Introduction
Cerebral ischemia is a leading cause of death and
disability in adults. Despite a large number of animal experiments
and clinical studies, there is no effective therapy for protecting
against ischemic damage in humans (1,2).
Cerebral ischemia reperfusion triggers an ischemic cascade in the
brain characterized by excitotoxicity, ionic imbalance, oxidative
stresses, endoplasmic reticulum (ER) stress and mitochondrial
disturbances. This ultimately results in programmed cell death and
necrosis (3–6). Previous studies showed that ER
stress plays a crucial role in mediating neuronal cell death
resulting from ischemia reperfusion (7,8)
and recent studies have suggested that attenuation of
ischemia-induced ER stress can protect neuron cells against
ischemia reperfusion injury (9,10).
The ER is an important subcellular organelle
responsible for proper folding and sorting of proteins (11–15). Reperfusion after ischemia causes a
cellular stress condition characterized by glucose deprivation,
depletion of ER Ca2+ stores, and exposure to free
radicals. This results in accumulation of unfolded proteins in the
ER lumen, a condition referred to as ER stress (11–15). This then triggers the unfolded
protein response to restore ER function via activation of three ER
transmembrane receptors including inositol requiring enzyme,
PRKR-like endoplasmic reticulum kinase, and activating
transcription factor 6 (16).
Activated PERK leads to phosphorylate eukaryotic translation
initiation factor 2 subunit α (eIF2α), resulting in inhibition of
protein synthesis (17,18). Furthermore, phosphorylated eIF2α
also causes increased translation of activating transcription
factor 4 (ATF4) and CCAAT/enhancer binding protein homologous
protein (CHOP) (19,20). Activated CHOP has been known to
play a key role in ER stress-induced apoptosis through
downregulation of anti-apoptotic factor B cell lymphoma-2 (Bcl-2)
and upregulation of reactive oxygen species (ROS) (8,21,22). Drugs which inhibit ER
stress-induced apoptosis provide neuroprotective effects in rats
after transient middle cerebral artery (MCA) occlusion (23–25). Therefore, attenuation of ER
stress-induced apoptosis may be a therapeutic strategy in the
treatment of stroke in the future.
Melatonin (N-acetyl-5-methoxy-tryptamine) provides
neuroprotective effects and protects the brain against ischemic
stroke in rats and mice (26–28). In the acute stage of stroke,
treatment with melatonin decreases ischemic infarct size (28–31), reduces DNA fragmentation (30,32), diminishes mitochondrial cytochrome
c release (33,34), and inhibits caspase-3 activity in
the brains of rats (33). Our
previous studies have demonstrated that melatonin reduces
intracerebral cellular inflammatory response (31) and oxidative damage, protects
against gray and white matter damage, as well as improves
neuroplasticity, neurobehavioral and electrophysiological outcomes
in rats following MCA occlusion (27,28,35,36). However, it was not previously
known whether melatonin mediated ER stress during ischemic
reperfusion. In this study, we investigated the modulation effect
of melatonin on ischemic reperfusion-induced ER stress in the
brain.
Materials and methods
Animals
Adult male Sprague-Dawley rats, weighing 240–290 g,
were procured from the University Laboratory Animal Center of
National Cheng Kung University (NCKU). Animal experiments were
conducted after approval and in accordance with the strict
guidelines of the Subcommittee on Research Animal Care of NCKU
University Medical Center, and the standards meet the guidelines of
the Taiwan National Institutes of Health.
Chemicals and reagents
All chemicals were purchased from Sigma-Aldrich (St.
Louis, MO, USA) unless otherwise indicated. Melatonin was dissolved
in polyethylene glycol 400 (PEG 400) or dimethylsulfoxide (DMSO)
(both from Sigma-Aldrich).
Neuronal cultures and oxygen and glucose
deprivation (OGD) induced neuronal cell injury
According to a previously described method, cultured
neurons were obtained from the cerebral cortices of 1-day-old
Sprague-Dawley rats (36,37). Experiments were performed on
cultured neurons between 7 and 10 days in vitro. OGD was
achieved by inducing hypoxia and aglycemia according to the method
(38,39). The OGD medium consisting of Hank's
Balanced Salt Solution (HBSS) lacking glucose. The HBSS solution
was bubbled with N2 for 30 min to deplete glucose and
oxygen from intracellular stores and extracellular space. Cultured
neurons were pretreated with melatonin (10–500 µM) or
control (0.1% DMSO) for 30 min, and then were subjected to OGD.
After the deprivation period, cultured neurons were incubated in
the culture medium under normal conditions (a humidified incubator
with 5% CO2 at 37°C). Each experiment consisted of three
samples. Four independent instances were carried out for each
experiment.
Transient MCA occlusion model and drug
administration
Focal cerebral ischemia was induced in the rats by
MCA occlusion using a modification of the intraluminal technique
(26,27,35,40–42). Recirculation/reperfusion of
cerebral blood flow was allowed by gently removing the monofilament
carefully after 90 min of ischemia. In the sham-operated animals,
all procedures except for the insertion of the nylon filament were
carried out.
Animals were administered intravenously with either
melatonin (5 mg/kg) or the same volume of PEG-saline at the start
of reperfusion. In the first series of experiments, the animals
treated with melatonin (5 mg/kg, n=5) or control (PEG-saline, n=6)
were euthanized after 24 h following the ischemic insult then
evaluated for brain infarction and neuronal damage. The second
series of animals received melatonin (5 mg/kg, n=15) or control
(n=15) upon reperfusion and were evaluated for ER stress-associated
proteins by western blotting at 1 and 24 h post-reperfusion. The
third series of animals received melatonin (5 mg/kg, n=19) or
control (n=12) upon reperfusion and were then assessed for ER
stress- and neuronal cell-associated proteins using
immunofluorescence staining at 1 and 24 h post-reperfusion.
Additionally, 15 animals received sham-operations to serve as
nonischemic controls.
Animal sacrifice and quantification of
ischemic damage
Rats subjected to MCA occlusion were perfused under
anesthesia with cold phosphate-buffered saline (PBS) and then with
4% paraformaldehyde prepared in 0.1 M PBS for internal fixation.
The subject brains were quickly removed, stored in the same
fixative for 24 h, and sequentially immersed in 15 and 30% sucrose
at 4°C for 48 h. They were then embedded in Optimal Cutting
Temperature compound (OCT; Miles Inc., Elkhart, IN, USA) and frozen
in liquid nitrogen. The brains were sectioned coronally on a
cryostat (HM-500O; Microm International GmbH, Walldorf, Germany).
Serial sections of 40 µm at eight preselected coronal levels
with 1-mm intervals from the stereotaxic coordinates of the Bregma
AP +2.22 to -4.78 mm were mounted on poly-L-lysine-coated slides
and dried overnight at 37°C.
Brain infarction was determined by staining
preselected brain slices with hematoxylin and eosin (H&E)
stain. Under light microscopy, areas of neuronal perikarya
displaying typical morphological features of ischemic damage were
delineated. Infarction volume was measured using a computerized
image analyzer (MCID Elite; Imaging Research Inc., St. Catharines,
ON, Canada) and expressed as a percentage of the contralateral
hemisphere volume (41).
Histological analysis
The brain sections and cultured neurons were
collected on poly-L-lysine coated slides for further processing.
For immunohistochemistry, sections were permeabilized using Triton
X-100 and sections were blocked in 1% serum for 1 h. After
blocking, sections were incubated with rabbit anti-p-PERK antibody
(1:200; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
rabbit anti-eIF2 antibody (1:200; Cell Signaling Technology, Inc.,
Danvers, MA, USA), mouse anti-neuronal nuclei (NeuN) monoclonal
antibody (1:1,000; eBioscience, Inc., San Diego, CA, USA), mouse
anti-microtubule-associated protein-2 (MAP-2) monoclonal antibody
(1:1,000; Santa Cruz Biotechnology, Inc.), and mouse anti-glial
fibrillary acidic protein (GFAP) monoclonal antibody (1:1,000;
eBioscience, Inc.) at 4°C overnight. Appropriate secondary antibody
conjugated with biotin (1:100) was then added, followed by
FITC-conjugated streptavidin and Texas red-conjugated streptavidin
(1:100) (both from Jackson ImmunoResearch, Inc., West Grove, PA,
USA). Sections were counterstained for nucleus with
4′,-6-diamidino-2-phenylindole (DAPI) and then visualized under a
fluorescence microscope (Olympus IX71; Olympus Optical Co., Ltd.,
Tokyo, Japan). The signals for FITC (green), Texas red (red), and
DAPI (blue) were superimposed using Image Pro Plus 5.1 software
(Media Cybernetics, Silver Spring, MD, USA).
