Introduction
Hepatic fibrosis is defined as the aberrant and
excess production of extracellular matrix (ECM) components, which
is involved in numerous human chronic liver diseases (CLDs)
(1). Although fibrogenesis is
considered a potentially reversible scarring response in the liver,
persistent fibrosis can lead to progressive loss of organ function
and subsequent liver failure (2).
Furthermore, immune dysregulation occurs in several liver diseases,
including autoimmune hepatitis, alcohol-related liver disease and
primary biliary cirrhosis (1).
Progress in determining the underlying mechanisms of immune-induced
liver fibrogenesis has given realistic hopes to patients with CLD;
however, scientific and clinical challenges remain (3).
Hepatic stellate cells (HSCs) have garnered
attention because they give rise to ~90% of ECM-generating
myofibroblasts during hepatic fibrosis (4). In addition to repetitive injury of
hepatocytes and inflammation following destructive stimulation, the
activation/proliferation of HSCs is a major mechanism that
contributes to hepatic fibrosis (5). There is a growing consensus that
HSCs may function as recipients of inflammatory signals, and thus
generate fibrogenic cytokines, including transforming growth factor
(TGF)-β (6). Persistently
activated TGF-β signaling leads to overproduction of ECM by HSCs in
the liver (7). Conversely,
inhibition of TGF-β signaling can attenuate
dimethylnitrosamine-induced liver fibrosis, but fails to completely
abrogate it (8,9), thus indicating that TGF-β may not be
the sole regulator in fibrotic diseases.
Activins are another important TGF-β-like group in
the TGF-β superfamily (10).
Similar to TGF-βs, activins, including activin A, B, AB and AC, are
secreted proteins formed by various activin subunits as homo- and
heterodimers (11). Activins
signal by binding to heteromeric complexes, which consist of type I
and II receptors (11). Activin A
receptor type 2A (ACVR2A, formerly known as ACTRII) is an essential
type II receptor for activin (10). Notably, increased expression of
activin A in cirrhotic and fibrotic liver tissues has previously
been reported (12). Inhibition
of activin A with its main biological inhibitor follistatin
mitigates carbon tetrachloride (CCl4)-induced hepatic
fibrosis (13). These studies
suggest a contributory role for activated activin signaling in
hepatic fibrosis. However, the precise mechanisms underlying
activin-mediated events in hepatic fibrosis, particularly with
regards to immune-mediated fibrosis, remain to be fully
elucidated.
In response to injurious stimuli, HSCs interact
closely with liver-resident cells such as hepatocytes, endothelial
cells and Kupffer cells, and with infiltrating immune cells, such
as cluster of differentiation (CD)4+ T lymphocytes
(14). Concanavalin A (Con A) is
a plant lectin, multiple injections of which induce T-cell-mediated
liver fibrosis in mice (15). In
addition, it has been suggested that Con A administration promotes
the infiltration and differentiation of T helper (Th)17 cells in
the liver (16). Interleukin
(IL)-17 is mainly generated by Th17 cells and has been reported to
accelerate liver fibrosis by activating HSCs (17). However, whether activin signaling
has a role in Th17-mediated HSC activation is unclear and requires
further investigation.
In the present study, T-cell-mediated liver fibrosis
was established in BALB/c mice following numerous injections of Con
A. To disrupt activin signaling delivery, recombinant adenoviruses
expressing ACVR2A short hairpin (sh)RNA were administered to
experimental animals and primary mouse HSCs (mHSCs). In addition,
IL-17A and IL-17F, two important Th17-family cytokines, were used
to stimulate mHSCs expressing normal or decreased ACVR2A. The
results indicated that inhibition of the activin signaling pathway
attenuated Con A-induced fibrotic injury in the liver and
suppressed IL-17-induced mHSC activation.
Materials and methods
Animal model
BALB/c mice (weight, 20–23 g; age, 8–10 weeks) were
purchased from Liaoning Changsheng Biotech Co., Ltd. (Benxi, China)
and maintained under a 12-h light/dark cycle with free access to
food and water. The present study was approved by the Institutional
Animal Care and Use committee of Mudanjiang Medical University
(Mudanjiang, China).
The whole experiment consisted of two parts. In
experiment part 1, a total of 16 mice were randomly divided into
two groups: i) Control (n=8) and ii) Con A (n=8) groups. For
establishment of the immune-associated hepatic fibrosis animal
model, mice in the Con A group were administered intravenous (i.v.)
injections of Con A (8 mg/kg/week; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) through the tail vein for up to 6 weeks
(18). The mice were anesthetized
by pentobarbital sodium (50 mg/kg body weight; intraperitoneal
injection; Xiya Reagent, Linshu, China), and the blood was obtained
from the retro-orbital plexus. The blood was then centrifuged at
850 × g for 10 min (4°C) to collect the serum. After sacrifice, the
liver was removed from each animal. The expression levels of
activin A, ACVR2A, IL-17A and IL-17F were subsequently
detected.
In experiment part 2, a total of 40 mice were
randomly divided into five groups (n=8 per group): i) Control, ii)
Con A, iii) ConA + Ad-negative control (NC) shRNA, iv) ConA +
Ad-ACVR2A shRNA, v) control + Ad-NC sh R NA. Recombinant
adenoviruses carrying ACVR2A shRNA (Ad-ACVR2A shRNA, target area:
5′-GGAGUGUCUUUUCUUUAAU-3′; NM_007396.4) (19) and NC shRNA (Ad-NC shRNA) (Shanghai
GenePharma Co., Ltd., Shanghai, China) were used in the present
study. Hepatic fibrosis was induced in mice in groups 2–4 as
aforementioned. At weeks 1 and 4, 1×109 plaque-forming
units Ad-ACVR2A shRNA or Ad-NC shRNA was administered to mice in
groups 3–5 via i.v. injection 24 h after Con A injection. Serum and
liver tissue samples were obtained from each mouse at the end of
week 6, and stored at −80°C or embedded into paraffin until
use.
Cell culture
Primary mHSCs were isolated from the fresh liver
samples of BALB/c mice according to previously reported protocols
(20–23) with minor modifications. Briefly,
the liver samples were initially perfused with PBS containing
heparin (2 U/ml) at 4°C for ~20 min, and were then digested with a
mix of pronase (1 mg/ml; Dalian Meilun Biotech Co., Ltd., Dalian,
China)/collagenase IV (0.4 mg/ml; Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA)/DNase I (0.2 mg/ml; Invitrogen;
Thermo Fisher Scientific, Inc.) at 37°C for ~30 min. Subsequently,
tissue samples were cut into small pieces and incubated with warm
PBS containing pronase (1 mg/ml)/collagenase IV (0.4 mg/ml)/DNase I
(0.2 mg/ml) at 37°C for additional 15 min. The digestion was
terminated by the addition of an equal volume of Dulbecco's
modified Eagle's medium (DMEM; Gibco; Thermo Fisher Scientific,
Inc.) containing 10% fetal bovine serum (FBS; HyClone; GE
Healthcare, Logan, UT, USA). The digested mix was filtered and
centrifuged at 160 × g at 4°C for 3 min, and the supernatant was
discarded. The cell pellet was resuspended in PBS, and centrifuged
again. Subsequently, cells suspended in DMEM were carefully added
to 5 ml 39.5% percoll solution (Beijing Solarbio Science &
Technology Co., Ltd., Beijing, China) and were centrifuged at 430 ×
g at 4°C for 20 min. The interface between DMEM and percoll
solution was collected, suspended in DMEM, and centrifuged at 160 ×
g at 4°C for 7 min to collect the final cell pellet. The cells were
then cultured with DMEM containing penicillin/streptomycin and 10%
FBS. The purity of HSCs was >90%, as determined by desmin
immunostaining (24). The first
passage of mHSCs was used in the present study. Recombinant murine
IL-17A and IL-17F (PeproTech, Inc., Rocky Hill, NJ, USA) at 10, 30
or 100 ng/ml were used to stimulate mHSCs for 1, 3, 6, 12, 24 or 48
h. For some experiments, the mHSCs were infected with Ad-ACVR2A
shRNA or Ad-NC shRNA (multiplicity of infection, both 50) for 24 h,
and were then stimulated with recombinant IL-17A (30 ng/ml) or
IL-17F (30 ng/ml) for additional 48 h.
