Introduction
Data from the World Health Organization (http:/www.who.int/cardiovascular_diseases/en/)
indicate that 17.5 million patients succumb to coronary vascular
diseases annually, an estimated 31% of all deaths worldwide. A
series of pharmacological and surgical therapies aimed at
revascularization may rapidly reverse myocardial ischemia (1). However, the following
ischemia̸reperfusion (I/R) injury paradoxically alleviates the
therapeutic benefits and results in further loss of cardiomyocytes
and cardiac microvascular endothelial cells (CMECs) (1,2).
Great efforts have been made to investigate the pathogenetic
processes underlying cardiomyocyte-associated I/R injury, while the
critical roles of CMECs and the underlying mechanisms have
attracted less attention (3).
CMECs constitute approximately one-third of all cardiac cells, and
act as first responders and central mediators in the regulation of
I/R injury (4–6). It is well known that CMEC injury
precedes cardiomyocyte lesions under I/R conditions (2,7).
I/R treatment predisposes to dysfunction of CMECs, mainly through
affecting their proliferation and migration, wherein inflammation-
and apoptosis-dependent signaling pathways are strongly involved
(2,4,8).
Moreover, there is close functional and physical crosstalk between
cardiomyocytes and CMECs. For example, one single cardiomyocyte is
surrounded by at least 3-4 myocardial capillaries (9); the apoptosis of CMECs and subsequent
release of pro-inflammatory and pro-apoptotic mediators promote
cardiomyocyte injury (10). These
key characteristics of CMECs have prompted the search for
interventional targets that may be directed at endogenous
mechanisms, thereby reducing the side effects of I/R.
Toll-like receptor 4 (TLR4) is a common upstream
sensor and acts as a pivotal initiator of multiple intracellular
pathways (11). Previous
investigations have highlighted the pleiotropic functions of TLR4
on regulating the malfunction of microvascular endothelial cells
during hypoxia/reoxygenation (H/R) (10,12,13). Its detrimental effects are mainly
due to the inflammation- and apoptosis-mediated cascade responses,
as well as the impairment of proliferation and migration, which are
in part modulated by the TLR4/nuclear factor (NF)-κB signaling
pathway (12–14). Furthermore, previous studies have
demonstrated that microvascular endothelial cells expressed
mitogen-activated protein kinases (MAPKs), including p38 MAPK,
c-Jun N-terminal kinase (JNK) and extracellular signal-regulated
kinase 1/2 (ERK1/2), which serve as key downstream signaling
molecules of TLR4 and are responsible for the initial inflammatory
and apoptotic activation under ischemic conditions (13,15). Of note, due to the abundant
expression of TLRs and adhesion molecules, particularly TLR4 and
intercellular adhesion melecule-1 (ICAM-1), CMECs act as 'sentinel'
cells that may be more sensitive to H/R stimulation through sensing
danger signals (12,16). These findings may prove meaningful
in the protection against I/R injury by regulating TLR4, although
the specific role of TLR4 and the more comprehensive molecular
mechanisms that are involved in H/R -related CMEC injury require
further elucidation.
The radioprotective 105 kDa protein (RP105) serves
as a TLR4 homologue, and is primarily expressed in mature
stereotypical immunocytes (17,18). Interestingly, RP105 may also be
detectable in resident cardiac cell types, such as cardiomyocytes,
vascular smooth muscle cells (VSMCs) and endothelial cells
(18–20). Due to lack of the Toll/interleukin
(IL)-1 receptor (TIR) domain, RP105 exerts its physiological
actions mainly through its direct interactions with TLRs (11,17). It is becoming increasingly clear
that the inactivation of TLR4-triggered signaling pathways
specifically regulated by RP105 was closely associated with the
pathological processes of vascular remodeling, neointima formation
and myocardial infarction (21,22). However, whether RP105 exerts a
direct effect on CMEC dysfunction under H/R conditions remains
unknown. The aim of the present study was to determine whether the
overexpression of RP105 attenuates H/R injury through limiting CMEC
inflammation and apoptosis, and by enhancing their proliferation
and migration abilities.
Materials and methods
CMEC isolation
CMECs were isolated from 60 male Sprague-Dawley (SD)
rats, as previously described (23). Briefly, the hearts of anesthetized
SD rats (weight, 80-100 g; age, 28 days) were quickly excised and
rinsed with phosphate-buffered saline (PBS) containing heparin.
After the atria, right ventricle and valves were removed, the left
ventricle was immersed in 75% ethanol for 30 sec, and one-third of
the outer free ventricular wall was discarded. The remaining
tissues were washed by PBS, and incubated in 0.2% collagenase for
10 min followed by 0.2% trypsin for 6 min at 37°C. The dissociated
cells were filtered using a 100-mm mesh filter and centrifuged at
111 × g for 10 min. The cells were re-suspended in Dulbecco's
modified Eagle's medium (DMEM) containing 20% fetal calf serum
(FCS), 1% endothelial cell growth factor, 1% endothelial cell
growth supplement, 0.5% penicillin/streptomycin and 0.12% heparin,
and then seeded onto dishes. The primary CMECs were cultured in an
incubator with 5% CO2 at 37°C. The identification of
CMECs was based on the morphological characteristics and typical
CD31 immunofluorescence assay, as previous depicted (2). All animal care and experimental
procedures were approved by the Institutional Animal Care and Use
Committee of Wuhan University, and conformed to the Guidelines for
the Care and Use of Laboratory Animals published by the US National
Institutes of Health.
Adenovirus transduction
Adenoviral vectors encoding RP105 (Ad-RP105) or
control Ad-green fluorescent protein (GFP) was designed as
previously demonstrated and provided by GeneChem (Shanghai, China)
(24). Prior to adenoviral
transfection, CMECs (1×106/ml) were seeded into 6-well
plates until they reached ~80% confluence. The cells were then
incubated with Ad-RP105 or Ad-GFP at various multiplicities of
infection (MOI) for 48 h. The cytotoxic effects of adenovirus on
CMECs were measured by 4% trypan blue dye, as reported previously
(25). Fluorescent microscopy was
used to observe GFP expression. The mRNA level of RP105 was further
measured to evaluate the effectiveness of adenoviral
transfection.
Experimental designations and H/R
model
Passage 2-3 CMECs were used for the following
experiments. Following synchronization for 6 h, the CMECs were
rinsed with PBS and randomly designated into four groups: i)
Control; ii) H/R; iii) Ad-GFP+ H/R; and iv) Ad-RP105+ H/R. At 48 h
post-adenoviral transfection, the CMECs were subjected to 4 h of
hypoxia followed by 2 h reoxygenation to simulate the in
vivo I/R injury model, as previously described (4,8).
Hypoxia was achieved by exposing CMECs to 94% N2-5%
CO2-1% O2 for 4 h at 37°C in Hanks' solution.
Subsequently, the buffer was replaced by fresh culture medium and
transferred into a normoxic incubator (95% air-5% CO2)
for 2 h to initiate reoxygenation. The control group was maintained
in a normoxic environment. All experiments were performed in
triplicate and repeated three times.
