Introduction
Alzheimer's disease (AD) is a prevalent type of
dementia, the morbidity of which is increasing in the elderly
population. Today, 47 million people live with dementia worldwide.
This number is projected to increase to more than 131 million by
2050, as populations age (1). AD
is a severe neurodegenerative disorder involving cognitive and
motor decline, which progresses from mild cognitive impairment
affecting the memory, concentration and speech, to late-stage
cognitive impairment where all aspects of behavior are affected
(2,3). Previous studies have suggested
various pathways for the pathogenesis of AD, including genetic
variations (4), internal and
external environmental factors (5), abnormal protein modification
(6,7), neural plasticity changes (8,9)
and oxidative stress (10,11).
However, the exact pathogenesis of AD remains unclear.
Mitochondria have been identified to serve a
critical role in the pathogenesis of AD. Postmortem results have
revealed abnormal mitochondrial ultrastructures and reduced
activities of mitochondrial localized enzymes (12,13), reduced mitochondrial numbers
(14) and decreased mitochondrial
oxygen consumption (15) in AD
patients. Furthermore, extracellular amyloid β (Aβ) deposition is
centrally associated with the pathogenesis of mitochondrial
dysfunction in AD. Cytoplasmic blebbing, mitochondrial calcium
dyshomeostasis, cytochrome c release, chromatin
condensation, nuclear damage and DNA fragmentation may be activated
locally following exposure to Aβ (16). In addition, increased
intracellular Aβ levels facilitate opening of the mitochondrial
permeability transition pores (17). Notably, defective mitochondrial
function has been implicated as an early event in the progression
of AD (18). Therefore, it
appears that mitochondria are a potential target for early-stage AD
treatment.
Statins, also known as hydroxymethyl glutaryl
coenzyme A reductase inhibitors, were first approved for use in
1987 (19). Simvastatin (SV) is a
statin that is widely used in the prevention and treatment of
atherosclerosis associated with cerebrovascular diseases. Notably,
clinical data have demonstrated that SV is able to ameliorate the
symptoms or delay the progression of cognitive impairment in AD
patients with mild cognitive impairment (20). Previous studies by the present
research team indicated that SV clearly improved the cognitive
function and increased cerebral blood flow in amyloid precursor
protein transgenic mice (21,22). In addition, SV has been shown to
enhance the human osteoblast proliferation associated with
mitochondrial energy generation (23), and protect human osteosarcoma
cells against oxidative stress-induced apoptosis through
mitochondrial mediated signaling (24). These data suggest that the anti-AD
effect of SV is potentially associated with the attenuation of
mitochondrial dysfunction. Therefore, the aim of the present study
was to evaluate the role of SV in mitochondrial dysfunction in AD
using an in vitro approach.
Materials and methods
Culture of SH-SY5Y cells
The study was conducted using SH-SY5Y human
neuroblastoma cells (a gift from Dr Zunji Ke, Institute for
Nutritional Sciences, Chinese Academy of Sciences, Shanghai,
China), who purchased the cells from American Type Culture
Collection (Manassas, VA, USA). The two laboratories made every
effort to keep the cell line pure and free from contamination. The
cells were cultured in minimum essential medium and Ham's F-12
nutrient mix (MEM/F12, 1:1), supplemented with 10% fetal bovine
serum (FBS) (both Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA), 100 U/ml penicillin and 100 U/ml streptomycin at 37°C in
a humidified atmosphere of 5% CO2 (Thermo Fisher
Scientific, Inc.). The cells were subcultured every 3 days.
Cell viability assay
The cells were plated at a density of
~3×104 viable cells/well in 96-well plates. The cells
were treated with 0, 1, 10, 100 and 1,000 nM concentrations of SV
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), or with 0, 1, 5
and 10 µM concentrations of human amyloid-β1-42 peptide
[Aβ1-42; Chinapeptide Co., Ltd., Shanghai, China) for 24
h. Combination treatments were also administered, where the cells
were pretreated with SV for 24 h prior to treatment with
Aβ1-42 for 24 h. The Aβ1-42 was dissolved in
distilled water, then diluted with phosphate-buffered saline (PBS)
and incubated at 37°C for 72 h prior to use. A
3-(4,5-dimethyl-thiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT;
Sigma-Aldrich; Merck KGaA) assay was performed to measure cell
viability using a 96-well plate reader (SpectraMax 190; Molecular
Devices, LLC, Sunnyvale, CA, USA). For the cell viability assay,
dimethylsulfoxide was used to dissolve the purple formazan after
MTT staining. The wavelength used to measure formazan was 570
nm.
Hoechst 33342 staining
Cells were stained with 10 µg/ml Hoechst
33342 (Invitrogen; Thermo Fisher Scientific, Inc.) for 15 min at
37°C. Nuclear morphological changes were analyzed using a
fluorescence microscope (Nikon Corporation, Tokyo, Japan).
Apoptosis assay by flow cytometry
Cells were tested with an Alexa Fluor®
488 Annexin V/Dead Cell Apoptosis kit with Alexa®
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. Briefly, following treatment, the cells
were harvested and stained with Annexin V and propidium iodide (PI)
fluorescent dyes. The stained cells were then analyzed by flow
cytometry (CFlow Plus 1.0.264.15; BD Biosciences, Franklin Lakes,
NJ, USA).
Intracellular reactive oxygen species
(ROS) determination
ROS levels were measured using the non-fluorescent
probe dichlorofluorescin diacetate (DCFH-DA; Beyotime Institute of
Biotechnology, Shanghai, China). The cells were incubated with
DCFH-DA at 37°C for 30 min, and the distribution of DCF
fluorescence was detected using a fluorescence microplate reader
(Spectra MaxGemini; Molecular Devices, LLC) at an excitation
wavelength of 488 nm and emission wavelength of 535 nm. The cells
were observed using a fluorescence microscope.
Assessment of mitochondrial membrane
potential (MMP)
A fluorescent dye,
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide (JC-1; Molecular Probes; Thermo Fisher Scientific, Inc.) was
used to monitor the MMP. The cells were incubated with 5
µg/ml JC-1 for 30 min at 37°C, then observed using a
fluorescence microscope. Fluorescence was quantified using a
fluorescence microplate reader (Spectra MaxGemini). Red
fluorescence intensity was detected at an excitation wavelength of
488 nm and emission wavelength of 595 nm and green fluorescence
intensity was detected at an excitation wavelength of 488 nm and
emission wavelength of 525 nm. When MMP was low, the JC-1
fluorescent dye existed as a monomer, which presented green
fluorescence; while it existed as polymer, which presented red
fluorescence when MMP was high.