Western blot analysis
Total proteins were isolated from liver tissues and
primary HSCs using radioimmunoprecipitation assay lysis buffer
(P0013B), and protein concentration was quantified using a
Bicinchoninic Acid Protein assay kit (P0009) (both from Beyotime
Institute of Biotechnology, Shanghai, China). Rabbit polyclonal
antibodies against ACVR2A (ab135634, 1:500; Abcam, Cambridge, UK),
type I α collagen (collagen I, BA0325, 1:400; Wuhan Boster
Biological Technology, Ltd., Wuhan, China), type IV α collagen
(collagen IV, BA2174, 1:400; Wuhan Boster Biological Technology,
Ltd.), Smad2 (bs-0718R, 1:500), phosphorylated
(p)-Smad2S465/467 (bs-3419R, 1:500; both from BIOSS,
Beijing, China) and a mouse monoclonal antibody against α-smooth
muscle actin (α-SMA, BM0002, 1:400; Wuhan Boster Biological
Technology, Ltd.) were used to detect the expression of
corresponding proteins. Briefly, equal protein samples (20
μg) mixed with loading buffer were separated by 10% SDS-PAGE
for 2.5 h and were then transferred onto polyvinylidene fluoride
membranes, which were blocked with 5% (M/V) non-fat milk (TTBS
buffered) for 1 h at room temperature. The membranes were then
incubated with primary antibodies at 4°C overnight, followed by
incubation with goat anti-rabbit or goat anti-mouse horseradish
peroxidase (HRP)-labeled-immunoglobulin G (IgG) secondary
antibodies (A0216 or A02087; 1: 5,000; Beyotime Institute of
Biotechnology) for 45 min at 37°C. Finally, the membranes were
visualized using an enhanced chemiluminescence reagent (Beyotime
Institute of Biotechnology) in the dark. β-actin served as the
control (sc-47778; 1:1,000; Santa Cruz Biotechnology, Inc., Dallas,
Texas, USA). The software used to semi-quantify the blots was
gel-pro analyzer (version 4).
Morphological structure analysis and
immunohistochemistry (IHC)
Staining reagents were purchased from Sinopharm
Chemical Reagent Co., Ltd. (Beijing, China) or Beijing Solarbio
Science & Technology Co., Ltd. Briefly, the liver tissues were
first fixed in 4% paraformaldehyde (Sinopharm Chemical Reagent Co.,
Ltd.) at room temperature for 24 h, and then rinsed with water for
4 h. After being dehydrated in an alcohol gradient (70, 80, 90 and
100%), the tissues were embedded into paraffin. The tissue block
was cut into 5-μm slices, slowly spread in water, and dried
at 60°C for ~40 min. Subsequently, the tissue sections were dewaxed
in xylene, rehydrated in an alcohol gradient (95, 85 and 75% for 2
min, respectively) and soaked in water for 2 min. The sections were
then treated with staining reagents accordingly to the
manufacturer's protocols. For hematoxylin and eosin (H&E)
staining, the prepared tissue sections were stained with
hematoxylin for 5 min and with eosin for 3 min. The collagen areas
were stained blue using Masson trichrome. In brief, the slices were
stained with acid ponceau/solferino for 1 min and then with aniline
blue for 5 min at room temperature. Cell nuclei were stained with
hematoxylin for 6 min. The main by-product of collagens,
hydroxyproline, was detected in fresh liver tissues using a
commercial detection kit (A030-1; Nanjing Jiancheng Bioengineering
Institute, Nanjing, China) according to the manufacturer's
protocol.
For IHC, tissue sections were incubated in citrate
buffer at 100°C for 10 min for antigen retrieval, and then with 3%
H2O2 at room temperature for 15 min to block
peroxidase activity. After washing, sections were treated with goat
serum (SL2-10; Beijing Solarbio Science & Technology Co., Ltd.)
for 15 min, and then with rabbit polyclonal antibodies against
α-SMA (BS-0189R, 1:200; BIOSS), collagen I (BA0325, 1:200) and
collagen IV (BA2174, 1:200) (both from Wuhan Boster Biological
Technology, Ltd.) at 4°C overnight under humid conditions. These
samples were then treated with biotin-labeled goat anti-rabbit IgG
(A0277; 1:200; Beyotime Institute of Biotechnology) at 37°C for 30
min, and then with HRP-labeled streptavidin (both from Beyotime
Institute of Biotechnology) for another 30 min. After visualization
with DAB (Beijing Solarbio Science & Technology Co., Ltd.),
images of the tissue sections were captured under a microscope
(BX53; Olympus, Tokyo, Japan).
Reverse transcription-quantitative
real-time polymerase chain reaction (RT-qPCR)
Primers used for detection of the mRNA expression
levels of ACVR2A, actin α2 (ACTA2), collagen type I α1 chain
(COL1A1) and collagen type IV α1 chain (COL4A1) genes are listed in
Table I. Total RNA was isolated
from liver samples and cells using the RNApure extraction kit
(RP1201), after which RNA was reverse transcribed into cDNA using
Super M-MLV reverse transcriptase (PR6502) (both from BioTeke
Corporation, Beijing, China) according to the manufacturer's
protocol. Subsequently, qPCR was performed in a mix containing
primer pairs (10 μM, 0.5 μl of each), cDNA (1
μl), SYBR-Green mastermix (10 μl, SY1020; Beijing
Solarbio Science & Technology Co., Ltd.) and ddH2O
(8 μl) on an Exicycler™ 96 real-time quantitative thermal
block (Bioneer Corporation, Daejeon, South Korea). The relative
mRNA expression levels of each gene were calculated using the
2−ΔΔCq method (25).
β-actin was used as a control. The thermocycling conditions were as
follows: 94°C for 5 min, 38 cycles at 94°C for 15 sec/60°C for 20
sec/72°C for 30 sec, 72°C for 150 sec, 40°C for 90 sec, melting
from 60°C to 94°C (every 1°C for 1 sec), 25°C for 60–120 sec.
 | Table ISequences of primers used in reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I
Sequences of primers used in reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene name | Sequence
(5′>3′) | Product (bp) | Gene ID |
|---|
| ACVR2A | F:
AAGATAAACGGCGACATTG | 232 | NM_007396.4 |
| R:
GTAACAGGATTTGAAGTGGG | | |
| ACTA2 | F:
CCGCAAATGCTTCTAAGTCCC | 195 | NM_007392.3 |
| R:
AATTGAATCAGTGTTGCTAGGC | | |
| COL1A1 | F:
GGCAAGACAGTCATCGAATACA | 137 | NM_007742.4 |
| R:
GAGGGAGTTTACACGAAGCAG | | |
| COL4A1 | F:
CGGCTATTCCTTCGTGATG | 206 | NM_009931.2 |
| R:
ATGGCGTGGGCTTCTTGA | | |
| β-actin | F:
CTGTGCCCATCTACGAGGGCTAT | 155 | NM_007393.5 |
| R:
TTTGATGTCACGCACGATTTCC | | |
Assessment of serum or cell supernatant
indices
The contents of activin A, IL-17A and IL-17F in the
serum, and the contents of collagen I and IV in cell supernatants
were determined using corresponding ELISA kits
(EK0302/EK0431/EK0796; Boster and SEA571Mu/SEA180Mu; Uscn Life
Sciences, Inc., Wuhan, China) according to the manufacturers'
protocols. The activities of aspartate aminotransferase (AST) and
alanine aminotransferase (ALT) were assessed using commercial
detection kits (C010-1/C009-1; Nanjing Jiancheng Bioengineering
Institute) according to the manufacturer's protocols.