Terminal deoxynucleotidyltransferase
(TdT)-mediated dUTP nick end labeling (TUNEL) assay
TUNEL assay was performed using an in situ
cell death detection kit (Calbiochem, Merk Biosciences, Bad Soden,
Germany) according to the manufacturer's protocol. Specifically,
sections were fixed with 4% paraformaldehyde prepared in 0.1 M PBS
and then incubated with 3% H2O2 at room
temperature for 5 min. The sections were digested with fresh
diluted proteinase K (1:200) at RT for 10 min. Then, TdT
Equilibration buffer was added to each section and allowed to
settle at RT for 30 min. Sections were incubated with TdT labeling
reaction mixture at 37°C for 2 h in the dark. The nuclei were
stained with DAPI. Sections were examined under the fluorescence
microscope (Olympus IX71; Olympus Optical Co., Ltd.). The positive
cells and total cells were counted for three fields at ×200
magnification. The results are presented as a ratio of positive
cells to total cells.
Western blot analysis
Samples were obtained from cultured neurons and from
the brain tissues of the contralateral, penumbral core, and
ischemic core regions which were quickly dissected on dry ice after
the animals were sacrificed. The 0.1 g of tissue was homogenized in
1 ml lysis buffer, containing 20 mM HEPES, 250 mM sucrose, 1 mM
EDTA (pH 7.5), 20 mM EGTA (pH 7.5), 10 mM KCl, 250 mM
MgCl2 and a complete protease inhibitor on ice for 30
min. Homogenates were centrifuged at 800 × g for 15 min at 4°C. The
nuclear extract pellets collected and stored at −80°C until used.
The supernatant was carefully transferred to another centrifuge
tube and was centrifuged at 100,000 × g for 1 h at 4°C. The
resultant supernatant containing cytoplasma extract and ER extract
pellets was harvested and stored at −80°C until used. After mixing
with sodium dodecyl sulfate buffer and heating 10 min at 100°C,
protein concentration was determined by bicinchoninic acid (BCA
protein assay kit; Thermo Fisher Scientific, Waltham, MA, USA). For
western blot analysis, an equal amount of protein (50 µg)
was loaded in each well and subjected to 10% sodium dodecyl
sulfate-polyacrylamide gels (SDS-PAGE). The separated proteins were
then transferred onto polyvinylidene fluoride (PVDF) microporous
membranes (IPVH00010; Millipore, Billerica, MA, USA) and blocked in
5% milk. The membranes were incubated with primary anti-p-PERK
(1:400; Santa Cruz Biotechnology, Inc.), anti-eIF2α (1:1,000; Cell
Signaling Technology, Inc.), ATF4 (1:300; Santa Cruz Biotechnology,
Inc.), CHOP (1:300) cleaved caspase-3 (1:1,000) (both from Cell
Signaling Technology, Inc.), actin (1:10,000; Chemicon
International), LaminA/C (1:1,000; Santa Cruz Biotechnology, Inc.)
antibodies, and finally incubated with horseradish
peroxidase-conjugated anti-rabbit/mouse IgG (1:5,000; Chemicon
International, Billerica, MA, USA). Bound antibodies were
visualized with the Amersham ECL system (GE Healthcare Biosciences
Corp., Piscataway, NJ, USA). A Luminescent Image Analyzer (Fujifilm
LAS-3000; Fuji Photo Film Co., Tokyo, Japan) was used to measure
optical densities.
Statistical analysis
Data are presented as the mean ± standard deviation
of the mean. The significance of difference between means was
assessed by Student's t-test (single comparisons) or by one-way
analysis of variance (one-way ANOVA) with Fisher's protected least
significant difference (LSD) post hoc comparison. P<0.05 was
selected for statistical significance.
Results
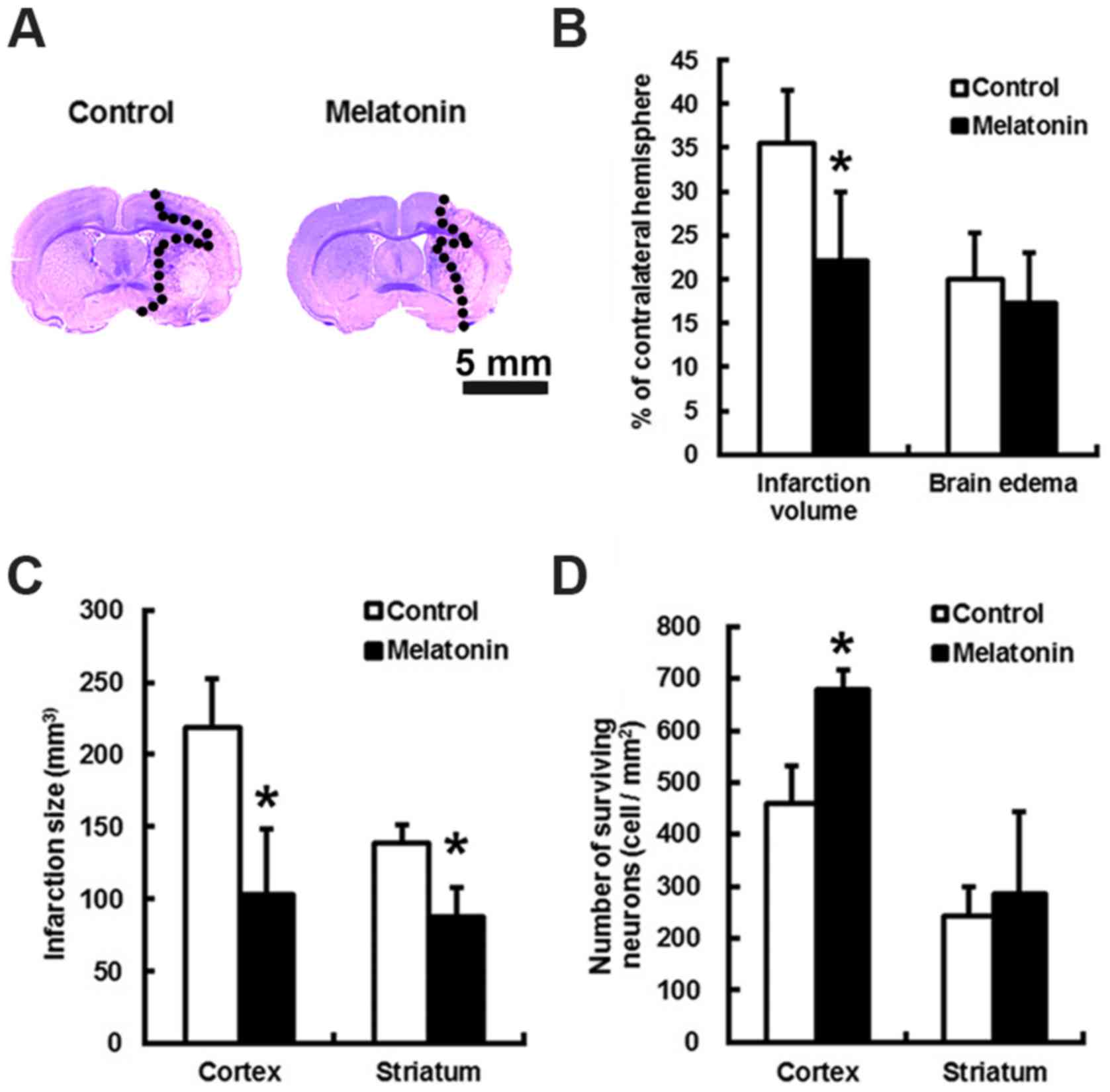
Melatonin reduced brain infarction
following MCA occlusion at 24 h
The animals treated with melatonin or control did
not have altered local cortical blood perfusion or core temperature
during the course of surgery (data not shown). Relative to control,
the brain infarct volumes of melatonin treated subjects were
reduced by 1.77-fold (P<0.05) at 24 h after stroke (Fig. 1A and B). This translates to a
melatonin-mediated decrease in cortex and striatum infract size by
2.12- and 1.58-fold, respectively, as well as an increase in the
number of the surviving cortex neurons by 1.47-folds (Fig. 1C and D; P<0.05).
