Introduction
Doxorubicin (Dox) is one of the effective
anthracycline anticancer drugs and is used extensively in clinical
practice (1). Despite its
effectiveness against cancer, its dose-dependent cardiotoxicity
restricts its long-term use in chemotherapy as it reduces the
quality of life of patients with cancer (2). Despite advances made over several
decades, the precise mechanism underlying Dox-induced
cardiomyopathy remains to be fully elucidated. Oxidative stress,
inflammation and apoptosis have been suggested as the mechanisms of
Dox-induced cardiotoxicity, and all these mechanisms lead to
apparent cardiomyocyte senescence, which can, at least partly,
result in cardiac remodeling and dysfunction (3,4).
However, a specific strategy is required to protect patients
against Dox-induced cardiotoxicity.
Previous studies have demonstrated that mesenchymal
stem cell (MSC) transplantation attenuates myocardial damage, such
as that present following myocardial infarction (5). Several studies have also shown
promising results with MSC transplantation in animal models of
Dox-induced heart failure (6,7).
The cardioprotective effects of MSCs involve several mechanisms,
including paracrine antifibrotic and anti-apoptotic factors, which
may contribute to the attenuation of cardiac fibrosis following Dox
treatment (6). MSCs also enhanced
the capillary density, attenuating the impaired contractility of
cardiomyocytes, in a model of Dox-induced heart failure (8). The mechanism underlying the
protective effects of MSCs against cardiomyocyte senescence remain
to be fully elucidated, although it is the main factor contributing
to Dox-related cardiac remodeling and dysfunction. The present
study investigated whether coculture with MSCs can reverse the
senescence of cardiomyocytes induced by Dox and the mechanism
involved.
The Notch signaling pathway is an evolutionarily
conserved signaling system, which regulates cell proliferation,
differentiation and fate determination in embryonic and adult
organs (9). Notch signaling is
also involved in the self-renewal and proliferation of adult cells
in several organs and cellular systems (10). Evidence has shown that the Notch
ligand, Jagged-1, can be used to inhibit senescence in long-term
sheet cultures of MSCs (9). Notch
signaling is crucial in cardiac development, guiding the cell fate
decisions that underlie myocyte and vessel differentiation
(11). In cardiac progenitor
cells, an increase in Notch activity reduces markers of senescence
(12). Therefore, the present
study examined whether modulating the Notch pathway through
coculture with MSCs can relieve Dox-induced cardiac injury.
In the cardiovascular system, transforming growth
factor (TGF)-β1 is involved in fibrotic cardiac remodeling
(13). Dox causes dose-dependent
dilated cardiomyopathy when used to treat patients with cancer,
which is associated with the inhibition of endothelial cell
proliferation, migration and angiogenesis by TGF-β1 (14). The TGF-β1-related apoptosis of
contractile cells in the heart, cardiomyocytes, has been implicated
in the cardiac damage caused by Dox (15). A previous study demonstrated that
MSC transplantation improved cardiac function following myocardial
infarction by inhibiting the TGF-β1/small mothers against
decapentaplegic (SMAD) signaling pathway (16). Therefore, it was hypothesized that
MSCs protect cardiomyocytes from Dox-related injury by inhibiting
TGF-β1.
In the present study, the antisenescence effects of
MSCs were investigated, and the involvement of the
Jagged-1/Notch-1/TGF-β1 signaling pathway in the protection
afforded by MSCs against Dox-induced cardiotoxicity was
examined.
Materials and methods
Animals
Male 6-month-old Sprague-Dawley (SD) rats weighing
60–80 g were cared for in accordance with the published Guide for
the Care and Use of Laboratory Animals of the United States
National Institutes of Health. Rats were purchased from the
Laboratory Animal Center of Wenzhou Medical University (Wenzhou,
China). They were housed in a 12 h light/dark cycle at 21±2°C, with
a relative humidity of 30–70% and free access to food and water.
All animal procedures were approved by the Institutional Animal
Care and Use Committee of Wenzhou Medical University (Wenzhou,
China).
Reagents
Dulbecco’s modified Eagle’s medium (DMEM) and fetal
bovine serum (FBS) were obtained from HyClone Laboratories; GE
Healthcare Life Sciences (Logan, UT, USA). The Transcriptor First
Strand cDNA Synthesis kit, X-tremeGENE™ HP DNA transfection
reagent, TeloTAGGG™ Telomerase PCR ELISAPLUS kit, and FastStart
Universal SYBR Green Master (Rox) were obtained from Roche
Diagnostics GmbH (Mannheim, Germany). Rabbit monoclonal antibodies
directed against Notch-1 (cat. no. 3608), Jagged-1 (cat. no. 70109)
and β-actin (cat. no. 4970) were purchased from Cell Signaling
Technology, Inc. (Danvers, MA, USA). Notch-1 and Jagged-1 small
interfering RNAs (siRNAs) were obtained from Thermo Fisher
Scientific, Inc. (Waltham, MA, USA). 3-(4,5-dimethyl
thiazolyl-2)-2,5-diphenyl tetrazolium bromide (MTT) and dimethyl
sulfoxide (DMSO) were purchased from Sigma-Aldrich; EMD Millipore
(Billerica, MA, USA). Recombinant TGF-β1 was purchased from Thermo
Fisher Scientific, Inc. The Rat VEGF ELISA kit and Rat TGF-β1 ELISA
kit was purchased from Abcam (Cambridge, UK).
Cell culture
Rat embryonic myoblasts (H9c2 cells) were obtained
from the American Type Culture Collection (Rockville, MD, USA). The
cells were cultured in DMEM supplemented with 10% FBS at 37°C with
5% CO2. All cells used in each assay were at passages
5–8 (17).
Bone-marrow MSCs were isolated from the femurs and
tibias of the SD rats, as described previously (18). Briefly, the bone-marrow cells were
flushed from the femur and tibia with 5 ml of DMEM/F12 medium. The
red blood cells were lysed and discarded, and the remaining cells
(5×105) were plated in a 25 cm2 flask in 6 ml
of DMEM/F12 supplemented with 10% FBS and 1%
penicillin/streptomycin. The cells were cultured at 37°C under 5%
CO2. Following 3 days in culture, the adherent MSCs were
maintained in culture and the medium was replaced every 3 days.
Once the culture reached 80–90% confluence, the cells were
trypsinized and passaged at a dilution of 2:3. All the cells used
in subsequent assays were in passages 3–5. The characteristics of
the MSCs were demonstrated by immunophenotyping, as described
previously (19).
Transwell cocultures of MSCs and H9c2
cells, and cell treatment
A Transwell system was used to prevent the MSCs from
directly contacting the H9c2 cells. The MSCs and H9c2 cells were
placed in the upper and lower chambers of the Transwell apparatus,
respectively, at a density of 1×106 cells/well. The H9c2
cells were pretreated with Dox (0.5 μM) for 1 h at 37°C, as
previously described (20) and,
prior to coculture, they were washed with PBS. The recombinant
TGF-β1 (10 ng/ml) or anti-VEGF receptor2 (anti-VEGFR2) antibody (25
μg/ml) was added to the culture medium of the H9c2 cells 1 h
prior to their coculture.
Cell proliferation assay
The rate of cell proliferation was estimated with
the Cell Counting Kit-8 (CCK-8) assay (HaiGene Technology, Harbin,
China), according to the manufacturer’s protocol. Briefly,
1×105 cells were grown in a 96-well plate and incubated
with the CCK-8 solution for 1 h at 37°C, following which the
absorbance of each well at 450 nm was recorded. Three replicate
experiments were performed.