Cell proliferation detection
Cell proliferation was detected by the Cell Counting
Kit-8 (CCK-8) assay (Dojindo, Tokyo, Japan) according to the
manufacturer's instructions (2).
The CMECs were seeded onto 96-well plates at a density of
1×104 cells/well and subjected to H/R injury as
described above, until reaching ~80% confluence. Subsequently, 10
μl CCK-8 reagents were added into each well and incubated
for 3 h in a 5% CO2 incubator. The absorbance at 450 nm
was measured using a microplate reader (Bio-Rad Laboratories,
Hercules, CA, USA). Cell viability (%) was expressed as the ratio
of mean absorbance in the test wells to mean absorbance in the
control well.
Cell migration measurement
Cell migration was measured using a 24-well
Transwell chamber with an 8-μm pore size (Millipore,
Billerica, MA, USA), as described by Wang et al (8). After H/R injury, CMECs were
suspended with DMEM supplemented with 5% FCS and added into the
upper chamber. The lower chamber was filled with DMEM containing
20% FCS. At 12 h after incubation (5% CO2 incubator at
37°C), the migrated cells were fixed by 4% paraformaldehyde for 30
min, and subsequently stained with 0.1% crystal violet. Migration
was quantitatively detected through counting cells in 5 random
microscopic fields (magnification, ×200).
Apoptosis analysis
Apoptosis of CMECs was determined by Annexin
V-APC/7-aminoactinomycin D (AV-APC/7-AAD) dual staining, based on
the manufacturer's recommendations (26). CMECs were collected after H/R and
stained with 7-AAD for 10 min in the dark at room temperature,
followed by more staining with AV-APC. The AV-APC-positive and
7-AAD-negative cells were considered to be apoptotic CMECs.
Evaluation of IL-6 and tumor necrosis
factor (TNF)-α secretion
The supernatants of cultured CMECs were collected
for ELISA (12). IL-6 and TNF-α
were measured by commercial ELISA kits in accordance with the
manufacturer's instructions (R&D Systems, Minneapolis, MN,
USA).
Reverse transcription quantitative
polymerase chain reaction (RT-qPCR) assay
Total RNA from CMECs was extracted by TRIzol reagent
(Invitrogen; Thermo Fisher Scientific, Carlsbad, CA, USA), and
reverse-transcribed into cDNA using a commercial synthesis kit
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) based on the
manufacturer's protocol (25).
RT-qPCR was performed using SYBR-Green (Fermentas, Waltham, MA,
USA) with the ABI Prism 7500 system. PCR was performed as follows
:50°C for 2 min, 95°C for 10 min, and 40 cycles of 95°C for 30 sec
and 60°C for 30 sec. Data were corrected to β-actin gene
expression. The primers used for amplification were as follows:
RP105, forward 5′-TGGGGACATTTGAGGACATT-3′; and reverse
5′-GCTGTTAGGTCCAGCTCCTG-3′.
Western blot analysis
Western blotting was performed as previously
described (25). The nuclear and
cytoplasmic proteins were extracted in accordance with the
manufacturer's protocol. Denatured proteins were separated by
electrophoresis on 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis gels and transferred onto polyvinylidene fluoride
membranes. Immunoblotting was probed by primary antibodies against
glyceraldehyde 3-phosphate dehydrogenase (GAPDH; (1:1,000 dilution;
sc-48166; Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA),
lamin B (1:1,000 dilution; sc-20682; Santa Cruz Biotechnology,
Inc.), RP105 (1:800 dilution; sc-27841; Santa Cruz Biotechnology,
Inc.), TLR4 (1:500 dilution; sc-10741; Santa Cruz Biotechnology,
Inc.), p38 MAPK (1:500 dilution; sc-535; Santa Cruz Biotechnology,
Inc.), p-p38 MAPK (1:500 dilution; sc-7973; Santa Cruz
Biotechnology, Inc.), JNK (1:500 dilution; sc-137020; Santa Cruz
Biotechnology, Inc.), p-JNK (1:600 dilution; sc-6254; Santa Cruz
Biotechnology, Inc.), ERK1/2 (1:300 dilution; sc-374239; Santa Cruz
Biotechnology, Inc.), p-ERK1/2 (1:400 dilution; sc-7976; Santa Cruz
Biotechnology, Inc.), NF-κB/p65 (1:800 dilution; sc-101752; Santa
Cruz Biotechnology, Inc.), ICAM-1 (1:400 dilution; sc-31724; Santa
Cruz Biotechnology, Inc.), IL-6 (1:800 dilution; sc-57315; Santa
Cruz Biotechnology, Inc.) and TNF-α (1:600 dilution; sc-8301; Santa
Cruz Biotechnology, Inc.). Protein bands were identified by
horseradish peroxidase-conjugated rabbit anti-rat IgG secondary
antibodies (1:800 dilution; bs-0346R-HRP; BIOSS, Boston, MA, USA).
The outcomes were then visualized and analyzed by an enhanced
chemiluminescence reagent kit (Thermo Fisher Scientific, Inc.).
GAPDH and lamin B were used as internal loading controls for
cytoplasmic proteins and nuclear NF-κB/p65, respectively.
Statistical analysis
The statistical data are presented as mean ±
standard deviation. Comparisons between groups were performed with
one-way analysis of variance and the Student-Newman-Keuls (SNK)-q
test. P-values <0.05 were considered to indicate statistically
significant differences.