Firefly luciferase adenosine triphosphate
(ATP) assay
ATP was quantified using the luciferin-luciferase
method. Cells were harvested following treatment and assayed for
ATP using an ATP assay kit (Beyotime Institute of Biotechnology).
In brief, cells were lysed by the addition of 100 µl
ATP-releasing reagent. The lysates were incubated with the
luciferin substrate and luciferase enzyme in the dark for 10 min to
stabilize the luminescent signal. A fluorescence microplate reader
was used to measure the bioluminescence intensity.
Oxygen consumption measurement
A Clark oxygen electrode (Oxygraph 1.01; Hansatech
Instruments, Ltd., King's Lynn, UK) was used to estimate the change
of oxygen consumption of the cells. A small stir bar maintained the
cells in suspension, and a Peltier heating block maintained the
temperature at 37°C. Data from the electrode were exported to a
computerized chart recorder, which calculated the rate of oxygen
consumption.
Assessment of mitochondrial mass
Cells were incubated with 1 µg/ml MitoTracker
Red FM (Molecular Probes; Thermo Fisher Scientific, Inc.) at 37°C
for 30 min, and the fluorescence intensity was detected using a
fluorescence microplate reader at an excitation wavelength of 581
nm and emission wavelength of 644 nm. Cells were observed using a
fluorescence microscope.
Western blot analysis
Following treatment, 1×106 cells were
collected and equal amounts of protein samples were subjected to
western blot analysis, as previously described (25). The release of cytochrome c
from the mitochondria to the cytosol was measured using western
blotting as reported by Piqué et al (26). Lysates were centrifuged at 12,000
× g at 4°C for 15 min to obtain supernatants (cytosolic extracts
free of mitochondria) and the pellets (the mitochondria-containing
fraction). The supernatants and pellets were subjected to western
blotting as described in the aforementioned references. The
following primary antibodies were used: Mouse anti-B-cell lymphoma
2 (Bcl-2) antibody (1:1,000; #15071; Cell Signaling Technology,
Inc., Danvers, MA, USA), rabbit anti-Bcl-2-associated X protein
(Bax) antibody (1:1,000; ab32503; Abcam, Cambridge, MA, USA), mouse
anti-peroxisome proliferator-activated receptor γ coactivator-1α
(PGC-1α) antibody (1:100; sc-518025; Santa Cruz Biotechnology,
Inc., Dallas, TX, USA), mouse anti-cytochrome c antibody
(1:100; #3026; BioVision, Inc., Miliptas, CA, USA), rabbit
anti-voltage-dependent anion channel (VDAC) antibody (1:100;
sc-98708; Santa Cruz Biotechnology, Inc.), rabbit anti-caspase-3
antibody (1:1,000; ab90437; Abcam) and mouse anti-β-actin antibody
(1:2,000; ab11003; Abcam). Visualization of the immunolabeled bands
was carried out using a chemiluminescence imaging system. The
chemiluminescence reagent was Immobilon Western chemiluminescent
HRP substrate (WBKLS0500; Millipore, Billerica, MA, USa). Image J
(version k 1.45) software was used for the quantification of the
western blots.
Statistical analysis
Data are expressed as the mean ± standard deviation
and were compared by one-way analysis of variance followed by
Newman-Keuls post-hoc multiple-comparison tests. The statistical
analysis was conducted using GraphPad Prism 5 software (GraphPad
Software, Inc., La Jolla, CA, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
SV protects against
Aβ1-42-induced cytotoxicity in SH-SY5Y cells
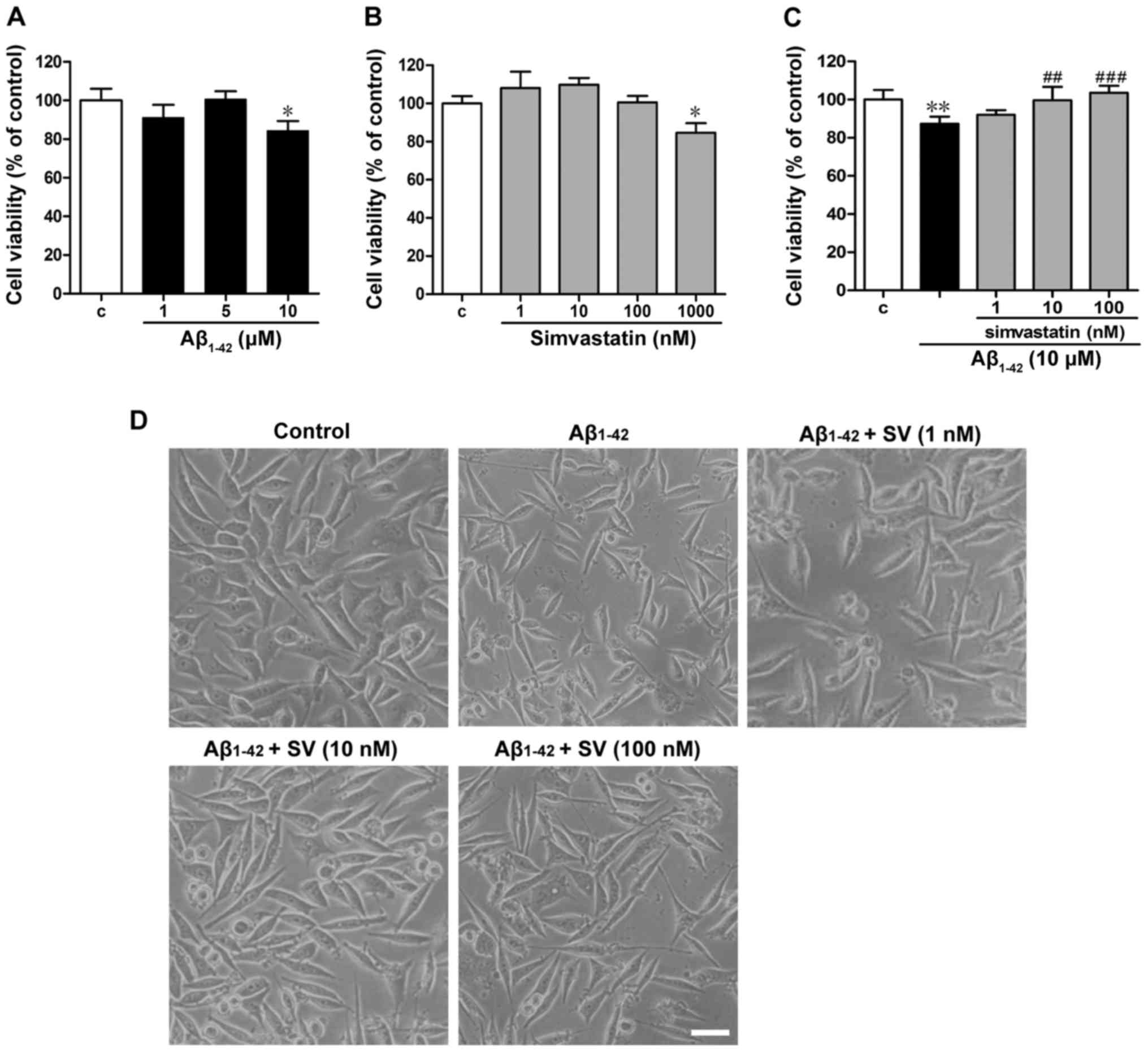
The effects of Aβ1-42 and SV on SH-SY5Y
cells were detected. The viability of the cells treated with 10
µM Aβ1-42 was significantly decreased compared
with that of the untreated control group (Fig. 1A). Low concentrations of SV (≤100
nM) exhibited no effect on the viability of SH-SY5Y cells, whilst
1,000 nM SV significantly reduced the viability of the cells
(Fig. 1B). Exposure to 10
µM Aβ1-42 for 24 h significantly decreased cell
viability compared with that of the control cells, and pretreatment
with 10 or 100 nM SV for 24 h significantly attenuated the
Aβ1-42-induced reduction in viability (Fig. 1C). Morphological observations
revealed that the cell number was clearly reduced when cells were
treated with 10 µM Aβ1-42 (Fig. 1D). The cell numbers of the groups
that were pretreated with SV were markedly increased when compared
with those in the Aβ1-42-treated group (Fig. 1D).