MTT assay
The MTT assay was used to determine cell viability.
Briefly, 300 μl of MTT reagent was added to each
cell-containing well 2 h prior to harvesting. The supernatant was
then removed and incubated with 400 μl of DMSO for 10 min.
The absorbance at 540 nm was recorded with an enzyme-linked
immunosorbent assay plate reader. Three replicate experiments were
performed.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
The expression levels of several genes were analyzed
by RT-qPCR analysis. The expression levels of p53, p16,
Notch1, Jagged-1 (Jag1), transforming growth
factor-β1(Tgfb1) and Gapdh were analyzed, in addition
to telomere length and telomerase activity. cDNA was amplified with
Power SYBR Green PCR Master mix and the appropriate primers in an
Applied Biosystems StepOnePlus Real-Time PCR system (Thermo Fisher
Scientific, Inc.), in a reaction system of 20 μl, containing
10 μl cDNA, 4.0 μl buffer and 1 μl primers at
95°C for 10 min, followed by 40 cycles of 15 sec at 95°C and 1 min
at 60°C. A melting curve was generated to examine the specificity
of the amplification. The relative expression levels were
calculated with the 2−ΔΔCq method (21), using Gapdh as the internal
control. The primer pairs used to detect the mRNA levels of the
target genes are listed in Table
I.
 | Table IPrimer sequences. |
Table I
Primer sequences.
| Gene | Sequence |
|---|
| p16 | F:
5′-GGTCACCGACAGGCATAACTTC-3′ |
| p53 | R:
5′-AAAGGAGGGCTGAGGCCTAA-3′ |
| Notch-1 | F:
5′-TCTGTCATCTTCCGTCCCTTCTC-3′ |
| Jagged-1 | R:
5′-CCGTGCACATAACAGACTTGGCT-3′ |
| Telomere
length | F:
5′-GATGACCTGGGCAAGTC-3′ |
| R:
5′-CCCTGTTGTTCTGCATATCT-3′ |
| F:
5′-GCTGACTTAGAATCCCTGTGTTA-3′ |
| R:
5′-AGGGTACTGTTGACTAGCTTT-3′ |
| F:
5′-GGTTTTTGAGGGTGAGGGTGAGGGTGAGGGTGA-3′ |
| R:
5′-TCCCGACTATCCCTATCCCTATCCCTATCCCTATCC-3′ |
| TGF-β1 | F:
5′-ATTCAAGTCAACTGTGGAGCAAC-3′ |
| GAPDH | R:
5′-CGAAAGCCCTGTATTCCGTCT-3′ |
| siRNA-Notch-1 | F:
5′-GGCTCTCTGCTCCTCCCTGTT-3′ |
| siRNA-Jagged-1 | R:
5′-GGCTCTCTGCTCCTCCCTGTT-3′ |
| siRNA-NT | AAG TGG GAC CTG CCT
GAA TGG |
| AAG GAG TAT CAG TCC
CGC GTC |
| AAT CGC ATA GCG TAT
GCC GTT |
Western blot analysis
Western blot analyses were performed, as previously
described (18). Briefly, the
cell samples were ruptured with lysis buffer. The lysates were
centrifuged for 5 min at 4°C and 12,000 × g prior to analysis of
the supernatant containing the cellular proteins. Protein
concentration was determined with a bicinchoninic acid protein
assay kit. For each sample, 20 μg of total protein was
resolved by 10% SDS-PAGE and transferred onto polyvinylidene
difluoride membranes. The membranes were incubated overnight at 4°C
with antibodies directed against Notch-1, Jagged-1 and β-actin
diluted at 1:1,000. The following day, the membranes were incubated
for 1 h at 37°C with horseradish peroxidase-conjugated goat
anti-rabbit IgG secondary antibody (cat. no. 7074; 1:2,000; Cell
Signaling Technology, Inc.), and developed with chemiluminescent
substrate. The stained protein bands were visualized with the
Bio-Rad ChemiDoc™ XRS system, and analyzed with the QuantityOne
software (version 4.5.2; Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
Enzyme-linked immunosorbent assays
(ELISAs)
The concentrations of VEGF and TGF-β1 secreted into
the cell culture media were measured with ELISA kits. The assays
were performed in 96-well microplates, according to the ELISA
manufacturer’s protocol. Three replicate experiments were
performed.
Gene knockdown with siRNA
The cells were transfected using X-tremeGENE™ HP DNA
transfection reagent, according to the manufacturer’s protocol. The
H9c2 cells were plated in a 6-well plate at a density of
1×106 cells and treated for 20 min with X-tremeGENE™ HP
DNA transfection reagent at a reagent volume to siRNA mass ratio of
3:1. The cells were then transfected with the mixture containing
100 nM siRNA and incubated in 2 ml of culture medium for 48 h at
37°C. A scrambled siRNA (siRNA-NT) was used as the control. siRNA
sequences were presented in Table
I. The transfection efficiencies of siRNA-Notch-1 and
siRNA-Jagged-1 were assessed with RT-qPCR analysis.
Measurement of relative telomere
length
The relative telomere lengths in the H9c2 cells were
quantified using RT-qPCR analysis, based on a previously
established method (22) and as
described above. and normalized to the Gapdh gene. The
primer pairs used to detect the telomere length are listed in
Table I.
Measurement of relative telomerase
activity
The telomerase activity of the H9c2 cells was
examined with the TeloTAGGG™ Telomerase PCR ELISAPLUS kit,
according to the manufacturer’s protocol. The cell lysates were
centrifuged for 20 min at 4°C and 12,000 × g, and 3 μl of
cell extract was used to amplify each telomeric repeat. Inactivated
cell lysate (3 μl) was used for the telomeric repeat
amplification protocol (TRAP) reaction, according to the
manufacturer’s protocol. An internal control from the kit was
amplified with each TRAP reaction to confirm the absence of any PCR
inhibitor. Using ELISA, the amplified products were immobilized on
streptavidin-coated microtiter plates via the biotin-streptavidin
interaction, and detected with an anti-digoxigenin antibody
conjugated with peroxidase for 30 min at 25°C (100 μl
working solution; included in the ELISAPLUS kit). Following the
addition of a peroxidase substrate
(3,3′,5,5′-tetramethylbenzidine), the TRAP products were quantified
by measuring the absorbance at 450 nm with a microplate reader.
Statistical analysis
Data are expressed as mean ± standard deviation.
Differences among groups were compared with one-way analysis of
variance followed by Tukey’s multiple comparisons test, and
comparisons between two groups were made with Student’s t-test in
the SPSS v19.0 package (IBM SPSS, Armonk, NY, USA). P<0.05 was
considered to indicate a statistically significant difference.
