 mmol/l PMSF) with protease inhibitors (1 mg/ml
pepstatin, 1 mg/ml aprotinin and 1 mg/ml leupeptin). Protein
concentration was determined using the Bradford assay kit (Bio-Rad
Laboratories, Hercules, CA, USA). Protein (20 μg) was boiled
for 10 min in 4X loading buffer (250 mM Tris-Cl [pH 6.8], 2% sodium
dodecyl sulfate, 10% glycerol, 20 mM dithiothreitol and 0.01%
bromophenol blue), electrophoresed on 10% SDS-PAGE gels, and
transferred by electroblotting onto nitrocellulose membranes.
Following overnight blocking with 4% non-fat milk at 4°C, the
membranes were incubated with rabbit anti-ghrelin antibody
(1:5,000; Phoenix Pharmaceuticals) and rabbit anti-β-actin
(1:1,000; bs-0061R; BIOSS, Beijing, China) for 2 h at room
temperature. After washing in Tris-buffered saline/Tween-20, the
membranes were incubated with an anti-rabbit secondary antibody
conjugated to horseradish peroxidase (1:10,000; sc-2040; Santa Cruz
Biotechnology, Inc.) for 1 h at room temperature. Cross-reactivity
was visualized using ECL western blot analysis detection reagents,
and analyzed through scanning densitometry by the Tanon Image
