Introduction
Acute lung injury (ALI) and acute respiratory
distress syndrome (ARDS) are life-threatening diseases induced by
severe sepsis, severe bacterial pneumonia, trauma and burns
(1,2). Although advances have been made in
understanding the pathophysiology of ALI and ARDS, there is no
effective therapeutic strategy for treating either (3). Various molecular mechanisms are
associated with the pathogenesis and progression of ALI, including
oxidative stress and inflammatory responses (4). ALI is characterized by extreme
inflammation, excessive neutrophil infiltration into the lung
tissues, release of pro-inflammatory cytokines, and lung
endothelial and epithelial injuries, resulting in edema and gas
exchange deterioration (5,6).
Reactive oxygen species (ROS)-induced oxidative
stress contributes to ALI development (7) via stimulating TXNIP/NLRP3, nuclear
factor (NF)-κB and mitogen-activated protein kinase (MAPK)
signaling pathways (8).
TXNIP/NLRP3 activation is linked to ALI and TXNIP blocks
antioxidant function; its overexpression has been observed in
diverse diseases (9,10). Accumulating evidence indicates
that the NLRP3 inflammasome initiates the innate immunity and
promotes inflammatory responses (11). Importantly, TXNIP links oxidative
stress to NLRP3 activation in an ROS-sensitive manner (12). The process accelerates the
secretion of pro-inflammatory cytokines, including tumor necrosis
factor (TNF)-α, interleukin (IL)-1β and IL-6, which are associated
with NF-κB activity, eventually contributing to ALI (13). MAPKs are implicated in cellular
inflammatory processes in response to oxidative stress (14).
LR (LR; acacetin-7-O-β-D-rutinoside) is a natural
flavonoid glycoside abundant in various vegetables and fruits
(15). Plants containing LR have
been reportedly used to treat inflammatory diseases, cancer and
hypertension (16,17). LR alleviates LPS-induced cytokine
production in a murine macrophage cell line (18). Recently, LR was reported to have
antioxidant activity associated with enhancement of nuclear
factor-erythroid 2-related factor 2 (Nrf2). LR activated Nrf2
activation to reduce ROS generation, inhibiting
ischemia-reperfusion injury (19). Thus, LR may have potential for use
in the treatment of ALI.
The current study reports that LPS induced ALI, as
evidenced by platelet activation, inflammation and oxidative stress
via activating NF-κB and TXNIP/NLRP3 pathways, was suppressed by
linarin partly via suppressing NF-κB and TXNIP/NLRP3, and by
potentiating Nrf2. Therefore, LR pretreatment may prevent
ALI-induced by LPS in vitr and in vivo.
Materials and methods
Animals and treatment
A total of 100 male C57BL/6 mice (6–8 weeks old,
weighing, 18–20 g) were purchased from The Animal Center of Nanjing
Medical University (Nanjing, China), and provided with food and
water ad libitu and kept in climate-controlled quarters with
a 12 h light/dark cycle with food and water in cages under the
germ-free conditions at a temperature of 25°C. The mice were housed
for a minimum of one week for environmental adaptation prior to
experimentation. All procedures were in accordance with the
Regulations of Experimental Animal Administration issued by the
Ministry of Science and Technology of the People's Republic of
China, and before the animal experiments were performed, the
procedures were approved by the Research Ethical Committee of
Huai'an First People's Hospital, Nanjing Medical University
(Nanjing, China).
C57BL/6 mice were randomly divided into 5 groups as
follows (n=20 in each group): i) Control (Con); ii) LPS treatment
(LPS); iii) Low dose of LR treatment (LPS treatment + oral gavage
of 12.5 mg/kg LR for 3 days, LD-LR); iv) Medium dose of LR
treatment (LPS treatment + oral gavage of 25 mg/kg LR for 3 days,
MD-LR); and v) High dose of LR treatment (LPS treatment + oral
gavage of 50 mg/kg LR for 3 days, HD-LR). The mice from the control
and LPS groups received an equal volume of normal saline (0.3 ml)
instead of LR. LR was purchased from Fanke Biotech Fanke Biotech
Co., Ltd. (http://fanketech.biomart.cn, FA-1701, HPLC ≥98%,
Shanghai, China). At 3 h following drug administration on the third
day, the mice were slightly anesthetized with inhalation of diethyl
ether (1 ml diethyl ether dipping cotton balls), and 10 µg LPS in
50 µl PBS was intranasally instilled to induce lung injury. The
mice in the control group were given 50 µl normal saline in the
absence of LPS. A total of 24 h later for LPS treatment, all mice
were sacrificed through an intraperitoneal injection of 50 mg/kg
pentobarbital (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and
bronchoalveolar lavage fluid (BALF) and lung tissue samples were
harvested for further researches. The ratios of lung wet/dry weight
were measured by dividing the wet weight by the dry weight. The
middle lobe of the right lung was excised and the wet weight was
recorded. Then, the lung was placed in an incubator for 24 h at
80°C to obtain the dry weight.
Cells and culture
Thehuman lung epithelial cell line, BEAS-2B, was
purchased from Shanghai Haoran Biological Technology Co., Ltd.
(Shanghai, China) and kept in DMEM/F12 containing 1%
penicillin/streptomycin and 10% FBS (Hyclone; GE Healthcare Life
Sciences, Chalfont, UK). Cells were then cultured in a humidified
atmosphere with 5% CO2 and 95% humidity at 37°C in an
incubator.
Biochemical assays
Superoxide dismutase (SOD, A001-3), catalase (CAT,
A007-1-1) and glutathione peroxidase (GPx, A005) activities, as
well as malondialdehyde (MDA, A003-1) levels in lung tissue sample
were determined by commercially available kits (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China) following the
manufacturer's instructions. H2O2 levels in
lung tissues were measured using a hydrogen peroxide assay kit,
(Beyotime Institute of Biotechnology, Haimen, China) following the
manufacturer's instructions. O2− in lung
tissues was measured through lucigenin chemiluminescence method.
Briefly, lung tissues of mice under different conditions were
weighed and homogenized in a homogenization buffer using HEPES and
EDTA. Following centrifugation (1,000 × g at 4°C for 10 min), an
aliquot of the supernatant was incubated with 5 µM lucigenin
in Krebs-HEPES buffer. Light emission was measured with a Tecan
Infinite 200 (Tecan Group AG, Männedorf, Switzerland). Specificity
for O2− was evaluated by adding SOD (350
U/ml) to the incubation medium. BCA Protein Quantitative Analysis
kit (Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used to
measure the protein concentration.
Assessment of arterial blood gas
A total of 40 male C57BL/6 mice (6–8-weeks old,
18–20 g weight) were pretreated with LR (0, 12.5, 25 and 50 mg/kg)
for 3 days by gavage orally. At 3 h following drug administration
on the third day, the mice were slightly anesthetized with
inhalation of diethyl ether (1 ml diethyl ether dipping cotton
balls), and 10 µg LPS in 50 µl PBS was instilled
intranasally to induce lung injury. The mice in the control group
were given 50 µl of normal saline without LPS. At 24 h
following LPS treatment, the mice were sacrificed by an
intraperitoneal injection of pentobarbital (50 mg/kg;
Sigma-Aldrich; Merck KGaA). The retro-orbital blood samples (1 ml)
from each mouse was collected and used to measure the parameters of
arterial blood gas, including blood hemoglobin concentration (Hgb),
pH, HCO3, partial pressure of oxygen (PO2),
partial pressure of carbon dioxide (PCO2), lactate (LAC)
and base excess (BE) using Hitachi 8500 automatic analyzer
(Hitachi, Ltd., Tokyo, Japan).
Immunofluorescence analysis
For measurement of CD41 using immunofluorescence
staining was performed. Briefly, the frozen sections of lung tissue
samples were washed with PBS twice and then were blocked with 1%
bovine serum albumin at room temperature for 30 min. Then the cells
and heart tissue samples were incubated overnight with rabbit
anti-CD41 primary antibody (ab63983, 1:50; Abcam, Cambridge, UK).
After being washed with PBS for three times, the samples were
performed with goat anti-rabbit secondary antibody conjugated to
Alexa Fluor 647 (A0468, 1:400) and goat anti-rabbit secondary
antibody conjugated to Alexa Fluor 488 (A0423, 1:400) (both from
Beyotime Institute of Biotechnology) for 1 h under dark conditions.
Then, the cells were counterstained with DAPI (Beyotime Institute
of Biotechnology) for 15 min, and the tissue sections were analyzed
using an immunofluorescence microscope. Three slides per
experimental condition are repeated three times.
Bronchoalveolar lavage fluid (BALF)
isolation and analysis
BALF was collected by flushing the left lung (0.4
ml, three times). Total BALF cells were measured using a
hemocytometer. A part of the BALF was centrifuged for 5 min at 92 ×
g by cytospin on a microscopic slide at 4°C. BALF cells were
stained using Giemsa at room temperature for 10 min. The rest of
BALF was filtrated through a 0.22 µm pore-size filter and
then stored at −80°C for protein and cytokine detection, while the
cell pellet was resuspended in PBS for counting the neutrophils.
The concentration of BALF protein was measured using BCA Protein
Assay kit.