Results
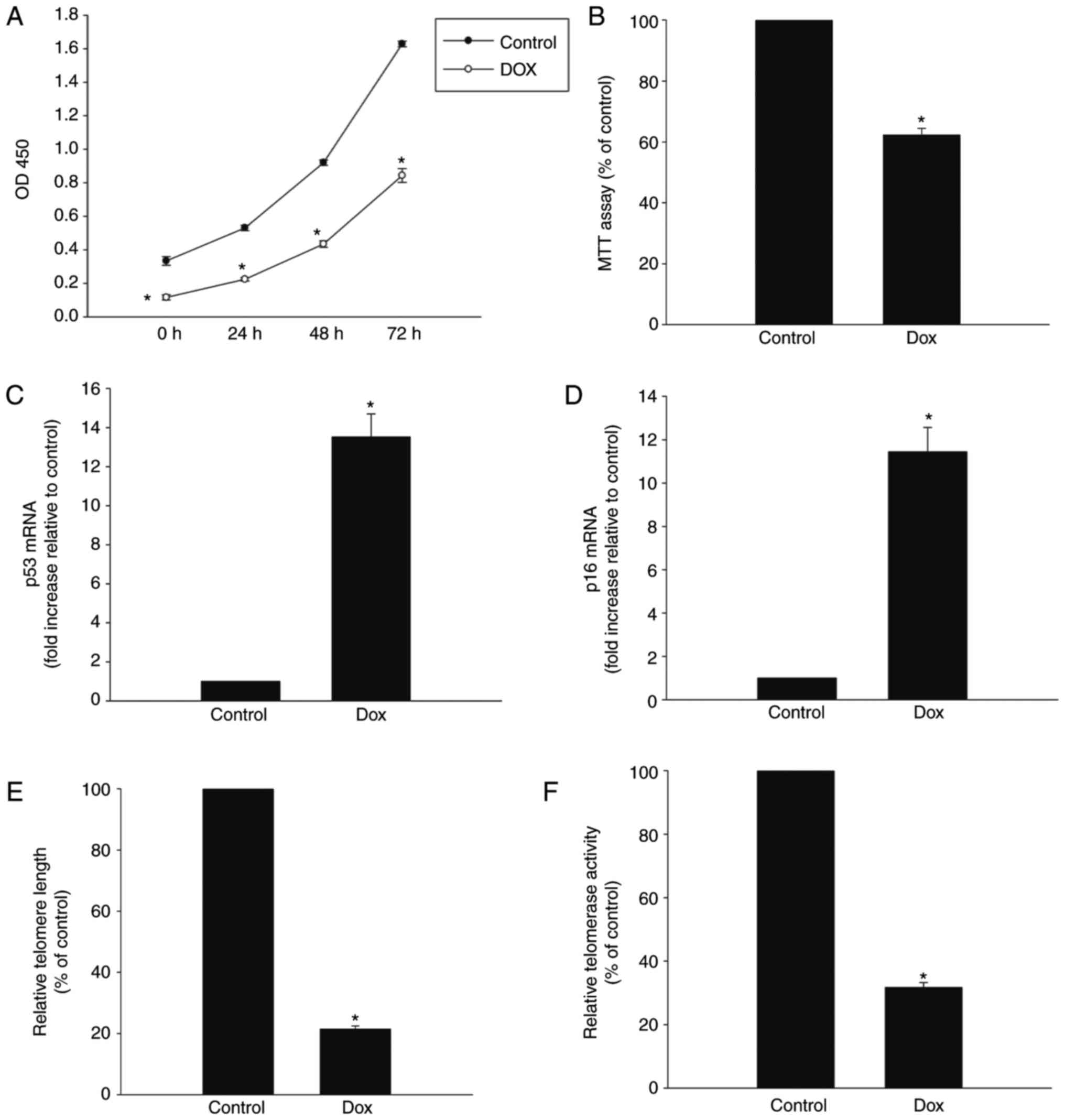
Dox induces senescence of H9c2 cells
Dox is cytotoxic towards cardiomyocytes (20), therefore, the present study
investigated whether Dox induced senescence in H9c2 cells. As shown
in Fig. 1A, when the H9c2 cells
were treated with Dox at a concentration of 0.5 μmol/l, the
proliferation rate decreased significantly immediately following
treatment and remained low for at least 72 h. Dox treatment also
significantly reduced the viability of the H9c2 cells, measured
with the MTT assay (Fig. 1B). The
expression levels of senescence-related genes p53 and p16 were
markedly increased in the Dox treatment group (Fig. 1C and D). Dox treatment also
induced telomere shortening, which was associated with a reduction
in telomerase activity (Fig. 1E and
F).
Coculture with MSCs restores
cardiomyocytes via the VEGF juxtacrine pathway
To examine the antisenescence effect of MSCs on
Dox-treated H9c2 cells, a Transwell coculture system were used.
MSCs have been shown to exert a cytoprotective effect through a
juxtacrine mechanism (23),
therefore, the present study examined whether MSCs restored H9c2
cells treated with Dox through a juxtacrine pathway. As expected,
coculture with the MSCs induced the release of VEGF (Fig. 2A). Cell viability and
proliferation were markedly higher in the Dox+MSC group, compared
with that in the group treated with Dox alone (Fig. 2B and C). In addition, the
expression levels of p53 and p16 were reduced in the cells exposed
to Dox+MSCs, compared with their expression levels in cells treated
with Dox alone (Fig. 2D and E).
The addition of MSCs to the coculture system reversed the
Dox-induced telomere shortening and reduced telomerase activity
(Fig. 2F and G). However, the
addition of anti-VEGFR2 antibody to the coculture system reversed
the MSC-induced antisenescence effects (Fig. 2B–G).
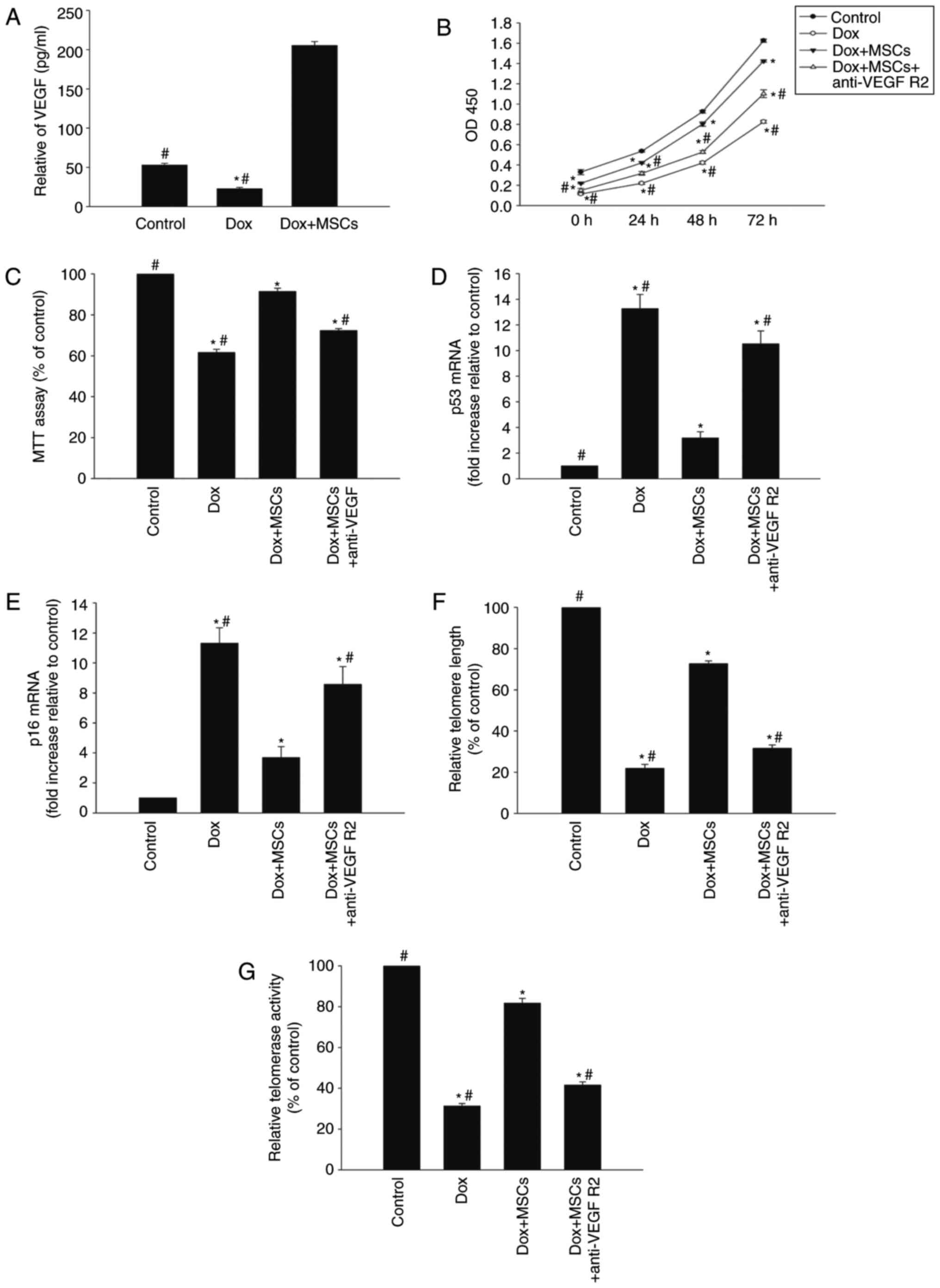 | Figure 2Coculture of cardiomyocytes with MSCs
restores the cells via a VEGF juxtacrine pathway. To examine the
antisenescence effects of MSCs on Dox-treated H9c2 cells, a
Transwell coculture system was used. (A) ELISA of secretion of VEGF
from H9c2 cells incubated with Dox or cocultured with MSCs in the