system. Proghrelin levels were expressed as ratios to β-actin.
Statistical analysis
Values are expressed as mean ± standard error of
mean. Data were analyzed using two-way analysis of variance (ANOVA)
[2×2: treatment (MSG/saline) × food deprivation/non-food
deprivation group] with Bonferroni post hoc tests, when
appropriate. Food intake was analyzed by using repeated measures
ANOVA (MSG/saline) with Bonferroni post hoc tests. The remaining
data were all analyzed by one-way ANOVA. P<0.05 was considered
to indicate statistically significant differences. Statistical
analyses were performed using SPSS for Windows, version 16.0 (SPSS,
Inc., Chicago, IL, USA).
Results
Effects of MSG on ARC and AP
neuroanatomy
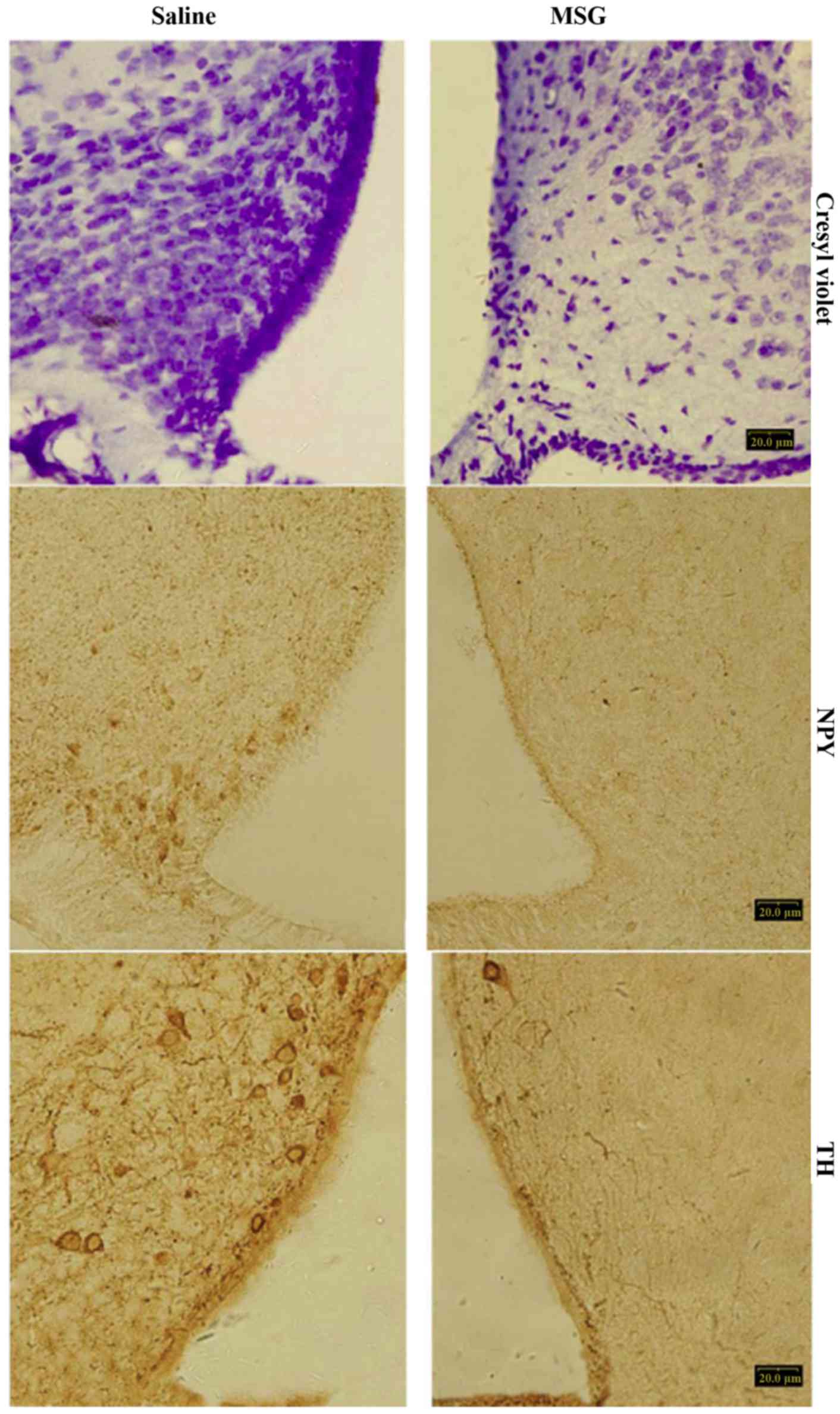
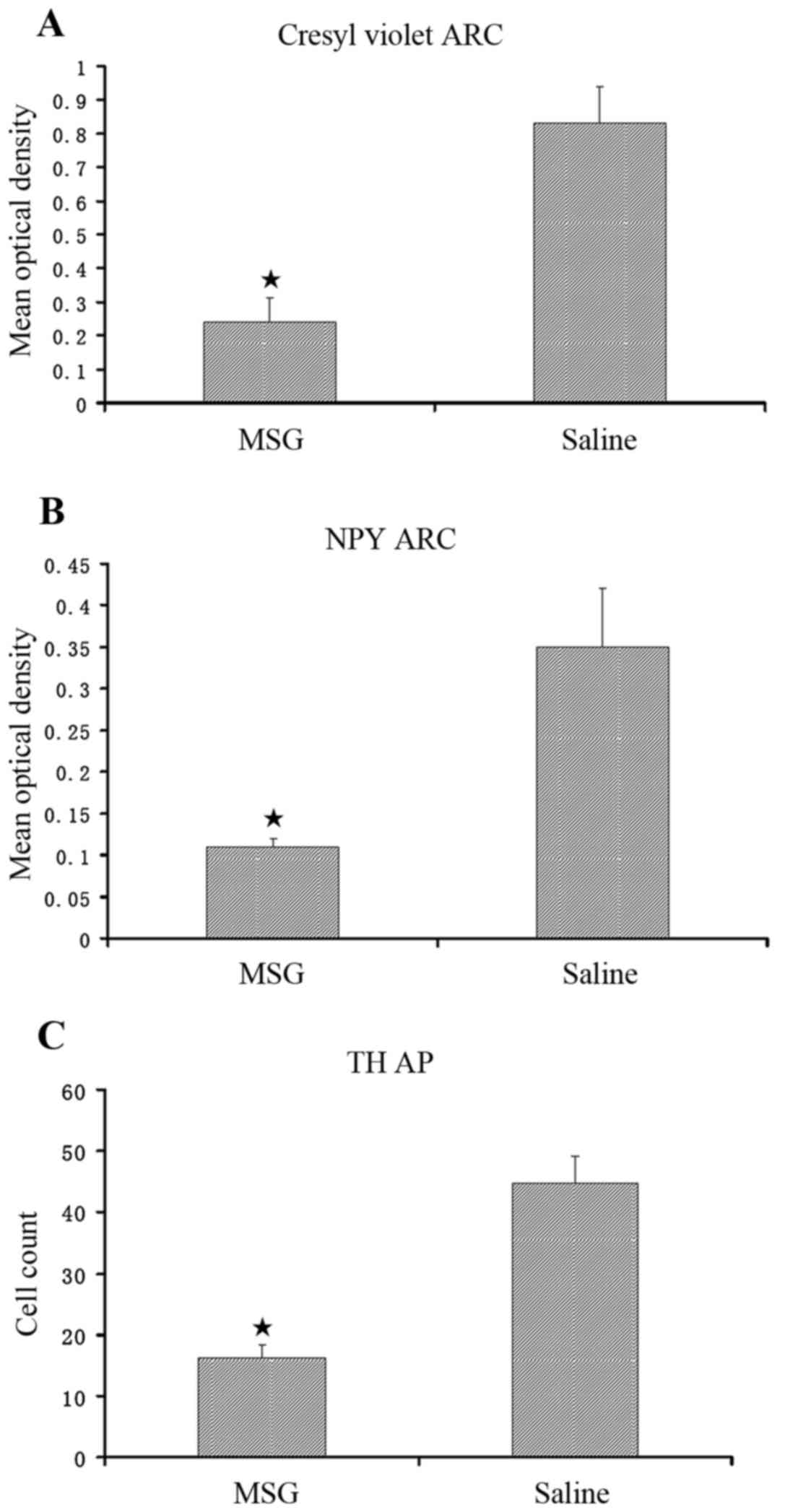
Compared with saline controls, MSG-treated mice
exhibited significantly decreased cresyl violet (Nissl) staining
(Fig. 1), NPY-IR cells and fibers
(Figs. 1, 2A and B), and TH-positive cells
(Figs. 1 and 2C) in the ARC (P<0.01). Furthermore,
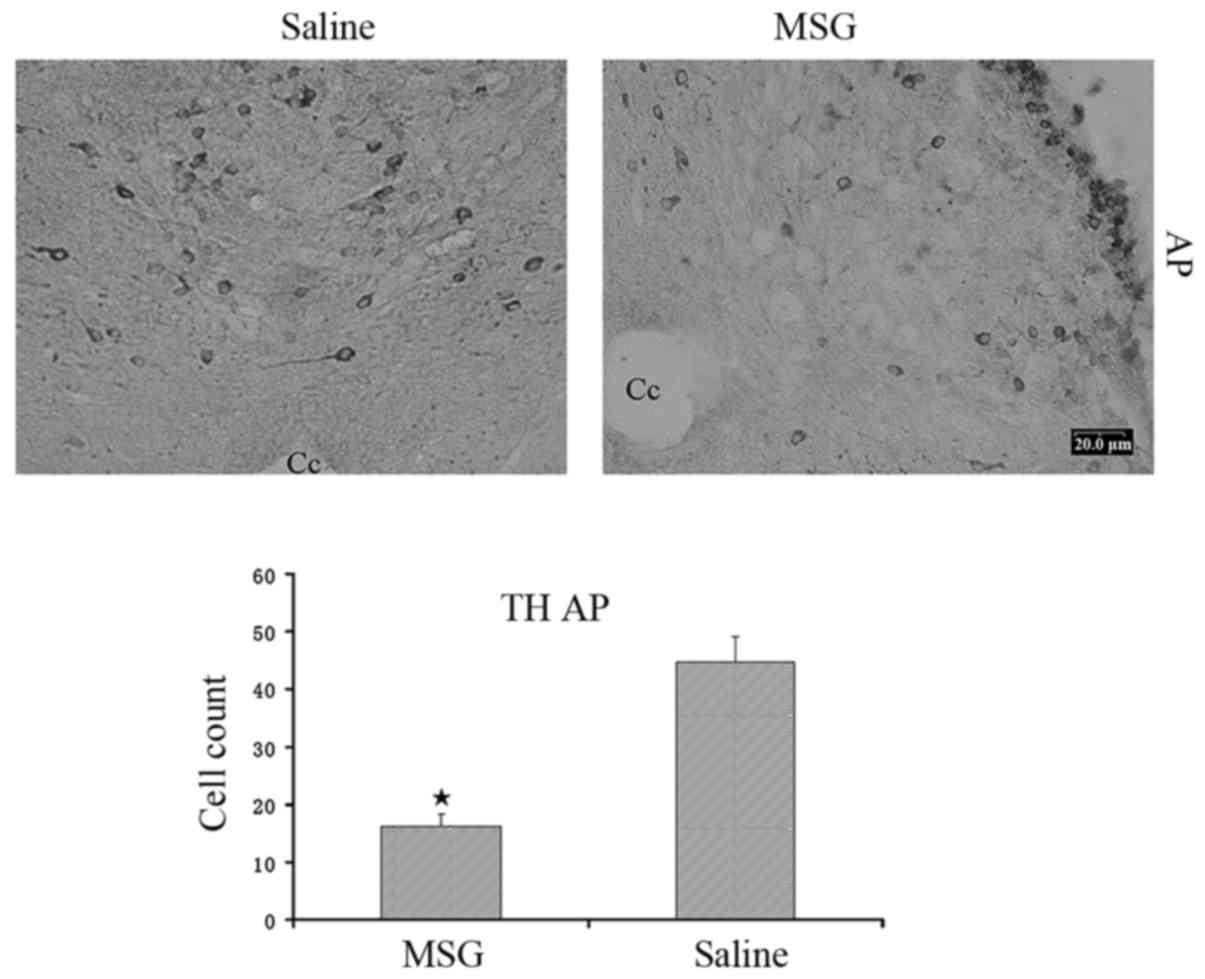
TH-positive cells were significantly reduced in the AP of
MSG-treated mice (P<0.01, Fig.