Measurement of cytokine levels in
BALF
IL-1β (cat. no. MLB00C), TNF-α (cat. no. MTA00B),
IL-10 (cat. no. DY417), IL-6 (cat. no. M6000B), cyclooxygenase
(COX)2 (cat. no. DYC4198-2), monocyte chemotactic protein 5 (MCP-5;
cat. no. MCC120), macrophage inflammatory protein-1α (MIP-1α) (cat.
no. MMA00), thromboxane B2 (TXB2; cat. no. KGE011) and
myeloperoxidase (MPO; cat. no. DY3667) levels in BALF were measured
using ELISA kits (R&D System Inc., Minneapolis, MN, USA). The
procedures were performed following the manufacturer's
instructions.
Immunohistochemical analysis
Histopathological evaluation was performed on mice
not subjected to BAL. Lungs were inflated and fixed with 10%
buffered formalin for 48 h at room temperature and embedded in
paraffin (Beyotime Institute of Biotechnology). Tissue sections
were cut at 4 µm thickness and stained with hematoxylin and
eosin (H&E) following the regular staining method. The sections
were deparaffinized in xylene for 10 min. Then, the slides were
re-hydrated in absolute alcohol for 5 min each, followed by 95%
alcohol for 2 min and 70% alcohol for 2 min and then washing with
water. Subsequently, the sections were stained in Harris
hematoxylin solution for 8 min at room temperature, followed by
washing under a running water for 5 min. The slides were then
differentiated in 1% acid alcohol for 30 sec, followed by washing
using running tap water for 1 min. Then, the sections were
subjected to bluing in 0.2% ammonia water or saturated lithium
carbonate solution for 30 sec at room temperature, followed by
washing under running water for 5 min and rinsing in 95% alcohol.
The sections were counterstained in eosin-phloxine solution for 30
sec at room temperature; the slides were dehydrated via 95% alcohol
for 5 min and cleared in xylene for 5 min. Finally, the sections
were mounted with xylene-based mounting medium. The severity of
injury was judged based on the following criteria: 0, no injury; 1,
injury to 25% of the field; 2, injury to 50% of the field; 3,
injury to 75% of the field; and 4, diffuse injury. The ultimate
score was obtained by adding the aforementioned scores.
Immunohistochemistry was performed using the paraffin-embedded
tissue sections at 4 µm thickness mounted on glass slides.
The slides were then deparaffinized and rehydrated. Then, the lung
sections were incubated with primary antibodies against mouse TXNIP
(ab210826, dilution: 1:200) and xanthine oxidase (XO, ab176165,
dilution: 1:200) (both from Abcam), and then with biotin secondary
antibodies (B3640; Sigma-Aldrich; Merck KGaA) at room temperature
for 30 min. The number of XO-, and TXNIP-positive cells in lung
sections was counted using Pax-it software (version 6; Paxcam,
Villa Park, IL, USA).
Flow cytometry assays
Whole blood samples from mice were collected and
maintained in acid citrate dextrose (Sigma-Aldrich; Merck KGaA).
Anti-CD11b eFluor450 (1:100), anti-CD41-allophycocyanin (1:100)
(both from eBioscience; Thermo Fisher Scientific, Inc.),
anti-Ly6C-PE (1:100; BD Biosciences, Franklin Lakes, NJ, USA) and
anti-Ly6G-phycoerythrin (1:100; BD Biosciences) were used to detect
leukocyte and platelets antigens. Samples were examined with an
LSRII flow cytometer (BD Biosciences) and analyzed using FCS
express software (version 3.0, De Novo Software, Glendale, CA,
USA). Neutrophils and monocytes were gated through their forward-
and side-scatter characteristics and through their
Ly-6G+/CD11b+ (neutrophil) and
Ly-6G−/CD11b+/Ly-6C+ (monocyte)
expression pattern. Platelet-neutrophil and platelet-monocyte
aggregates were calculated using CD41 antibody staining.
ROS generation
Intracellular ROS generation regulated by LPS, ATP
and N-Acetylcysteine (NAC) (both from Sigma-Aldrich; Merck KGaA)
was assessed by calculating the fluorescence intensity of
2′,7′-dichlorofluorescein diacetate oxidized product (Molecular
Probes; Thermo Fisher Scientific, Inc.). The relative fluorescence
intensity of oxidized product of 2′,7′-dichlorofluorescein
diacetate, was tested at an excitation wavelength of 485 nm and an
emission wavelength of 530 nm using a Quantus™ Fluorometer (Promega
Corporation, Madison, WI, USA). All cells after various treatments
were harvested for ROS assessment through ROS Fluorescent Probe-DCF
(Vigorous Biotechnology Beijing Co., Ltd., Beijing,
China;http://www.vigorousbiol.com/)
according to the manufacturer's instructions.
Reverse transcription-quantitative
(RT-qPCR) analysis
Lung samples were immediately acquired from mice
following sacrifice and were used in the extraction of total RNA
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
Subsequently, the RNA was purified using the RNeasy mini kit
following the manufacturer's instructions (Qiagen, Inc., Valencia,
CA, USA). cDNA was synthesized using SuperScript II RNase H-Reverse
Transcriptase (Invitrogen; Thermo Fisher Scientific, Inc.).
SYBR-Green Master Mix (Applied Biosystems; Thermo Fisher
Scientific, Inc.; 10 µl Master Mix, 0.5 µl forward
primer, 0.5 µl reverse primer, 1 µl cDNA and 8
µl ddH2O) was used, and qPCR was performed in
accordance with the manufacturer's protocol. The 7000 Sequence
Detection system (Applied Biosystems; Thermo Fisher Scientific,
Inc.) was applied to perform qPCR analysis. The specific sequences
of primers used in the study for gene amplification were shown in
Table I. The cycling conditions
(33 cycles) were exhibited as followings: 95°C for 5 min, 95°C for
10 sec, 60°C for 30 sec, 95°C for 15 sec, 60°C for 60 sec and 95°C
for 15 sec. The expression levels of targeting mRNAs were
normalized to that of housekeeping gene of GAPDH using the
2−ΔΔCq method (20).
 | Table IPrimer sequences of RT-PCR test. |
Table I
Primer sequences of RT-PCR test.
| Gene | Forward primers
(5′-3′) | Reverse primers
(5′-3′) |
|---|
| GAPDH C |
GGTCCATAGCAAAACGAGG |
TACACTACGCAGCCAACACACC |
| iNOS |
TTGGTGGAAGGCAGTTGAGG |
CAGCAGGTAGACAAACCATTCA |
| MCP-5 |
ATGCCGAGGAACAGATATCTA |
CGACCATATCGCACGAGTGCA |
| TNF-α |
AGCACACAAGTGGCACAACG |
CATGCGGTCGTAGTCCATAAT |
| MIP-1α C |
AGCAGGCAGCACGTGTCGC |
CTTCAACATATGTTAACAGTAC |
| IL-1β |
GAAGTATGCTTAGCCAGTAAG |
GAGCGTCGCAATTGTTGTGG |
| IL-10 C |
GATCTGCTATCCCACGGA |
CTGACCAGCCATAGCCAGA |
| SOD1 |
GGAGCACTGAACGCACAGA |
CTACGGAGATTCAAACTCGTG |
| COX2 |
CCGCATTCTGGTATAACGAACA |
ACCGAGAAGCCAGCACCAT |
| IL-6 |
ATTCGGCAACCGAGATATAGA |
TGACAACTAGAACGATCCAACC |
| SOD2 |
GCATAACTCGAGAACTTACGC |
ACTACATTATCGGGTCGGAGTA |
| CAT |
GAGGACTATATCTTCTCGAC |
GAATGGTCCCTCGCACTCG |
| GCLC |
GTGACCCTTAACTTGAGAG |
AGGACGCAGATCGGACCAACG |
| Ly6G |
GCATCTACGTACAGGACCG |
AGTCGGTGATGACAGTATGAG |
| CD41 |
ATATCGGCCTATACTCGAT |
CAGGATGCTAGTCGTAATGTA |
Western blot analysis
The total protein of lung tissue samples and cells
was extracted by addition of cold lysis buffer [10 mM Tris-HCl, 1
mM EDTA, and 250 mM sucrose, pH 7.4, containing 15 µg/ml
aprotinin, 5 µg/ml leupeptin, 0.1 mM phenylmeth-anesulfonyl
fluoride (PMSF), 1 mM NaF, and 1 mM Na3VO4
(Beyotime Institute of Biotechnology)] for 50 min at 4°C. Then, the
lysates were centrifuged at 12,000 × g for 20 min at 4°C. The
soluble protein concentrations in the lysates were assessed using a
BCA Protein Assay kit. Next, 40 µg total protein was
separated on a 12% SDS-PAGE gel and was transferred onto a
polyvinylidene difluoride membrane. Then, the membrane was blocked
in 5% dried milk for 2 h at room temperature and was incubated with
specific primary antibodies at 4°C overnight. The membrane was then
washed with TBS Tween-20 (TBST, containing 0.1% Tween-20) for three
times, followed by incubation with a horseradish
peroxidase-conjugated secondary antibody (A0208; Beyotime Institute
of Biotechnology) at room temperature for 2 h. Following another
round of washing with TBST, the membrane was then developed using
enhanced chemiluminescence (Thermo Fisher Scientific, Inc.). The
antibodies used in the study are shown below. The staining
intensity of the bands was quantitated by densitometry through
ImageJ software (version 1.49, National Institute of Health,
Bethesda, MD, USA). Triplicate experiments with at least triplicate
samples were performed. The primary antibodies: Rabbit anti-NLRP3
(1:1,000, cat. no. 15101), rabbit anti-ASC (1:1,000, cat. no.