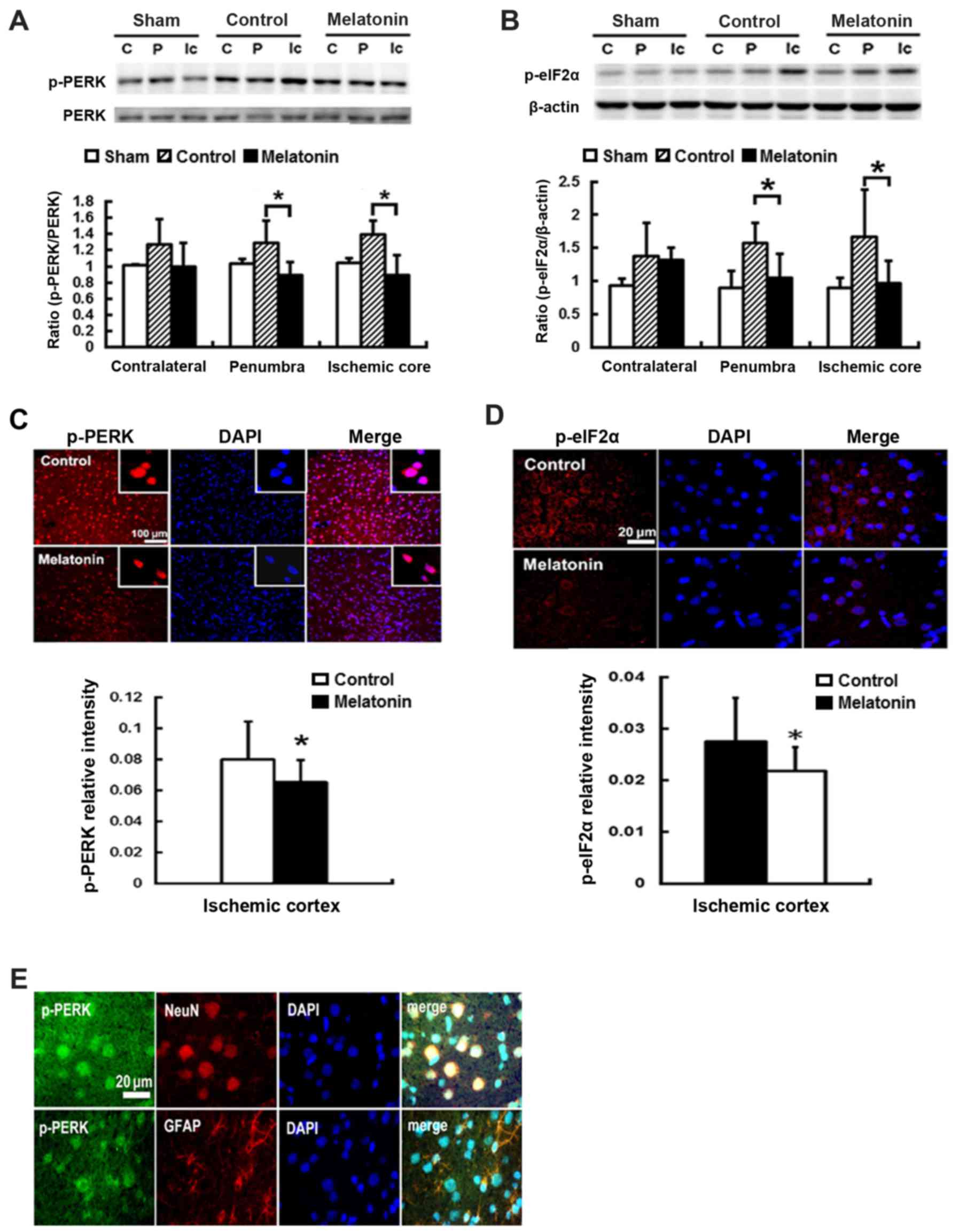
Effects of melatonin on the expression
protein of p-PERK- p-elF2α-ATF4-CHOP signaling pathway in rats
subjected to MCA occlusion
To study the effect of melatonin-inhibited ER stress
resulting from ischemia reperfusion, we evaluated the expression of
ER stress-associated proteins including p-PERK, p-eIF2α, ATF4 and
CHOP in the contralateral, penumbra cortex and ischemic core, of
rats after post-insult. Post-ischemic expression of p-PERK and
p-eIF2α protein was induced in ischemic brain 1 h after reperfusion
onset and then declined (data not shown). The results revealed that
ischemia reperfusion increased p-PERK and p-eIF2α expression at
penumbral and ischemic core regions, while the rats treated with
melatonin showed lower levels in p-PERK and p-eIF2α, as compared
with control groups (Fig. 2A and
B). For the melatonin treated animals, there was a significant
reduction of 31 and 36% p-PERK expression of the penumbral and
ischemic core regions, respectively, as compared to the control
groups (P<0.05) (Fig. 2A).
Effective inhibition of p-eIF2α levels at the penumbral and
ischemic core regions by 33 and 43%, respectively, was found in
melatonin-treated animals compared to control groups (P<0.05)
(Fig. 2B). Additionally, the
reduction of p-PERK and p-eIF2α of 18 and 21% intensity,
respectively, was found in the ischemia cortex of the
melatonin-treatment animals compared to controls (P<0.05)
(Fig. 2C and D). The
immunofluorescence staining revealed the expression of p-PERK was
localized on neuronal cells (NeuN positive cells) and glial cells
(GFAP positive cells) in the ischemic brains (Fig. 2E).