3).
Body and WAT pad masses, and food
intake
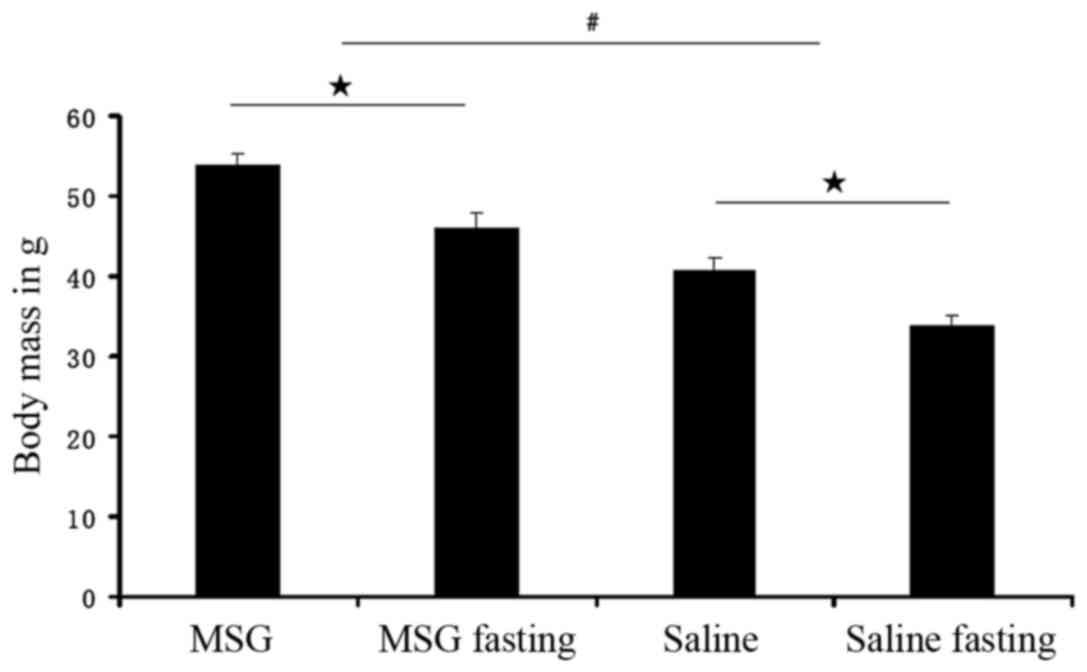
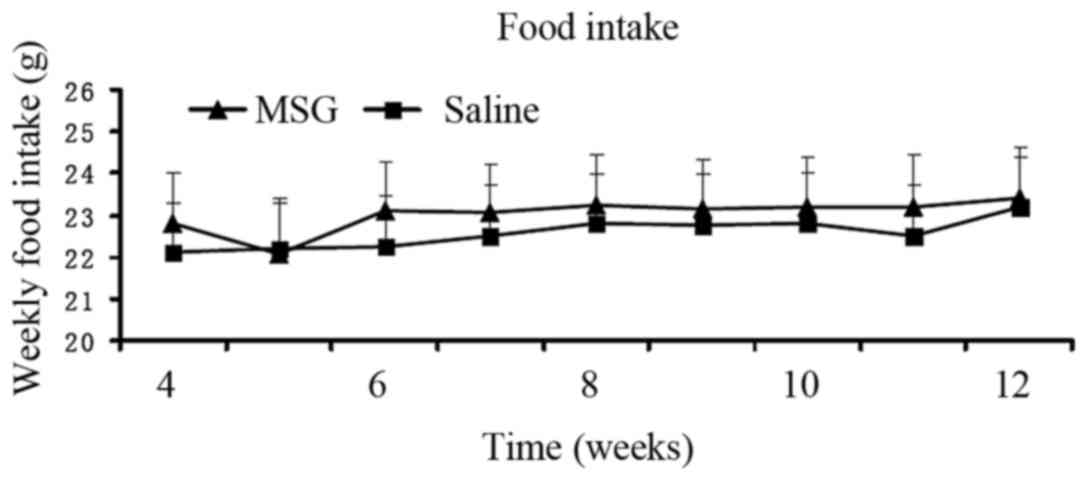
Body mass index was significantly increased in
MSG-treated 3-month-old mice (P<0.01) compared with saline
controls (Fig. 4), but the
difference in food intake from 1- to 3-month-old mice between the
two groups was not statistically significant (Fig. 5).