67824) (both from Cell Signaling Technology, Inc., Danvers, MA,
USA), rabbit anti-caspase-1 (1:1,000, ab1872), rabbit anti-Nrf2
(1:1,000, ab31163) (both from Abcam), rabbit anti-HO-1 (1:1,000,
cat. no. 70081), rabbit anti-p-ERK1/2 (1:1,000, cat. no. 4370; Cell
Signaling Technology, Inc.), rabbit anti-ERK1/2 (1:1,000, ab17942;
Abcam), rabbit anti-p-JNK (1:1,000, cat. no. 9255), rabbit anti-JNK
(1:1,000, #9252) (both from Cell Signaling Technology, Inc.),
rabbit anti-XO (1:1,000, ab109235), rabbit anti-TXNIP (1:1,000,
ab188865) (both from Abcam), rabbit anti-p-p38 (1:1,000, cat. no.
9211), rabbit anti-p38 (1:1,000, cat. no. 8690), rabbit anti-p-IKKα
(1:1,000, cat. no. 2697), rabbit anti-IKKα (1:1,000, cat. no. 2682)
(all from Cell Signaling Technology, Inc.), anti-NF-κB (1:1,000,
ab19870), rabbit anti-p-NF-κB (1:1,000, ab86299), rabbit anti-IκBα
(1:1,000, ab32518), rabbit anti-p-IκBα (1:1,000, ab24783) (all from
Abcam), rabbit and anti-GAPDH (1:500, sc-293335, Santa Cruz
Biotechnology, Inc., Dallas, TX, USA). Immunoreactive bands were
visualized by ECL Immunoblot Detection system (Pierce; Thermo
Fisher Scientific, Inc.) and exposed to Kodak (Rochester, NY, USA)
X-ray film. Every protein expression levels will be defined as grey
value (ImageJ, Version 1.4.2b, National Institutes of Health) and
standardized to housekeeping gene of GAPDH and expressed as a fold
of control. All experiments were performed in triplicate and done
three times independently.
Statistical analysis
Data are expressed as the means ± standard error of
the mean. Treated cells, tissues and the corresponding controls
were compared using GraphPad Prism (GraphPad Software, Inc., La
Jolla, CA, USA) by a one-way analysis of variance with Dunn's least
significant difference tests. P<0.05 was considered to indicate
a statistically significant difference.
Results
LR attenuates LPS-induced ALI in
mice
Platelets were counted and pulmonary edema was
measured with a ratio of lung wet to dry weight. Fig. 1A presents an increased wet/dry
ratio in the LPS-treated group and LR reduced this in a
dose-dependent manner. In addition, as shown in Table II, PO2,
HCO3, Hgb, BE and PH increased in the LPS-treated group
and these were reduced in LR-pretreated groups. In contrast, LR
pretreatment upregulated LAC reduced by LPS but PCO2 was
not significantly different in mice treated as indicated. Arterial
blood gas analysis indicated that LR may be protective in
LPS-induced ALI in mice. Thromboxane A2 is a marker of platelet
activation, and is rapidly hydrolyzed to its inactive stable
metabolite, TXB2 (21). Plasma
TXB2 were upregulated in mice challenged with LPS and LR treatment
dramatically diminished TXB2 expression (Fig. 1B). H&E staining scores were
high following LPS treatment indicating lung damage, but this was
reversed by LR administration in a dose-dependent manner (Fig. 1C). CD41, a specific platelet
marker, can be measured to confirm sequestration of platelets in
pulmonary capillaries (22).
Platelet activation is involved in ALI (23,24) and immunofluorescence analysis
revealed that LR dose-dependently reduced CD41 expression in lung
tissues of mice treated with LPS (Fig. 1D). Moreover, RT-qPCR analysis
verified downregulated CD41 in LPS-treated lung tissue samples of
mice with ALI (Fig. 1E). Thus, LR
may have protective effects against LPS-induced ALI.
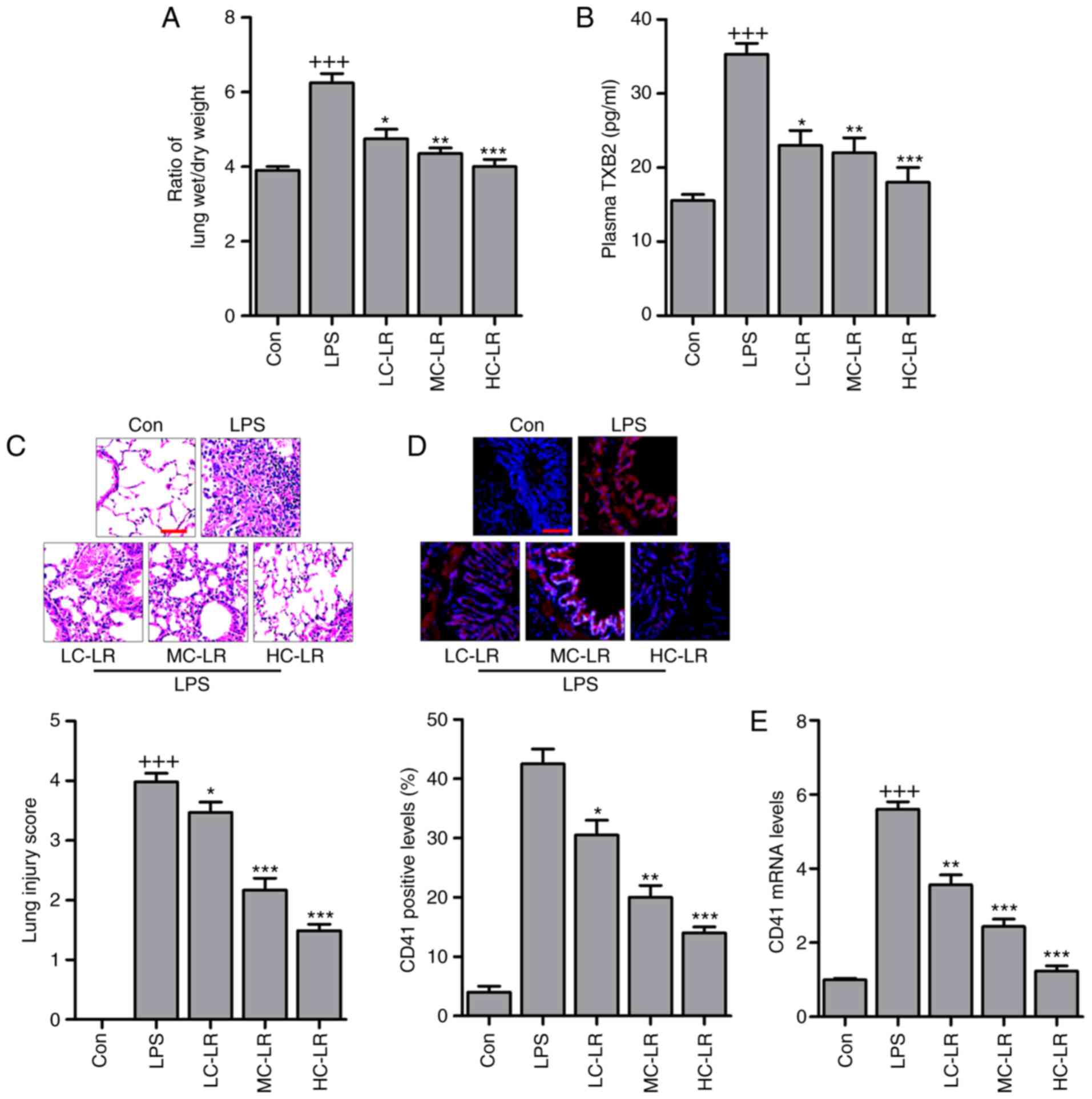 | Figure 1LR ameliorates the acute lung injury
of mice with LPS induction. (A) The ratio of lung wet to dry weight
was measured. (B) TXB2 levels in plasma were evaluated using ELISA
method. (C) Representative images of hematoxylin and eosin staining
of LPS-induced lung tissues treated with or without LR. Scale bar,
100 µm. (D) The immunofluorescence analysis of CD41 positive
levels in lung tissue samples treated under various conditions, and
the images were displayed. Scale bar, 100 µm. (E) Reverse
transcription-quantitative polymerase chain reaction analysis was
used to determine CD41 expression levels in LPS-induced lung
tissues of mice administered with different doses of LR. Data are
represented as mean ± standard error of the mean of three
independent experiments (n=6). +++P<0.001 vs. the Con
group in the absence of any treatments; *P<0.05,
**P<0.01 and ***P<0.001 vs. the LPS
groups. LPS, lipopolysaccharide; LR, linarin; Con, control; LC-LR,
low concentration linarin; MC-LR, medium concentration linarin;
HC-LR, high concentration linarin. |
| Table IIBlood gas analysis in mice. |
Table II
Blood gas analysis in mice.