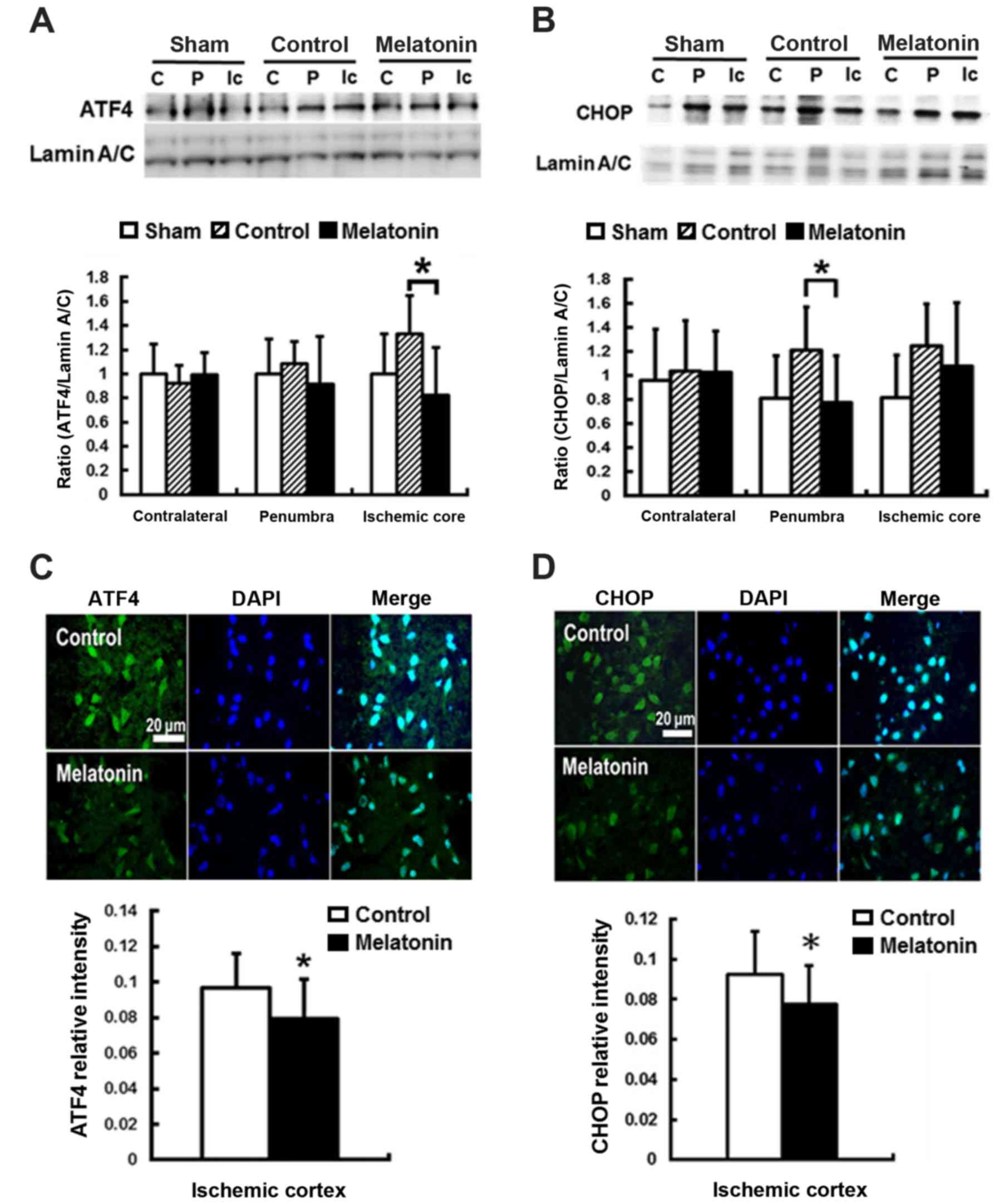
For ER stress-induced apoptosis, the pro-apoptotic
transcription factor CHOP plays a key role (43). CHOP is the downstream signal of
the PERK-p-eIF2α-ATF4 pathway in unfolded protein response
(44). The results revealed that
post-ischemic expression of ATF4 and CHOP protein was induced in
ischemic brain 1 h and reached peak at 24 h after reperfusion onset
(data not shown). Compared with the control groups, significantly
decreased ATF4 expression at ischemic core and CHOP expression at
penumbra by 38 and 36%, respectively, were found in the
melatonin-treated animals at 24 h after ischemia reperfusion
(P<0.05) (Fig. 3A and B). The
immunofluorescent assay results showed lower ATF4 and CHOP
intensity in the ischemia cortex by 12 and 14%, respectively, in
the melatonin-treated animals at 24 h after reperfusion onset
(P<0.05) (Fig. 3C and D).
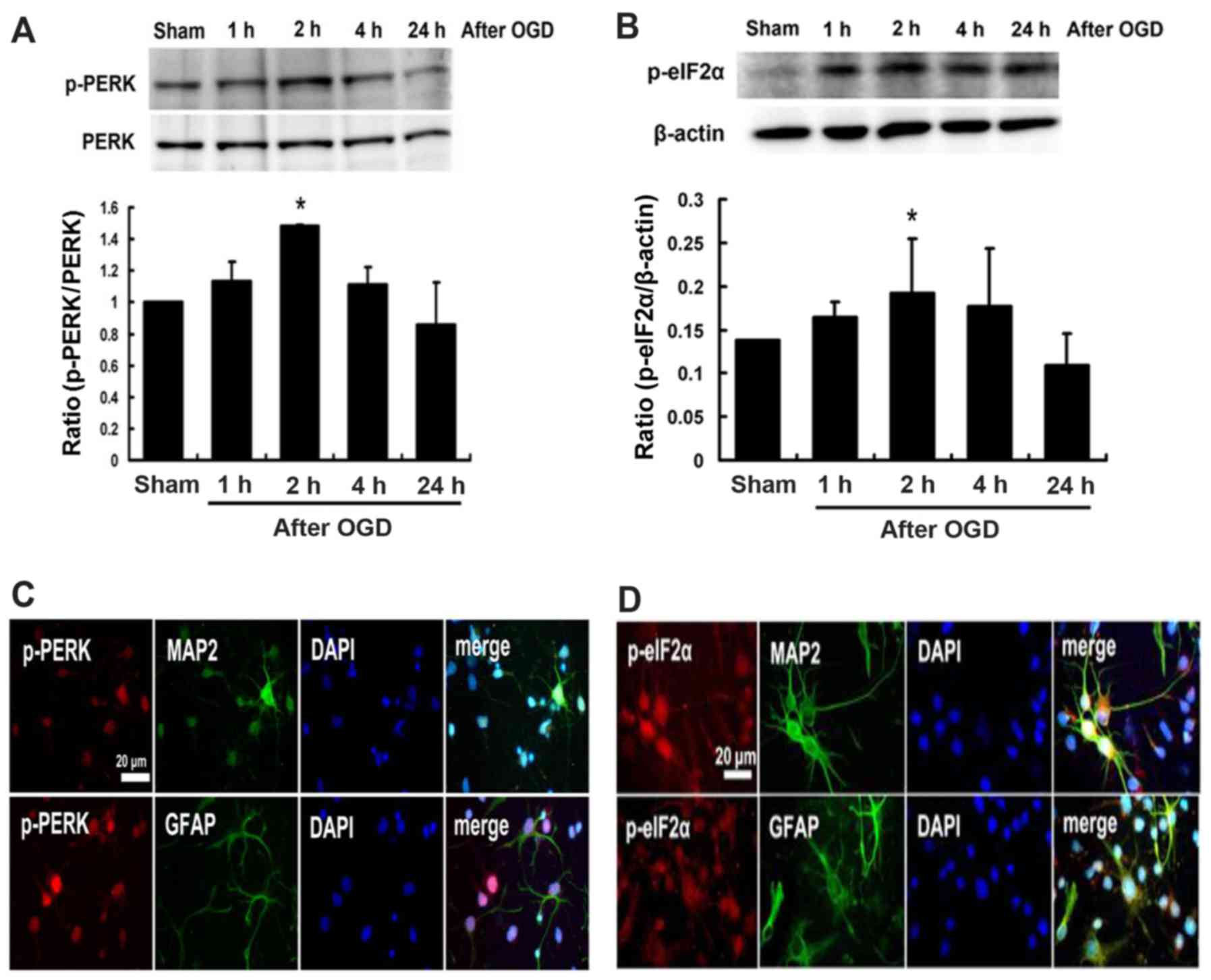
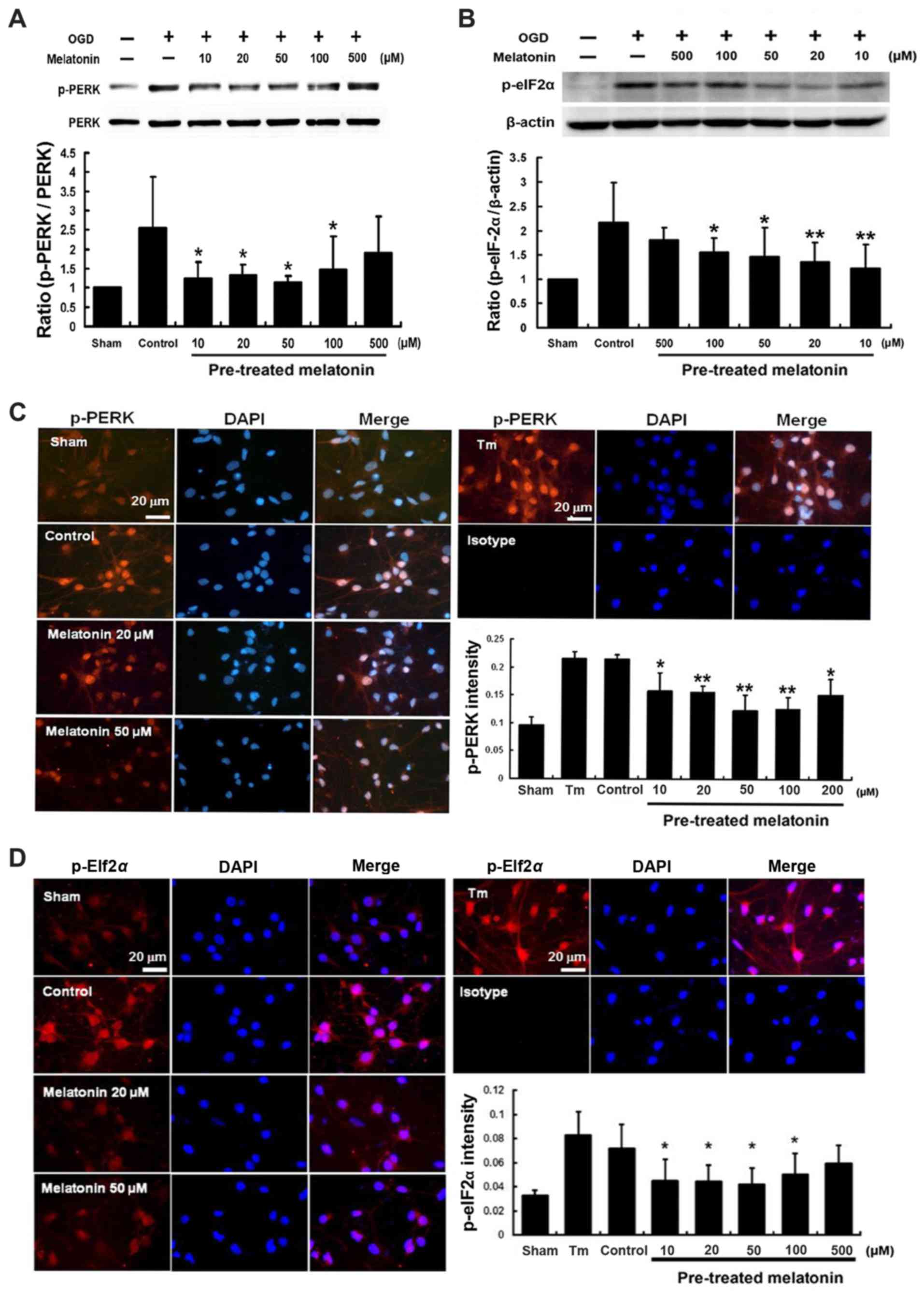
Effects of melatonin on the expression of
p-PERK and p-elF2α proteins in cultured neurons after OGD
injury
The western blot analysis showed that the expression
of p-PERK and p-eIF2α proteins induced at 1 h following cultured
neurons exposed to OGD, peaked at 2 h and then declined (Fig. 4A and B). Expressions of p-PERK and
p-eIF2α were localized on neuronal cells (MAP2 positive cells) and