SV decreases the
Aβ1-42-induced apoptosis of SH-SY5Y cells
Hoechst 33342 staining demonstrated that the cells
treated with 10 µM Aβ1-42 underwent chromatin
condensation and nuclear fragmentation (indicated by arrows;
Fig. 2A). This suggested that the
cell death induced by Aβ1-42 involved apoptosis. The
cell apoptotic rate was detected by flow cytometry with Annexin
V/PI-staining of the cells. The apoptotic rate in the
Aβ1-42 treatment group was 15.6%, which was ~2-fold
greater than that of the control group. The apoptotic rates in the
SV treatment groups (those treated with SV and Aβ1-42,)
were significantly decreased compared with those in the group
treated with Aβ1-42 alone. Notably, the apoptosis level
of the 100 nM SV treatment group was 7.7%, and was almost restored
to the level of the control group (Fig. 2B). To further examine the pathway
of apoptosis, caspase-3 content in the cytosol, which is the final
executor of the apoptotic process (27), was examined using western blot
analysis. When compared with the control group, cleaved-caspase-3
protein levels were significantly increased in the 10 µM
Aβ1-42 group, suggesting that Aβ1-42 induced
apoptosis through a caspase-dependent pathway (Fig. 2C). Following pretreatment with 10
and 100 nM SV, the protein levels of cleaved-caspase-3 was
significantly decreased compared with those in the group treated
with Aβ1-42 alone. In addition, the Bcl-2/Bax protein
ratio was significantly reduced following treatment with
Aβ1-42 compared with that in the control group. However,
pretreatment with 10 and 100 nM SV significantly increased the
Bcl-2/Bax protein ratio, which suggests an anti-apoptotic effect
(Fig. 2D). Furthermore,
Aβ1-42 induced the release of cytochrome c from
the mitochondria to the cytoplasm, and SV inhibited this release of
cytochrome c (Fig.
2E).
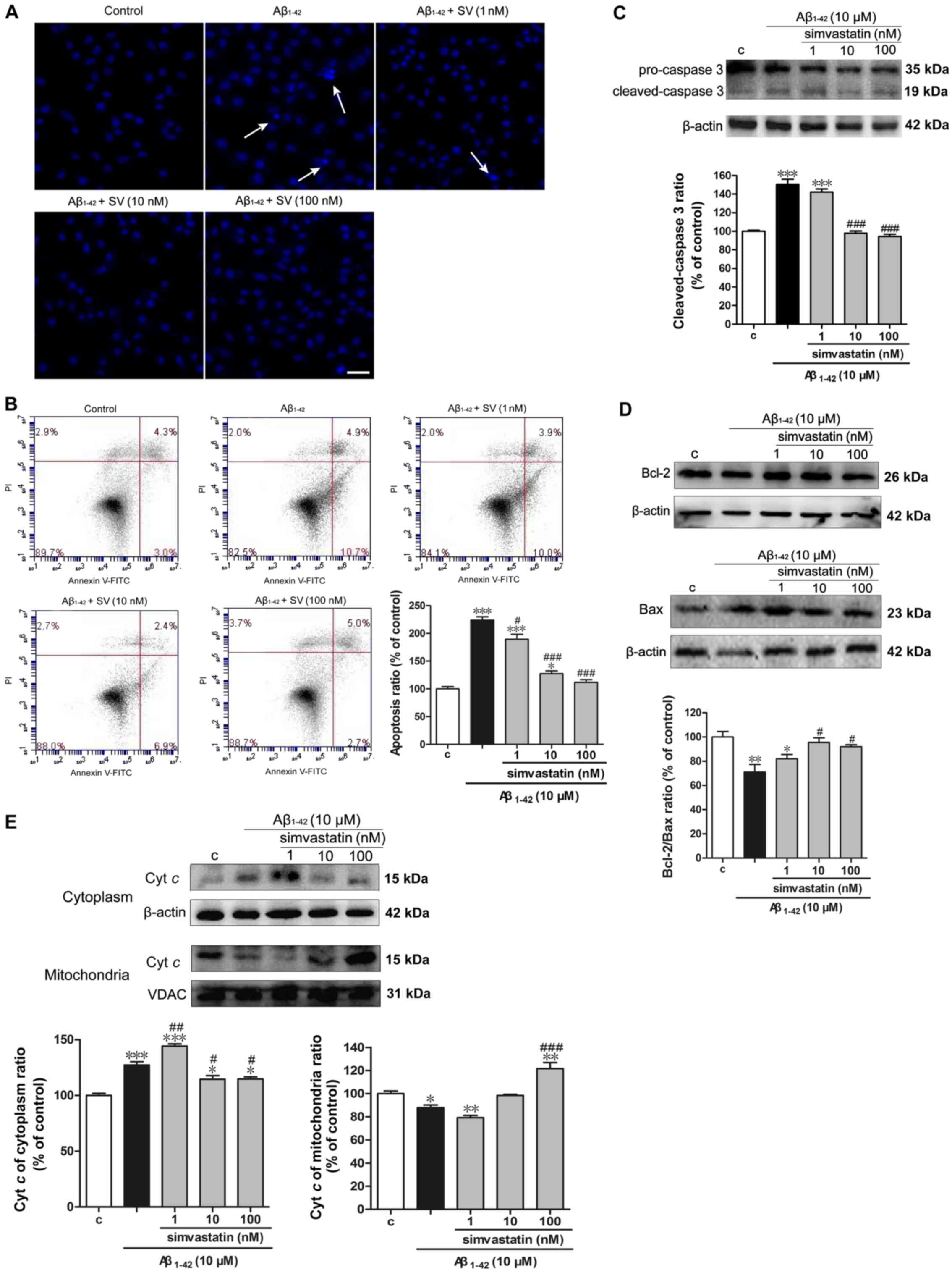 | Figure 2Simvastatin decreases
Aβ1-42-induced apoptosis in SH-SY5Y cells. (A) Cells
stained with Hoechst 33342 observed using fluorescence microscopy
exhibit apoptotic changes (arrows). Scale bar, 100 µm. (B)
Analysis of cells stained with Annexin V/PI by flow cytometry.