Statistical analysis
The experiments were repeated for 3 times. Data are
expressed as the means ± standard deviation or standard error. SPSS
20.0 software (IBM Corp., Armonk, NY, USA) was used to analyze the
results. Data with equal variance between two groups were analyzed
using Mann-Whitney test, whereas those with unequal variance were
analyzed using a Student's t-test. Data over three groups were
analyzed via one-way analysis of variance (ANOVA) and Bonferroni
post hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
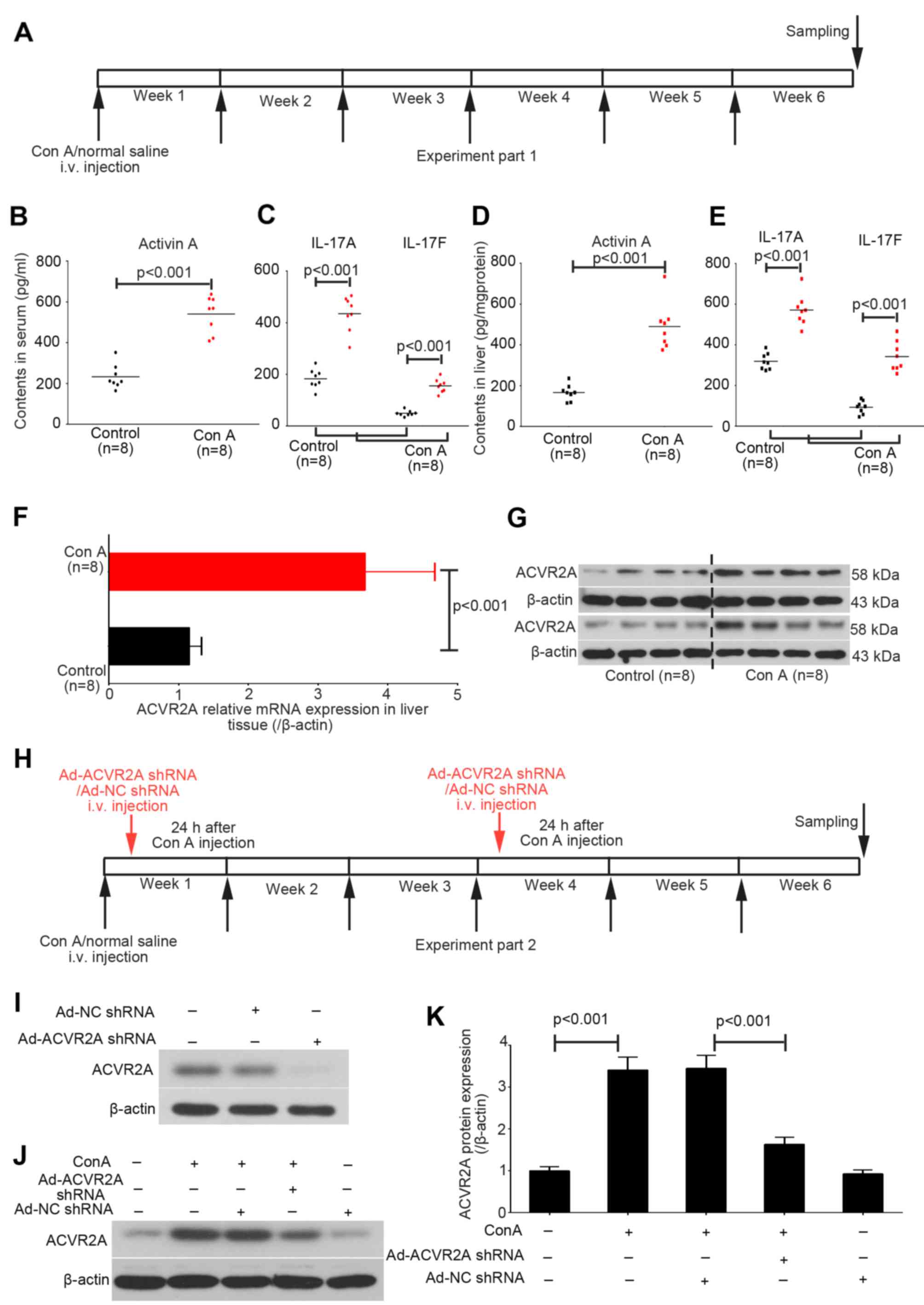
Con A administration stimulates
activation of activin and IL-17 signaling in the liver
In the present study, liver and serum samples were
collected from the control and chronic Con A-treated mice at the
end of week 6 (Fig. 1A). The
contents of activin A, and IL-17A and IL-17F in the serum and liver
tissue samples were detected using corresponding ELISA kits.
Activin A levels were increased in Con A-treated mice compared with
in the control mice (Fig. 1B).
Increases in serum IL-17A and IL-17F levels were also detected in
Con A-treated mice (Fig. 1C).
Similar increases in expression were detected for these three
indices in liver tissues (Fig. 1D and
E).
The mRNA and protein expression levels of ACVR2A
were also upregulated in liver tissues obtained from Con A-treated
mice, as determined by RT-qPCR and western blot analysis (Fig. 1F and G). Subsequently, adenovirus
particles containing ACVR2A shRNA or NC shRNA were intravenously
injected into mice in the Con A group (Fig. 1H). The results demonstrated that
Ad-NC shRNA did not affect the expression of ACVR2A in mice with or
without liver fibrosis, whereas Ad-ACVR2A shRNA significantly
inhibited ACVR2A expression in fibrotic liver tissue (Fig. 1I).