presence of Dox. In H9c2 cells incubated with Dox, cocultured with
MSCs in the presence of Dox, or cocultured with MSCs in the
presence of Dox and anti-VEGFR2antibody, (B) cell proliferation was
determined with the Cell Counting Kit-8 assay, and (C) cell
viability was analyzed with the MTT assay. mRNA levels of (D) p53
and (E) p16 and (F) telomere length were analyzed with reverse
transcription-quantitative polymerase chain reaction analysis. (G)
Relative telomerase activity was measured. Data are presented as
the mean ± standard deviation of three independent experiments;
*P<0.05, vs. control; #P<0.05, vs. Dox+MSCs. Dox,
doxorubicin; MSCs, mesenchymal stem cells; VEGF, vascular
endothelial growth factor; VEGF R2, VEGF receptor 2; MTT,
3-(4,5-dimethyl thiazolyl-2)-2,5-diphenyl tetrazolium bromide. |
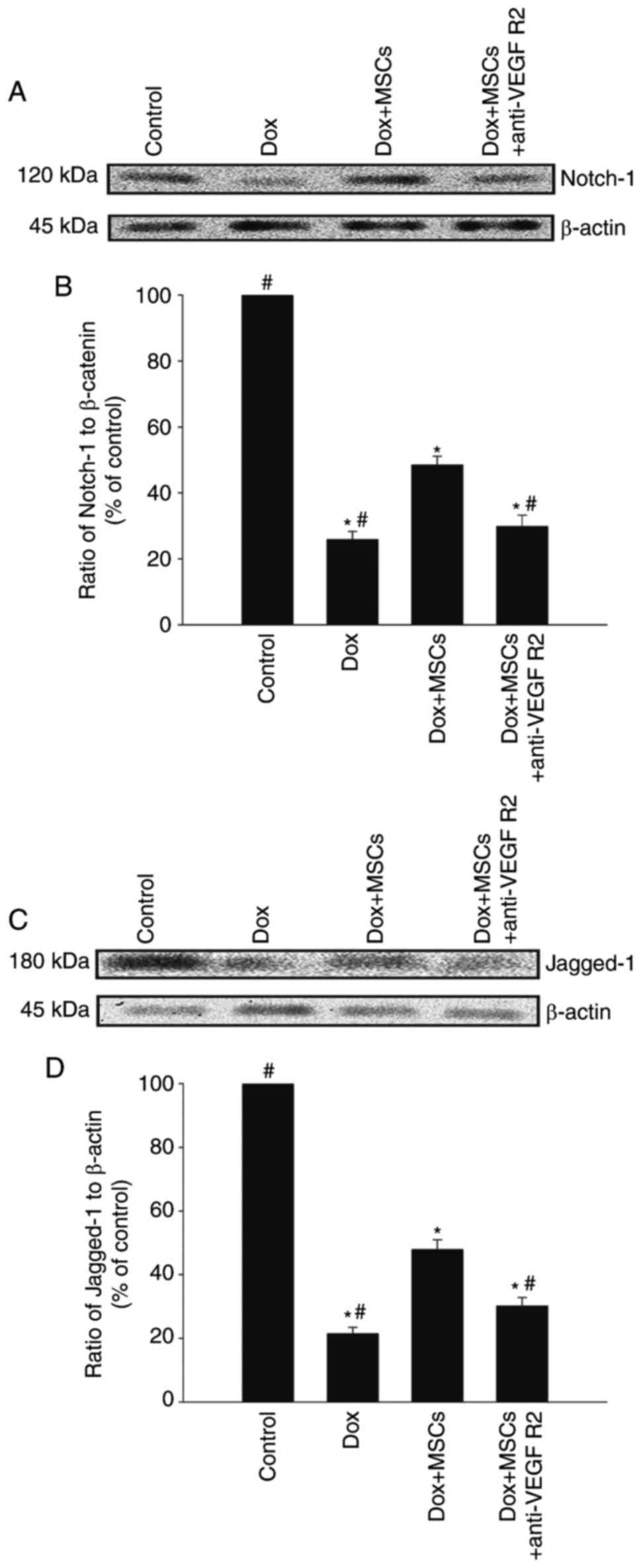
Jagged-1/Notch-1 signaling pathway
dynamically regulates the MSC-induced antisenescence effect
The Jagged-1/Notch-1 signaling pathway is reported
to be an important regulator of rejuvenation in several cell types
(24). Therefore, the present
study investigated whether this pathway mediates the antisenescence
effects of MSCs. As shown in Fig.
3A–D, treatment with Dox reduced the expression levels of
Notch-1 and Jagged-1, which were restored by coculture with MSCs.
However, when the H9c2 cells were treated with an anti-VEGFR2
antibody, the restorative effects of the MSCs were eliminated.
To confirm the role of the Jagged-1/Notch-1 pathway
in the antisenescence effect of MSCs, the expression of Notch-1 or
Jagged-1wassilencedwith siRNA and the senescence of H9c2 cells in
the presence of Dox were examined. As shown in Fig. 4A and B, the knockdown of the
Notch1 or Jag1 gene with siRNA reduced the expression
of Notch-1 and Jagged-1respectively. Silencing Notch1 or
Jag1 significantly attenuated the antisenescence effect of
MSCs, shown by the reduced proliferation and viability of the H9c2
cells (Fig. 4C and D), and
increased the expression levels of p53 and p16 (Fig. 4E and F). Silencing Notch1
or Jag1 also reduced the telomere length and telomerase
activity in the H9c2 cells (Fig. 4G
and H). By contrast, siRNA-NT had no significant effect.