Results
Efficiency of adenoviral transduction
into CMECs
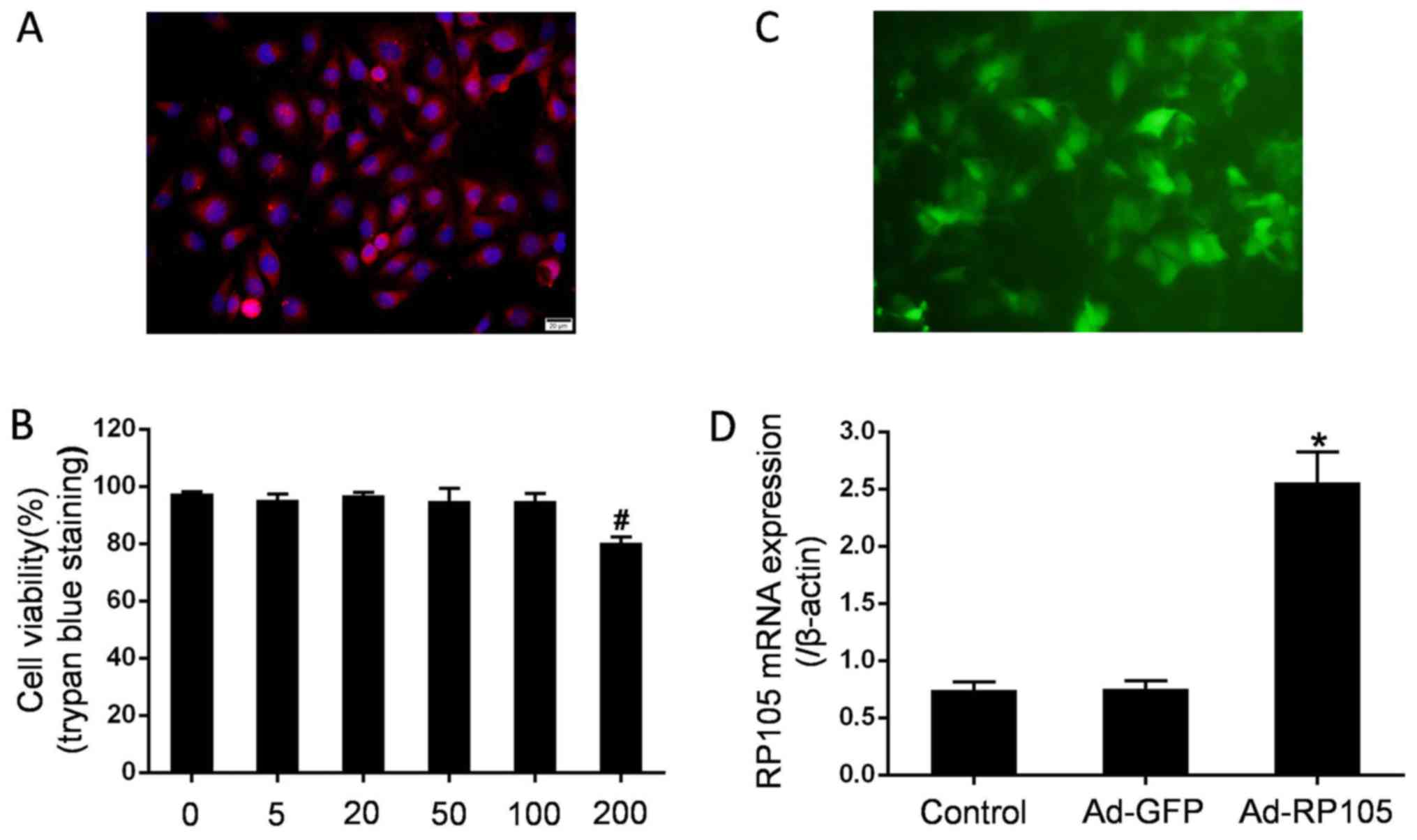
The confluence of the CMEC monolayer exhibited a
cobblestone appearance under an inverted microscope. The typical
CD31 immunofluorescence staining for CMECs identification was
demonstrated (red, CD31; blue, nuclei dyed by DAPI) (Fig. 1A). At 48 h after the viruses were
transduced at different MOI, the cytotoxic adenoviral effects were
measured using 4% trypan blue dye. As shown in Fig. 1B, the adenovirus exerted no effect
on CMEC viability at various MOI up to 100, but exhibited a 80.3%
decrease with an MOI of 200. Thus, the MOI of 100 was utilized for
the following experiments. Fluorescent microscopy and RT-qPCR
analysis were performed to evaluate the success of the adenoviral
transfection. Emission green fluorescence was highly displayed in
CMECs transduced with Ad-GFP (Fig.
1C). Moreover, CMEC transduction with Ad-RP105 markedly
promoted the expression of RP105 mRNA compared with that in control
cells and Ad-GFP-treated cells (P<0.05) (Fig. 1D). These observations indicated
the effectiveness of adenoviral transfection.
RP105 is minimally expressed in rat CMECs
in response to H/R
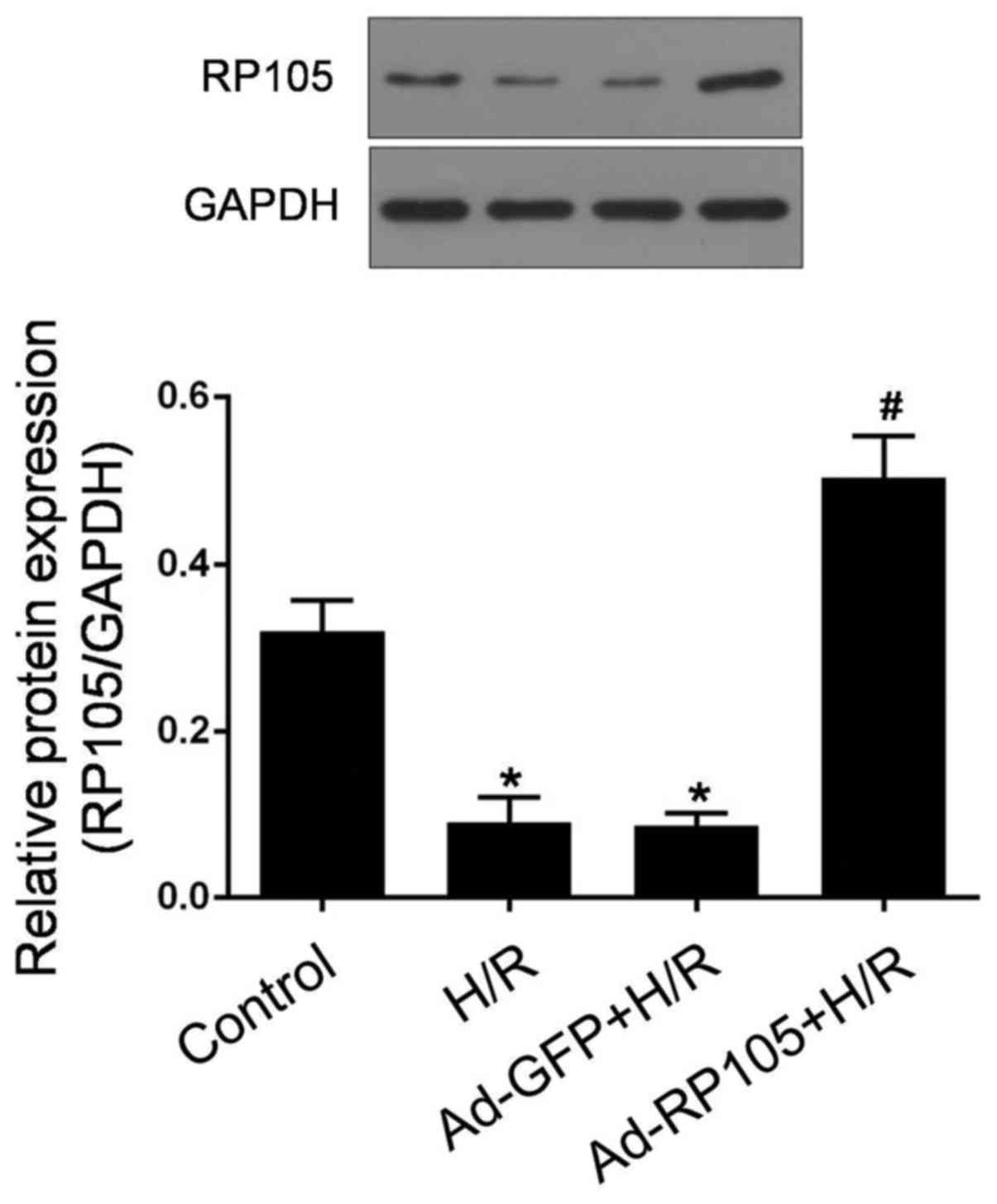
As shown in Fig.