| Indexes | Con | LPS | LC-LR | MC-LR | HC-LR |
|---|
| PO2
(mmHg) | 71.3±1.2 | 88.9±2.4b | 85.1±1.7 | 80.3±1.9d | 78.5±2.2d |
| PCO2
(mmHg) | 37.6±1.3 | 37.9±1.9 | 38.1±1.6 | 38.4±1.5 | 38.1±1.8 |
| HCO3
(mmol) | 16.8±0.4 | 22.9±1.0a | 20.1±0.8 | 18.6±0.9d | 17.8±0.6d |
| LAC (mM) | 3.8±0.2 | 2.4±0.4b | 2.9±0.2d | 3.1±0.3d | 3.4±0.4e |
| Hgb (g/dl) | 121.81±3.8 | 148.36±4.2c | 135.85±4.8 | 130.66±4.6d | 125.78±2.8e |
| BE (mEq/l) | −7.6±0.8 | −3.3±0.6b | −4.1±0.9 | −5.6±1.0d | −6.6±0.8d |
| PH | 7.08±0.02 | 7.36±0.03a | 7.28±0.01 | 7.13±0.03 | 7.10±0.04d |
LR inhibits LPS-induced inflammation
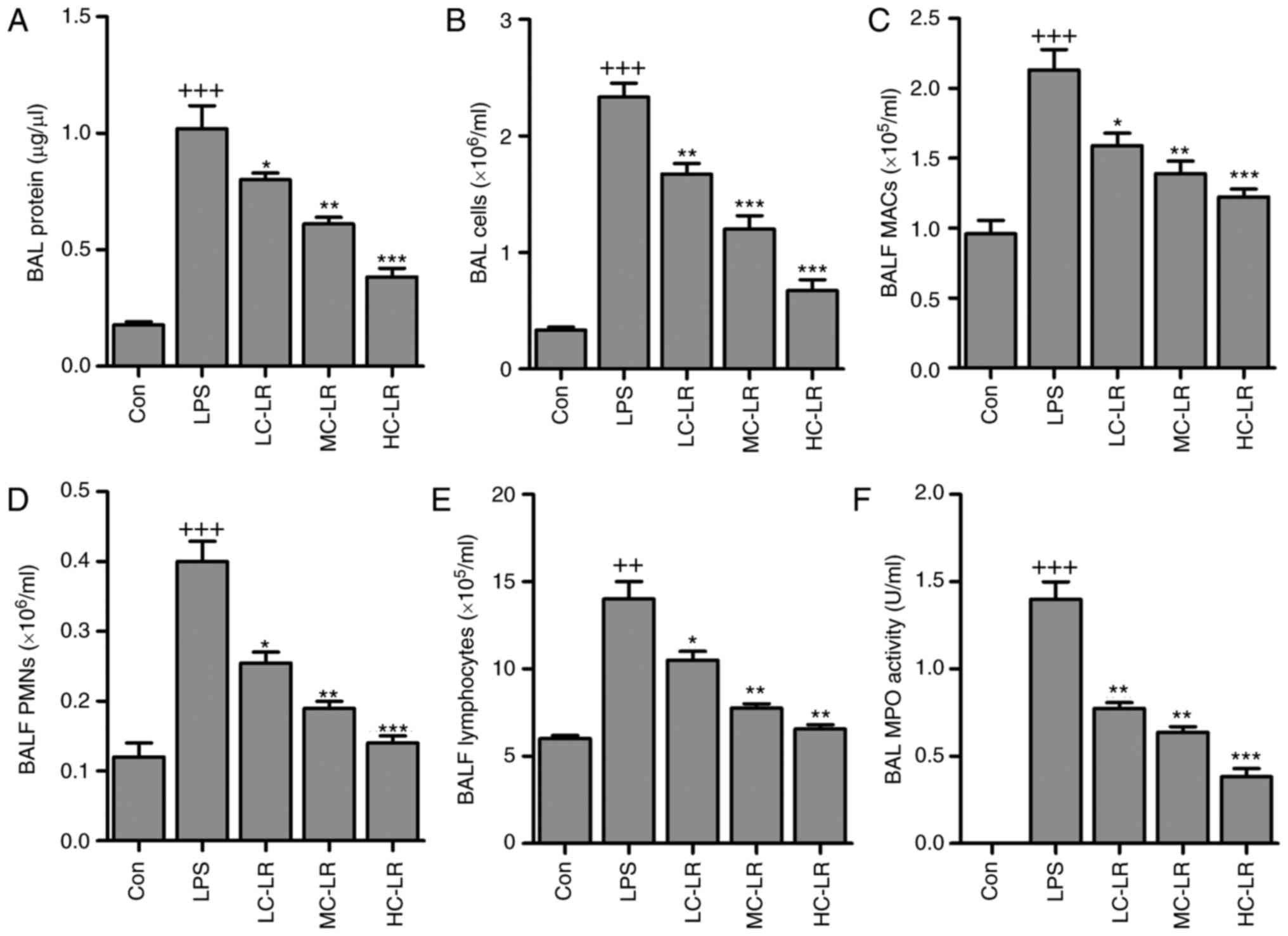
infiltration in BALF
BALF cytology data show that alveoli undergo
dramatic shifts after ALI (25).
BAL total protein calculated as an indicator of lung permeability
was elevated after LPS instillation (26). Mice given LR had reduced BAL
protein compared to the LPS group (Fig. 2A). BAL cells were increased,
indicating lung permeability, was markedly increased by LPS
treatment, which was reduced by LR administration (Fig. 2B). In addition, BALF macrophages
(MACs), BALF polymorphonuclear leukocytes (PMNs) and lymphocytes
were increased with LPS induction. However, LR treatment reduced
MACs, PMNs and lymphocytes in BALF (Fig. 2C–E). MPO activity, a marker of
neutrophil activation (Fig. 2F)
was greater in LPS-treated samples compared to controls. MPO
activity was reduced in LR-treated groups.
LR inactivates platelets in LPS-induced
ALI
Antibodies against leukocyte-specific surface
markers of Ly6G, Ly6C and CD11b were used to measure
neutrophil-platelet aggregates (NPA) and monocyte-platelet
aggregates (MPAs), defined as
Ly6G+/CD11b+/CD41+ and
Ly6C+/CD11b+/CD41+, respectively.
Whole blood extracted from mice had more NPA and MPA after LPS
treatment and this was time-dependent. LR (50 mg/kg) considerably
reduced the proportion of NPAs and MPAs after LPS-treatment for 48
h (Fig. 3A and B). NPAs in BAL
were elevated after LPS induction and then reduced by LR (Fig. 3C). Furthermore, TXB2 in BAL was
greater after LPS treatment and this was downregulated by LR
dose-dependently (Fig. 3D). High
expression of Ly6G induced by LPS was abolished following LR
administration (Fig. 3E). Thus,
LR can alleviate LPS-induced ALI via inactivating platelets. LPS
treatment increased serum and BALF TNF-α, IL-1β, IL-6, MCP-5,
MIP-1α COX2 and IL-10 (Fig. 4)
and LR administration reduced all of these except IL-10, which was
enhanced, indicating an anti-inflammatory effect of LR.