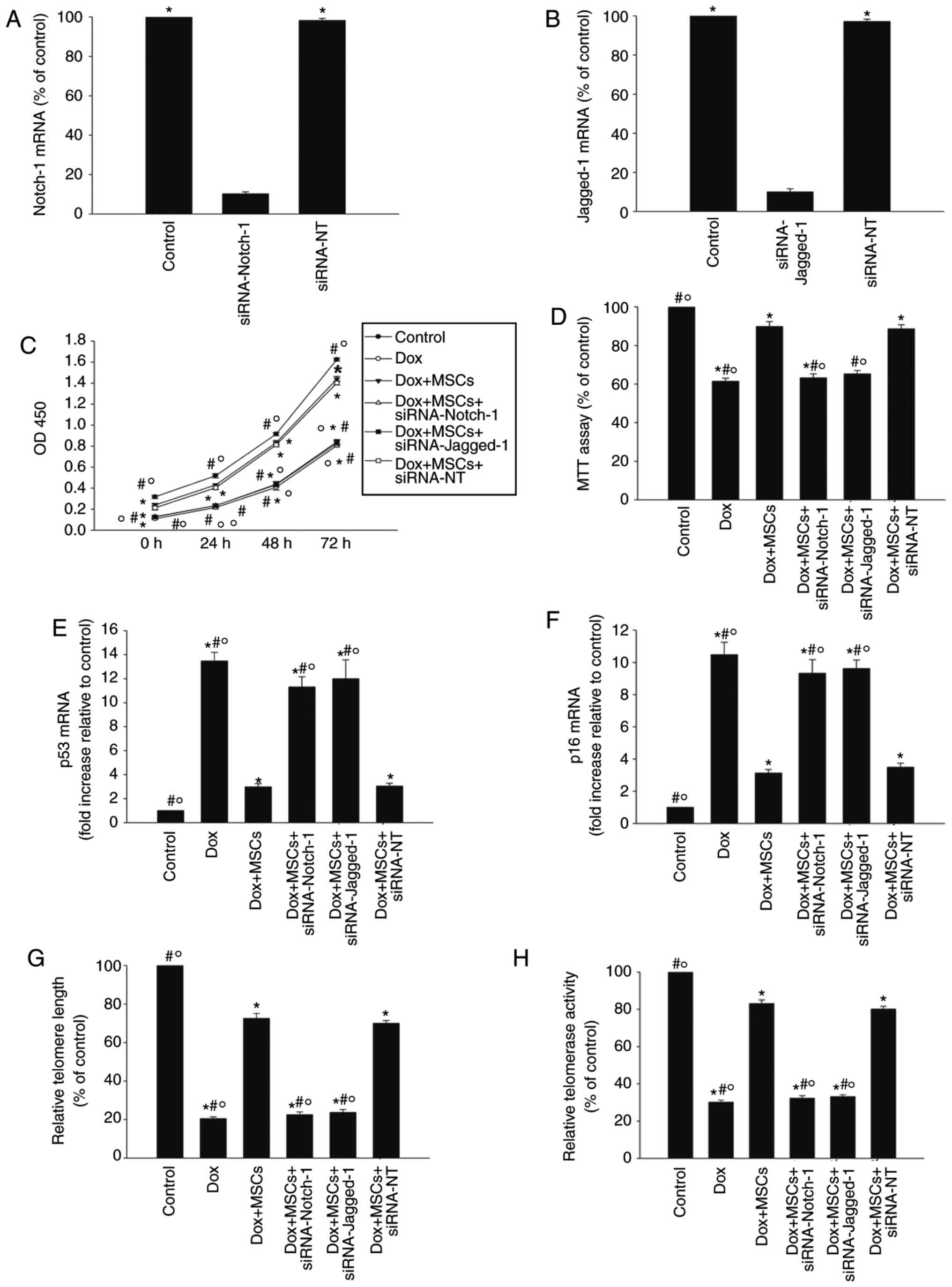 | Figure 4Jagged-1/Notch-1 signaling pathway
dynamically regulates MSC antisenescence effects. (A) H9c2 cells
were transfected with siRNA directed against Notch-1 or with
siRNA-NT as the control. siRNA-mediated transfection efficiency was
determined with RT-qPCR. Each column represents the mean ± standard
deviation of three independent experiments; *P<0.05,
vs. siRNA-Notch-1. (B) H9c2 cells were transfected with siRNA
directed against Jagged-1 or with siRNA-NT as the control.
siRNA-mediated transfection efficiency was determined with RT-qPCR.
Each column represents the mean ± standard deviation of three
independent experiments; *P<0.05, vs. siRNA-Jagged-1.
To determine whether the Jagged-1/Notch-1 signaling pathway is
involved in the antisenescence actions of MSCs, H9c2 cells were
transfected with siRNA against Notch-1 or Jagged-1, or with
siRNA-NT as the control, and then cocultured with MSCs in the
presence of Dox. In parallel experiments, the cells were treated
with Dox alone or cocultured with MSCs in the presence of Dox. H9c2
cells under normal culture conditions were used as the control. (C)
Growth (proliferation) curves of H9c2 cells determined with a Cell
Counting Kit-8 assay. (D) Cellular viability was analyzed with an
MTT assay. (E) p53, (F) p16 and (G) telomere length were analyzed
with RT-qPCR analysis. (H) Relative telomerase activity was
measured. Data are presented as the mean ± standard deviation of
three independent experiments; *P<0.05, vs. control;
#P<0.05, vs. Dox+MSCs; ○P<0.05, vs.
Dox+MSCs+siRNA-NT. Dox, doxorubicin; MSCs, mesenchymal stem cells;
siRNA, small interfering RNA; NT, scramble control; MTT,
3-(4,5-dimethyl thiazolyl-2)-2,5-diphenyl tetrazolium bromide;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction. |
Jagged-1/Notch-1 signaling pathway
reciprocally regulates TGF-β1 to reverse Dox-induced
senescence
TGF-β1, one of several Jagged-1/Notch-1 signaling
pathway targets, is important in the cellular senescence process.
In the presence of Dox, the basal mRNA expression of Tgfb1
and the release of TGF-β1 protein from H9c2 cells were increased,
whereas coculture with MSCs reduced the expression and release of
TGF-β1. However, the addition of anti-VEGFR2 antibody, or the
silencing of Notch1 or Jag1 with siRNA reduced the
effects of MSCs on the expression and release of TGF-β1 (Fig. 5A and B).