2, RP105 was highly expressed in rat CMECs under normoxic
conditions, whereas the expression of RP105 at the protein level
was considerably lower in response to H/R (P<0.05 vs. control).
Of note, compared with a coincident decrease in Ad-GFP-pretreated
CMECs after H/R, a marked upregulation of RP105 was observed in
Ad-RP105-transduced CMECs post- H/R (P<0.05). These results
indicated the potential role of RP105 in the pathogenesis of
CMEC-mediated H/R injury.
RP105 enhances proliferation and
migration of H/R -treated CMECs
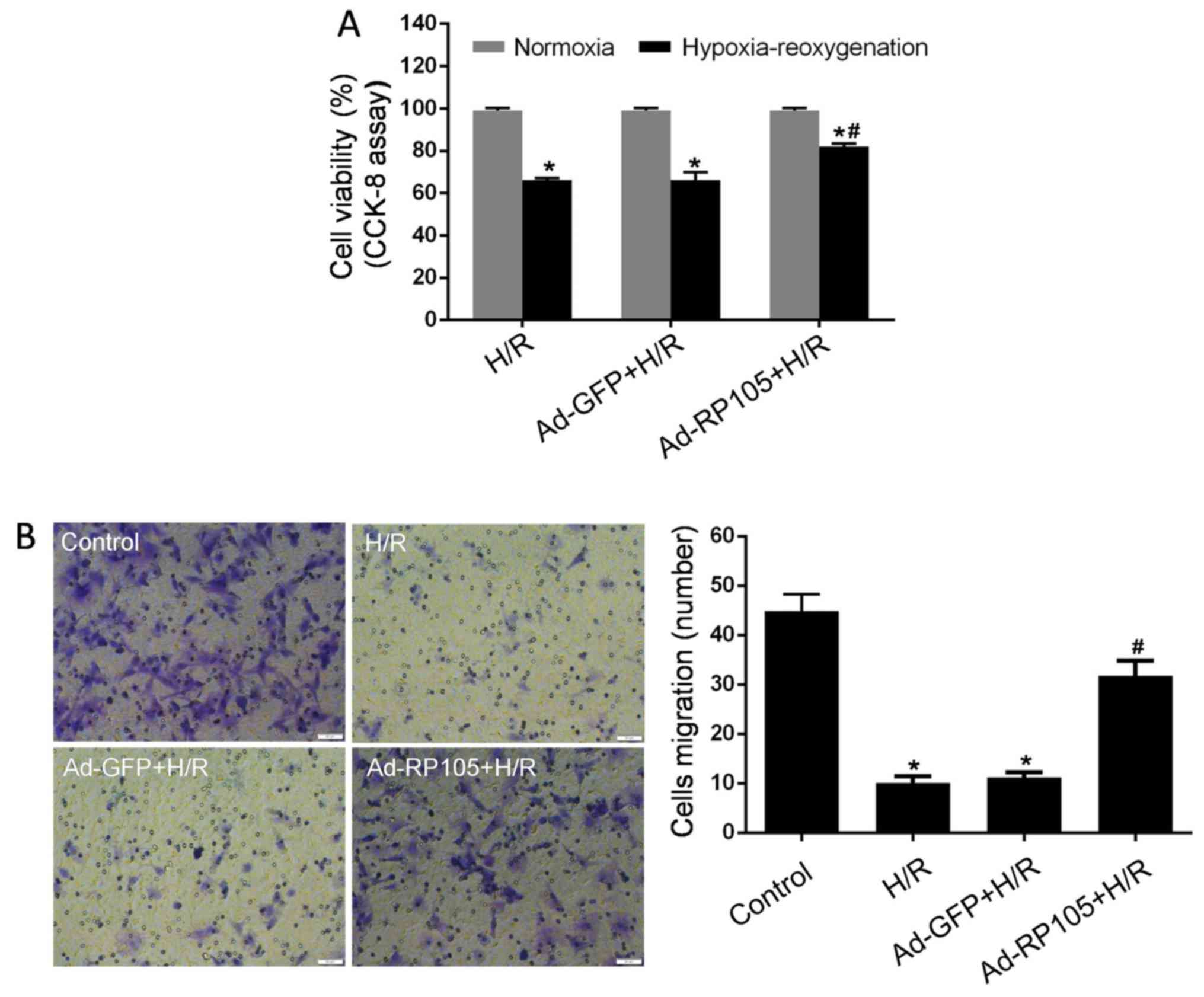
Impairments in cell proliferation and migration
ability are widely considered as important indices of CMEC
dysfunction caused by H/R injury (4,8,10).
To evaluate the effects of RP105 on CMEC malfunction, CCK-8 and
Transwell chamber assays were performed after H/R injury. As seen
in Fig. 3A, cell proliferation in
the H/R group was markedly alleviated compared with the control
group (P<0.05). However, pretreatment with Ad-RP105 in CMECs
strongly reversed this decrease post- H/R (P<0.05 vs. H/R
group). In line with the results on proliferation, CMECs under H/R
conditions exhibited impaired migration ability as evidenced by
decreased movement to the bottom surface of the Transwell chamber
compared with control cells (Fig.
3B). Overexpression of RP105 contributed to the significantly
enhanced migration of CMECs after H/R (P<0.05 vs. H/R group).
Moreover, proliferation and migration were not affected by Ad-GFP
transfection in H/R CMECs (P>0.05 vs. H/R group). These results
provide evidence that RP105 markedly ameliorates the dysfunction of
CMECs induced by H/R injury.