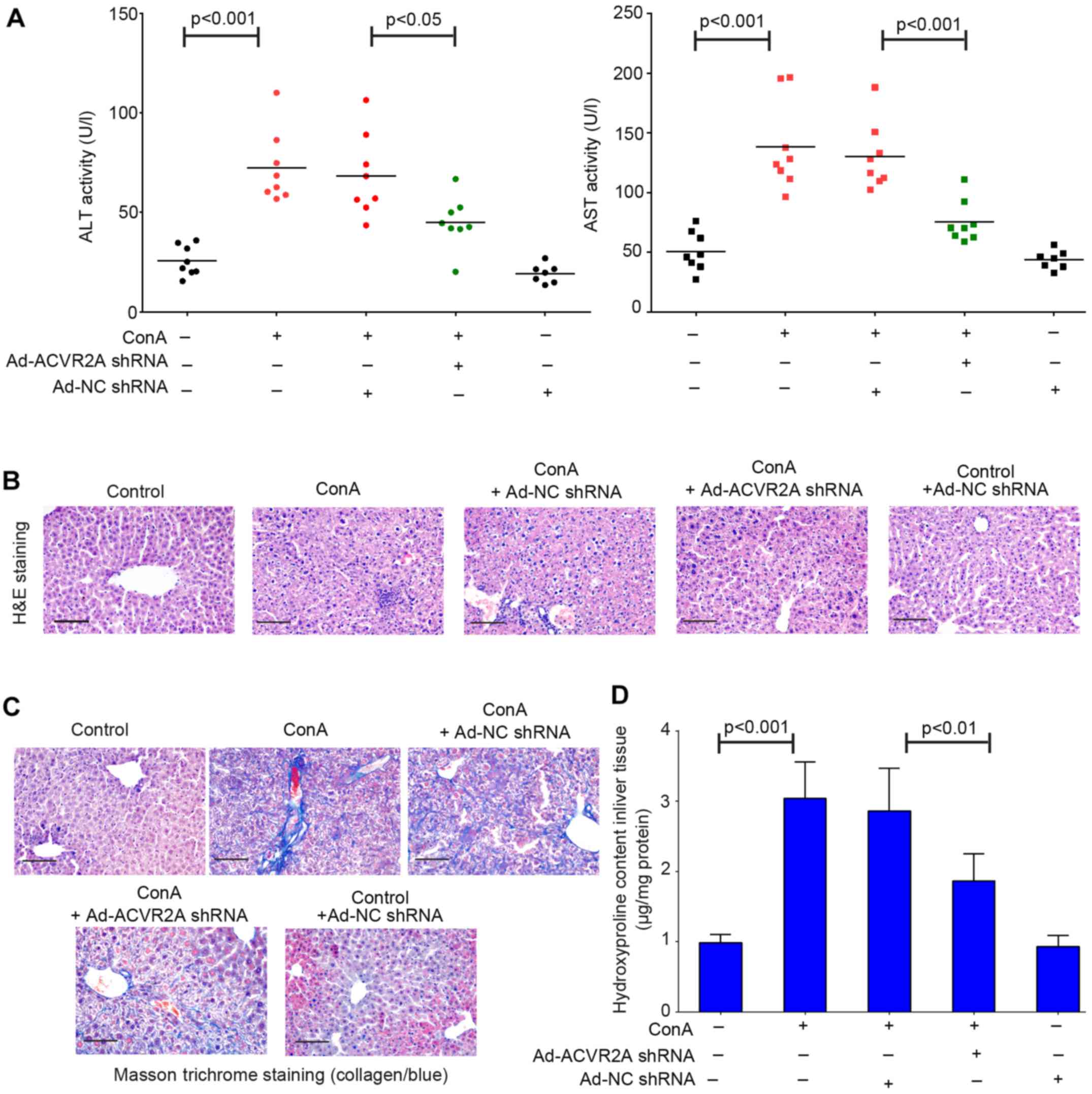
Adenovirus-mediated inhibition of ACVR2A
attenuates Con A-induced liver fibrosis
To detect whether inhibition of ACVR2A may preserve
liver function of Con A-treated mice, the activities of ALT and
AST, which are transaminases critical for liver function, were
tested in serum samples using the corresponding kits. The results
indicated that ALT and AST activities were increased in mice
treated with Con A compared with in the control group (Fig. 2A). Conversely, treatment with
Ad-ACVR2A shRNA, but not Ad-NC shRNA, partly rescued the impaired
liver function, as evidenced by decreased ALT and AST activities
(Fig. 2A). The results of H&E
staining demonstrated that mice in the control group exhibited
normal and clear lobular architecture, and the hepatic cells were
arranged in neat rows (Fig. 2B).
However, apparent pathological alterations were detected in the
livers of Con A-treated mice. The hepatic cord was disarranged and
inflammatory cells abnormally infiltrated into the liver (Fig. 2B). The pathological situation was
not improved by treatment with Ad-NC shRNA; however, it was partly
improved by Ad-ACVR2A shRNA treatment, although not completely
(Fig. 2B). Furthermore, liver
fibrosis was detected using Masson trichrome staining; evident
collagen deposition was observed in liver samples from Con
A-treated mice (Fig. 2C).
Adenovirus-mediated blockade of activin signaling had an
antifibrotic effect in the liver (Fig. 2C). In addition, downregulation of
hydroxyproline in Ad-ACVR2A shRNA-treated mice also confirmed the
reduced liver fibrosis (Fig. 2D).
Notably, in one of the control groups, mice received two injections
of Ad-NC shRNA, in order to investigate whether the adenoviruses
used in the present study had any adverse effects on the mice.
There were no significant alterations in liver structure and
function between normal control mice and those treated with Ad-NC
shRNA. Collectively, these results indicated that abnormal
activation of the activin/ACVR2A signaling pathway may contribute
to immune-associated fibrosis.
Inhibition of activin/ACVR2A signaling
suppresses Con A-induced activation of HSCs in vivo
Excessive collagen deposition indicates abnormal
fibrosis in the liver. The majority of ECM-generating
myofibroblasts in the liver are derived from activated HSCs, and
α-SMA is a molecular marker for activated HSCs (26). In the present study, the mRNA
expression levels of ACTA2, COL1A1 and COL4A1 were determined using
RT-qPCR, and the expression levels of their respective proteins,
α-SMA, collagens I and IV, were detected using western blot
analysis and IHC. The results demonstrated that in Con A-treated
mice, the mRNA expression levels of ACTA2, COL1A1 and COL4A1 were
increased in the liver, whereas mice treated with Ad-ACVR2A shRNA
exhibited decreased expression(Fig.
3A). Similar alterations were detected in α-SMA, collagen I and
IV protein expression (Fig. 3B and
C). Furthermore, the enhanced phosphorylation of Smad2 in
fibrotic liver tissues was suppressed when activin/ACVR2A signaling
was blocked (Fig. 3D).
Collectively, these data demonstrated that activation of HSCs was
decreased in mice treated with Ad-ACVR2A shRNA.