glial cells (GFAP positive cells) (Fig. 4C and D). Furthermore, pretreatment
with melatonin at 10–100 µM effectively inhibited expression
of both p-PERK and p-eIF2α by 51–42 and 43–28%, respectively, after
OGD-induced cultured neuron injury (P<0.05) (Fig. 5A and B). Immunofluorescent
staining indicated that pretreatment with melatonin at 10–100
µM also significantly reduced the intensity of p-PERK and
p-eIF2α by 26–41 and 37–31%, respectively, in the cultured neurons
after OGD injury (P<0.05) (Fig.
5C and D).
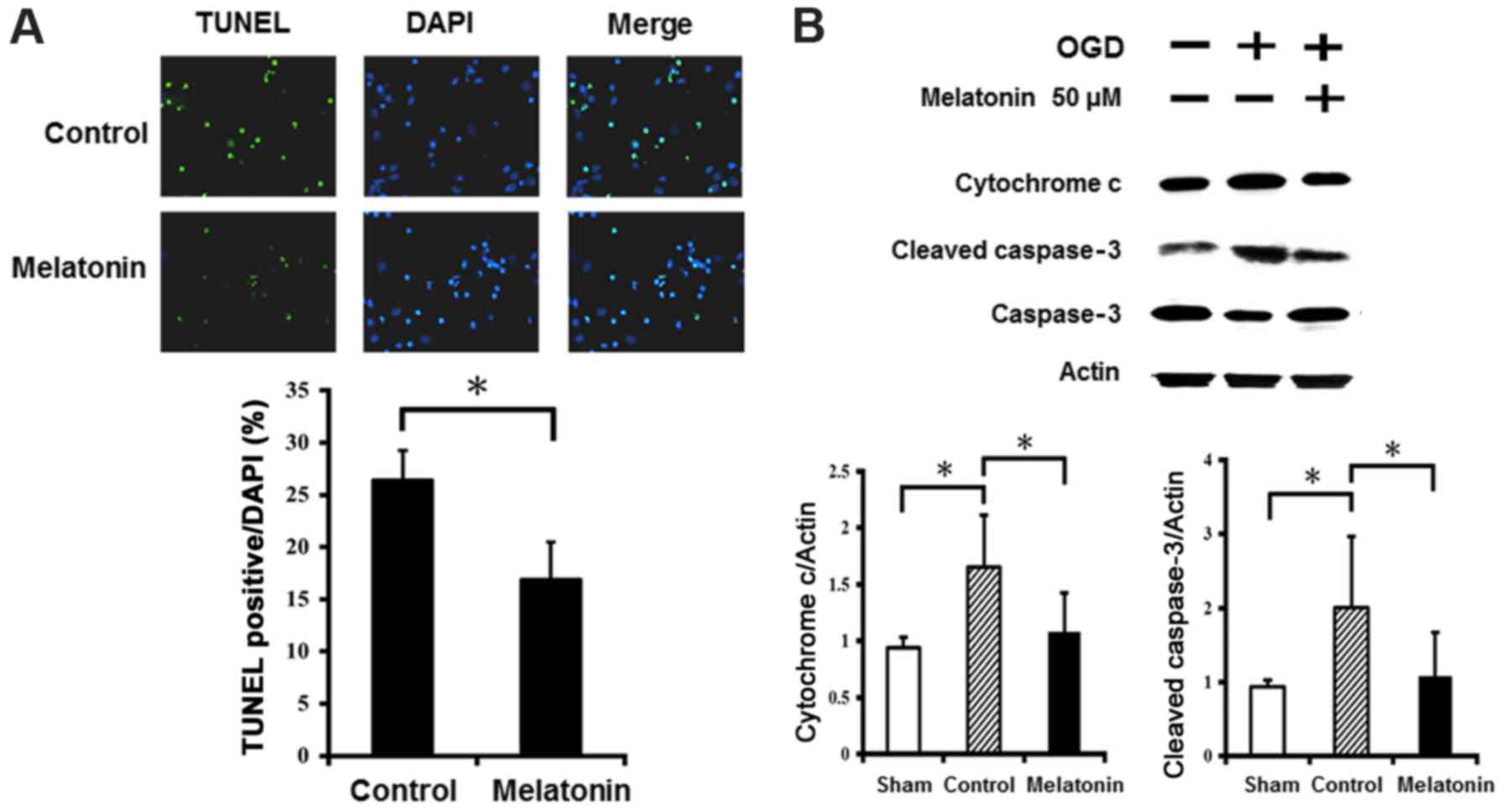
The anti-apoptotic effects of melatonin
in ischemic reperfusion
The results of TUNEL staining showed abundance of
TUNEL-positive neurons in cultured neurons after OGD injury
(Fig. 6A). Pretreatment with
melatonin at 50 µM significantly reduced the number of
TUNEL-positive neurons in the cultured neurons after OGD injury.
Western blot analysis revealed that OGD injury markedly increased
expression of cytochrome c and cleaved caspase-3 in the
cultured neurons, while the neurons treated with melatonin at 50
µM showed significantly reduced cytochrome c and
cleaved caspase-3 levels compared to control (Fig. 6B). These results indicate
melatonin prevented neuronal apoptosis by decreasing cytochrome
c and cleaved caspase-3 levels after OGD-induced injury.