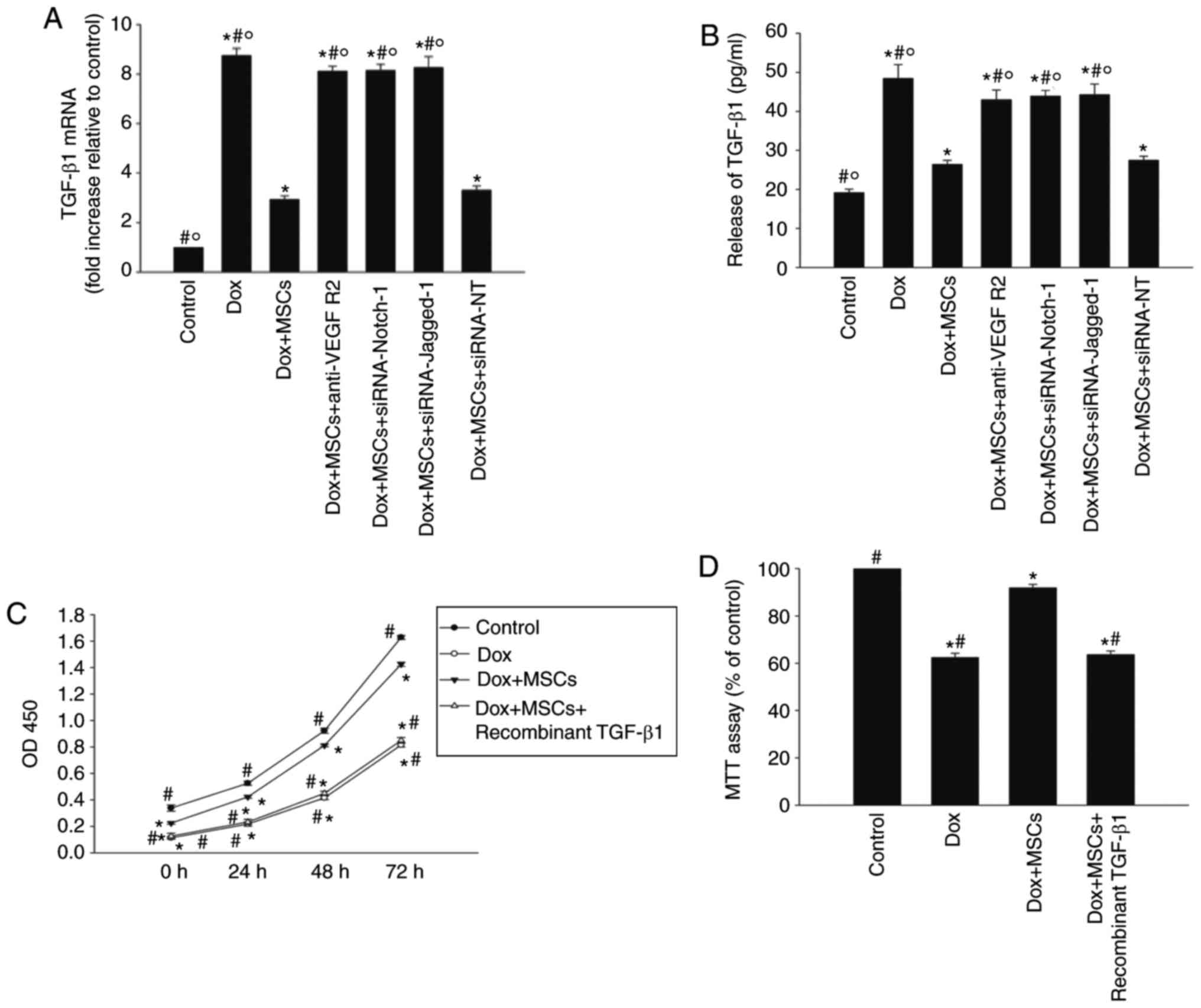 | Figure 5Jagged-1/Notch-1 signaling pathway
reciprocally regulates TGF-β1 to reverse Dox-induced senescence.
H9c2 cells were transfected with siRNA directed against Notch-1 or
Jagged-1, or with siRNA-NT as the control, and then cocultured with
MSCs in the presence of Dox. In parallel experiments, H9c2 cells
were incubated with Dox, cocultured with MSCs in the presence of
Dox, or cocultured with MSCs in the presence of Dox and anti-VEGFR2
antibody. H9c2 cells under normal culture conditions were used as
the control. (A) RT-qPCR analysis of mRNA levels of TGF-β1. (B)
Concentrations of TGF-β1 in the culture medium were analyzed with
an enzyme-linked immunosorbent assay. Each column represents the
mean ± standard deviation of three independent experiments;
*P<0.05, vs. control; #P<0.05, vs.
Dox+MSCs; ○P<0.05, vs. Dox+MSCs+siRNA-NT. In H9c2
cells incubated with Dox, cocultured with MSCs in the presence of
Dox, or cocultured with MSCs in the presence of Dox and recombinant
TGF-β1, (C) cell proliferation was determined with the Cell
Counting Kit-8 assay, and (D) viability was analyzed with an MTT
assay. (E) p53, (F) p16 and (G) telomere length were analyzed with
RT-qPCR analysis. (H) Relative telomerase activity was measured.
Data are presented as the mean ± standard deviation of three
independent experiments; *P<0.05, vs. Dox;
#P<0.05, vs. Dox+MSCs+recombinant TGF-β1. Dox,
doxorubicin; MSCs, mesenchymal stem cells; TGF-β1, transforming
growth factor-β1; siRNA, small interfering RNA; NT, scramble
control; MTT, 3-(4,5-dimethyl thiazolyl-2)-2,5-diphenyl tetrazolium
bromide; RT-qPCR, reverse transcription-quantitative polymerase
chain reaction. |
Subsequently, recombinant TGF-β1 was used to examine
the effect of TGF-β1. Recombinant TGF-β1 reduced H9c2 cell
proliferation from day 1 of treatment, and this effect was
maintained for at least 3 days. It also reduced cell viability
(Fig. 5C and D), compared with
that in the cells cocultured with MSCs only. Recombinant TGF-β1
also rescued the previously observed reduction in p53 and p16
caused by coculture with MSCs (Fig.
5E and F), and eliminated the antisenescence effects of MSCs on
telomere length and activity (Fig. 5G
and H).
Discussion
Dox is one of the most widely used chemotherapeutic
agents and is effective against a wide range of tumors (25). Despite its beneficial effects
against cancer, the clinical use of Dox has the serious
disadvantage of cardiotoxicity, which seriously limits its
oncological therapeutic applicability (26). The mechanism of Dox-induced
cardiotoxicity has been investigated extensively but remains
controversial, although cellular senescence is considered to be a
possible important mechanism (27). Modulating cellular senescence may
allow the development of cardioprotective strategies, avoid the
interruption or discontinuation of necessary cancer treatments, and
reduce early and late cardiovascular and oncological morbidity and
mortality rates (28,29). The data in the present study
suggested that Dox-induced cardiotoxicity developed from the
specific accumulation of cellular senescence, accompanied by the
elevated expression of senescence-related genes, reduced telomere
length and reduced telomerase activity.
Previous studies have reported that the
administration of autologous bone-marrow-derived MSCs improves the
left ventricular ejection fraction and other cardiac functional
parameters in models of Dox-induced dilated cardiomyopathy
(30). However, the mechanism of
this therapeutic approach remains to be fully elucidated. MSCs have
previously been shown to significantly reduce ischemic cardiac
damage through a paracrine pathway (31) and, in a previous study, the
culture medium of MSCs antagonized the senescence and apoptosis of
cardiomyocytes and cardiac progenitor cells, two major processes in
Dox-induced cardiotoxicity (32).
The results of the present study suggested that MSCs exert an
antisenescence effect by triggering the secretion of VEGF, and that
this effect is reduced by an anti-VEGFR2 antibody. Asa previous
study found that H9c2 cells also express VEGF (33), the present study used RT-qPCR
analysis to examine the mRNA level of VEGF in the H9c2 cells,
however the results of the RT-qPCR analysis showed no difference
between the H9c2 cells cocultured with MSCs and H9c2 cells alone.