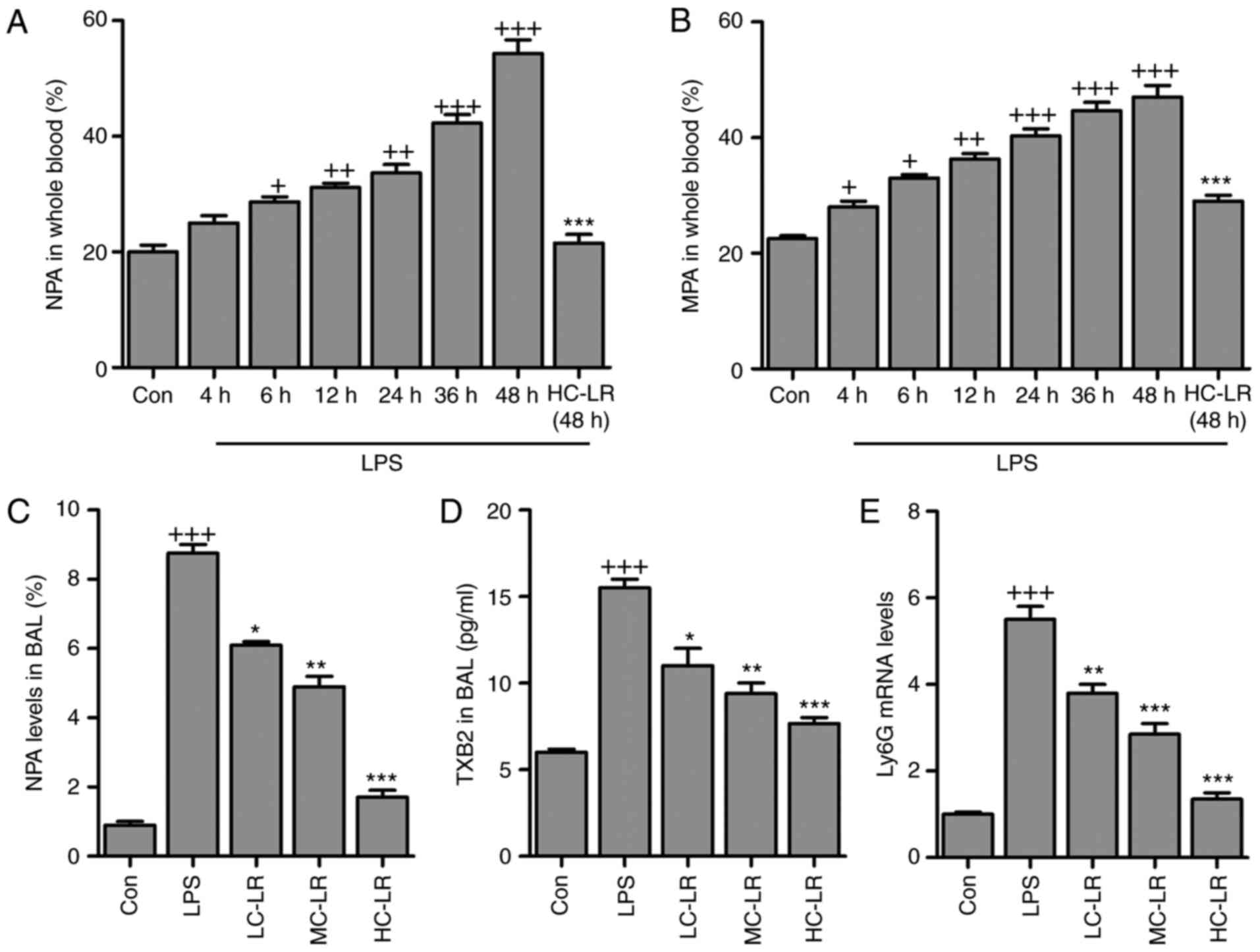 | Figure 3LR inactivates platelets in
LPS-challenged mice with acute lung injury. (A) The percentage of
neutrophil-platelet aggregates (NPA) and (B) percentage of
monocyte-platelet aggregates (MPAs) in whole blood of mice at
different time points after LPS instillation and the effects of LR
treatment at described times. (C) NPA quantification and (D) TXB2
levels in BAL were measured. (E) Reverse transcription-quantitative
polymerase chain reaction analysis was used to determine Ly6G
expression levels in LPS-induced lung tissues of mice administered
with different doses of LR. Data are represented as mean ± standard
error of the mean of three independent experiments (n=6).
+P<0.05, ++P<0.01 and
+++P<0.001 vs. the Con group in the absence of any
treatments. *P<0.05, **P<0.01 and
***P<0.001 vs. the LPS groups. LR, linarin; LPS,
lipopolysaccharide; Con, control; LC-LR, low concentration linarin;
MC-LR, medium concentration linarin; HC-LR, high concentration
linarin. NPA, neutrophil-platelet aggregates; MPAs,
monocyte-platelet aggregates. |
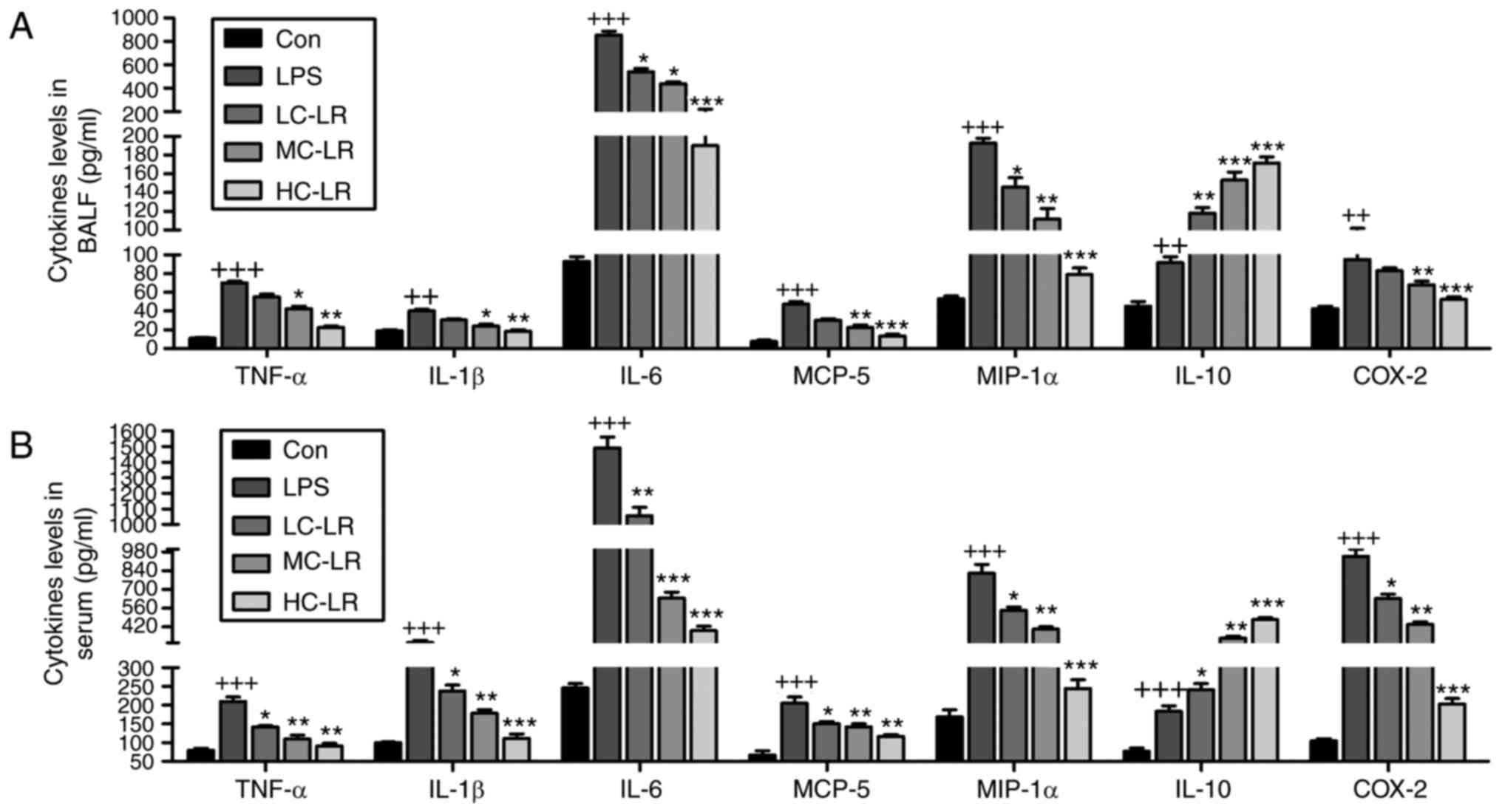 | Figure 4LR reduces LPS-induced
pro-inflammatory cytokines release in BALF and serum. TNF-α, IL-1β,
IL-6, MCP-5, MIP-1α, IL-10 and COX2 levels in (A) BALF and (B)
serum were measured using ELISA methods. Data are represented as
mean ± standard error of the mean of three independent experiments
(n=6). ++P<0.01 and +++P<0.001 vs. the
Con group in the absence of any treatments. *P<0.05,
**P<0.01 and ***P<0.001 vs. the LPS
groups. LR, linarin; LPS, lipopolysaccharide; BALF, bronchoalveolar
lavage fluid; TNF-α, tumor necrosis factor-α; IL, interleukin;
MCP-5, monocyte chemotactic protein 5; MIP-1α, macrophage
inflammatory protein-1α; COX2, cyclooxygenase 2; Con, control;
LC-LR, low concentration linarin; MC-LR, medium concentration
linarin; HC-LR, high concentration linarin. |
MDA levels induced LPS were reduced after LR
treatment (Fig. 5A). In addition,
oxidative stress induced by LPS, as evidenced by elevated levels of
H2O2 and O2. in lung tissue
samples was suppressed by LR treatment, and SOD, CAT and GPx
activity in lung tissue samples (Fig.
5B) were elevated by LPS, and LR treatment increased these
levels even further suggesting antioxidant activity. RT-qPCR assays
demonstrated that SOD2, SOD1, CAT and glutamate-cysteine ligase
(GCLC) expression were enhanced with LPS induction and increased
more with LR (Fig. 5C).
LPS-triggered increased Nrf2 and HO-1 was augmented by LR
dose-dependently (Fig. 5D). Thus,
LR exerts an antioxidant effect against LPS-induced ALI in
vivo.