RP105 attenuates H/R -related CMEC
injury
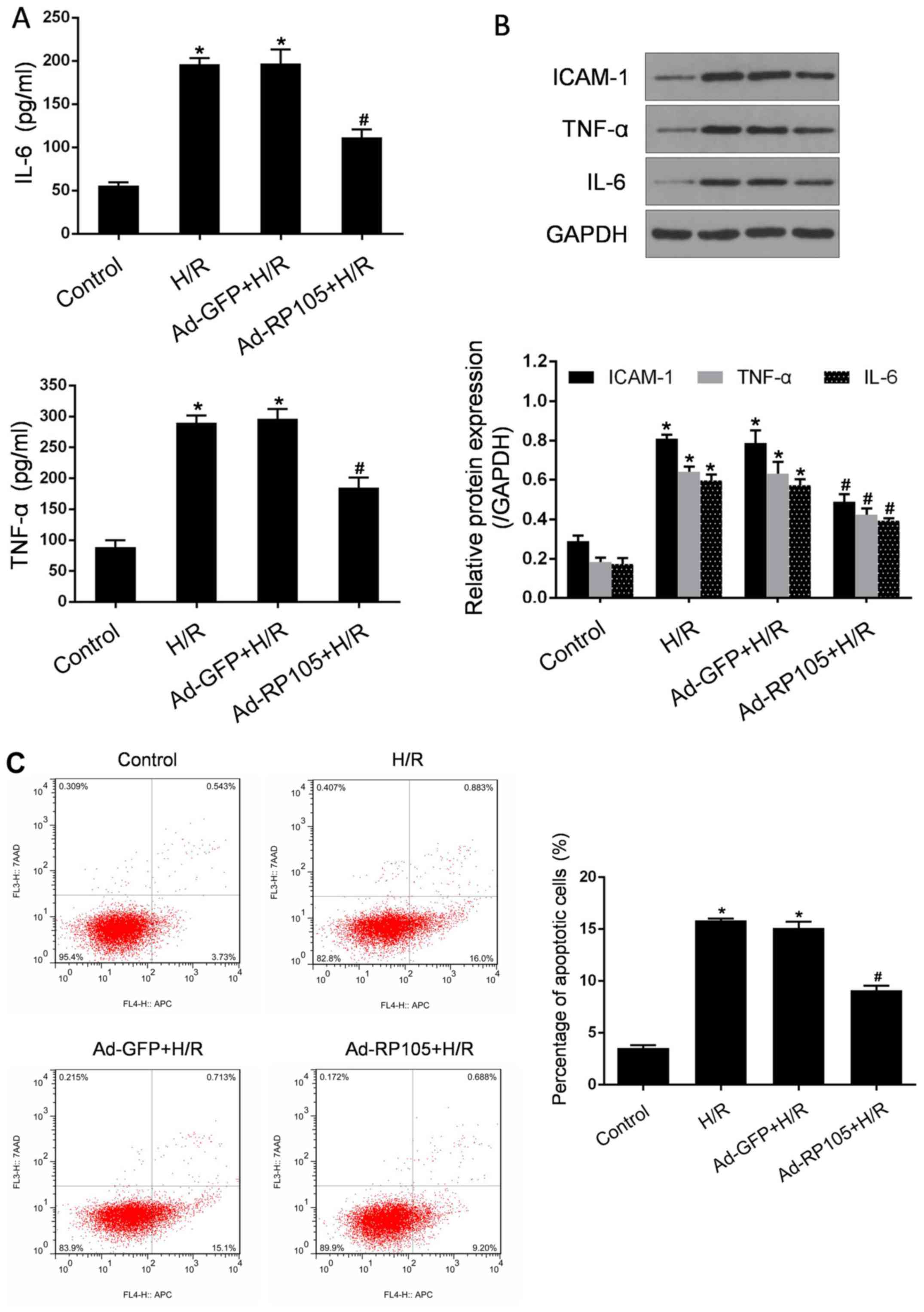
Inflammation and apoptosis are considered as
reliable indicators and crucial mechanisms for H/R -induced CMEC
injury (4,8,10).
To further investigate the involvement of RP105 in the protection
of CMECs against H/R injury, its effects on these parameters were
evaluated. As regards the inflammatory response, a markedly higher
secretion of IL-6 and TNF-α was triggered after H/R, consistent
with significantly increased expression levels of IL-6, TNF-α and
ICAM-1 proteins in CMECs exposed to H/R (P<0.05 vs. control;
Fig. 4A and B). The transduction
of Ad-RP105 resulted in a significant reduction of the
abovementioned pro-inflammatory mediators compared with CMECs
treated with H/R (P<0.05). In addition, similar patterns were
observed in H/R -related activation of apoptosis (Fig. 4C). Using flow cytometric analysis,
the number of apoptotic CMECs was found to be markedly increased
following H/R injury compared with control cells (P<0.05).
However, overexpression of RP105 by Ad-RP105 led to a significant
decrease of the apoptotic rate (P<0.05 vs. H/R group).
Transduction with Ad-GFP in H/R CMECs exerted no obvious effect on
the abovementioned injury parameters (P>0.05 vs. H/R group).
These findings indicate that RP105 possesses pleiotropic
CMEC-protective effects against H/R injury.
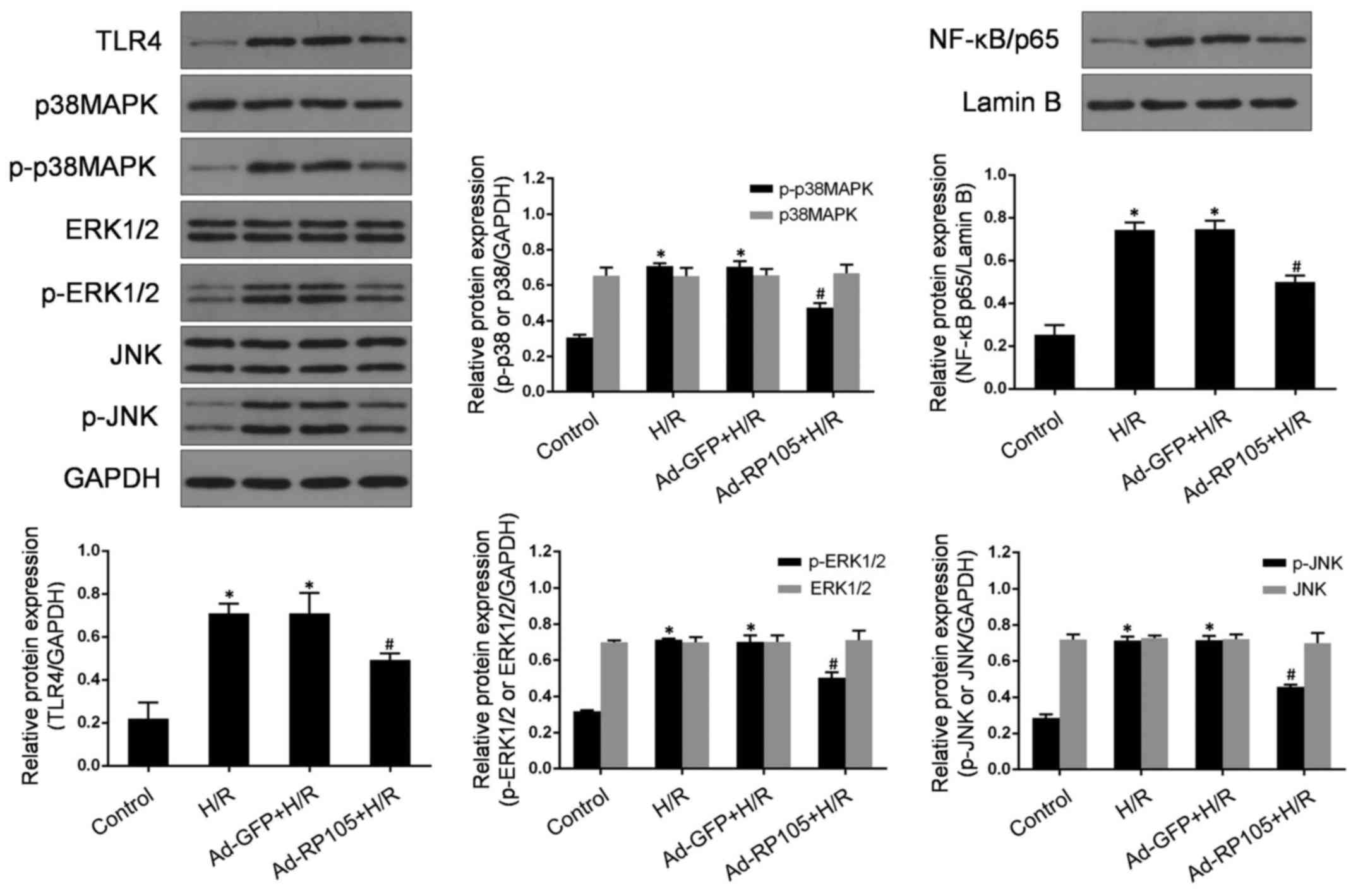
RP105 represses the expression of
TLR4/MAPKs/NF-κB after H/R injury
TLR4, MAPKs and NF-κB have been demonstrated to
participate, in combination and/or separately, in the pathological
process of microvascular endothelial cell injury under ischemic
conditions (10,27). TLR4 acts as a common upstream
sensor and stimulates MAPKs̸NF-κB signaling, which may constitute
the original mechanism underlying H/R -related CMEC injury. To
detect whether RP105 regulates the activities of MAPKs and NF-κB
through antagonizing TLR4 in CMECs post- H/R, western blotting was
used to measure the expressions of TLR4, p38 MAPK, p-p38 MAPK, JNK,
p-JNK, ERK1/2, p-ERK1/2 and nuclear NF-κB/p65 (Fig. 5). As expected, there was a
markedly increased expression of TLR4, p-p38 MAPK, p-JNK, p-ERK1/2
and nuclear NF-κB/p65 proteins in CMECs following H/R injury
compared with control cells (P<0.05). However, H/R -induced
activation of TLR4/MAPKs/NF-κB signaling was significantly
abolished by Ad-RP105 pretreatment in CMECs (vs. H/R, P<0.05).