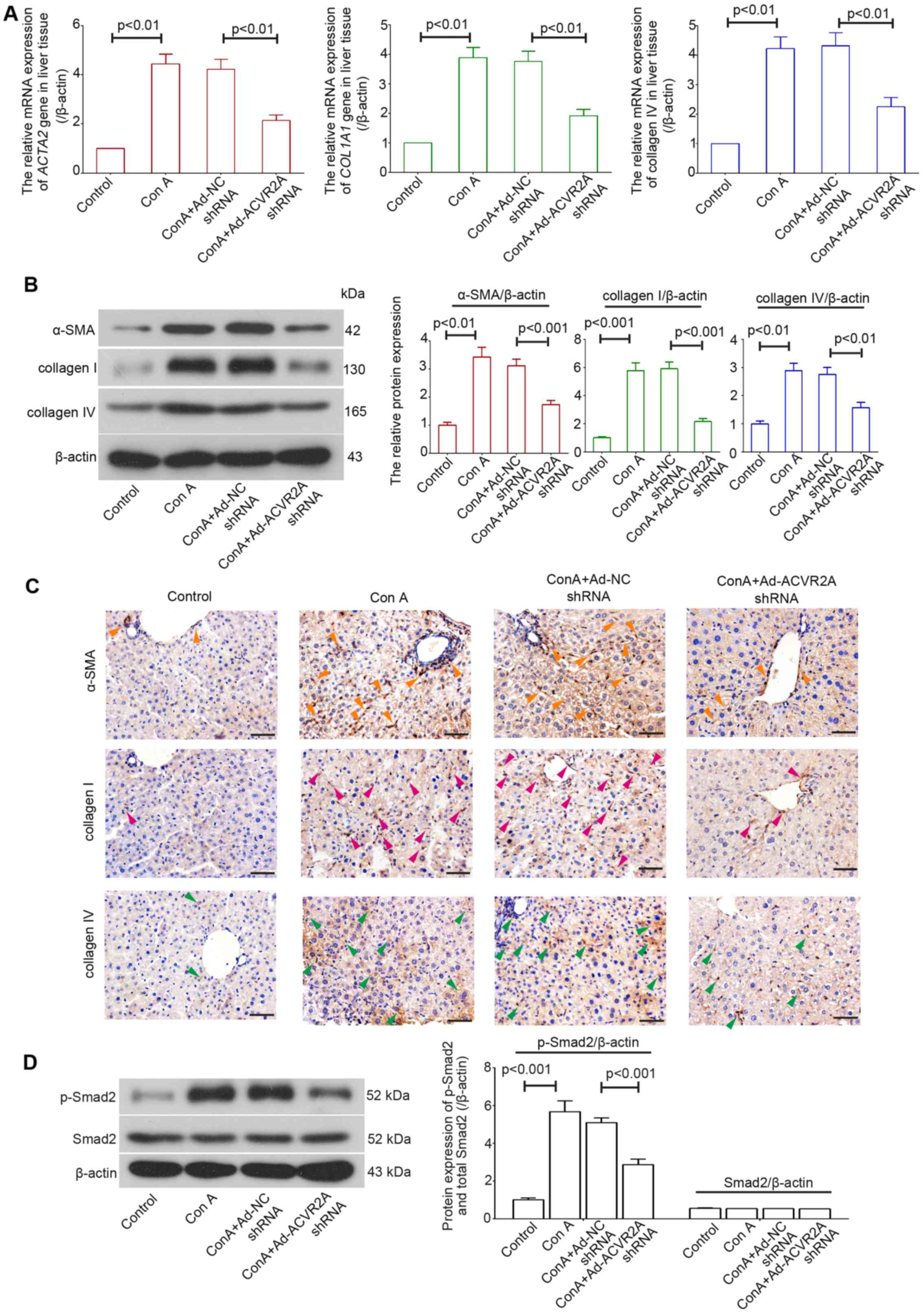 | Figure 3Knockdown of ACVR2A suppresses Con
A-induced activation of hepatic stellate cells in vivo. (A)
Relative mRNA and (B) protein expression levels of ACTA2, COL1A1
and COL4A1 were determined using reverse transcription-quantitative
polymerase chain reaction and western blot analysis, respectively.
(C) Immunohistochemistry was performed to detect the protein
expression levels of α-SMA, collagen I and IV. Orange arrowheads,
representative α-SMA-positive cells; pink arrowheads,
representative collagen I-positive cells; green arrowheads,
representative collagen IV-positive cells. Scale bars, 50
μm. (D) Protein expression levels of p-Smad2 and total Smad2
in liver tissues were detected using western blot analysis. Data
are expressed as the means ± standard error (n=5/group). α-SMA,
α-smooth muscle actin; ACVR2A, activin A receptor type 2A; Con A,
concanavalin A; ; NC, negative control; p-, phosphorylated; shRNA,
short hairpin RNA. |
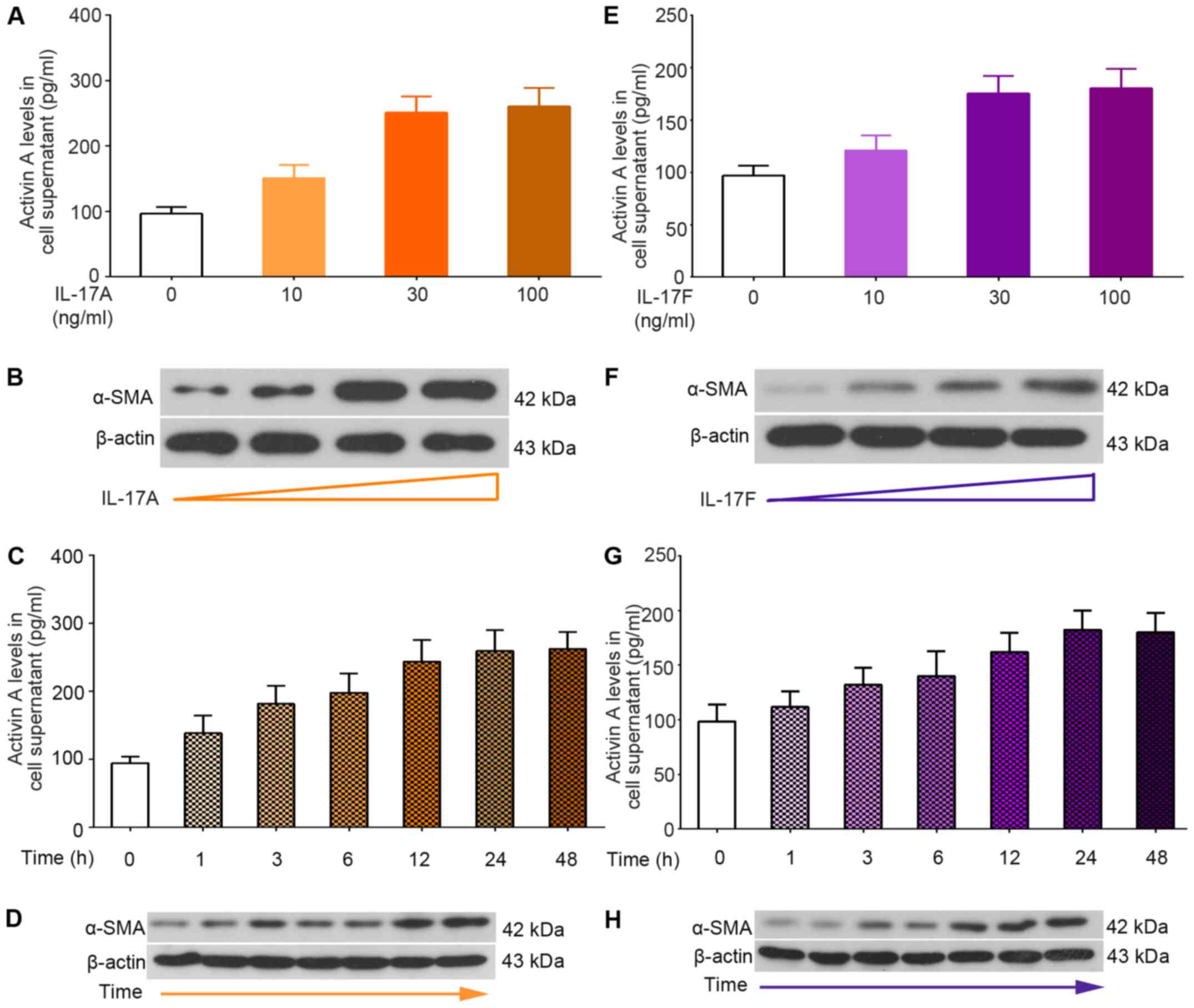
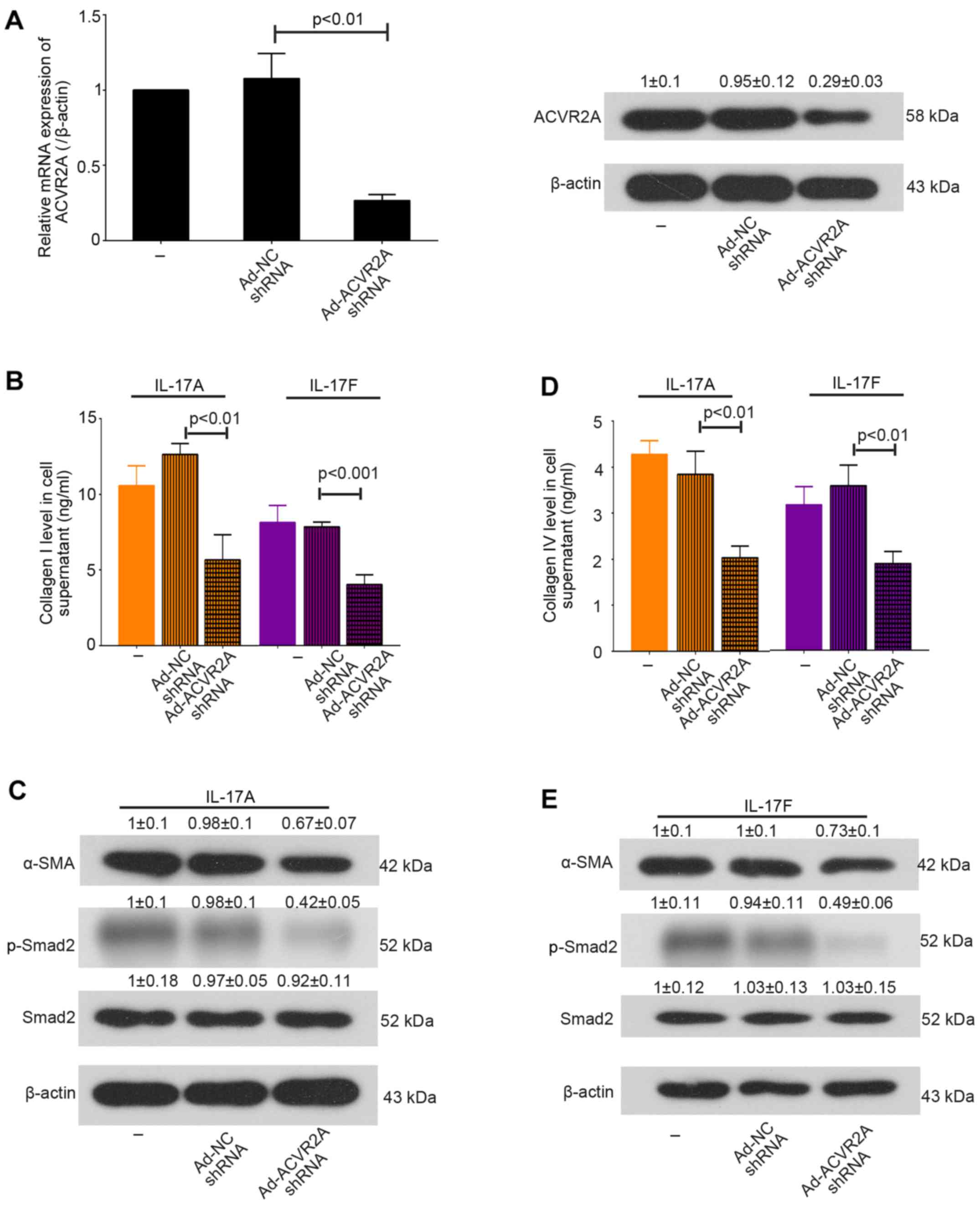
Ad-ACVR2A shRNA suppresses IL-17-induced
activation of primary mHSCs in vitro
In order to determine the role of the
activin/ACVR2A/Smad2 pathway in immune-associated liver fibrosis,
primary mHSCs were isolated from normal liver samples and were
treated with IL-17A and IL-17F (Fig.
4). Following stimulation with IL-17A or IL-17F, mHSCs produced
more activin A, as evidenced by ELISA (Fig. 4A, C, E and G). In addition, an
increase in α-SMA was detected in mHSCs in response to IL-17
treatment (Fig. 4B, D, F and H).
Furthermore, in order to disrupt activin/ACVR2A signaling in
primary mHSCs, cells were infected with Ad-ACVR2A shRNA or Ad-NC
shRNA for 24 h, and were then stimulated with IL-17A or IL-17F for
an additional 48 h. The results demonstrated that IL-17-induced
overproduction of collagens was suppressed when ACVR2A was
inhibited. In addition, the expression levels of α-SMA and p-Smad2
were decreased in mHSCs infected with Ad-ACVR2A shRNA (Fig. 5). Taken together, these results
suggested that knockdown of ACVR2A may inhibit IL-17-induced mHSC
activation in vitro.
Discussion
The natural inhibitor of activin, follistatin, has
been reported to attenuate CCl4-induced liver fibrosis
by reducing HSC proliferation and activation (27), thus suggesting that activated
activin signaling may contribute to liver fibrosis. An immune