Western blotting shows that simvastatin (C) decreased
cleaved-caspase-3 levels, (D) increased the Bcl-2/Bax protein ratio
and (E) inhibited the release of cyt c from mitochondria to
cytoplasm. Data are presented as mean ± standard deviation (n=3).
*P<0.05, **P<0.01 and
***P<0.001 vs. control; #P<0.05,
##P<0.01 and ###P<0.001 vs. only
Aβ1-42 treatment. Aβ1-42, amyloid
Aβ1-42; SV, simvastatin; c, control; Bcl-2, B-cell
lymphoma 2; Bax, anti-Bcl-2-associated X protein; PI, propidium
iodide; Cyt c, cytochrome c. |
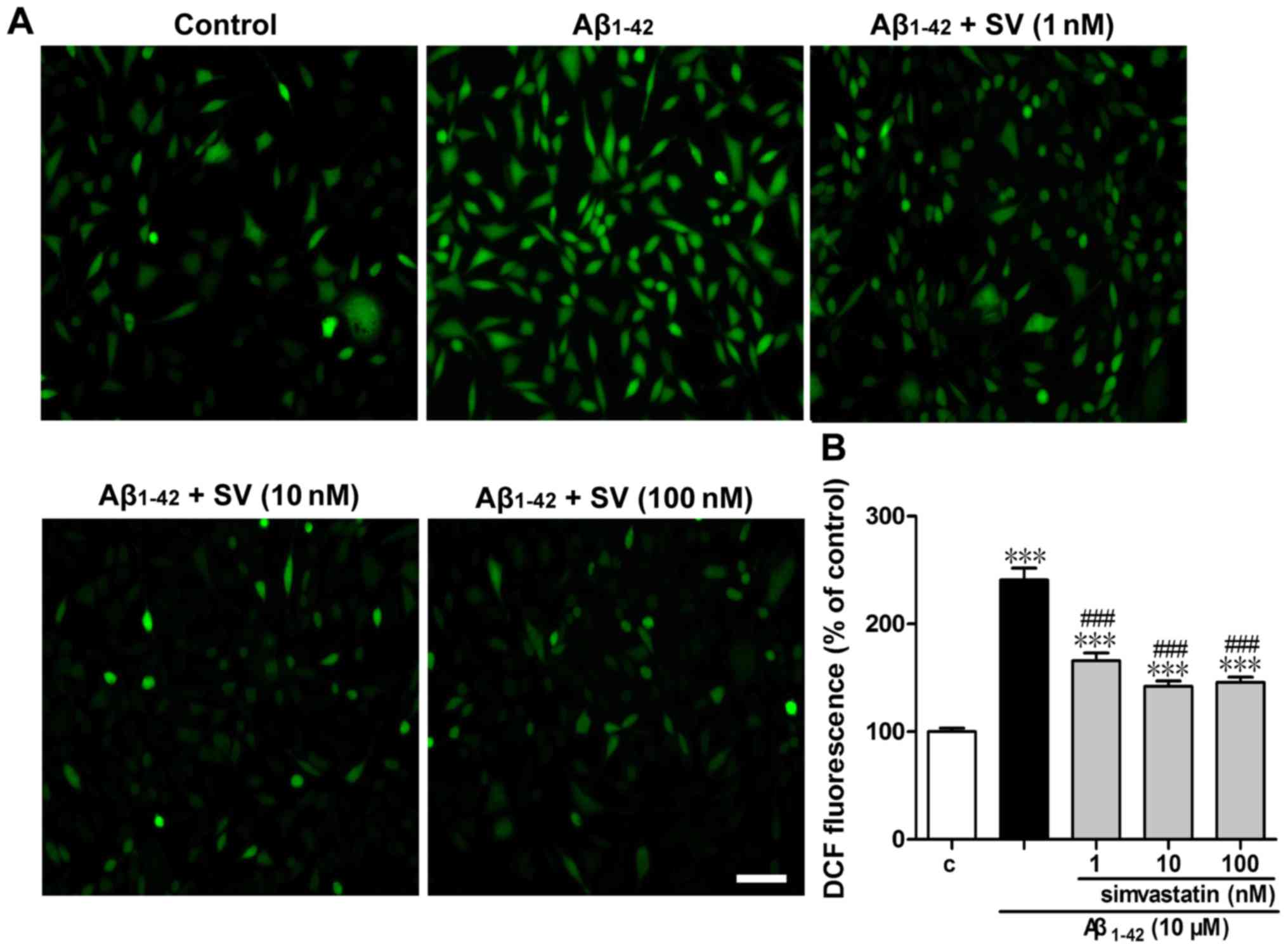
SV protects against
Aβ1-42-induced intracellular ROS elevation
To determine whether Aβ1-42 increased the
intracellular ROS level, cells were labeled with DCFH-DA and then
examined under a fluorescence microscope and using a fluorescence
microplate reader (Fig. 3).
Treatment with 10 µM Aβ1-42 for 24 h led to a
significant increase in fluorescence intensity compared with that
of the control group (Fig. 3B).
In comparison with the 10 µM Aβ1-42 treatment
group, SV significantly attenuated ROS accumulation at all three
concentrations. Representative fluorescence photomicrographs
(Fig. 3A) are consistent with the
quantitative results.