Moreover, the levels of total p38 MAPK, ERK1/2 and JNK exhibited no
obvious differences among the four groups (P>0.05). Ad-GFP
transduction exerted no significant effect on the expression of
those proteins (vs. H/R group, P>0.05). Taken together, these
findings indicate that the protective effects of RP105 may be
closely correlated with the blockade of TLR4/MAPKs/NF-κB-mediated
H/R injury in CMECs.
Discussion
Although the cardiomyocyte-protective role of RP105
has been previously demonstrated, its involvement and causal
properties in ameliorating H/R injury of coronary CMECs remain
unclear. To the best of our knowledge, the present study is the
first to elucidate the effects of RP105 in CMEC H/R injury. The
results indicated that RP105 is minimally expressed in H/R -treated
CMECs. Overexpression of RP105 via adenoviral vectors significantly
contributed to a reduction of IL-6, TNF-α and ICAM-1 expression, as
well as fewer apoptotic CMECs relative to those in un-transduced
and Ad-GFP-transduced CMECs subjected to H/R injury. Moreover, the
proliferation and migration of H/R -treated CMECs were also
strongly promoted by Ad-RP105 transduction. Mechanistic studies
reported that RP105 overexpression blunts the expression of TLR4,
p-ERK1/2, p-p38 MAPK, p-JNK and nuclear NF-κB/p65. Therefore, RP105
may be used as a therapeutic approach against CMEC H/R injury.
Inflammatory responses have been widely investigated
due to their impact on the initiation and expansion of myocardial
I/R injury (12). Damaging
inflammatory cells, including neutrophils, macrophages and
monocytes, are recruited to the ischemic regions, and the
concomitant cytotoxic outcomes comprise the central mechanisms
causing acute losses and delayed remodeling of cardiomyocytes and
adjacent vascular endothelial cells in case of I/R (1,4).
CMECs, in particular, act as central mediators in detecting
damage-associated molecular patterns and determine cardiac
responses to I/R injury (8),
among which the activation of TLR4 and the upregulation of ICAM-1
synergistically induce the recruitment of inflammatory cells
(2,12). Inflammatory stimulation leads to
the upregulation of ICAM-1, an increase in cytokine production, as
well as the activation of apoptosis (12,28). In addition, recent studies
identified a potential correlation between TLR4 inhibition and
repressed synthesis of ICAM-1 in endothelial cells exposed to high
glucose stimulation, and TLR4 is likely to be a key modulator in
the regulation of ICAM-1 expression under inflammatory conditions
(29,30). The present study demonstrated a
previously unknown CMEC-protective role of RP105 against H/R injury
by suppressing the expression and secretion of pro-inflammatory
cytokines (IL-6 and TNF-α), and by limiting ICAM-1 expression,
which are paralleled by TLR4 downregulation. In terms of the
importance of ICAM-1 and TLR4 in initiating and amplifying
inflammatory responses in CMECs, RP105 may play a key role in
repressing H/R injury.
Previous studies have demonstrated that the
activation of TLR4̸NF-κB signaling has a detrimental effect in the
dysfunction of CMECs in response to H/R (10). However, the exact mechanisms that
sensitize CMECs to H/R injury remain incompletely understood and
the endogenous mechanisms that may regulate the disassociation
between TLR4 and CMEC H/R injury are not known. Previous studies
revealed that the activity of p-ERK1/2 was involved in the
inflammatory injury of brain microvascular endothelial cells caused
by mock ischemic treatment (31),
and its participation in inducing apoptosis under hypoxic
conditions was also verified (32,33). Moreover, the pro-inflammatory and
-apoptotic patterns of p-p38 MAPK and p-JNK activation in the
ischemic endothelium have also been extensively investigated
(10–15,27,34). Upon stimulation in the
endothelium, TLR4 triggers the inflammation and apop-tosis
cascades, partly through phosphorylation of MAPKs and NF-κB
activation, all of which may be abolished by TLR4 and/or MAPKs
inhibition (10,12–15,27,33). However, whether MAPKs and NF-κB
participate in the multifarious pathogenesis of CMEC H/R injury via
TLR4-mediated mechanisms has not been fully elucidated. Our study
demonstrated that TLR4/MAPKs/NF-κB signaling was significantly
upregulated in CMECs subjected to H/R injury, followed by promotion
of inflammation and activation of apoptosis. Therefore, we
hypothesized that RP105, known for its TLR4-inhibitory properties,
was able to abolish the causal association between TLR4/MAPKs/NF-κB
signaling and CMEC H/R injury. Our findings, therefore, indicate
that RP105, particularly its CMEC-protective properties, may
provide a promising therapeutic approach to ameliorating H/R
injury.
Of note, the role of RP105 in cardiovascular disease
has been extensively investigated. A recent study reported the
protective properties of RP105 in vascular remodeling and
myocardial infarction via TLR4-inhibitory mechanisms (21,22). Our previous findings also
demonstrated the cardiomyocyte-protective properties of RP105
against I/R injury through downregulation of TLR4 (11). Although RP105 acted as a
structural and functional inhibitor of TLR4, its role may not be
merely confined to TLR4-dependent approaches (11). For example, researchers observed
facilitated vein graft lesions in RP105−/− mice
resulting from the enhancement of chemoattractant chemokine
ligand-2 production and inflammatory response (20). RP105 deficiency alleviated
atherosclerotic plaque development through downregulation of C-C
chemokine receptor-2 expression (35). In addition, RP105 plays a major
regulatory role in adipose tissue inflammation independently of
TLR4-mediated pathways (36). It
is becoming increasingly clear that the biological actions of RP105
are mediated through TLR4-independent pathways, and one mechanism
is possibly associated with the RP105/PI3K/Akt axis (11). The findings of the present study
established that RP105 attenuated CMEC H/R injury in a
TLR4-dependent manner. Whether other causal mechanisms are involved
and RP105 modulation may provide pleiotropic protective effects
requires further elucidation.