The increase of free VEGF in the H9c2 MSC coculture may result from
the MSCs only.
Unlike several other agonist/receptor systems, Notch
signaling relies on non-diffusible ligands (δ, δ-like, and Jagged),
which are integral membrane proteins that engage with and activate
the surface receptors on immediately adjacent cells (9). Notch is involved in embryonic
development, cell fate determination and cancer. It has also been
linked to senescence; in oxygen-tension-regulated self-renewal, the
Notch signaling pathway was activated in MSCs (34). The Notch signaling pathway is
closely associated with Dox-induced cardiac injury, and a previous
study detected an initial reduction in the expression of Notch
pathway genes on day 3 following Dox treatment, corresponding to
cardiomyocyte death by apoptosis and necrosis (35). The data from the present study
showed that the Jagged-1/Notch-1 signaling pathway was
significantly inhibited when the H9c2 cells were treated with Dox,
whereas coculture with MSCs activated the Jagged-1/Notch-1
signaling pathway, exerting a restorative effect, which was
eliminated by silencing of the Notch1 or Jag1
gene.
Numerous compounds with prosenescence effects have
been assessed in models of Dox-induced cardiotoxicity. TGF-β is
associated with Dox-induced cardiotoxicity (36). In the cardiovascular system, TGF-β
is involved in fibrotic cardiac remodeling, and a previous study
suggested that inhibition of the TGF-β pathway alleviates left
ventricular remodeling and systolic and diastolic dysfunction, thus
alleviating the detrimental effects of Dox on endothelial cells
(37). It has also been
demonstrated that the inhibition of TGF-β attenuates diastolic
dysfunction by reducing cardiac fibrosis in a model of
anthracycline-induced cardiomyopathy (38). A previous study indicated that
TGF-β can induce cellular senescence through repressing the
telomerase reverse transcriptase gene in tumor cells (39), coincident with the results in the
present study, in which Dox induced the elevation of TGF-β, leading
to shortening telomere length and impairing the telomerase
activity. MSC transplantation has been shown to significantly
inhibit cardiac fibrosis following myocardial infarction and
mediate a reduction in the expression of TGF-β/SMAD2 (40). In the present study, increased
expression of TGF-β was detected in the Dox-treated H9c2 cells,
which was reduced by coculture of the cells with MSCs. It was found
that recombinant TGF-β1 decreased the antisenescence effects of the
MSCs.
In conclusion, the present study demonstrated that
the attenuation of cardiomyocyte senescence during coculture with
MSCs may have important therapeutic implications for Dox-induced
cardiomyopathy. The results of the present study suggested that
MSCs protected H9c2 cells from Dox-induced senescence by
stimulating the secretion of VEGF, leading to activation of the
Notch-1/Jagged-1 signaling pathway, which then inhibited the
secretion of TGF-β1. The coculture of cardiomyocytes with MSCs may
provide a unique therapeutic opportunity targeting cell senescence,
in a useful strategy for the treatment of Dox-induced
cardiomyopathy.
Acknowledgments
Not applicable.
References
|
1
|
Octavia Y, Tocchetti CG, Gabrielson KL,
Janssens S, Crijns HJ and Moens AL: Doxorubicin-induced
cardiomyopathy: From molecular mechanisms to therapeutic
strategies. J Mol Cell Cardiol. 52:1213–1225. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Carvalho C, Santos RX, Cardoso S, Correia
S, Oliveira PJ, Santos MS and Moreira PI: Doxorubicin: The good,
the bad and the ugly effect. Curr Med Chem. 16:3267–3285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Stěrba M, Popelová O, Vávrová A, Jirkovský
E, Kovaříková P, Geršl V and Simůnek T: Oxidative stress, redox
signaling, and metal chelation in anthracycline cardiotoxicity and
pharmacological cardioprotection. Antioxid Redox Signal.
18:899–929. 2013. View Article : Google Scholar
|
|
4
|
Singh P, Sharma R, McElhanon K, Allen CD,
Megyesi JK, Beneš H and Singh SP: Sulforaphane protects the heart
from doxorubicin-induced toxicity. Free Radic Biol Med. 86:90–101.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Guarita-Souza LC, Teixeira de Carvalho KA,
Francisco JC, Simeoni R and Faria-Neto JR: Cellular transplantation
for the treatment of non-ischaemic dilated cardiomyopathies. Eur
Heart J Suppl. 10:K7–K10. 2008. View Article : Google Scholar
|
|
6
|
Mohammadi Gorji S, Karimpor Malekshah AA,
Hashemi-Soteh MB, Rafiei A, Parivar K and Aghdami N: Effect of
mesenchymal stem cells on doxorubicin-induced fibrosis. Cell J.
14:142–151. 2012.
|
|
7
|
Ezquer F, Gutiérrez J, Ezquer M, Caglevic
C, Salgado HC and Calligaris SD: Mesenchymal stem cell therapy for
doxorubicin cardiomyopathy: Hopes and fears. Stem Cell Res Ther.
6:1162015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Garbade J, Dhein S, Lipinski C, Aupperle
H, Arsalan M, Borger MA, Barten MJ, Lehmann S, Walther T and Mohr
FW: Bone marrow-derived stem cells attenuate impaired contractility
and enhance capillary density in a rabbit model of
doxorubicin-induced failing hearts. J Card Surg. 24:591–599. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tian Y, Xu Y, Xue T, Chen L, Shi B, Shu B,
Xie C, Max Morandi M, Jaeblon T, Marymont JV and Dong Y: Notch
activation enhances mesenchymal stem cell sheet osteogenic
potential by inhibition of cellular senescence. Cell Death Dis.
8:e25952017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Iglesias-Bartolome R and Gutkind JS:
Signaling circuitries controlling stem cell fate: To be or not to
be. Curr Opin Cell Biol. 23:716–723. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Towbin JA, Lorts A and Jefferies JL: Left
ventricular non-compaction cardiomyopathy. Lancet. 386:813–825.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Gude N, Joyo E, Toko H, Quijada P,
Villanueva M, Hariharan N, Sacchi V, Truffa S, Joyo A, Voelkers M,
et al: Notch activation enhances lineage commitment and protective
signaling in cardiac progenitor cells. Basic Res Cardiol.
110:292015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ieronimakis N, Hays AL, Janebodin K,
Mahoney WM Jr, Duffield JS, Majesky MW and Reyes M: Coronary
adventitial cells are linked to perivascular cardiac fibrosis via
TGFβ1 signaling in the mdx mouse model of duchenne muscular
dystrophy. J Mol Cell Cardiol. 63:122–134. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Castañares C, Redondo-Horcajo M,
Magán-Marchal N, Ten Dijke P, Lamas S and Rodríguez-Pascual F:
Signaling by ALK5 mediates TGF-beta-induced ET-1 expression in
endothelial cells: A role for migration and proliferation. J Cell
Sci. 120:1256–1266. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Konorev EA, Vanamala S and Kalyanaraman B:
Differences in doxorubicin-induced apoptotic signaling in adult and
immature cardiomyocytes. Free Radic Biol Med. 45:1723–1728. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hou J, Wang L, Hou J, Guo T, Xing Y, Zheng
S, Zhou C, Huang H, Long H, Zhong T, et al: Peroxisome
proliferator-activated receptor gamma promotes mesenchymal stem
cells to express connexin43 via the inhibition of tgf-β1/smads
signaling in a rat model of myocardial infarction. Stem Cell Rev.