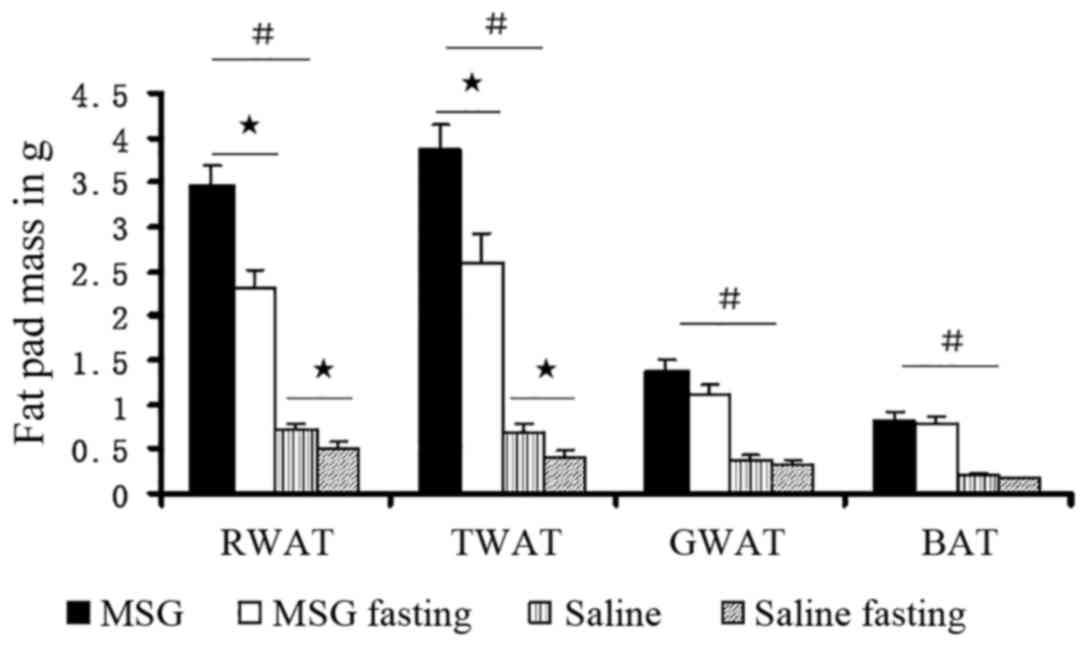
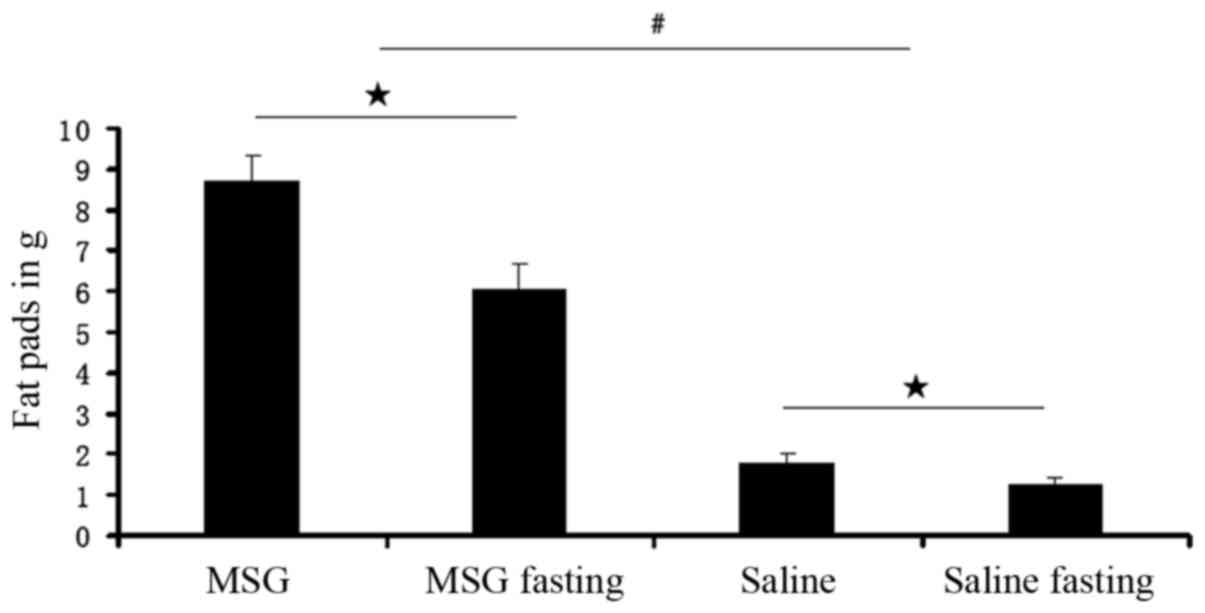
Compared with saline controls, the WAT pad mass was
significantly increased in MSG-treated mice for all depots assayed
(IWAT, RWAT, and GWAT; P<0.01; Fig. 7), as well as the total dissected
WAT (P<0.01, Fig. 6). Both
MSG- and saline-treated animals exhibited significant food
deprivation-induced decreases in the three depots (IWAT, RWAT and
GWAT; P<0.05), compared with their respective ad libitum
fed counterparts (Fig. 7). As all
depot assayed masses (IWAT, RWAT and GWAT) decreased (Figs. 6 and 7), neonatal MSG administration did not
prevent food deprivation-induced lipid mobilization.
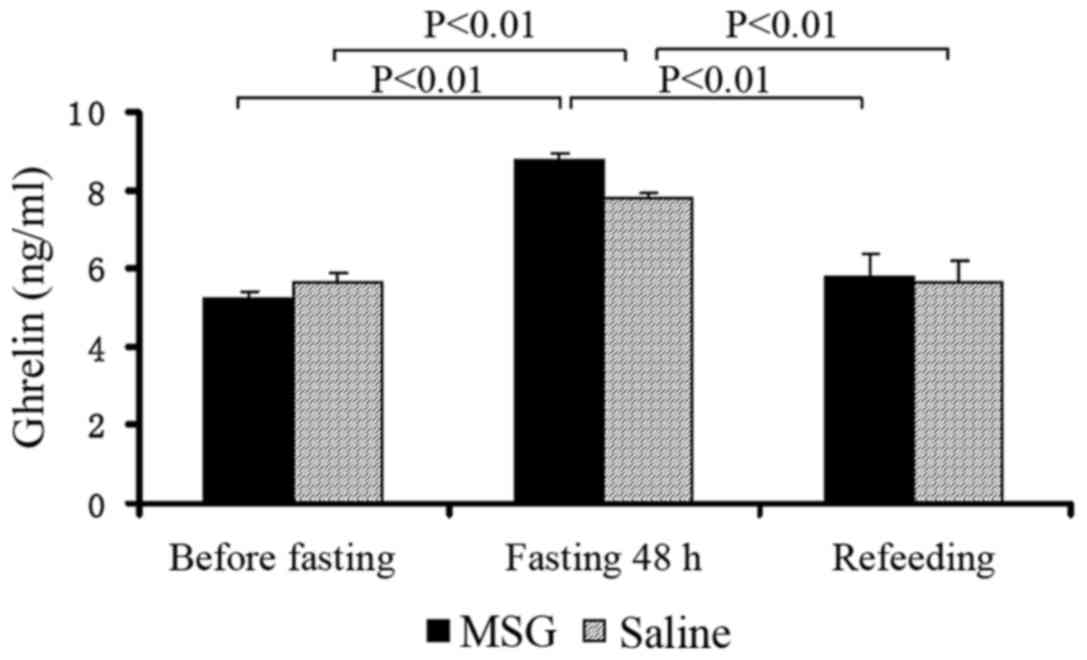
Plasma ghrelin concentrations
Plasma ghrelin levels were significantly increased
in MSG- and saline-treated mice that were fasted for 48 h, compared
with the levels before fasting and after re-feeding (P<0.01);
however, there was no significant difference between MSG- and
saline-treated mice before and after fasting for 48 h, and during
re-feeding (Fig. 8).