The protective properties of RP105 in the setting of
I/R injury were primarily ascribed to be targeted on cardiomyocytes
(11,18). In our previous studies, regional
transduction of Ad-RP105 significantly limited
cardiomyocyte-related inflammatory, apoptotic and autophagic
responses, along with limiting infarct size and attenuation of
cardiac function in I/R injury (11,18). However, the contribution of RP105
to CMEC H/R injury remains largely unknown. Recently, Karper et
al demonstrated that RP105 was implicated in the process of
VSMC-mediated neointima formation and post-interventional vascular
remodeling (21). RP105
deficiency may disturb migration of monocytes in atherosclerotic
plaque formation (35). These
observations suggest that RP105 may play important roles in
cardiovascular pathological conditions that are not limited to
cardiomyocytes. In the present study, it was demonstrated that
RP105 ameliorated CMEC inflammation and apoptosis caused by H/R.
The effects of RP105 on cell proliferation and migration were also
expounded for the first time. Our findings identified CMECs as the
target of the microvascular endothelium-protective actions of
RP105, as well as expanded its beneficial effects beyond I/R
-treated cardiomyocytes. Interestingly, previous studies revealed
the dual role of RP105 in regulating TLR4 and TLR2, with RP105
inhibiting the activity of TLR4 and its downstream signaling
molecules and, conversely, facilitating TLR2-induced inflammatory
response (17). The detrimental
effects of TLR2 activation on coronary vascular endothelial cells
during I/R has been previously investigated (37). However, whether RP105 regulates
CMEC-associated H/R injury through a TLR2-dependent mechanism
requires further confirmation.
Taken together, the findings presented herein
highlight the importance of RP105 and its role in CMEC protection
from H/R injury. The underlying mechanism at least partly involves
the inactivation of TLR4/MAPKs/NF-κB signaling. Therefore,
interventional strategies that selectively regulate RP105 may be
potential candidates for the mitigation of H/R injury. Future
investigations are required, focusing on endogenous targets, such
as specific microRNAs, lncRNAs and/or circRNAs in CMECs, by means
of alternatively regulating RP105 followed by TLR4 modulation,
which may represent a novel modality for protecting cells against
I/R injury.
Abbreviations:
|
RP105
|
radioprotective 105 kDa protein
|
|
CMECs
|
cardiac microvascular endothelial
cells
|
|
H/R
|
hypoxia/reoxygenation
|
|
I/R
|
ischemia/reperfusion
|
|
TLRs
|
Toll-like receptors
|
|
MAPKs
|
mitogen activated protein kinases
|
|
TIR
|
Toll-IL-1 receptor
|
Acknowledgments
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81200156, 81200088,
81470387 and 81570331) and the Fundamental Research Funds for the
Central Universities (grant no. 20120141120079).
References
|
1
|
Ibanez B, Heusch G, Ovize M and Van de
Werf F: Evolving therapies for myocardial ischemia̸reperfusion
injury. J Am Coll Cardiol. 65:1454–1471. 2015. View Article : Google Scholar
|
|
2
|
Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T,
Ma Q, Han T, Zhang Y, Tian F, et al: Liraglutide protects cardiac
microvascular endothelial cells against hypoxia/reoxygenation
injury through the suppression of the SR-Ca(2+)-XO-ROS axis via
activation of the GLP-1R̸PI3K̸Akt̸survivin pathways. Free Radic
Biol Med. 95:278–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cui H, Li X, Li N, Qi K, Li Q, Jin C,
Zhang Q, Jiang L and Yang Y: Induction of autophagy by Tongxinluo
through the MEK̸ERK pathway protects human cardiac microvascular
endothelial cells from hypoxia/reoxygenation injury. J Cardiovasc
Pharmacol. 64:180–190. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu Y, Lian K, Zhang L, Wang R, Yi F, Gao
C, Xin C, Zhu D, Li Y, Yan W, et al: TXNIP mediates NLRP3
inflammasome activation in cardiac microvascular endothelial cells
as a novel mechanism in myocardial ischemia/reperfusion injury.
Basic Res Cardiol. 109:4152014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Li JM, Mullen AM and Shah AM: Phenotypic
properties and characteristics of superoxide production by mouse
coronary microvascular endothelial cells. J Mol Cell Cardiol.
33:1119–1131. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhou Y, Zhang Y, Gao F, Guo F, Wang J, Cai
W, Chen Y, Zheng J and Shi G: N-n-butyl haloperidol iodide protects
cardiac microvascular endothelial cells from hypoxia/reoxygenation
injury by downregulating Egr-1 expression. Cell Physiol Biochem.
26:839–848. 2010. View Article : Google Scholar
|
|
7
|
Qi XF, Li YJ, Chen ZY, Kim SK, Lee KJ and
Cai DQ: Involvement of the FoxO3a pathway in the
ischemia/reperfusion injury of cardiac microvascular endothelial
cells. Exp Mol Pathol. 95:242–247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang J, Hong Z, Zeng C, Yu Q and Wang H:
NADPH oxidase 4 promotes cardiac microvascular angiogenesis after
hypoxia/reoxygenation in vitro. Free Radic Biol Med. 69:278–288.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Coulombe KL, Bajpai VK, Andreadis ST and
Murry CE: Heart regeneration with engineered myocardial tissue.
Annu Rev Biomed Eng. 16:1–28. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang Z, Li W, Sun D, Zhao L, Zhang R,
Wang Y, Zhou X, Wang H and Cao F: Toll-like receptor 4 signaling in
dysfunction of cardiac microvascular endothelial cells under
hypoxia/reoxygenation. Inflamm Res. 60:37–45. 2011. View Article : Google Scholar
|
|
11
|
Guo X, Jiang H and Chen J: RP105-PI3K-Akt
axis: A potential therapeutic approach for ameliorating myocardial
ischemia̸reperfusion injury. Int J Cardiol. 206:95–96. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hinkel R, Lange P, Petersen B, Gottlieb E,
Ng JK, Finger S, Horstkotte J, Lee S, Thormann M, Knorr M, et al:
Heme oxygenase-1 gene therapy provides cardioprotection via control
of post-ischemic inflammation: An experimental study in a pre-
clinical pig model. J Am Coll Cardiol. 66:154–165. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zanotti G, Casiraghi M, Abano JB, Tatreau
JR, Sevala M, Berlin H, Smyth S, Funkhouser WK, Burridge K, Randell
SH, et al: Novel critical role of Toll-like receptor 4 in lung
ischemia-reperfusion injury and edema. Am J Physiol Lung Cell Mol
Physiol. 297:L52–L63. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Chen Y, Huang XJ, Yu N, Xie Y, Zhang K,
Wen F, Liu H and Di Q: HMGB1 contributes to the expression of
P-Glycoprotein in mouse epileptic brain through Toll-Like receptor
4 and receptor for advanced glycation end products. PLoS One.