SV protects against Aβ1-42
induced mitochondrial dysfunction by increasing MMP, intracellular
ATP level and the cell respiration rate
To determine whether Aβ1-42 affects
mitochondrial function, the changes of MMP were investigated.
Treatment of the cells with 10 µM Aβ1-42 resulted
in increased green fluorescence intensity and reduced red
fluorescence intensity of JC-1 compared with that in the control
group, which indicates that the MMP was decreased (Fig. 4A). Pretreatment with SV increased
the red fluorescence intensity and reduced the green fluorescence
intensity, indicating that SV protected against the depolarization
of the mitochondrial membrane induced by Aβ1-42. The
quantified red/green fluorescence ratios (Fig. 4B) were consistent with the
fluorescence photomicrographs. MMP is the driving force of ATP
synthesis, and the loss of MMP is expected to result reduce the ATP
level in cells; thus, the cellular ATP level is an important
determinant of cell death (28).
As demonstrated in Fig. 4C, 10
µM Aβ1-42 induced a significant reduction in
cellular ATP content compared with that in the control group.
Intracellular ATP levels increased following pretreatment with all
three concentrations of SV. As the intracellular ATP level is
associated with the cell respiration rate, a Clark oxygen electrode
was used to determine the cell respiration rate. As demonstrated in
Fig. 4D, compared with the
control group, treatment with 10 µM Aβ1-42 led to
a significant reduction in cell respiration rate, which was
significantly attenuated by pretreatment with 10 or 100 nM SV.
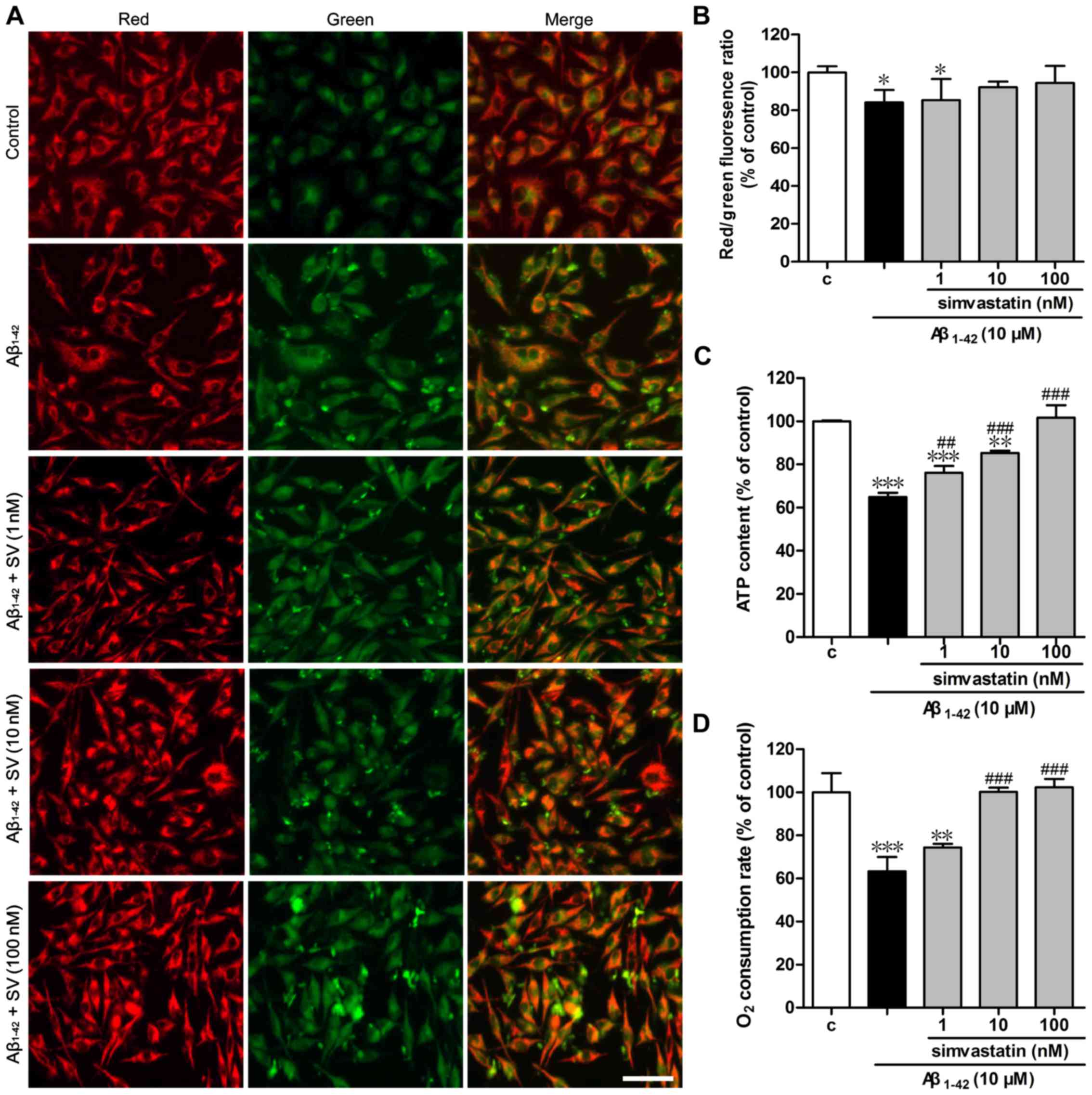 | Figure 4Simvastatin protects against
Aβ1-42-induced mitochondrial dysfunction. Changes of
mitochondria stained with JC-1 were (A) visualized using
fluorescence microscopy and (B) detected using a fluorescence
microplate reader. Scale bar, 50 µm. (C) ATP levels were
measured with the luciferin-luciferase assay. (D) Cell respiration
rates were determined using a Clark oxygen electrode. Data are
presented as mean ± standard deviation; (B) n=5 and (C and D) n=3.
*P<0.05, **P<0.01 and
***P<0.001 vs. control; ##P<0.01 and
###P<0.001 vs. only Aβ1-42 treatment.