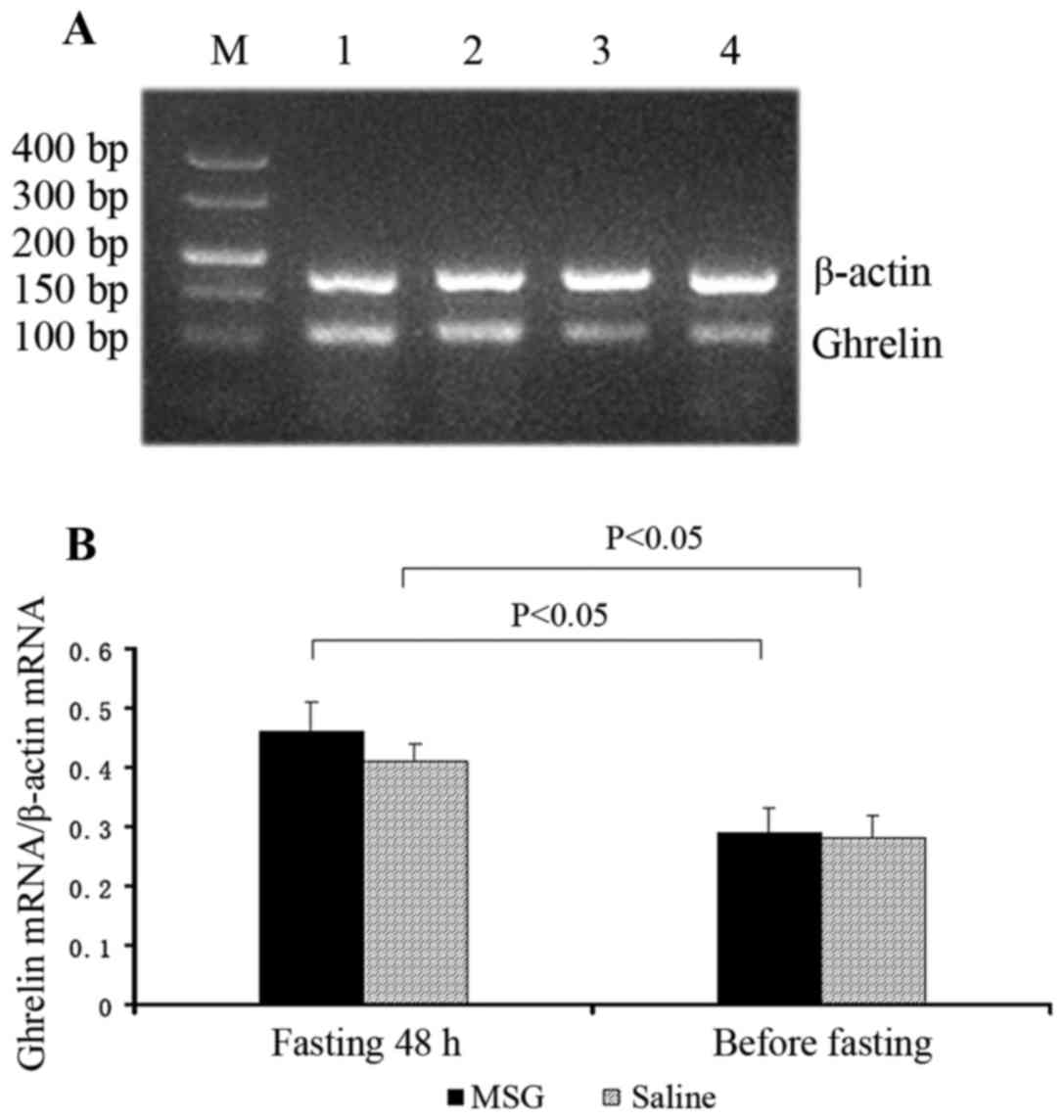
Ghrelin mRNA expression
Ghrelin mRNA expression in the stomach significantly
increased in MSG- and saline-treated mice that were fasted for 48
h, compared with their respective counterparts before fasting
(P<0.05). However, there was no significant difference in
ghrelin mRNA expression in the stomach between MSG- and
saline-treated mice before and after fasting for 48 h (Fig. 9).
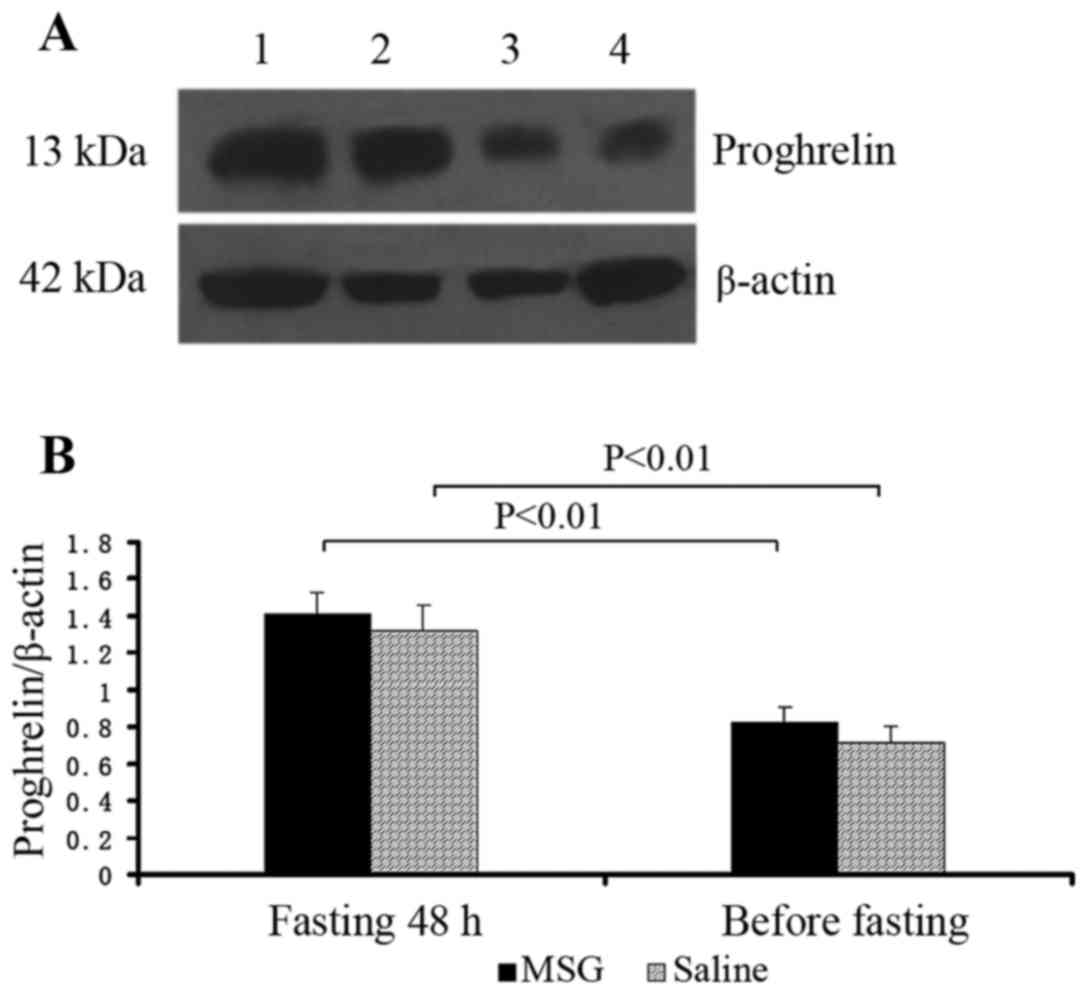
Proghrelin protein expression
Proghrelin protein expression in the stomach
significantly increased in MSG- and saline-treated mice that were
fasted for 48 h, compared with their respective counterparts before
fasting (P<0.01); however, there was no significant difference
in proghrelin protein expression in the stomach between MSG-and
saline-treated mice before and after fasting for 48 h (Fig. 10).