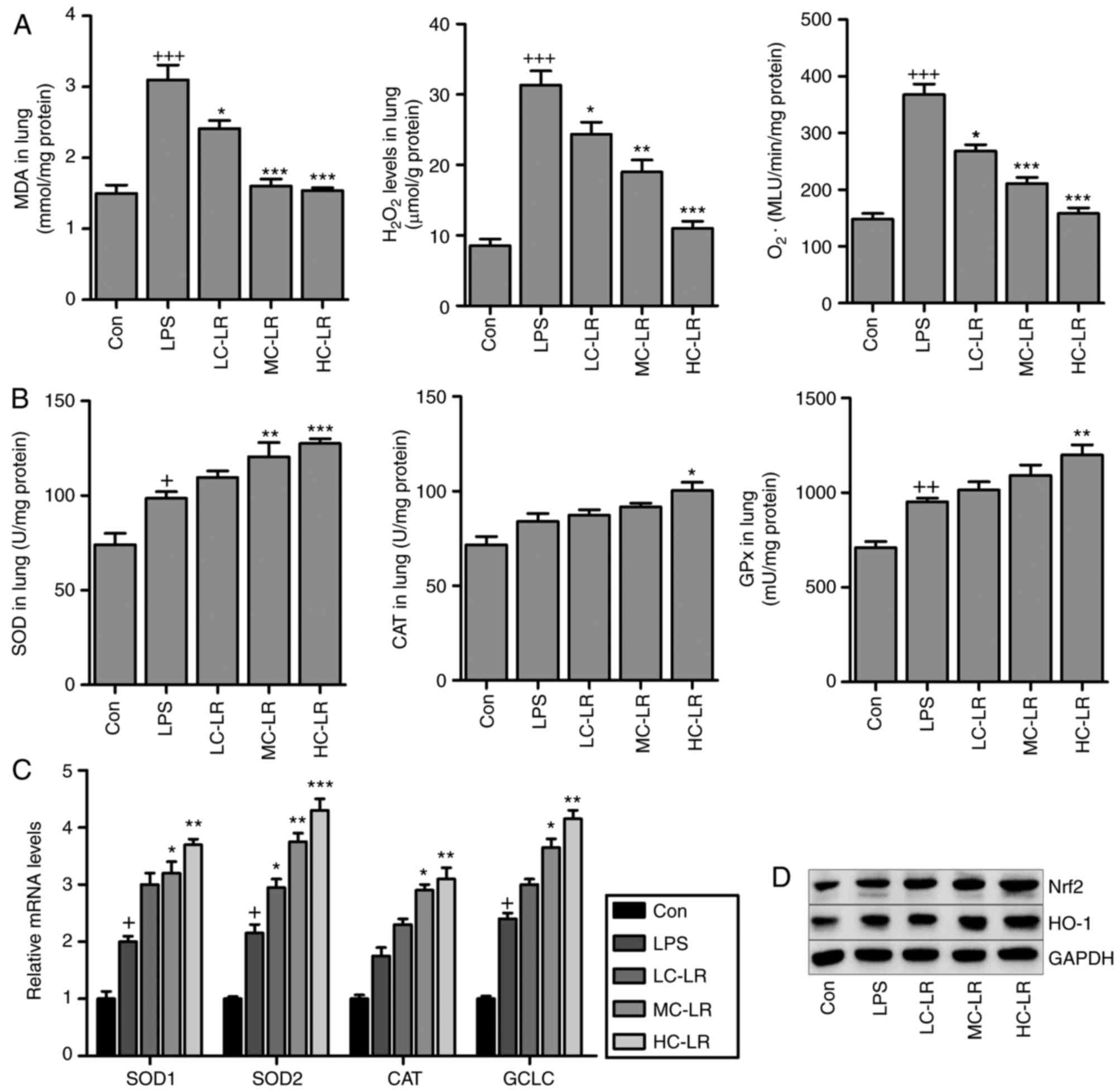 | Figure 5LR suppresses oxidative stress
induced by LPS in the lung tissues of mice. (A) MDA,
H2O2 and O2− levels in
the lung tissue samples were calculated. (B) SOD activity, CAT
activity, and GPx activity in the lung tissues were measured. (C)
Reverse transcription-quantitative polymerase chain reaction
analysis of SOD2, SOD1, CAT and glutamate-cysteine ligase (GCLC).
(D) Nrf2 and heme oxygenase-1 (HO-1) expression levels were
evaluated using western blot analysis. Data are represented as mean
± standard error of the mean of three independent experiments
(n=6). +P<0.05, ++P<0.01 and
+++P<0.001 vs. the Con group in the absence of any
treatments. *P<0.05, **P<0.01 and
***P<0.001 vs. the LPS groups. LR, linarin; LPS,
lipopolysaccharide; MDA, malondialdehyde; SOD, superoxide
dismutase; CAT, catalase; GPX, glutathione peroxidase; Nrf2,
nuclear factor-erythroid 2-related factor 2; Con, control; LC-LR,
low concentration linarin; MC-LR, medium concentration linarin;
HC-LR, high concentration linarin. |
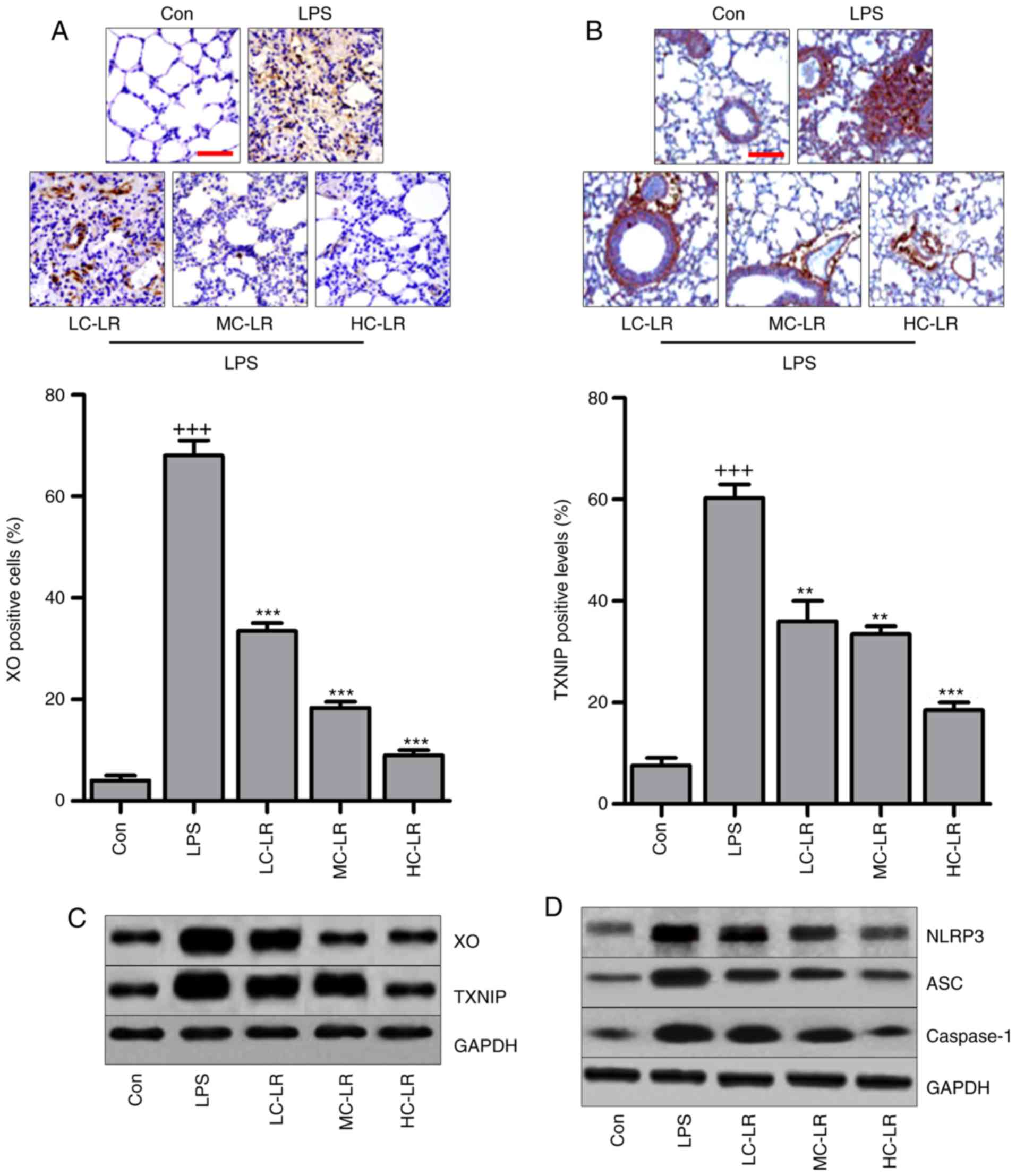
LR inhibits the TXNIP/NLRP3 signaling
pathway in the lung tissues of mice exposed to LPS
The TXNIP/NLRP3 signaling pathway is involved in the
progression of oxidative stress (27,28). Therefore, immunohistochemical
analysis was used to calculate XO and TXNIP in lung tissue samples.
Fig. 6A and B show that XO and
TXNIP were significantly induced by LPS compared to controls and LR
administration reduced expression of XO and TXNIP. Fig. 6C also shows that LR can reduce
LPS-induced increased expression of XO and TXNIP. Western blotting
indicated that NLRP3, ASC and caspase-1 protein were markedly
increased by LPS treatment, and decreased following LR
administration (Fig. 6D). Thus,
the TXNIP/NLRP3 pathway are involved in LR-modulated ALI induced by
LPS.