Aβ1-42, amyloid Aβ1-42; SV, simvastatin; c,
control; JC-1,
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide; ATP, adenosine triphosphate. |
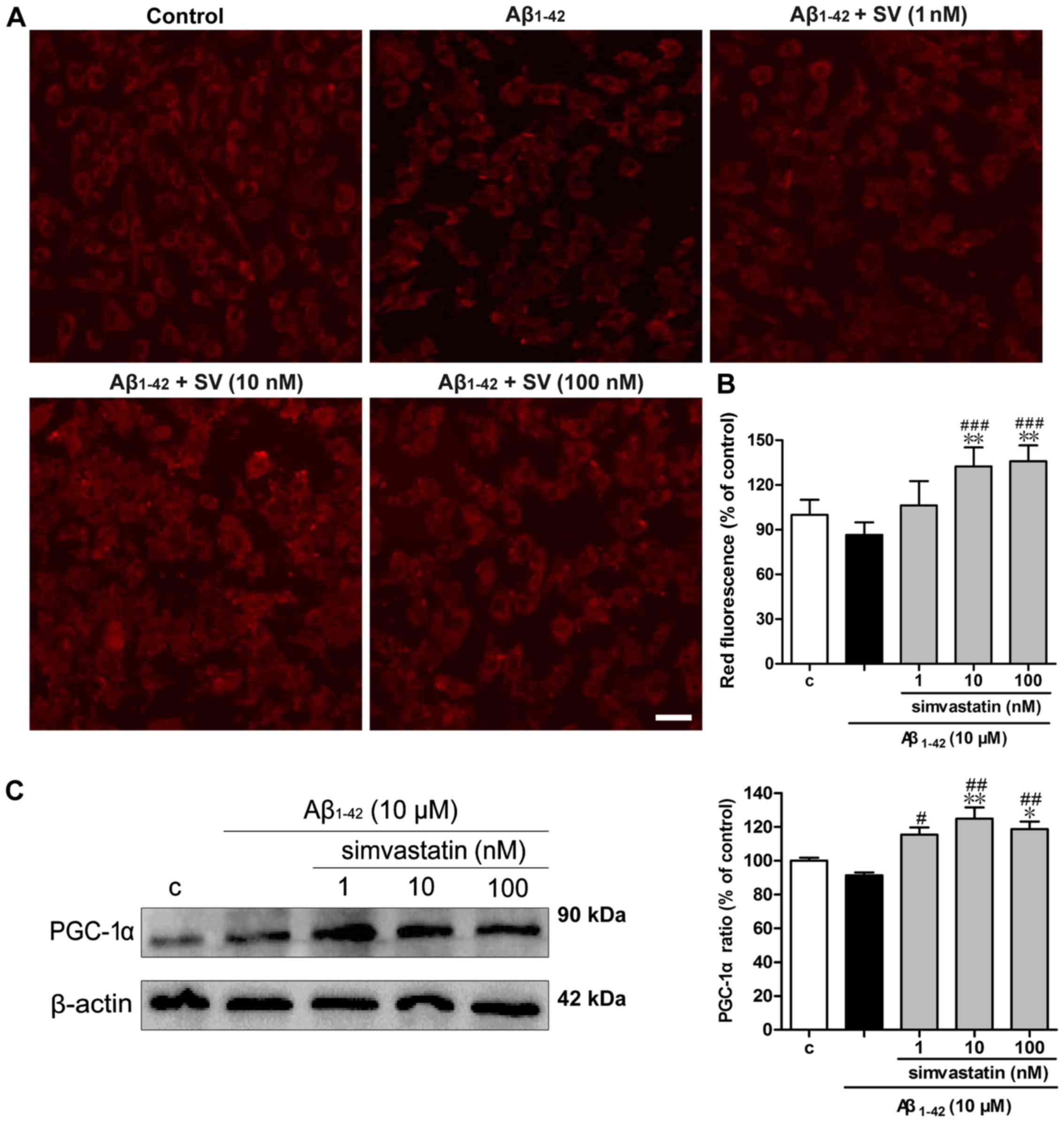
SV protects against Aβ1-42
induced reductions in mitochondrial number
MitoTracker Red staining was used to examine the
numbers of mitochondria. As demonstrated in Fig. 5A, the red fluorescence of the
Aβ1-42 treatment group was weak compared with that of
the control group, whilst pretreatment with SV increased the
fluorescence intensity compared with that of the Aβ1-42
treatment group. The quantitative results (Fig. 5B) were consistent with the
fluorescence photomicrographs. As PGC-1α has been demonstrated to
be associated with mitochondrial biogenesis (29), the expression of PGC-1α was
detected by western blotting. As demonstrated in Fig. 5C, 10 and 100 nM SV significantly
increased PGC-1α protein levels.
Discussion
In the present study, the role of SV in
Aβ1-42-induced injury was investigated in vitro,
and the results provide novel evidence that modulation of
mitochondrial function is an important mechanism by which SV
attenuates Aβ1-42-induced SH-SY5Y cell insult.
Aβ is considered to serve a central role in the
pathogenesis of AD. Mei et al (30) revealed that the viability of
SH-SY5Y cells significantly decreased in a concentration-dependent
manner following treatment with Aβ1-42. The findings of
the present study are consistent with these results. The 10
µM concentration of Aβ1-42 was chosen for testing
in subsequent experiments.
The mitochondrial pathway is a major apoptosis
signaling pathway. The mitochondrial pathway of apoptosis is
regulated by the Bcl-2 family proteins consisting of two subgroups
according to structural homology: Anti-apoptotic proteins such as
Bcl-2 and pro-apoptotic proteins such as Bax (31). Furthermore, anti-apoptotic Bcl-2
has been indicated to inhibit ROS production (32), while pro-apoptotic Bax induces ROS
production. In addition, increased intracellular Aβ levels
facilitate the opening of mitochondrial permeability transition
pores resulting in the release of cytochrome c into the
cytoplasm and activation of caspase-3, which induces cell apoptosis
(33). In the present study,
Annexin V/PI staining data indicated that SV significantly
inhibited the Aβ1-42 induced SH-SY5Y cell apoptosis. To
further study the molecular mechanisms underlying the
anti-apoptotic activity of SV, the expression levels of Bcl-2, Bax,
cytochrome c and cleaved-caspase-3 were evaluated using
western blot analysis. The results demonstrated that SV inhibited
the release of cytochrome c from the mitochondria to the
cytoplasm, downregulated the protein levels of Bax and
cleaved-caspase-3, and upregulated Bcl-2 protein levels in
Aβ1-42-treated SH-SY5Y cells, indicating that SV
inhibited Aβ1-42-induced apoptosis partially through
regulation of the mitochondrial apoptosis pathway.
Oxidative stress is considered to have an
association with AD (34).
Notably, oxidative stress may be an early event in disease
progression, as a previous study indicated that Aβ binds to
Aβ-binding alcohol dehydrogenase, resulting in an increase in ROS
generation and reduction in ATP production (35). In the present study, ROS
generation in the Aβ treatment group was increased significantly
compared with that in the control cells. However, in the SV
treatment groups, the Aβ1-42-induced ROS generation was
decreased significantly compared with that in the Aβ treatment
group, which indicates that SV inhibited the intracellular ROS
generation induced by Aβ.