imbalance is involved in liver fibrosis (1); however, the mechanisms underlying
activin signaling in immune-induced liver fibrosis remain unclear.
In the present study, adenovirus-mediated knockdown of ACVR2A was
performed in mice treated with Con A and in primary mHSCs
stimulated with IL-17. The results indicated that the suppression
of activin signaling attenuated chronic Con A
administration-induced hepatic fibrosis, in addition to preserving
liver function. Collagen production in IL-17-stimulated mHSCs was
also inhibited in response to ACVR2A knockdown.
Patella et al reported that serum levels of
activin A were much higher in human subjects with chronic viral
hepatitis (28), thus revealing a
link between increased activin A and liver disorders. Similar to
TGF-β, activins initiate signaling by binding to heterodimers,
which consist of the two type II receptors: ACVR2A and activin A
receptor type 2B (10). Our
previous study reported that the mRNA expression levels of activin
β and ACVR2A may be increased in the liver of mice following a
single injection of CCl4, and that blockade of activin
signaling using a specific blocking antibody may preserve liver
function (29). This previous
study revealed an involvement of activin signaling in
repair/regeneration-related acute liver injury. The present study
aimed to further explore the role of this signaling pathway in
immune-associated fibrotic liver injury.
The present study detected increased levels of
activin A in the serum, and of its receptor ACVR2A in liver tissues
of mice with T-cell-mediated fibrosis. Suppression of activin
signaling was achieved using adenoviruses containing ACVR2A shRNA.
The results indicated that systematic delivery of Ad-ACVR2A shRNA
effectively inhibited ACVR2A expression in fibrotic liver tissues.
Furthermore, liver function was partly restored in response to
Ad-ACVR2A shRNA, and the activation of HSCs and collagen deposition
were also inhibited. George et al demonstrated that the
TGF-β receptor antagonist is able to partly block fibrogenesis
induced by ligation of the common bile duct in rats (30). These findings indicated that TGF-β
may be a determinant for liver fibrosis; however, it may not be the
only one. The present study suggested a pathogenic role for
activated activin signaling in experimental liver fibrosis,
particularly in immune-induced liver fibrosis.