Discussion
Recent studies revealed that ER stress is an
essential signaling event for neuronal injury resulting from
ischemia reperfusion (6,8,23,45,46). In this study, we demonstrated that
melatonin effectively mediates ER stress-induced neuron cell death
in rat brains and cultured neurons after ischemic reperfusion. Our
results showed that melatonin treatment provided effective
neuroprotection by increasing the number of surviving neurons and
reducing infarct size in ischemia-reperfusion rats. Melatonin
treatment can also significantly modulate protein levels by
attenuating p-PERK, p-eIF2α, ATF4 and CHOP expression in both rat
and cultured neurons after ischemia reperfusion. Concurrently, the
number of TUNEL-positive neurons was reduced and expression of
cytochrome c and cleaved caspase-3 was restrained. These
results suggest that melatonin protects neuron cells against
ischemia-reperfusion injury by decreasing ER stress-induced
apoptosis.
Previous research indicated ER stress plays a major
role in mediating ischemic reperfusion damage in brains (6,8).
Similar to previous studies, our results also demonstrate that
ischemia reperfusion injury increased the expression of ER
stress-associated proteins such as p-PERK, p-eIF2α, ATF4 and CHOP
in the penumbra and ischemia cortex (23). However, melatonin-treatment
significantly reduced the expression of p-PERK, p-eIF2α, ATF4 and
CHOP in both rat brains and cultured neurons after ischemia
reperfusion injury. Moreover, the levels of apoptosis markers
cytochrome c and cleaved caspase-3 resulting from ischemia
reperfusion were reduced by melatonin treatment. We also found that
pretreatment with melatonin for inhibiting p-PERK and p-eIF2α
expression was dose-dependent. The optimal dose of melatonin for
p-PERK and p-eIF2α inhibition was 20–50 µM for the
OGD-induced cultured neurons. Consistent with our previous studies
(38,47), a melatonin dose at 20–50 µM
showed the best radical scavenging performance as detected by DPPH
and ATBS assay. Therefore, we suggest that melatonin attenuates
ischemic-induced ER stress by decreasing free radicals in the brain
after ischemia reperfusion.
Oxidative stress and ROS generation are part of
ischemic brain pathogenesis and are also integral components of ER
stress (14,31,48). When oxidative stress and ROS are
induced in the ischemic brain after ischemia reperfusion, oxidative
damage to ER organelles may lead to ischemic neuronal cell death.
Upon ER stress, activated PERK not only phosphorylates eIF2α to
suppress protein synthesis but also increases translation of
transcription factors ATF4 and CHOP, finally resulting in cell
death-signal activation (49). It
has been reported that ATF4 is an oxidative stress-inducible,
prodeath transcription factor for neurons after stroke (50). Indeed, mice lacking ATF4 show
significantly smaller infarcts, improved behavioral outcome and
resistance to neuronal cell death compared with wild mice subjected
to ischemic stroke (50). Thus,
the increased expression of ATF4 plays an important pro-death role
after ischemia reperfusion injury. After ischemia reperfusion, our
previous studies (26–28) found that melatonin reduces
neuronal cell death in the ischemic brain via its excellent
free-radical scavenge and antioxidative effects. This study confers
evidence that melatonin attenuated upregulated ATF4 by inhibiting
oxidative stress, contributing to anti-apoptosis effects.
CHOP, a transcription factor regulated by the
PERK-elF2α-ATF4 pathway under ER stress, has been reported to be
involved in ER stress-induced apoptosis after ischemia reperfusion
(23,51). Indeed, CHOP-deficiency provides
resistance to ER stress-induced cell death both in vitro and
in vivo (44,52,53). Previous studies reported that CHOP
mediated ER stress induced-apoptosis by reducing the expression of
anti-apoptotic factor Bcl-2 and increasing the expression of ROS
(54,55). Our results found that ischemia
reperfusion injury increased expression of the CHOP protein and
apoptosis of cells in the ischemic core and cultured neurons.
Moreover, melatonin treatment not only attenuated the expression of
the CHOP protein in the ischemic brain but also significantly
reduced the number of TUNEL-positive cells and the level of cleaved
caspase-3 in the cultured neurons exposed to OGD. Additionally, our
previous studies indicated that melatonin could protect neurons
against ischemia reperfusion injury by reducing the oxidative
stress, lipid peroxidation and ROS generation (26–28). Therefore, we hypothesize that some
molecules of the CHOP pathway, such as Bcl-2 and ROS, may
contribute to decreased apoptosis via melatonin treatment.
In this study, we found that the levels of p-PERK,
p-elF2α and CHOP increased in the contralateral in rats with
ischemic stroke. Previous studies indicated that acute brain damage
often reveals decreases in blood flow and metabolism in areas
unaffected by the lesion. This phenomenon, diaschisis, is caused by
pathological deficits in brain areas remote from the initial
ischemic lesion (56,57). The sites of the originally damaged
and diaschisis areas are connected to each other by neurons
(58,59). The damaged structure disrupts the
function of other intact systems and causes a physiological
imbalance. The injury is produced by an acute focal disturbance in
an area of the ischemic brain. Therefore, ischemia reperfusion
induced ER-stress occurs contralaterally due to diaschisis.
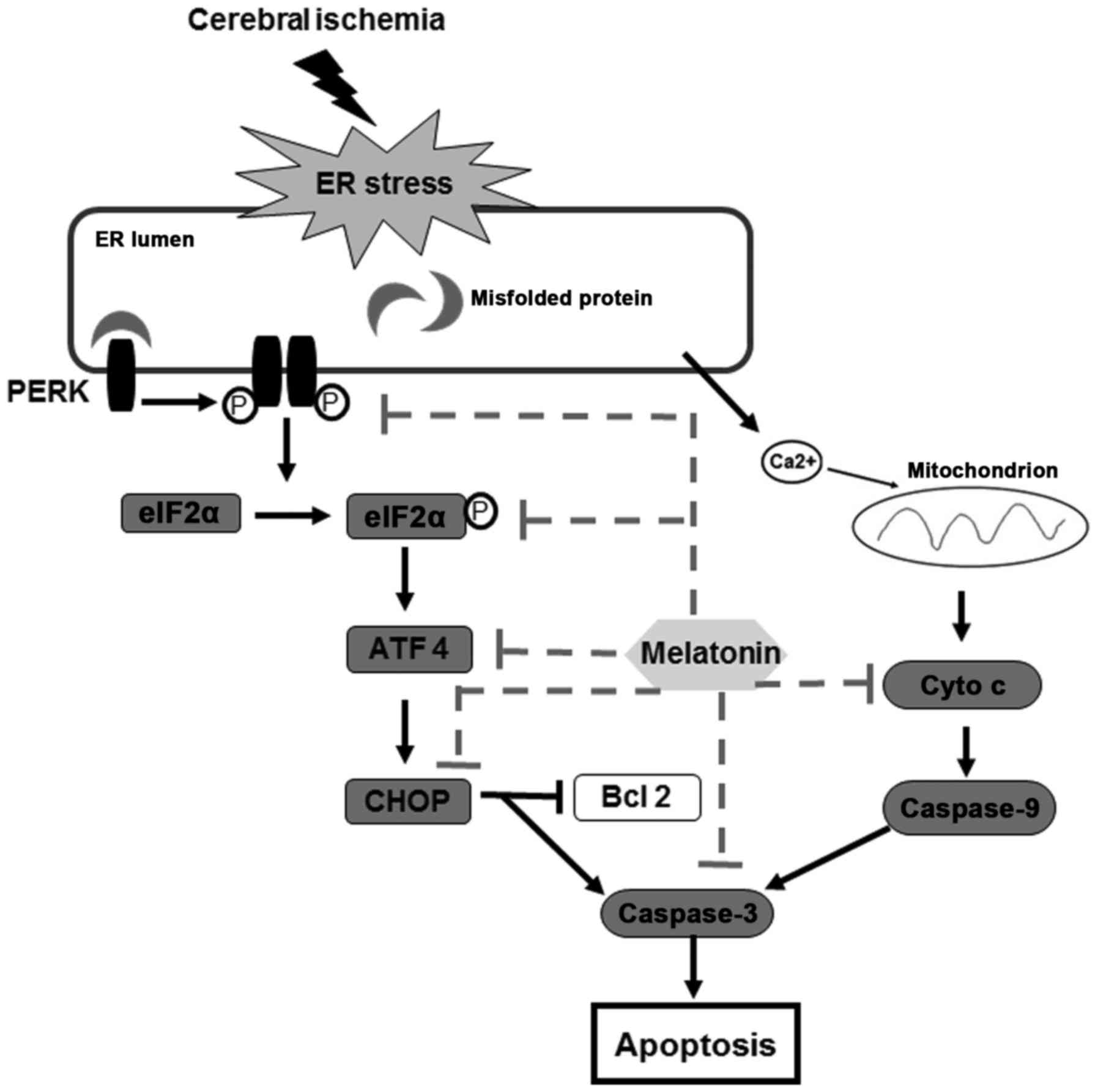
This study provides experimental evidence that
melatonin protects against cerebral ischemia reperfusion injury,
and the mechanisms for neuroprotection is related to the
attenuation of PERK-elF2α-CHOP pathway and inhibition of ER
stress-induced apoptosis (Fig.