11:885–899. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Witek P, Korga A, Burdan F, Ostrowska M,
Nosowska B, Iwan M and Dudka J: The effect of a number of H9C2 rat
cardiomyocytes passage on repeatability of cytotoxicity study
results. Cytotechnology. 68:2407–2415. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Xia W, Zhang F, Xie C, Jiang M and Hou M:
Macrophage migration inhibitory factor confers resistance to
senescence through cd74-dependent ampk-foxo3a signaling in
mesenchymal stem cells. Stem Cell Res Ther. 6:822015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hou M, Liu J, Liu F, Liu K and Yu B: C1q
tumor necrosis factor-related protein-3 protects mesenchymal stem
cells against hypoxia- and serum deprivation-induced apoptosis
through the phosphoinositide 3-kinase/Akt pathway. Int J Mol Med.
33:97–104. 2014. View Article : Google Scholar
|
|
20
|
Piegari E, Russo R, Cappetta D, Esposito
G, Urbanek K, Dell’Aversana C, Altucci L, Berrino L, Rossi F and De
Angelis A: MicroRNA-34a regulates doxorubicin-induced
cardiotoxicity in rat. Oncotarget. 7:62312–62326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2-(Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Xia W and Hou M: Macrophage migration
inhibitory factor rescues mesenchymal stem cells from
doxorubicin-induced senescence though the PI3K-Akt signaling
pathway. Int J Mol Med. 41:1127–1137. 2018.
|
|
23
|
Kourembanas S: Exosomes: Vehicles of
intercellular signaling, biomarkers, and vectors of cell therapy.
Annu Rev Physiol. 77:13–27. 2015. View Article : Google Scholar
|
|
24
|
Xu LL, Fu HX, Zhang JM, Feng FE, Wang QM,
Zhu XL, Xue J, Wang CC, Chen Q, Liu X, et al: Impaired function of
bone marrow mesenchymal stem cells from immune thrombocytopenia
patients in inducing regulatory dendritic cell differentiation
through the Notch-1/Jagged-1 signaling pathway. Stem Cell Dev.
26:1648–1661. 2017. View Article : Google Scholar
|
|
25
|
Cardinale D, Colombo A, Bacchiani G,
Tedeschi I, Meroni CA, Veglia F, Civelli M, Lamantia G, Colombo N,
Curigliano G, et al: Early detection of anthracycline
cardiotoxicity and improvement with heart failure therapy.
Circulation. 131:1981–1988. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carver JR, Shapiro CL, Ng A, Jacobs L,
Schwartz C, Virgo KS, Hagerty KL, Somerfield MR and Vaughn DJ; ASCO
Cancer Survivorship Expert Panel: American society of clinical
oncology clinical evidence review on the ongoing care of adult
cancer survivors: Cardiac and pulmonary late effects. J Clin Oncol.
25:3991–4008. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Du WW, Yang W, Chen Y, Wu ZK, Foster FS,
Yang Z, Li X and Yang BB: Foxo3 circular RNA promotes cardiac
senescence by modulating multiple factors associated with stress
and senescence responses. Eur Heart J. 38:1402–1412. 2017.
|
|
28
|
Jackson JG, Pant V, Li Q, Chang LL,
Quintás-Cardama A, Garza D, Tavana O, Yang P, Manshouri T, Li Y, et
al: p53-mediated senescence impairs the apoptotic response to
chemotherapy and clinical outcome in breast cancer. Cancer Cell.
21:793–806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Baar MP, Brandt RMC, Putavet DA, Klein
JDD, Derks KWJ, Bourgeois BRM, Stryeck S, Rijksen Y, van
Willigenburg H, Feijtel DA, et al: Targeted apoptosis of senescent
cells restores tissue homeostasis in response to chemotoxicity and
aging. Cell. 169:132–147. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mao C, Hou X, Wang B, Chi J, Jiang Y,
Zhang C and Li Z: Intramuscular injection of human umbilical
cord-derived mesenchymal stem cells improves cardiac function in
dilated cardiomyopathy rats. Stem Cell Res Ther. 8:182017.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Bollini S, Cheung KK, Riegler J, Dong X,
Smart N, Ghionzoli M, Loukogeorgakis SP, Maghsoudlou P, Dubé KN,
Riley PR, et al: amniotic fluid stem cells are cardioprotective
following acute myocardial infarction. Stem Cells Dev.
20:1985–1994. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lazzarini E, Balbi C, Altieri P, Pfeffer
U, Gambini E, Canepa M, Varesio L, Bosco MC, Coviello D, Pompilio
G, et al: The human amniotic fluid stem cell secretome effectively
counteracts doxorubicin-induced cardiotoxicity. Sci Rep.
6:299942016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou N, Fu Y, Wang Y, Chen P, Meng H, Guo
S, Zhang M, Yang Z and Ge Y: p27kip1 haplo-insufficiency improves
cardiac function in early-stages of myocardial infarction by
protecting myocardium and increasing angiogenesis by promoting IKK
activation. Sci Rep. 4:59782014. View Article : Google Scholar :
|
|
34
|
Rios C, D’Ippolito G, Curtis KM, Delcroix
GJ, Gomez LA, El Hokayem J, Rieger M, Parrondo R, de Las Pozas A,
Perez-Stable C, et al: Low oxygen modulates multiple signaling
pathways increasing self-renewal while decreasing differentiation,
senescence and apoptosis in stromal miami cells. Stem Cells Dev.
25:848–460. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Packard RRS, Baek KI, Beebe T, Jen N, Ding
Y, Shi F, Fei P, Kang BJ, Chen PH, Gau J, et al: Automated
segmentation of light-sheet fluorescent imaging to characterize
experimental doxorubicin-induced cardiac injury and repair. Sci
Rep. 7:86032017. View Article : Google Scholar
|
|
36
|
Chua S, Lee FY, Chiang HJ, Chen KH, Lu HI,
Chen YT, Yang CC, Lin KC, Chen YL, Kao GS, et al: The
cardioprotective effect of melatonin and exendin-4 treatment in a
rat model of cardiorenal syndrome. J Pineal Res. 61:438–456. 2016.
View Article : Google Scholar
|
|
37
|
Sun Z, Schriewer J, Tang M, Marlin J,
Taylor F, Shohet RV and Konorev EA: The TGF-β pathway mediates
doxorubicin effects on cardiac endothelial cells. J Mol Cell
Cardiol. 90:129–138. 2016. View Article : Google Scholar
|
|
38
|
Cappetta D, Esposito G, Piegari E, Russo
R, Ciuffreda LP, Rivellino A, Berrino L, Rossi F, De Angelis A and
Urbanek K: SIRT1 activation attenuates diastolic dysfunction by
reducing cardiac fibrosis in a model of anthracycline
cardiomyopathy. Int J Cardiol. 205:99–110. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Li H, Xu D, Toh BH and Liu JP: TGF-beta
and cancer: Is Smad3 a repressor of hTERT gene? Cell Res.
16:169–173. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chen P, Wu R, Zhu W, Jiang Z, Xu Y, Chen
H, Zhang Z, Chen H, Zhang L, Yu H, et al: Hypoxia preconditioned
mesenchymal stem cells prevent cardiac fibroblast activation and
collagen production via leptin. PloS one. 9:e1035872014. View Article : Google Scholar : PubMed/NCBI
|