Discussion
The role of ARC cells in altering energy intake is
indisputable, with increasing emphasis placed on their involvement
in energy expenditure (20,21). The activation of ARC neurons by
energy-related stimuli is clear: ARC NPY (22) and AgRP gene expression (22,23) increases with food deprivation,
while ARC pro-opiomelanocortin and cocaine- and
amphetamine-regulated transcript gene expression decreases
(22,24). In the present study, ARC crystal
violet staining was significantly decreased in MSG-treated mice,
indicating overall cell loss, as well as significantly decreased
ARC, NPY-IR and TH-IR neurons. TH-IR cells were significantly
decreased in the AP (Fig. 3).
Therefore, there were distinct MSG-induced ARC and AP lesions. Our
results revealed that the body mass index significantly increased
in 3-month-old MSG-treated mice, compared with saline controls;
however, there were no significant differences in food intake in 1-
to 3-month-old mice between the two groups. The previously reported
MSG-induced obesity without overeating (25) was similar to our results. The
precise reason for the MSG-induced obesity in the present study is
unknown, although one possible reason may be the decrease in energy
expenditure. The ARC was revealed by transneuronal viral tract
tracing using the pseudo-rabies virus (26-29) as a component of the sympathetic
nervous system outflow circuit to WAT. Thus, ARC peptide systems
appear to be likely candidates not only for the modulation of
energy intake and expenditure, but may also participate in other
energy-related responses, such as lipid mobilization. However, that
MSG was found to induce the destruction of ARC, and food
deprivation-induced lipid mobilization did not differ from that in
saline controls. These data suggest that the ARC is not essential
for food deprivation-induced lipid mobilization, but that the
central nervous system contains sufficient neurocircuitry for such
responses.
Ghrelin, a recently discovered peptide hormone, has
been described as a 'hunger signal'. Ghrelin increases food intake
when injected into either the forebrain or hindbrain ventricles and
has been well-established to stimulate food consumption in both
lean and obese humans (30), as
well as food intake upon peripheral and brain injection in various
naive animal species (31). In
addition to regulating food consumption, ghrelin is also involved
in body weight modulation. Chronic administration of this peptide
leads to body weight gain in rodents, not only through increasing
appetite, but also more prominently by promoting fat storage in WAT
(32,33). Furthermore, total ghrelin levels
are inversely correlated with body mass index, as these levels
increase in anorexic and cachectic patients, and decrease under
conditions of obesity (33,34). In the present study, it was
observed that the body mass index significantly increased in
3-month-old MSG-treated mice compared with saline-treated controls;
however, the plasma ghrelin levels were not significantly different
between MSG-treated and saline-treated mice. Furthermore, there
were no significant differences in food intake in 1- to 3-month-old
mice between the two groups. Since there were no changes in plasma
ghrelin levels, there were no changes in appetite in the two
groups. Chronic alterations of ghrelin signaling pathways more
prominently affect energy expenditure rather than food intake,
although adaptive and compensatory regulatory mechanisms may also
take place under conditions of chronically altered ghrelin
signaling by genetic modifications (35). These data strongly support the
hypothesis that the cause of obesity in MSG-treated mice is the
decrease in energy expenditure. Hence, larger studies are required
to confirm this hypothesis.
The ARC is strongly implicated in the regulation of
food intake (8); ghrelin is also
produced centrally in the ARC of the hypothalamus (6) and in neurons adjacent to the third
ventricle (7). NPY and AgRP are
orexigenic neuropeptides (9)
regulated by ghrelin. Ghrelin, which is detected in neurons of the
ARC, sends projections to NPY/AgRP neurons (7,36).
Circulating ghrelin levels increase prior to a meal and decline
postprandially in experimental animals and humans (37). The postprandial suppression of
plasma ghrelin has been considerably more extensively investigated
compared with the preprandial peak. The brain mechanism of the
preprandial peak in plasma ghrelin remains unknown. If ARC does
perform such a function, it would affect the preprandial peak of
plasma ghrelin when destroyed. However, contrary to our hypothesis
in the present study, plasma ghrelin levels significantly increased
in MSG-treated mice that were fasted for 48 h, compared with levels
prior to fasting and after re-feeding; however, the prepran-dial
peak of plasma ghrelin continued to exist in MSG-treated mice. A
recent study by Luquet et al (38) demonstrated that neonatal ablation
of NPY/AgRP neurons had minimal effects on feeding, while their
ablation in adults caused rapid starvation. Their results suggest
that network-based compensatory mechanisms may develop following
ablation of NPY/AgRP neurons in neonates, but these do not readily
occur when these neurons become essential in adults. Luquet et
al (39) also reported that
the ablation of NPY/AgRP neurons in neonatal mice did not affect
feeding in response to glucoprivation, while the feeding response
to the ghrelin receptor agonist was completely abrogated. Their
findings demonstrate that NPY/AgRP neurons are not necessary for
generating or mediating the orexigenic response to glucose
deficiency, but these neurons are essential for the feeding
response to ghrelin and re-feeding on standard chow after fasting.
Tamura et al (40)
reported that the ICV administration of 1 μg ghrelin
significantly increased 4 h food intake in normal controls, while
this peptide did not increase food intake in MSG-treated rats. This
indicates that feeding response to ghrelin requires an intact ARC.
The primary action of ghrelin on appetite control is via the ARC,
although it may act on another type of GHS-R, besides GHS-R1a.