7).
Abbreviations:
|
ER
|
endoplasmic reticulum
|
|
OGD
|
oxygen and glucose deprivation
|
|
MCA
|
middle cerebral artery
|
|
p-PERK
|
phosphorylation of PRKR-like
endoplasmic reticulum kinase
|
|
p-eIF2α
|
phosphorylation of eukaryotic
translation initiation factor 2α
|
|
ATF4
|
activating transcription factor 4
|
|
CHOP
|
C/EBP homologous protein
|
|
TUNEL
|
deoxynucleotidyl transferase-mediated
dUTP nick end labeling
|
|
Bcl-2
|
anti-apoptotic factor B cell
lymphoma-2
|
|
ROS
|
reactive oxygen species
|
|
MAP-2
|
microtubule-associated protein-2
|
|
GFAP
|
glial fibrillary acidic protein
|
Acknowledgments
Not applicable.
References
|
1
|
Cheung K and Kaufmann P: Efficiency
perspectives on adaptive designs in stroke clinical trials. Stroke.
42:2990–2994. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mantz J, Degos V and Laigle C: Recent
advances in pharmacologic neuroprotection. Eur J Anaesthesiol.
27:6–10. 2010. View Article : Google Scholar
|
|
3
|
Roussel BD, Kruppa AJ, Miranda E, Crowther
DC, Lomas DA and Marciniak SJ: Endoplasmic reticulum dysfunction in
neurological disease. Lancet Neurol. 12:105–118. 2013. View Article : Google Scholar
|
|
4
|
Paschen W: Endoplasmic reticulum
dysfunction in brain pathology: Critical role of protein synthesis.
Curr Neurovasc Res. 1:173–181. 2004. View Article : Google Scholar
|
|
5
|
Chen X, Kintner DB, Luo J, Baba A, Matsuda
T and Sun D: Endoplasmic reticulum Ca2+ dysregulation
and endoplasmic reticulum stress following in vitro neuronal
ischemia: Role of Na+-K+-Cl−
cotransporter. J Neurochem. 106:1563–1576. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nakka VP, Gusain A, Mehta SL and Raghubir
R: Molecular mechanisms of apoptosis in cerebral ischemia: Multiple
neuroprotective opportunities. Mol Neurobiol. 37:7–38. 2008.
View Article : Google Scholar
|
|
7
|
Ferri KF and Kroemer G: Organelle-specific
initiation of cell death pathways. Nat Cell Biol. 3:E255–E263.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tajiri S, Oyadomari S, Yano S, Morioka M,
Gotoh T, Hamada JI, Ushio Y and Mori M: Ischemia-induced neuronal
cell death is mediated by the endoplasmic reticulum stress pathway
involving CHOP. Cell Death Differ. 11:403–415. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Qi X, Hosoi T, Okuma Y, Kaneko M and
Nomura Y: Sodium 4-phenylbutyrate protects against cerebral
ischemic injury. Mol Pharmacol. 66:899–908. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sokka AL, Putkonen N, Mudo G, Pryazhnikov
E, Reijonen S, Khiroug L, Belluardo N, Lindholm D and Korhonen L:
Endoplasmic reticulum stress inhibition protects against
excitotoxic neuronal injury in the rat brain. J Neurosci.
27:901–908. 2007. View Article : Google Scholar
|
|
11
|
Paschen W and Doutheil J: Disturbances of
the functioning of endoplasmic reticulum: A key mechanism
underlying neuronal cell injury. J Cereb Blood Flow Metab. 19:1–18.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ma Y and Hendershot LM: The unfolding tale
of the unfolded protein response. Cell. 107:827–830. 2001.
View Article : Google Scholar
|
|
13
|
DeGracia DJ, Kumar R, Owen CR, Krause GS
and White BC: Molecular pathways of protein synthesis inhibition
during brain reperfusion: Implications for neuronal survival or
death. J Cereb Blood Flow Metab. 22:127–141. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hayashi T, Saito A, Okuno S, Ferrand-Drake
M, Dodd RL and Chan PH: Damage to the endoplasmic reticulum and
activation of apoptotic machinery by oxidative stress in ischemic
neurons. J Cereb Blood Flow Metab. 25:41–53. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Boyce M and Yuan J: Cellular response to
endoplasmic reticulum stress: A matter of life or death. Cell Death
Differ. 13:363–373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: Disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Prostko CR, Brostrom MA and Brostrom CO:
Reversible phosphorylation of eukaryotic initiation factor 2 alpha
in response to endoplasmic reticular signaling. Mol Cell Biochem.
127–128:255–265. 1993. View Article : Google Scholar
|
|
18
|
Ron D: Translational control in the
endoplasmic reticulum stress response. J Clin Invest.
110:1383–1388. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Harding HP, Novoa I, Zhang Y, Zeng H, Wek
R, Schapira M and Ron D: Regulated translation initiation controls
stress-induced gene expression in mammalian cells. Mol Cell.
6:1099–1108. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lu PD, Harding HP and Ron D: Translation
reinitiation at alternative open reading frames regulates gene
expression in an integrated stress response. J Cell Biol.
167:27–33. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Oida Y, Shimazawa M, Imaizumi K and Hara
H: Involvement of endoplasmic reticulum stress in the neuronal
death induced by transient forebrain ischemia in gerbil.
Neuroscience. 151:111–119. 2008. View Article : Google Scholar
|
|
22
|
Zhao H, Yenari MA, Cheng D, Sapolsky RM
and Steinberg GK: Biphasic cytochrome c release after transient
global ischemia and its inhibition by hypothermia. J Cereb Blood
Flow Metab. 25:1119–1129. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu CX, Liu R, Gao M, Zhao G, Wu S, Wu CF
and Du GH: Pinocembrin protects brain against ischemia/reperfusion
injury by attenuating endoplasmic reticulum stress induced
apoptosis. Neurosci Lett. 546:57–62. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu X, Wang M, Chen H, Guo Y, Ma F, Shi F,
Bi Y and Li Y: Hypothermia protects the brain from transient global
ischemia/reperfusion by attenuating endoplasmic reticulum
response-induced apoptosis through CHOP. PLoS One. 8:e534312013.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang Z, Zhang H, Xu X, Shi H, Yu X, Wang
X, Yan Y, Fu X, Hu H, Li X, et al: bFGF inhibits ER stress induced
by ischemic oxidative injury via activation of the PI3K/Akt and
ERK1/2 pathways. Toxicol Lett. 212:137–146. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lee EJ, Wu TS, Lee MY, Chen TY, Tsai YY,
Chuang JI and Chang GL: Delayed treatment with melatonin enhances
electrophysiological recovery following transient focal cerebral
ischemia in rats. J Pineal Res. 36:33–42. 2004. View Article : Google Scholar
|
|
27
|
Chen HY, Hung YC, Chen TY, Huang SY, Wang
YH, Lee WT, Wu TS and Lee EJ: Melatonin improves presynaptic
protein, SNAP-25, expression and dendritic spine density and
enhances functional and electrophysiological recovery following
transient focal cerebral ischemia in rats. J Pineal Res.