10:e01409182015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mountain DJ, Singh M and Singh K:
Downregulation of VEGF-D expression by interleukin-1beta in cardiac
microvascular endothelial cells is mediated by MAPKs and
PKCalpha̸beta1. J Cell Physiol. 215:337–343. 2008. View Article : Google Scholar
|
|
16
|
Liu RR, Li J, Gong JY, Kuang F, Liu JY,
Zhang YS, Ma QL, Song CJ, Truax AD, Gao F, et al: MicroRNA-141
regulates the expression level of ICAM-1 on endothelium to decrease
myocardial ischemia-reperfusion injury. Am J Physiol Heart Circ
Physiol. 309:H1303–H1313. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu B, Zhang N, Liu Z, Fu Y, Feng S, Wang
S, Cao Y, Li D, Liang D, Li F, et al: RP105 involved in activation
of mouse macrophages via TLR2 and TLR4 signaling. Mol Cell Biochem.
378:183–193. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li X, Yang J, Yang J, Dong W, Li S, Wu H
and Li L: RP105 protects against myocardial ischemia-reperfusion
injury via suppressing TLR4 signaling pathways in rat model. Exp
Mol Pathol. 100:281–286. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hijiya N, Miyake K, Akashi S, Matsuura K,
Higuchi Y and Yamamoto S: Possible involvement of toll-like
receptor 4 in endothelial cell activation of larger vessels in
response to lipo-polysaccharide. Pathobiology. 70:18–25. 2002.
View Article : Google Scholar
|
|
20
|
Wezel A, de Vries MR, Maassen JM, Kip P,
Peters EA, Karper JC, Kuiper J, Bot I and Quax PH: Deficiency of
the TLR4 analogue RP105 aggravates vein graft disease by inducing a
pro-inflammatory response. Sci Rep. 6:242482016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Karper JC, Ewing MM, de Vries MR, de Jager
SC, Peters EA, de Boer HC, van Zonneveld AJ, Kuiper J, Huizinga EG,
Brondijk TH, et al: TLR accessory molecule RP105 (CD180) is
involved in post-interventional vascular remodeling and soluble
RP105 modulates neointima formation. PLoS One. 8:e679232013.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Louwe MC, Karper JC, de Vries MR, Nossent
AY, Bastiaansen AJ, van der Hoorn JW, Willems van Dijk K, Rensen
PC, Steendijk P, Smit JW, et al: RP105 deficiency aggravates
cardiac dysfunction after myocardial infarction in mice. Int J
Cardiol. 176:788–793. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xia JB, Liu GH, Chen ZY, Mao CZ, Zhou DC,
Wu HY, Park KS, Zhao H, Kim SK, Cai DQ, et al: Hypoxia̸ischemia
promotes CXCL10 expression in cardiac microvascular endothelial
cells by NFkB activation. Cytokine. 81:63–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yang J, Guo X, Yang J, Ding JW, Li S, Yang
R, Fan ZX and Yang CJ: RP105 protects against apoptosis in
ischemia/reperfusion-induced myocardial damage in rats by
suppressing TLR4-mediated signaling pathways. Cell Physiol Biochem.
36:2137–2148. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang J, Chen L, Ding J, Zhang J, Fan Z,
Yang C, Yu Q and Yang J: Cardioprotective effect of miRNA-22 on
hypoxia/reoxygenation induced cardiomyocyte injury in neonatal
rats. Gene. 579:17–22. 2016. View Article : Google Scholar
|
|
26
|
Choi HS, Kim MK, Choi YK, Shin YC, Cho SG
and Ko SG: Rhus verniciflua Stokes (RVS) and butein induce
apoptosis of paclitaxel-resistant SKOV-3̸PAX ovarian cancer cells
through inhibition of AKT phosphorylation. BMC Complement Altern
Med. 16:1222016. View Article : Google Scholar
|
|
27
|
Ye EA and Steinle JJ: miR-146a attenuates
inflammatory pathways mediated by TLR4̸NF-κB and TNFα to protect
primary human retinal microvascular endothelial cells grown in high
glucose. Mediators Inflamm. 2016:39584532016. View Article : Google Scholar
|
|
28
|
Shi J, Zhou J and Zhang M: Microcystins
induces vascular inflammation in human umbilical vein endothelial
cells via activation of NF-κB. Mediators Inflamm.
942159:20152015.
|
|
29
|
Mudaliar H, Pollock C, Ma J, Wu H, Chadban
S and Panchapakesan U: The role of TLR2 and 4-mediated inflammatory
pathways in endothelial cells exposed to high glucose. PLoS One.
9:e1088442014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sawa Y, Ueki T, Hata M, Iwasawa K, Tsuruga
E, Kojima H, Ishikawa H and Yoshida S: LPS-induced IL-6, IL-8,
VCAM-1, and ICAM-1 expression in human lymphatic endothelium. J
Histochem Cytochem. 56:97–109. 2008. View Article : Google Scholar
|
|
31
|
Li F, Li W, Li X, Li F, Zhang L, Wang B,
Huang G, Guo X, Wan L, Liu Y, et al: Geniposide attenuates
inflammatory response by suppressing P2Y14 receptor and downstream
ERK1/2 signaling pathway in oxygen and glucose deprivation-induced
brain microvascular endothelial cells. J Ethnopharmacol. 185:77–86.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cheng F, Lan J, Xia W, Tu C, Chen B, Li S
and Pan W: Folic acid attenuates vascular endothelial cell injury
caused by hypoxia via the inhibition of ERK1̸2̸NOX4̸ROS pathway.
Cell Biochem Biophys. 74:205–211. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Li J, Zhang Z, Lv L, Qiao H, Chen X and
Zou C: (−)-Epigallocatechin gallate inhibits asymmetric
dimethylarginine-induced injury in human brain microvascular
endothelial cells. Neurochem Res. 41:1868–1876. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Wang LW, Chang YC, Chen SJ, Tseng CH, Tu
YF, Liao NS, Huang CC and Ho CJ: TNFR1-JNK signaling is the shared
pathway of neuroinflammation and neurovascular damage after
LPS-sensitized hypoxic-ischemic injury in the immature brain. J
Neuroinflammation. 11:2152014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Wezel A, van der Velden D, Maassen JM,
Lagraauw HM, de Vries MR, Karper JC, Kuiper J, Bot I and Quax PH:
RP105 deficiency attenuates early atherosclerosis via decreased
monocyte influx in a CCR2 dependent manner. Atherosclerosis.
238:132–139. 2015. View Article : Google Scholar
|
|
36
|
Nagai Y, Watanabe Y and Takatsu K: The TLR
family protein RP105̸MD-1 complex: A new player in obesity and
adipose tissue inflammation. Adipocyte. 2:61–66. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jin C, Cleveland JC, Ao L, Li J, Zeng Q,
Fullerton DA and Meng X: Human myocardium releases heat shock
protein 27 (HSP27) after global ischemia: The proinflammatory
effect of extracellular HSP27 through toll-like receptor (TLR)-2
and TLR4. Mol Med. 20:280–289. 2014. View Article : Google Scholar : PubMed/NCBI
|