Faulconbridge et al (41)
demonstrated that fourth ventricle ghrelin (150 pmol) injections
increased Fos expression only in the nucleus of the solitary tract,
but not in the ARC or PVN. This indicates that the ingestive
response to caudal brainstem ghrelin administration does not depend
on the activation of neurons in the PVN or ARC. Tamura et al
(40) revealed that the ablation
of ARC neurons by neonatal MSG treatment resulted in the loss of
the appetite-stimulating effects of ghrelin, as well as the double
knockout of the potent orexigenic neurotransmitters NPY and AgRP
(42). Therefore, although ARC
neurons are not essential for rodents to respond to food
deprivation, this does not mean that they do not contribute to food
sensing under normal conditions, since compensatory mechanisms may
have emerged after the ablation of ARC neurons. The neonatal
ablation of ARC neurons allows alternative mechanisms to develop,
in order for rodents not to depend on these neurons for
survival.
Ghrelin-positive X/A-like cells distributed
throughout the gastric oxyntic mucosa (2,3)
are the main source of circulating ghrelin (43), as demonstrated by the sharp
decline in ghrelin levels following gastrectomy (5). We also investigated changes in
ghrelin mRNA and preprotein levels in the stomach in the two groups
of mice. In our results, ghrelin mRNA and preprotein (proghrelin)
expression in the stomach significantly increased in MSG- and
saline-treated mice that were fasted for 48 h, compared with their
respective counterparts before fasting; however, there were no
significant differences in ghrelin mRNA and preprotein expression
in the stomach between MSG-treated and saline-treated mice before
and after fasting for 48 h. Ghrelin mRNA and proghrelin expression
in the stomach significantly increased in the two groups of mice
that were fasted for 48 h. Hence, the plasma ghrelin level was
increased, and the preprandial peak of plasma ghrelin persisted in
MSG-treated mice. After re-feeding, plasma ghrelin levels decreased
in these two groups of mice. These data suggest that ARC is not
essential for the preprandial ghrelin peak and postprandial
suppression in MSG-treated mice.
In conclusion, our results demonstrated that MSG
induced the destruction of the ARC, but food deprivation-induced
lipid mobilization did not different from that in saline controls,
and the preprandial peak of plasma ghrelin persisted in MSG-treated
mice. Hence, ARC is not essential for normal food
deprivation-induced preprandial ghrelin peak and lipid mobilization
in MSG-treated mice. However, these findings do not mean that ARC
neurons do not contribute to food sensing and lipid mobilization
under normal conditions, as compensatory mechanisms may emerge
following ablation of ARC neurons.
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 30670679 and
30870816 to Z.Y. Jiang) and the Medical Science and Technology
Development Program of Shandong Province (grant no. 2011QZ002 to
Q.C. Li).
References
|
1
|
Kojima M, Hosoda H, Date Y, Nakazato M,
Matsuo H and Kangawa K: Ghrelin is a growth-hormone-releasing
acylated peptide from stomach. Nature. 402:656–660. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Date Y, Kojima M, Hosoda H, Sawaguchi A,
Mondal MS, Suganuma T, Matsukura S, Kangawa K and Nakazato M:
Ghrelin, a novel growth hormone-releasing acylated peptide, is
synthesized in a distinct endocrine cell type in the
gastrointestinal tracts of rats and humans. Endocrinology.
141:4255–4261. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Mizutani M, Atsuchi K, Asakawa A, Matsuda
N, Fujimura M, Inui A, Kato I and Fujimiya M: Localization of acyl
ghrelin- and des-acyl ghrelin-immunoreactive cells in the rat
stomach and their responses to intragastric pH. Am J Physiol
Gastrointest Liver Physiol. 297:G974–G980. 2009. View Article : Google Scholar
|
|
4
|
Tschöp M, Weyer C, Tataranni PA,
Devanarayan V, Ravussin E and Heiman ML: Circulating ghrelin levels
are decreased in human obesity. Diabetes. 50:707–709. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jeon TY, Lee S, Kim HH, Kim YJ, Son HC,
Kim DH and Sim MS: Changes in plasma ghrelin concentration
immediately after gastrectomy in patients with early gastric
cancer. J Clin Endocrinol Metab. 89:5392–5396. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lu S, Guan JL, Wang QP, Uehara K, Yamada
S, Goto N, Date Y, Nakazato M, Kojima M, Kangawa K, et al:
Immunocytochemical observation of ghrelin-containing neurons in the
rat arcuate nucleus. Neurosci Lett. 321:157–160. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cowley MA, Smith RG, Diano S, Tschöp M,
Pronchuk N, Grove KL, Strasburger CJ, Bidlingmaier M, Esterman M,
Heiman ML, et al: The distribution and mechanism of action of
ghrelin in the CNS demonstrates a novel hypothalamic circuit
regulating energy homeostasis. Neuron. 37:649–661. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Schwartz MW, Woods SC, Porte D Jr, Seeley
RJ and Baskin DG: Central nervous system control of food intake.
Nature. 404:661–671. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Abizaid A and Horvath TL: Brain circuits
regulating energy homeostasis. Regul Pept. 149:3–10. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Wang L, Saint-Pierre DH and Taché Y:
Peripheral ghrelin selectively increases Fos expression in
neuropeptide Y-synthesizing neurons in mouse hypothalamic arcuate
nucleus. Neurosci Lett. 325:47–51. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kamegai J, Tamura H, Shimizu T, Ishii S,
Sugihara H and Wakabayashi I: Chronic central infusion of ghrelin
increases hypothalamic neuropeptide Y and Agouti-related protein
mRNA levels and body weight in rats. Diabetes. 50:2438–2443. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tschöp M, Flora DB, Mayer JP and Heiman
ML: Hypophysectomy prevents ghrelin-induced adiposity and increases
gastric ghrelin secretion in rats. Obes Res. 10:991–999. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Cummings DE, Purnell JQ, Frayo RS,
Schmidova K, Wisse BE and Weigle DS: A preprandial rise in plasma
ghrelin levels suggests a role in meal initiation in humans.
Diabetes. 50:1714–1719. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tschöp M, Wawarta R, Riepl RL, Friedrich
S, Bidlingmaier M, Landgraf R and Folwaczny C: Post-prandial
decrease of circulating human ghrelin levels. J Endocrinol Invest.
24:RC19–RC21. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Williams DL and Cummings DE: Regulation of
ghrelin in physiologic and pathophysiologic states. J Nutr.
135:1320–1325. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Olney JW: Brain lesions, obesity, and
other disturbances in mice treated with monosodium glutamate.