LR impedes MAPK and NF-κB pathways in
LPS-exposed mice with ALI
MAPKs (p38-MAPK, ERK1/2-MAPK and JNK-MAPK) are
associated with oxidative stress (29). Fig.
7A shows that LPS increased phosphorylated p38, ERK1/2 and JNK,
and LR considerably reduced this dose-dependently. Inflammation is
key to LPS-induced ALI, so western blotting was used to measure the
NF-κB pathway, which is important for regulating the secretion of
pro-inflammatory cytokines. Fig.
7B data show that LR reduced LPS-induced phosphorylation of
IKK-α, IκBα and NF-κB, indicating LR can be used to reduce
inflammation by inactivating the NF-κB signaling pathway.
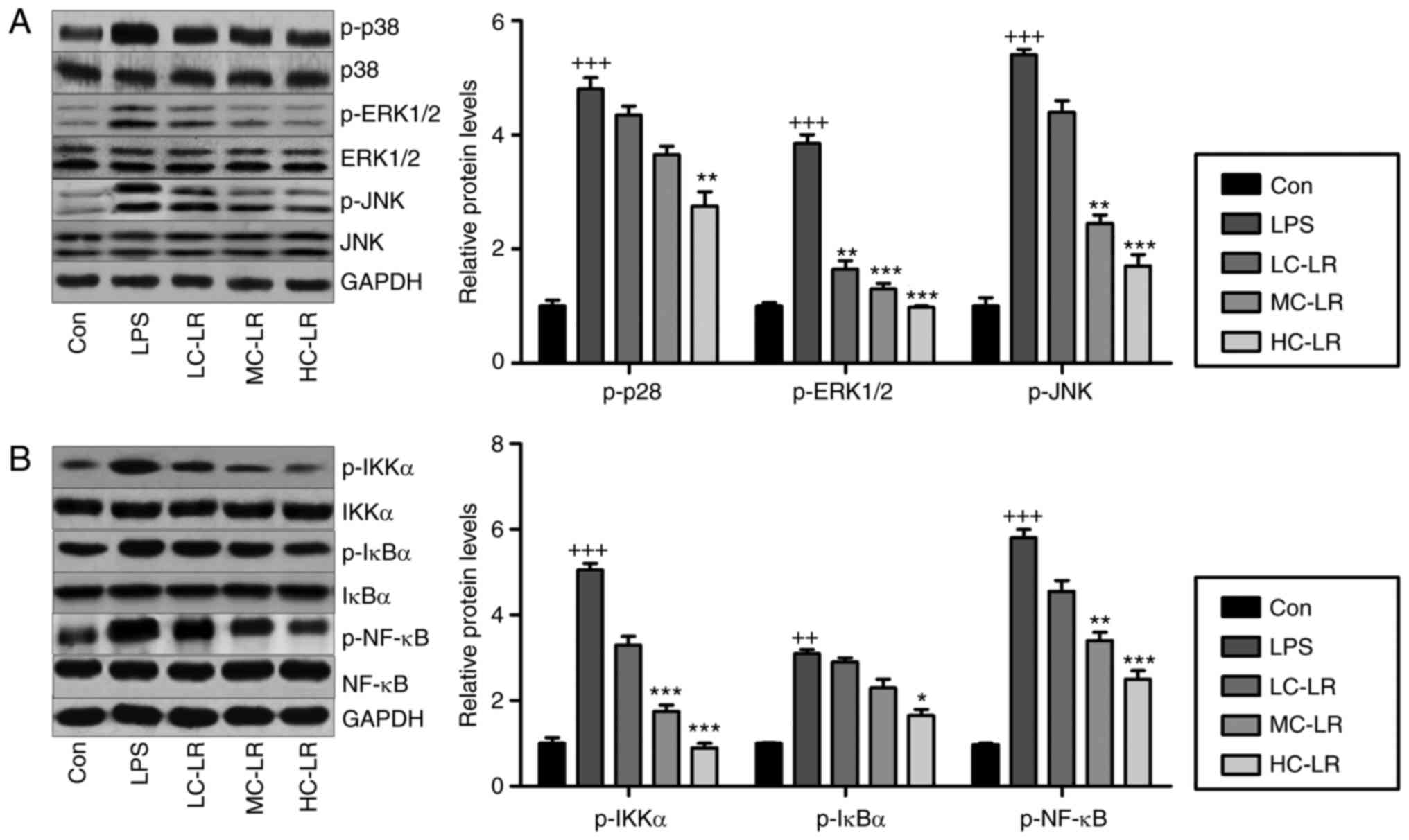 | Figure 7LR blocks mitogen-associated protein
kinase and NF-κB pathways in LPS-induced mice with ALI. (A)
Phosphorylated p38, phosphorylated ERK1/2 and phosphorylated JNK
levels were examined by use of western blot analysis. (B) IKK-α,
IκBα and NF-κB phosphorylation were tested through western blot
analysis. Data are represented as mean ± standard error of the mean
of three independent experiments (n=6). ++P<0.01 and
+++P<0.001 vs. the Con group in the absence of any
treatments. *P<0.05, **P<0.01 and
***P<0.001 vs. the LPS groups. LR, linarin; NF-κB,
nuclear factor-κB; ERK, extracellular signal-regulated kinase; JNK,
c-Jun N-terminal kinase; IKK-α, IκB kinase-α; Con, control; LC-LR,
low concentration linarin; MC-LR, medium concentration linarin;
HC-LR, high concentration linarin. |
LR reduces inflammation and oxidative
stress by suppressing NF-κB and TXNIP/NLRP3 pathways in vitro
LR may be involved in ameliorating LPS-induced ALI
in mice. Fig. 8A presents that
pro-inflammatory cytokines TNF-α, IL-1β, IL-6, MCP-5, COX2,
inducible nitric oxide synthase (iNOS) and MIP-1α were
significantly induced by LPS exposure, and in LR-pretreated groups,
these cytokines were downregulated in a dose-dependent manner.
IL-10 was stimulated by LPS and enhanced by LR. Western blot
analysis indicated that LPS exposure stimulated IKK-α, IκBα and
NF-κB phosphorylation (Fig. 8B)
and LR dose-dependently reduced NF-κB pathway activation.
Therefore, LR had an anti-inflammatory role in LPS-induced BEAS-2B
cells.
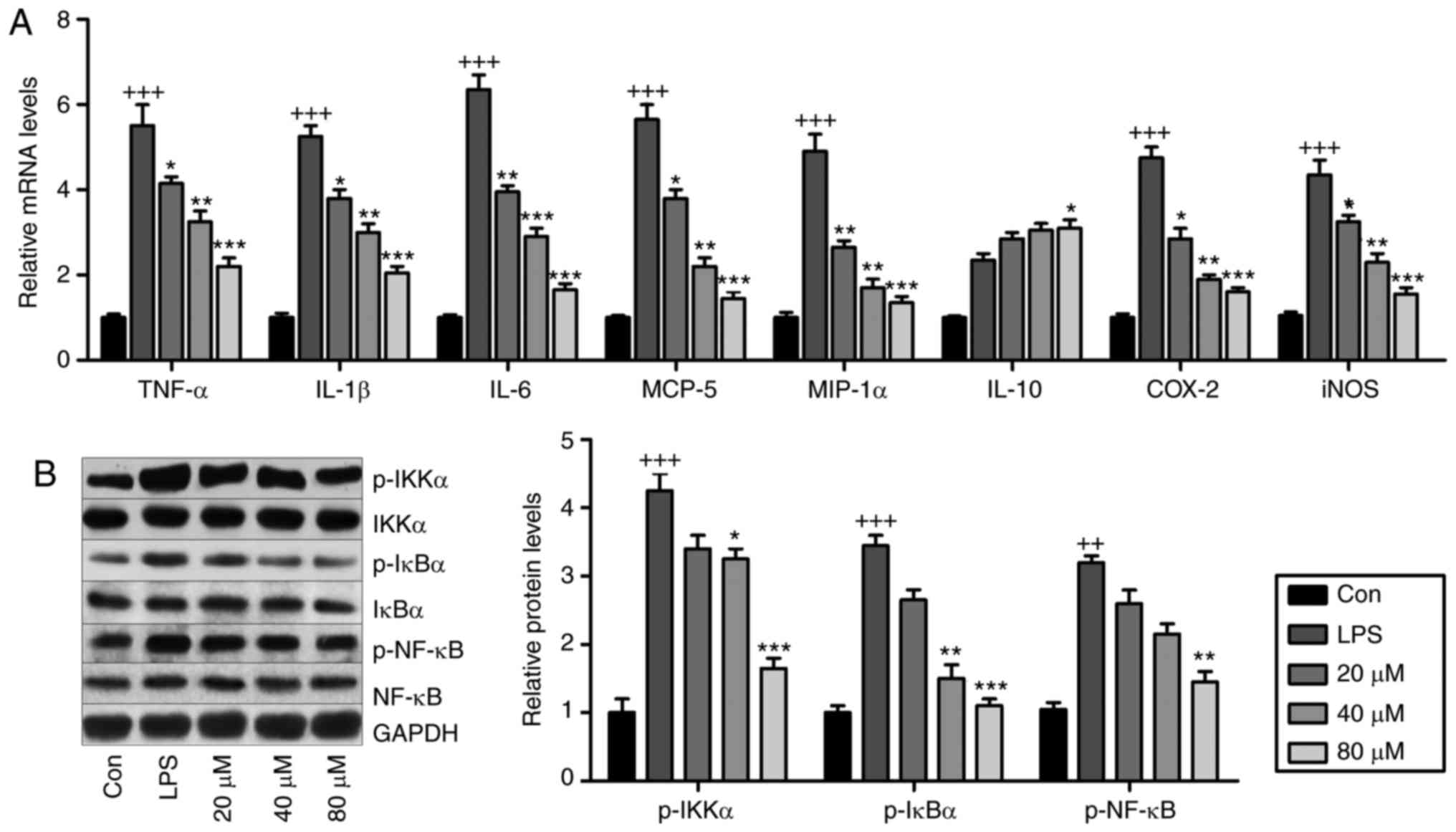 | Figure 8LR reduces inflammation through
suppressing the NF-κB pathway in vitro. The lung epithelia
cells of BEAS-2B were pretreated with different concentrations of
LR (0, 20, 40 and 80 µM) for 24 h, followed by LPS exposure
for another 1 h. Then, all cells were harvested for the following
research. (A) TNF-α, IL-1β, IL-6, MCP-5, MIP-1α, IL-10, iNOS and
COX2 gene levels in cells treated under various conditions were
measured using Reverse transcription-quantitative polymerase chain
reaction analysis. (B) IKK-α, IκBα and NF-κB activation were
examined via western blot analysis in vitro. Data are
represented as mean ± standard error of the mean of three
independent experiments (n=6). +P<0.05,
++P<0.01 and +++P<0.001 vs. the Con
group in the absence of any treatments. *P<0.05,
**P<0.01 and ***P<0.001 vs. the LPS
groups. LR, linarin; NF-κB, nuclear factor-κB; LPS,