Fibrotic autoimmune diseases, including primary
biliary cirrhosis and systemic sclerosis, are associated with an
inflammatory process in which Th17 lymphocytes serve a critical
role (31). IL-17 is a
proinflammatory and fibrogenic cytokine that is mainly produced by
Th17 cells. In a previous study, IL-17 elevation was detected in
animals with Con A-induced acute liver injury (32); similarly, in the present study,
IL-17A and IL-17F levels were upregulated in mice chronically
treated with Con A. HSCs are considered potential target cells for
IL-17, because they also express IL-17 receptors (33,34). Previous studies have indicated
that IL-17s strongly stimulate the activation of HSCs; however,
these studies have focused on TGF-β signaling (17,21). The production of activin A is
reported to be mediated by numerous immune-related cytokines,
including interferon-γ (IFN-γ) and IL-10 (35,36). Notably, in a Con A-induced liver
fibrosis model, the expression levels of IFN-γ and IL-10 were
increased in serum and liver samples (15). In order to exclusively study
whether IL-17s affect activin signaling transduction in HSCs, and
whether the interaction between IL-17 and activin signaling impacts
HSC activation, recombinant IL-17A and IL-17F were used to
stimulate primary mHSCs with normal or reduced ACVR2A expression
in vitro.
The present results were in agreement with a
previous study (21), thus
indicating that IL-17 may induce activation of mHSCs. TGF-β and
activin A are autocrine factors, which stimulate each other's
expression, thus triggering amplified fibrotic signaling in HSCs
(37). Notably, in addition to
TGF-β, the present study demonstrated that mHSCs generated more
activin A in response to IL-17 stimulation. Furthermore, mHSCs with
reducedACVR2A expression produced less collagens in the presence of
IL-17A or IL-17F; α-SMA expression was also decreased. Wada et
al previously reported that exogenous activin A activates HSCs
and increases their collagen production and α-SMA expression
(37). The present findings
supported the hypothesis that activated activin signaling is
involved in IL-17-induced HSC activation.
Smad proteins are known as intracellular mediators
for signaling transduction of TGF-β family members (38). The receptor-regulated Smads, such
as Smad2, can be recruited and phosphorylated by heteromeric
complexes, including activins and their receptors, and further form
heterotrimers with Smad4 to relocate into the nucleus, thus
regulating transcription via various promoters (10). Reportedly, exogenous activin A
induces phosphorylation on Smad2 at Ser465 and Ser467 in hepatic
progenitor cells (39).
Phosphorylation of Smad2 in the C terminus is considered a marker
of fibrogenic signaling (40).
Ser465 and Ser467 are located in the C terminus of Smad2, and the
phosphorylation of these sites may provide a recognition site for
interaction with Smad4 (41).
Therefore, due to the critical role of p-Ser465/Ser467 in Smad2
signaling activation, the present study detected p-Smad2
(Ser465/Ser467) expression. The results demonstrated that
phosphorylation of these two sites induced by Con A in liver and by
IL-17 in mHSCs were attenuated when activin signaling was
blocked.
Notably, besides Th17 cells, other polarized
CD4+ T-cells, including Th1, Th2 and regulatory T-cells,
are critical regulators of the immune response during fibrosis
(42). Further study is required
to explore whether the activin A/ACVR2A/Smad2 signaling pathway is
mediated by other CD4+ T-cells in hepatic fibrosis.
Furthermore, in the presence of proinflammatory factors, including
IL-1β and tumor necrosis factor-α, exogenous TGF-β1 treatment is
able to promote the conversion of naive CD4+ T-cells
into Th17 cells (43). Activin A
has a synergic role with TGF-β1 (44). Notably, Ihn et al
demonstrated that transcription of the ACVR2A gene is induced
during Th17 differentiation, but not in Th1 or Th2 cells, thus
suggesting that the activin/ACVR2A signaling pathway may serve a
role in Th17 polarization (45).
The present study focused on Th17 cell-mediated activation of HSCs;
however, to fully reveal the role of activin A/ACVR2A signaling in
immune-associated liver fibrosis, it will be interesting to
investigate whether activated HSCs in turn mediate the polarization
of CD4+ T-cells through this signaling pathway.
In conclusion, the present study demonstrated that
activated activin A/ACVR2A/Smad2 signaling may contribute to Con
A-induced hepatic fibrosis and IL-17-mediated HSC activation.
Further studies are required to explore the role of this signaling
pathway in the interaction between immune cells and HSCs in liver
fibrosis.
Acknowledgments
Not applicable.
References
|
1
|
Pellicoro A, Ramachandran P, Iredale JP
and Fallowfield JA: Liver fibrosis and repair: Immune regulation of
wound healing in a solid organ. Nat Rev Immunol. 14:181–194. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nanthakumar CB, Hatley RJ, Lemma S,
Gauldie J, Marshall RP and Macdonald SJ: Dissecting fibrosis:
Therapeutic insights from the small-molecule toolbox. Nat Rev Drug
Discov. 14:693–720. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lee YA, Wallace MC and Friedman SL:
Pathobiology of liver fibrosis: A translational success story. Gut.
64:830–841. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mederacke I, Hsu CC, Troeger JS, Huebener
P, Mu X, Dapito DH, Pradere JP and Schwabe RF: Fate tracing reveals
hepatic stellate cells as dominant contributors to liver fibrosis
independent of its aetiology. Nat Commun. 4:28232013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen RJ, Wu HH and Wang YJ: Strategies to
prevent and reverse liver fibrosis in humans and laboratory
animals. Arch Toxicol. 89:1727–1750. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Seki E and Schwabe RF: Hepatic
inflammation and fibrosis: Functional links and key pathways.
Hepatology. 61:1066–1079. 2015. View Article : Google Scholar :
|
|
7
|
Bissell DM, Roulot D and George J:
Transforming growth factor beta and the liver. Hepatology.
34:859–867. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakamura T, Sakata R, Ueno T, Sata M and
Ueno H: Inhibition of transforming growth factor beta prevents
progression of liver fibrosis and enhances hepatocyte regeneration
in dimethylnitrosamine-treated rats. Hepatology. 32:247–255. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qi Z, Atsuchi N, Ooshima A, Takeshita A
and Ueno H: Blockade of type beta transforming growth factor
signaling prevents liver fibrosis and dysfunction in the rat. Proc
Natl Acad Sci USA. 96:2345–2349. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Weiss A and Attisano L: The TGFbeta
superfamily signaling pathway. Wiley Interdiscip Rev Dev Biol.
2:47–63. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kreidl E, Oztürk D, Metzner T, Berger W
and Grusch M: Activins and follistatins: Emerging roles in liver
physiology and cancer. World J Hepatol. 1:17–27. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sugiyama M, Ichida T, Sato T, Ishikawa T,
Matsuda Y and Asakura H: Expression of activin A is increased in
cirrhotic and fibrotic rat livers. Gastroenterology. 114:550–558.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gold EJ, Francis RJ, Zimmermann A, Mellor
SL, Cranfield M, Risbridger GP, Groome NP, Wheatley AM and Fleming
JS: Changes in activin and activin receptor subunit expression in
rat liver during the development of CCl4-induced
cirrhosis. Mol Cell Endocrinol. 201:143–153. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Fujita T, Soontrapa K, Ito Y, Iwaisako K,
Moniaga CS, Asagiri M, Majima M and Narumiya S: Hepatic stellate
cells relay inflammation signaling from sinusoids to parenchyma in
mouse models of immune-mediated hepatitis. Hepatology.
63:1325–1339. 2016. View Article : Google Scholar
|
|
15
|
Louis H, Le Moine A, Quertinmont E, Peny
MO, Geerts A, Goldman M, Le Moine O and Devière J: Repeated
concanavalin A challenge in mice induces an interleukin
10-producing phenotype and liver fibrosis. Hepatology. 31:381–390.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tu CT, Li J, Wang FP, Li L, Wang JY and
Jiang W: Glycyrrhizin regulates CD4+ T cell response during liver
fibrogenesis via JNK, ERK and PI3K/AKT pathway. Int
Immunopharmacol. 14:410–421. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meng F, Wang K, Aoyama T, Grivennikov SI,
Paik Y, Scholten D, Cong M, Iwaisako K, Liu X, Zhang M, et al:
Interleukin-17 signaling in inflammatory, Kupffer cells, and
hepatic stellate cells exacerbates liver fibrosis in mice.