Science. 164:719–721. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Perez VJ and Olney JW: Accumulation of
glutamic acid in the arcuate nucleus of the hypothalamus of the
infant mouse followng subcutaneous administration of monosodium
glutamate. J Neurochem. 19:1777–1782. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Takasaki Y: Studies on brain lesion by
administration of monosodium L-glutamate to mice. I. Brain lesions
in infant mice caused by administration of monosodium L-glutamate.
Toxicology. 9:293–305. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Nikoletseas MM: Obesity in exercising,
hypophagic rats treated with monosodium glutamate. Physiol Behav.
19:767–773. 1977. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Small CJ, Liu YL, Stanley SA, Connoley IP,
Kennedy A, Stock MJ and Bloom SR: Chronic CNS administration of
Agouti-related protein (Agrp) reduces energy expenditure. Int J
Obes Relat Metab Disord. 27:530–533. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yasuda T, Masaki T, Kakuma T and
Yoshimatsu H: Centrally administered ghrelin suppresses sympathetic
nerve activity in brown adipose tissue of rats. Neurosci Lett.
349:75–78. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Mercer JG, Moar KM, Ross AW, Hoggard N and
Morgan PJ: Photoperiod regulates arcuate nucleus POMC, AGRP, and
leptin receptor mRNA in Siberian hamster hypothalamus. Am J Physiol
Regul Integr Comp Physiol. 278:R271–R281. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ebihara K, Ogawa Y, Katsuura G, Numata Y,
Masuzaki H, Satoh N, Tamaki M, Yoshioka T, Hayase M, Matsuoka N, et
al: Involvement of agouti-related protein, an endogenous antagonist
of hypothalamic melanocortin receptor, in leptin action. Diabetes.
48:2028–2033. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Kristensen P, Judge ME, Thim L, Ribel U,
Christjansen KN, Wulff BS, Clausen JT, Jensen PB, Madsen OD, Vrang
N, et al: Hypothalamic CART is a new anorectic peptide regulated by
leptin. Nature. 393:72–76. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bunyan J, Murrell EA and Shah PP: The
induction of obesity in rodents by means of monosodium glutamate.
Br J Nutr. 35:25–39. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bamshad M, Aoki VT, Adkison MG, Warren WS
and Bartness TJ: Central nervous system origins of the sympathetic
nervous system outflow to white adipose tissue. Am J Physiol.
275:R291–R299. 1998.PubMed/NCBI
|
|
27
|
Bowers RR, Festuccia WTL, Song CK, Shi H,
Migliorini RH and Bartness TJ: Sympathetic innervation of white
adipose tissue and its regulation of fat cell number. Am J Physiol
Regul Integr Comp Physiol. 286:R1167–R1175. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shi H and Bartness TJ: Neurochemical
phenotype of sympathetic nervous system outflow from brain to white
fat. Brain Res Bull. 54:375–385. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Song CK and Bartness TJ: CNS sympathetic
outflow neurons to white fat that express MEL receptors may mediate
seasonal adiposity. Am J Physiol Regul Integr Comp Physiol.
281:R666–R672. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Druce MR, Wren AM, Park AJ, Milton JE,
Patterson M, Frost G, Ghatei MA, Small C and Bloom SR: Ghrelin
increases food intake in obese as well as lean subjects. Int J
Obes. 29:1130–1136. 2005. View Article : Google Scholar
|
|
31
|
Wren AM, Small CJ, Ward HL, Murphy KG,
Dakin CL, Taheri S, Kennedy AR, Roberts GH, Morgan DG, Ghatei MA,
et al: The novel hypothalamic peptide ghrelin stimulates food
intake and growth hormone secretion. Endocrinology. 141:4325–4328.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Davies JS, Kotokorpi P, Eccles SR, Barnes
SK, Tokarczuk PF, Allen SK, Whitworth HS, Guschina IA, Evans BA,
Mode A, et al: Ghrelin induces abdominal obesity via
GHS-R-dependent lipid retention. Mol Endocrinol. 23:914–924. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tschöp M, Smiley DL and Heiman ML: Ghrelin
induces adiposity in rodents. Nature. 407:908–913. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cummings DE, Weigle DS, Frayo RS, Breen
PA, Ma MK, Dellinger EP and Purnell JQ: Plasma ghrelin levels after
diet-induced weight loss or gastric bypass surgery. N Engl J Med.
346:1623–1630. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Stengel A, Goebel M, Wang L and Taché Y:
Ghrelin, des-acyl ghrelin and nesfatin-1 in gastric X/A-like cells:
Role as regulators of food intake and body weight. Peptides.
31:357–369. 2010. View Article : Google Scholar
|
|
36
|
Guan JL, Wang QP, Kageyama H, Takenoya F,
Kita T, Matsuoka T, Funahashi H and Shioda S: Synaptic interactions
between ghrelin- and neuropeptide Y-containing neurons in the rat
arcuate nucleus. Peptides. 24:1921–1928. 2003. View Article : Google Scholar
|
|
37
|
Cummings DE, Purnell JQ, Frayo RS,
Schmidova K, Wisse BE and Weigle DS: A preprandial rise in plasma
ghrelin levels suggests a role in meal initiation in humans.
Diabetes. 50:1714–1719. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Luquet S, Perez FA, Hnasko TS and Palmiter
RD: NPY/AgRP neurons are essential for feeding in adult mice but
can be ablated in neonates. Science. 310:683–685. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Luquet S, Phillips CT and Palmiter RD:
NPY/AgRP neurons are not essential for feeding responses to
glucoprivation. Peptides. 28:214–225. 2007. View Article : Google Scholar
|
|
40
|
Tamura H, Kamegai J, Shimizu T, Ishii S,
Sugihara H and Oikawa S: Ghrelin stimulates GH but not food intake
in arcuate nucleus ablated rats. Endocrinology. 143:3268–3275.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Faulconbridge LF, Grill HJ, Kaplan JM and
Daniels D: Caudal brainstem delivery of ghrelin induces fos
expression in the nucleus of the solitary tract, but not in the
arcuate or paraventricular nuclei of the hypothalamus. Brain Res.
1218:151–157. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen HY, Trumbauer ME, Chen AS, Weingarth
DT, Adams JR, Frazier EG, Shen Z, Marsh DJ, Feighner SD, Guan XM,
et al: Orexigenic action of peripheral ghrelin is mediated by
neuropeptide Y and agouti-related protein. Endocrinology.
145:2607–2612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ariyasu H, Takaya K, Tagami T, Ogawa Y,
Hosoda K, Akamizu T, Suda M, Koh T, Natsui K, Toyooka S, et al:
Stomach is a major source of circulating ghrelin, and feeding state
determines plasma ghrelin-like immunoreactivity levels in humans. J
Clin Endocrinol Metab. 86:4753–4758. 2001. View Article : Google Scholar : PubMed/NCBI
|