Researchers have focused on a potential link between
AD and abnormal pathophysiological events in mitochondria (36–38). The MMP maintains its regular
oxidative phosphorylation state for the continuous production of
ATP (39) and its dissipation is
regarded as the early landmark of cell apoptosis (40). It has been reported that Aβ
strongly influences mitochondrial respiratory function, ROS
production and MMP (41,42). In the present study,
Aβ1-42 was used to simulate mitochondrial dysfunction,
in order to explore the neuroprotective effects of SV. To the best
of our knowledge, the present study is the first to verify that SV
pretreatment is able to prevent reductions in MMP and maintains the
MMP at levels comparable with those of normal control cells. The
results of the present study also indicate that SV reversed
Aβ1-42-induced mitochondrial dysfunction by increasing
intracellular ATP production, oxygen consumption and mitochondrial
number. These results suggest that SV reversed the
Aβ1-42-induced neurotoxicity partially through the
regulation of mitochondrial function.
In conclusion, the present study demonstrated for
the first time that SV exerts a neuroprotective effect on
Aβ1-42-induced neurotoxicity in SH-SY5Y cells, partially
through regulation of the mitochondrial apoptosis pathway and
mitochondrial function. These results suggest that the
neuroprotective effects observed for SV in patients with AD are
potentially associated with the ability of SV to protect the
mitochondria against Aβ-induced neurotoxicity. However, the
mechanism by which SV improves cognitive function requires further
investigation in studies using animals.
Glossary
Abbreviations
Abbreviations:
|
AD
|
Alzheimer's disease
|
|
Aβ
|
amyloid β peptide
|
|
SV
|
simvastatin
|
|
MTT
|
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide
|
|
ROS
|
reactive oxygen species
|
|
DCFH-DA
|
dichlorofluorescin diacetate
|
|
JC-1
|
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazol-carbocyanine
iodide
|
|
MMP
|
mitochondrial membrane potential
|
|
Cyt c
|
cytochrome c
|
|
PGC-1
|
peroxisome proliferator-activated
receptor γ coactivator-1
|
Acknowledgments
Not applicable.
Notes
[1]
Funding
The present study was supported by the National
Natural Science Foundation of China and Quebec Medical Research
Foundation (grant no. 81361120247) and the National Natural Science
Foundation of China (grant no. 81373400).
[2] Availability
of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
[3] Authors'
contributions
JG, XL and YL designed the study. YL, JS and JW
performed majority of experiments and analyzed data. YL, QL and JG
wrote and generated final draft of the manuscript. All authors read
and approved the final manuscript.
[4] Ethics
approval and consent to participate
Not applicable.
[5] Consent for
publication
Not applicable.
[6] Competing
interests
The authors declare that they have no competing
interests.
References
|
1
|
Prince M, Comas-Herrera A, Knapp M,
Guerchet M and Karagiannidou M: World Alzheimer Report 2016 -
Improving healthcare for people living with dementia: Coverage,
quality and costs now and in the future. Alzheimer's Disease
International; London:
|
|
2
|
Salmon DP: Neuropsychological features of
mild cognitive impairment and preclinical Alzheimer's disease. Curr
Top Behav Neurosci. 10:187–212. 2012. View Article : Google Scholar
|
|
3
|
Förstl H and Kurz A: Clinical features of
Alzheimer's disease. Eur Arch Psychiatry Clin Neurosci.
249:288–290. 1999. View Article : Google Scholar
|
|
4
|
Cong L and Jia J: Promoter polymorphisms
which regulate ADAM9 transcription are protective against sporadic
Alzheimer's disease. Neurobiol Aging. 32:54–62. 2011. View Article : Google Scholar
|
|
5
|
Tong Z, Zhang J, Luo W, Wang W, Li F, Li
H, Luo H, Lu J, Zhou J, Wan Y, et al: Urine formaldehyde level is
inversely correlated to mini mental state examination scores in
senile dementia. Neurobiol Aging. 32:31–41. 2011. View Article : Google Scholar
|
|
6
|
Tong Z, Han C, Luo W, Wang X, Li H, Luo H,
Zhou J, Qi J and He R: Accumulated hippocampal formaldehyde induces
age-dependent memory decline. Age (Dordr). 35:583–596. 2013.
View Article : Google Scholar
|
|
7
|
He R, Lu J and Miao J: Formaldehyde
stress. Sci China Life Sci. 53:1399–1404. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang X and Poo MM: Progress in neural
plasticity. Sci China Life Sci. 53:322–329. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Luo Z: Synapse formation and remodeling.
Sci China Life Sci. 53:315–321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang M, Zhao Z, He L and Wan C: A
meta-analysis of oxidative stress markers in schizophrenia. Sci
China Life Sci. 53:112–124. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sun P, Zhang Q, Han J, Tian Y and Zhang J:
TLR4 signaling induced TLR2 expression in the process of mimic
cerebral ischemia/reperfusion in vitro. Sci China Life Sci.
53:223–228. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Perry EK, Perry RH, Tomlinson BE, Blessed
G and Gibson PH: Coenzyme A-acetylating enzymes in Alzheimer's
disease: Possible cholinergic 'compartment' of pyruvate
dehydrogenase. Neurosci Lett. 18:105–110. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gibson GE, Sheu KF, Blass JP, Baker A,
Carlson KC, Harding B and Perrino P: Reduced activities of
thiamine-dependent enzymes in the brains and peripheral tissues of
patients with Alzheimer's disease. Arch Neurol. 45:836–840. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hirai K, Aliev G, Nunomura A, Fujioka H,
Russell RL, Atwood CS, Johnson AB, Kress Y, Vinters HV, Tabaton M,
et al: Mitochondrial abnormalities in Alzheimer's disease. J
Neurosci. 21:3017–3023. 2001.PubMed/NCBI
|
|
15
|
Sims NR, Finegan JM, Blass JP, Bowen DM
and Neary D: Mitochondrial function in brain tissue in primary
degenerative dementia. Brain Res. 436:30–38. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eckert A, Keil U, Marques CA, Bonert A,
Frey C, Schüssel K and Müller WE: Mitochondrial dysfunction,
apoptotic cell death, and Alzheimer's disease. Biochem Pharmacol.
66:1627–1634. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Parks JK, Smith TS, Trimmer PA, Bennett JP
Jr and Parker WD Jr: Neurotoxic Abeta peptides increase oxidative
stress in vivo through NMDA-receptor and nitric-oxide-synthase
mechanisms, and inhibit complex IV activity and induce a
mitochondrial permeability transition in vitro. J Neurochem.