Gastroenterology. 143:765–76. e1–3. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Song E, Lee SK, Wang J, Ince N, Ouyang N,
Min J, Chen J, Shankar P and Lieberman J: RNA interference
targeting Fas protects mice from fulminant hepatitis. Nat Med.
9:347–351. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu PB, Beppu H, Kawai N, Li E and Bloch
KD: Bone morphogenetic protein (BMP) type II receptor deletion
reveals BMP ligand-specific gain of signaling in pulmonary artery
smooth muscle cells. J Biol Chem. 280:24443–24450. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu WH, Hu HG, Tian Y, Wang SZ, Li J, Li
JZ, Deng X, Qian H, Qiu L, Hu ZL, et al: Bioactive compound reveals
a novel function for ribosomal protein S5 in hepatic stellate cell
activation and hepatic fibrosis. Hepatology. 60:648–660. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tan Z, Qian X, Jiang R, Liu Q, Wang Y,
Chen C, Wang X, Ryffel B and Sun B: IL-17A plays a critical role in
the pathogenesis of liver fibrosis through hepatic stellate cell
activation. J Immunol. 191:1835–1844. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Kim BG, Qian S, Letterio JJ, Fung
JJ, Lu L and Lin F: Hepatic stellate cells inhibit T cells through
active TGF-β1 from a cell surface-bound latent TGF-β1/GARP complex.
J Immunol. 195:2648–2656. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu MC, Chen CH, Liang X, Wang L, Gandhi
CR, Fung JJ, Lu L and Qian S: Inhibition of T-cell responses by
hepatic stellate cells via B7-H1-mediated T-cell apoptosis in mice.
Hepatology. 40:1312–1321. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Weiskirchen S, Tag CG, Sauer-Lehnen S,
Tacke F and Weiskirchen R: Isolation and culture of primary murine
hepatic stellate cells. Methods Mol Biol. 1627:165–191. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Chang J, Lan T, Li C, Ji X, Zheng L, Gou
H, Ou Y, Wu T, Qi C, Zhang Q, et al: Activation of Slit2-Robo1
signaling promotes liver fibrosis. J Hepatol. 63:1413–1420. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Patella S, Phillips DJ, Tchongue J, de
Kretser DM and Sievert W: Follistatin attenuates early liver
fibrosis: Effects on hepatic stellate cell activation and
hepatocyte apoptosis. Am J Physiol Gastrointest Liver Physiol.
290:G137–G144. 2006. View Article : Google Scholar
|
|
28
|
Patella S, Phillips DJ, de Kretser DM,
Evans LW, Groome NP and Sievert W: Characterization of serum
activin-A and follistatin and their relation to virological and
histological determinants in chronic viral hepatitis. J Hepatol.
34:576–583. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wang DH, Wang YN, Ge JY, Liu HY, Zhang HJ,
Qi Y, Liu ZH and Cui XL: Role of activin A in carbon
tetrachloride-induced acute liver injury. World J Gastroenterol.
19:3802–3809. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
George J, Roulot D, Koteliansky VE and
Bissell DM: In vivo inhibition of rat stellate cell activation by
soluble transforming growth factor beta type II receptor: A
potential new therapy for hepatic fibrosis. Proc Natl Acad Sci USA.
96:12719–12724. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fenoglio D, Bernuzzi F, Battaglia F,
Parodi A, Kalli F, Negrini S, De Palma R, Invernizzi P and Filaci
G: Th17 and regulatory T lymphocytes in primary biliary cirrhosis
and systemic sclerosis as models of autoimmune fibrotic diseases.
Autoimmun Rev. 12:300–304. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chen Y, Peng H, Chen Y, Wei H, Sun R and
Tian Z: CD49a promotes T-cell-mediated hepatitis by driving T
helper 1 cytokine and interleukin-17 production. Immunology.
141:388–400. 2014. View Article : Google Scholar :
|
|
33
|
Kolls JK and Lindén A: Interleukin-17
family members and inflammation. Immunity. 21:467–476. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang Q, Zhou J, Zhang B, Tian Z, Tang J,
Zheng Y, Huang Z, Tian Y, Jia Z, Tang Y, et al: Hepatitis B virus
induces IL-23 production in antigen presenting cells and causes
liver damage via the IL-23/IL-17 axis. PLoS Pathog. 9:e10034102013.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Seeger P, Bosisio D, Parolini S, Badolato
R, Gismondi A, Santoni A and Sozzani S: Activin A as a mediator of
NK-dendritic cell functional interactions. J Immunol.
192:1241–1248. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
González-Domínguez É, Domínguez-Soto Á,
Nieto C, Flores-Sevilla JL, Pacheco-Blanco M, Campos-Peña V,
Meraz-Ríos MA, Vega MA, Corbí ÁL and Sánchez-Torres C: Atypical
activin A and IL-10 production impairs human CD16+ monocyte
differentiation into anti-inflammatory macrophages. J Immunol.
196:1327–1337. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wada W, Kuwano H, Hasegawa Y and Kojima I:
The dependence of transforming growth factor-beta-induced collagen
production on autocrine factor activin A in hepatic stellate cells.
Endocrinology. 145:2753–2759. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Macias MJ, Martin-Malpartida P and
Massagué J: Structural determinants of Smad function in TGF-β
signaling. Trends Biochem Sci. 40:296–308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Chen L, Zhang W, Liang HF, Zhou QF, Ding
ZY, Yang HQ, Liu WB, Wu YH, Man Q, Zhang BX, et al: Activin A
induces growth arrest through a SMAD- dependent pathway in hepatic
progenitor cells. Cell Commun Signal. 12:182014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yoshida K, Murata M, Yamaguchi T,
Matsuzaki K and Okazaki K: Reversible human TGF-β signal shifting
between tumor suppression and fibro-carcinogenesis: Implications of
Smad phospho-isoforms for hepatic epithelial-mesenchymal
transitions. J Clin Med. 5:52016. View Article : Google Scholar
|
|
41
|
Souchelnytskyi S, Tamaki K, Engström U,
Wernstedt C, ten Dijke P and Heldin CH: Phosphorylation of Ser465
and Ser467 in the C terminus of Smad2 mediates interaction with
Smad4 and is required for transforming growth factor-beta
signaling. J Biol Chem. 272:28107–28115. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wynn TA: Fibrotic disease and the
T(H)1/T(H)2 paradigm. Nat Rev Immunol. 4:583–594. 2004. View Article : Google Scholar
|
|
43
|
Veldhoen M, Hocking RJ, Atkins CJ,
Locksley RM and Stockinger B: TGFbeta in the context of an
inflammatory cytokine milieu supports de novo differentiation of
IL-17-producing T cells. Immunity. 24:179–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kang JO, Lee JB and Chang J: Cholera toxin
promotes Th17 cell differentiation by modulating expression of
polarizing cytokines and the antigen-presenting potential of
dendritic cells. PLoS One. 11:e01570152016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Ihn HJ, Kim DH, Oh SS, Moon C, Chung JW,
Song H and Kim KD: Identification of Acvr2a as a Th17 cell-specific
gene induced during Th17 differentiation. Biosci Biotechnol
Biochem. 75:2138–2141. 2011. View Article : Google Scholar : PubMed/NCBI
|