47:260–270. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee EJ, Lee MY, Chen HY, Hsu YS, Wu TS,
Chen ST and Chang GL: Melatonin attenuates gray and white matter
damage in a mouse model of transient focal cerebral ischemia. J
Pineal Res. 38:42–52. 2005. View Article : Google Scholar
|
|
29
|
Cuzzocrea S, Costantino G, Gitto E, Mazzon
E, Fulia F, Serraino I, Cordaro S, Barberi I, De Sarro A and Caputi
AP: Protective effects of melatonin in ischemic brain injury. J
Pineal Res. 29:217–227. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun FY, Lin X, Mao LZ, Ge WH, Zhang LM,
Huang YL and Gu J: Neuroprotection by melatonin against ischemic
neuronal injury associated with modulation of DNA damage and repair
in the rat following a transient cerebral ischemia. J Pineal Res.
33:48–56. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee MY, Kuan YH, Chen HY, Chen TY, Chen
ST, Huang CC, Yang IP, Hsu YS, Wu TS and Lee EJ: Intravenous
administration of melatonin reduces the intracerebral cellular
inflammatory response following transient focal cerebral ischemia
in rats. J Pineal Res. 42:297–309. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kilic E, Kilic U, Yulug B, Hermann DM and
Reiter RJ: Melatonin reduces disseminate neuronal death after mild
focal ischemia in mice via inhibition of caspase-3 and is suitable
as an add-on treatment to tissue-plasminogen activator. J Pineal
Res. 36:171–176. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Andrabi SA, Sayeed I, Siemen D, Wolf G and
Horn TF: Direct inhibition of the mitochondrial permeability
transition pore: A possible mechanism responsible for
anti-apoptotic effects of melatonin. FASEB J. 18:869–871. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jou MJ, Peng TI, Reiter RJ, Jou SB, Wu HY
and Wen ST: Visualization of the antioxidative effects of melatonin
at the mitochondrial level during oxidative stress-induced
apoptosis of rat brain astrocytes. J Pineal Res. 37:55–70. 2004.
View Article : Google Scholar
|
|
35
|
Lee EJ, Lee MY, Chang GL, Chen LH, Hu YL,
Chen TY and Wu TS: Delayed treatment with magnesium: Reduction of
brain infarction and improvement of electrophysiological recovery
following transient focal cerebral ischemia in rats. J Neurosurg.
102:1085–1093. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Juan WS, Huang SY, Chang CC, Hung YC, Lin
YW, Chen TY, Lee AH, Lee AC, Wu TS and Lee EJ: Melatonin improves
neuroplasticity by upregulating the growth-associated protein-43
(GAP-43) and NMDAR postsynaptic density-95 (PSD-95) proteins in
cultured neurons exposed to glutamate excitotoxicity and in rats
subjected to transient focal cerebral ischemia even during a
long-term recovery period. J Pineal Res. 56:213–223. 2014.
View Article : Google Scholar
|
|
37
|
Lee WT, Lin MH, Lee EJ, Hung YC, Tai SH,
Chen HY, Chen TY and Wu TS: Magnolol reduces glutamate-induced
neuronal excitotoxicity and protects against permanent focal
cerebral ischemia up to 4 hours. PLoS One. 7:e399522012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee EJ, Chen HY, Hung YC, Chen TY, Lee MY,
Yu SC, Chen YH, Chuang IC and Wu TS: Therapeutic window for
cinnamophilin following oxygen-glucose deprivation and transient
focal cerebral ischemia. Exp Neurol. 217:74–83. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Cimarosti H, Rodnight R, Tavares A, Paiva
R, Valentim L, Rocha E and Salbego C: An investigation of the
neuroprotective effect of lithium in organotypic slice cultures of
rat hippocampus exposed to oxygen and glucose deprivation. Neurosci
Lett. 315:33–36. 2001. View Article : Google Scholar
|
|
40
|
Hung YC, Chen TY, Lee EJ, Chen WL, Huang
SY, Lee WT, Lee MY, Chen HY and Wu TS: Melatonin decreases matrix
metalloproteinase-9 activation and expression and attenuates
reperfusion-induced hemorrhage following transient focal cerebral
ischemia in rats. J Pineal Res. 45:459–467. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen TY, Tai SH, Lee EJ, Huang CC, Lee AC,
Huang SY and Wu TS: Cinnamophilin offers prolonged neuroprotection
against gray and white matter damage and improves functional and
electrophysiological outcomes after transient focal cerebral
ischemia. Crit Care Med. 39:1130–1137. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen TY, Lin MH, Lee WT, Huang SY, Chen
YH, Lee AC, Lin HW and Lee EJ: Nicotinamide inhibits nuclear
factor-kappa B translocation after transient focal cerebral
ischemia. Crit Care Med. 40:532–537. 2012. View Article : Google Scholar
|
|
43
|
Ma Y, Brewer JW, Diehl JA and Hendershot
LM: Two distinct stress signaling pathways converge upon the CHOP
promoter during the mammalian unfolded protein response. J Mol
Biol. 318:1351–1365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Zhang K and Kaufman RJ: From
endoplasmic-reticulum stress to the inflammatory response. Nature.
454:455–462. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Nakka VP, Gusain A and Raghubir R:
Endoplasmic reticulum stress plays critical role in brain damage
after cerebral ischemia/reperfusion in rats. Neurotox Res.
17:189–202. 2010. View Article : Google Scholar
|
|
46
|
Xin Q, Ji B, Cheng B, Wang C, Liu H, Chen
X, Chen J and Bai B: Endoplasmic reticulum stress in cerebral
ischemia. Neurochem Int. 68:18–27. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Tai SH, Hung YC, Lee EJ, Lee AC, Chen TY,
Shen CC, Chen HY, Lee MY, Huang SY and Wu TS: Melatonin protects
against transient focal cerebral ischemia in both reproductively
active and estrogen-deficient female rats: The impact of
circulating estrogen on its hormetic dose-response. J Pineal Res.
50:292–303. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Bhandary B, Marahatta A, Kim HR and Chae
HJ: An involvement of oxidative stress in endoplasmic reticulum
stress and its associated diseases. Int J Mol Sci. 14:434–456.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
DeGracia DJ and Montie HL: Cerebral
ischemia and the unfolded protein response. J Neurochem. 91:1–8.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Lange PS, Chavez JC, Pinto JT, Coppola G,
Sun CW, Townes TM, Geschwind DH and Ratan RR: ATF4 is an oxidative
stress-inducible, prodeath transcription factor in neurons in vitro
and in vivo. J Exp Med. 205:1227–1242. 2008. View Article : Google Scholar :
|
|
51
|
Lin JH, Li H, Zhang Y, Ron D and Walter P:
Divergent effects of PERK and IRE1 signaling on cell viability.
PLoS One. 4:e41702009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kohno K, Higuchi T, Ohta S, Kohno K, Kumon
Y and Sakaki S: Neuroprotective nitric oxide synthase inhibitor
reduces intracellular calcium accumulation following transient
global ischemia in the gerbil. Neurosci Lett. 224:17–20. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
McCullough KD, Martindale JL, Klotz LO, Aw
TY and Holbrook NJ: Gadd153 sensitizes cells to endoplasmic
reticulum stress by downregulating Bcl2 and perturbing the cellular
redox state. Mol Cell Biol. 21:1249–1259. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hung YC, Chou YS, Chang CH, Lin HW, Chen
HY, Chen TY, Tai SH and Lee EJ: Early reperfusion improves the
recovery of contralateral electrophysiological diaschisis following
focal cerebral ischemia in rats. Neurol Res. 32:828–834. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
White BC, Sullivan JM, DeGracia DJ, O'Neil
BJ, Neumar RW, Grossman LI, Rafols JA and Krause GS: Brain ischemia
and reperfusion: Molecular mechanisms of neuronal injury. J Neurol
Sci. 179:1–33. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Reinecke S, Lutzenburg M, Hagemann G,
Bruehl C, Neumann-Haefelin T and Witte OW: Electrophysiological
transcortical diaschisis after middle cerebral artery occlusion
(MCAO) in rats. Neurosci Lett. 261:85–88. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Enager P, Gold L and Lauritzen M: Impaired
neurovascular coupling by transhemispheric diaschisis in rat
cerebral cortex. J Cereb Blood Flow Metab. 24:713–719. 2004.
View Article : Google Scholar : PubMed/NCBI
|