76:1050–1056. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hauptmann S, Scherping I, Dröse S, Brandt
U, Schulz KL, Jendrach M, Leuner K, Eckert A and Müller WE:
Mitochondrial dysfunction: An early event in Alzheimer pathology
accumulates with age in AD transgenic mice. Neurobiol Aging.
30:1574–1586. 2009. View Article : Google Scholar
|
|
19
|
Cucchiara B and Kasner SE: Use of statins
in CNS disorders. J Neurol Sci. 187:81–89. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Simons M, Schwärzler F, Lütjohann D, von
Bergmann K, Beyreuther K, Dichgans J, Wormstall H, Hartmann T and
Schulz JB: Treatment with simvastatin in normocholesterolemic
patients with Alzheimer's disease: A 26-week randomized,
placebo-controlled, double-blind trial. Ann Neurol. 52:346–350.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tong XK, Lecrux C, Rosa-Neto P and Hamel
E: Age-dependent rescue by simvastatin of Alzheimer's disease
cerebrovascular and memory deficits. J Neurosci. 32:4705–4715.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tong XK, Nicolakakis N, Fernandes P,
Ongali B, Brouillette J, Quirion R and Hamel E: Simvastatin
improves cerebrovascular function and counters soluble
amyloid-beta, inflammation and oxidative stress in aged APP mice.
Neurobiol Dis. 35:406–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chuang SC, Liao HJ, Li CJ, Wang GJ, Chang
JK and Ho ML: Simvastatin enhances human osteoblast proliferation
involved in mitochondrial energy generation. Eur J Pharmacol.
714:74–82. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhao XH, Xu ZR, Zhang Q and Yang YM:
Simvastatin protects human osteosarcoma cells from oxidative
stress-induced apoptosis through mitochondrial-mediated signaling.
Mol Med Rep. 5:483–488. 2012.
|
|
25
|
Xu MF, Xiong YY, Liu JK, Qian JJ, Zhu L
and Gao J: Asiatic acid, a pentacyclic triterpene in Centella
asiatica, attenuates glutamate-induced cognitive deficits in mice
and apoptosis in SH-SY5Y cells. Acta Pharmacol Sin. 33:578–587.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Piqué M, Barragán M, Dalmau M, Bellosillo
B, Pons G and Gil J: Aspirin induces apoptosis through
mitochondrial cytochrome c release. FEBS Lett. 480:193–196. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Danial NN and Korsmeyer SJ: Cell death:
Critical control points. Cell. 116:205–219. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zamzami N, Marchetti P, Castedo M,
Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B and
Kroemer G: Sequential reduction of mitochondrial transmembrane
potential and generation of reactive oxygen species in early
programmed cell death. J Exp Med. 182:367–377. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
St-Pierre J, Drori S, Uldry M, Silvaggi
JM, Rhee J, Jäger S, Handschin C, Zheng K, Lin J, Yang W, et al:
Suppression of reactive oxygen species and neurodegeneration by the
PGC-1 transcriptional coactivators. Cell. 127:397–408. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mei Z, Yan P, Situ B, Mou Y and Liu P:
Cryptotanshinione inhibits β-amyloid aggregation and protects
damage from β-amyloid in SH-SY5Y cells. Neurochem Res. 37:622–628.
2012. View Article : Google Scholar
|
|
31
|
Ma L and Li W: Emodin inhibits LOVO
colorectal cancer cell proliferation via the regulation of the
Bcl-2/Bax ratio and cytochrome c. Exp Ther Med. 8:1225–1228. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ricci JE, Gottlieb RA and Green DR:
Caspase-mediated loss of mitochondrial function and generation of
reactive oxygen species during apoptosis. J Cell Biol. 160:65–75.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Camilleri A, Zarb C, Caruana M, Ostermeier
U, Ghio S, Högen T, Schmidt F, Giese A and Vassallo N:
Mitochondrial membrane permeabilisation by amyloid aggregates and
protection by polyphenols. Biochim Biophys Acta. 1828:2532–2543.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang X, Wang W, Li L, Perry G, Lee HG and
Zhu X: Oxidative stress and mitochondrial dysfunction in
Alzheimer's disease. Biochim Biophys Acta. 1842:1240–1247. 2014.
View Article : Google Scholar
|
|
35
|
Santos RX, Correia SC, Wang X, Perry G,
Smith MA, Moreira PI and Zhu X: Alzheimer's disease: Diverse
aspects of mitochondrial malfunctioning. Int J Clin Exp Pathol.
3:570–581. 2010.PubMed/NCBI
|
|
36
|
García-Escudero V, Martín-Maestro P, Perry
G and Avila J: Deconstructing mitochondrial dysfunction in
Alzheimer disease. Oxid Med Cell Longev. 2013:1621522013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Swerdlow RH, Burns JM and Khan SM: The
Alzheimer's disease mitochondrial cascade hypothesis: Progress and
perspectives. Biochim Biophys Acta. 1842:1219–1231. 2014.
View Article : Google Scholar :
|
|
38
|
Cadonic C, Sabbir MG and Albensi BC:
Mechanisms of mitochondrial dysfunction in Alzheimer's disease. Mol
Neurobiol. 53:6078–6090. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rhein V, Song X, Wiesner A, Ittner LM,
Baysang G, Meier F, Ozmen L, Bluethmann H, Dröse S, Brandt U, et
al: Amyloid-β and tau synergistically impair the oxidative
phosphorylation system in triple transgenic Alzheimer's disease
mice. Proc Natl Acad Sci USA. 106:20057–20062. 2009. View Article : Google Scholar
|
|
40
|
Gottlieb E, Armour SM, Harris MH and
Thompson CB: Mitochondrial membrane potential regulates matrix
configuration and cytochrome c release during apoptosis. Cell Death
Differ. 10:709–717. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hansson Petersen CA, Alikhani N, Behbahani
H, Wiehager B, Pavlov PF, Alafuzoff I, Leinonen V, Ito A, Winblad
B, Glaser E, et al: The amyloid beta-peptide is imported into
mitochondria via the TOM import machinery and localized to
mitochondrial cristae. Proc Natl Acad Sci USA. 105:13145–13150.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Picone P, Nuzzo D, Caruana L, Scafidi V
and Di Carlo M: Mitochondrial dysfunction: Different routes to
Alzheimer's disease therapy. Oxid Med Cell Longev. 2014:7801792014.
View Article : Google Scholar : PubMed/NCBI
|