lipopolysaccharide; TNF-α, tumor necrosis factor-α; IL,
interleukin; MCP-5, monocyte chemotactic protein 5; MIP-1α,
macrophage inflammatory protein-1α; iNOS, inducible nitric oxide
synthase; COX2, cyclooxygenase 2; Con, control. |
2′-7′-Dichlorofluorescein (DCF) analysis indicated
that LPS triggered high production of ROS, which was reduced by LR
in a dose-dependent manner (Fig. 9A
and B). In addition, in vitro, LR suppressed
LPS-triggered high expression of XO and TXNIP (Fig. 9C). In addition, LR reduced NLRP3,
ASC and caspase-1 protein expression induced by LPS. Finally, p38,
ERK1/2 and JNK phosphorylation were enhanced by LPS treatment, and
this was blunted by LR pretreatment in a dose-dependent manner
(Fig. 9D).
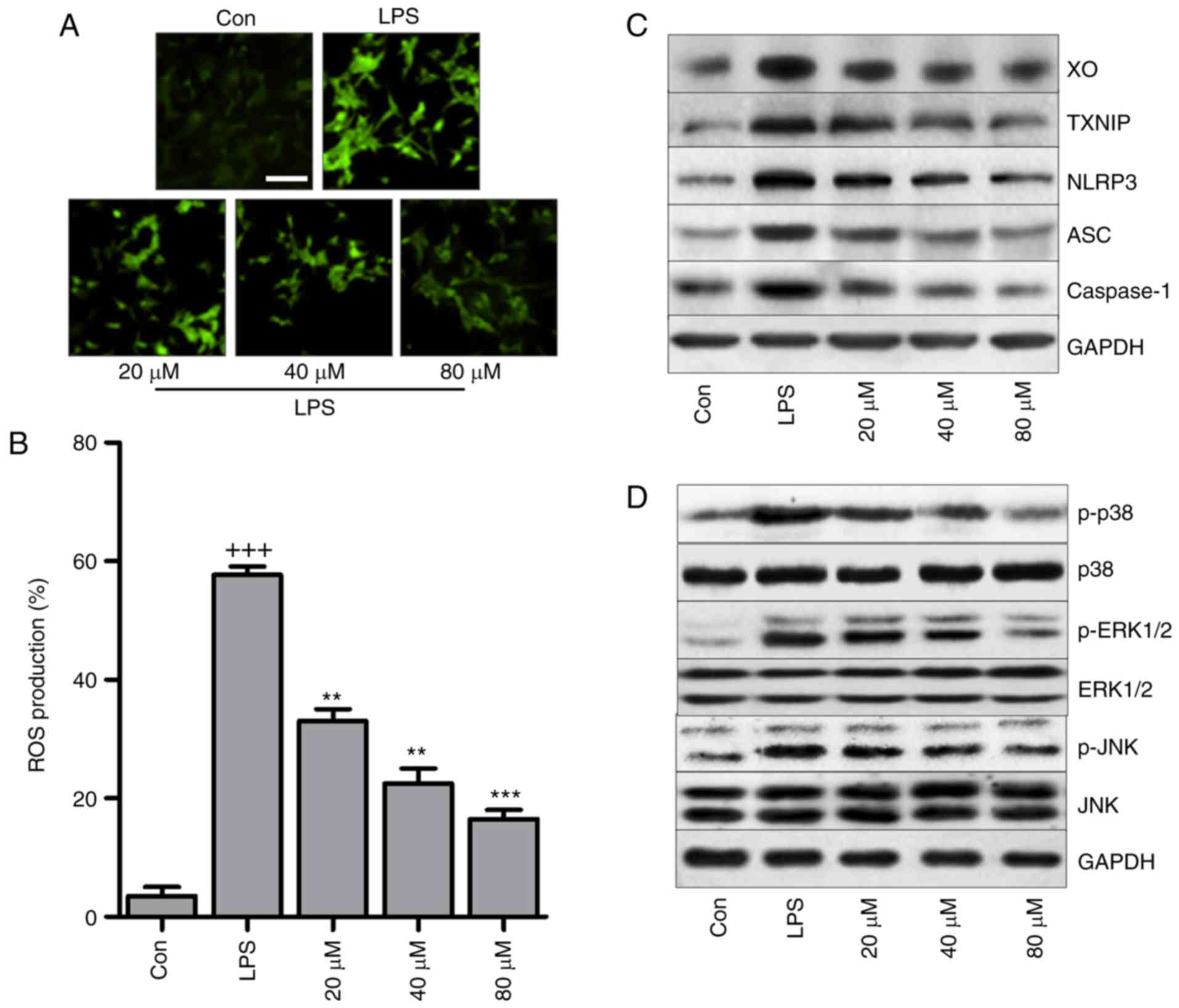 | Figure 9LR reduces oxidative stress through
suppressing the TXNIP/NLRP3 pathway in LPS-treated cells. The lung
epithelia cells of BEAS-2B were pretreated with different
concentrations of LR (0, 20, 40 and 80 µM) for 24 h,
followed by LPS exposure for 1 h. Then, all cells were harvested
for the following research. (A) 2′-7′-Dichlorofluorescein (DCF)
analysis was used to calculate ROS generation, and the
representative images were displayed. The scale bar is 50
µm. (B) The quantification of ROS production was exhibited.
(C) Western blot analysis of XO, TXNIP, LPRP3, apoptosis-associated
speck-like protein containing a C-terminal caspase recruitment
domain (ASC) and caspase-1 in LPS-treated cells in the presence or
absence of LR. (D) p-p38, p-ERK1/2 and p-JNK levels were measured
through western blot analysis. Data are represented as mean ±
standard error of the mean of three independent experiments (n=6).
+++P<0.001 vs. the Con group in the absence of any treatments.
**P<0.01 and ***P<0.001 vs. the LPS
groups. LR, linarin; LPS, lipopolysaccharide; XO, xanthine oxidase;
ERK, extracellular signal-regulated kinase; JNK, c-Jun N-terminal
kinase; ROS, reactive oxygen species; Con, control. |
Oxidative stress and inflammatory
activity of LR depend on ROS production in LPS-treated cells
Fig. 10A
indicates that LR reduced LPS- and ATP-induced ROS production
compared to LPS- and ATP-single treatment groups. NAC, an ROS
scavenger, reduced LPS-induced production of ROS. LR was similar to
NAC in suppressing ROS generation. Fig. 10B shows that high expression of
XO, TXNIP, p-p38, p-ERK1/2 and p-JNK induced by LPS were
downregulated by LR following ATP addition, indicating that
reducing oxidative stress-related signaling pathway expression was
implicated in LR-ameliorated lung injury. In addition, LR treatment
suppressed activation of IKK-α, IκBα and NF-κB in LPS and ATP
co-treatment groups (Fig. 10C).
Finally, pro-inflammatory cytokines TNF-α, IL-1β, IL-6, MCP-5,
COX2, iNOS and MIP-1α were induced by LPS exposure, and this agrees
with data in Fig. 8A. Moreover,
after ATP addition, the anti-inflammatory role of LR, especially at
the highest dose of 40 µM, was observed compared to the LPS
group (Fig. 10D). Thus, LR can
reduce LPS-induced ROS, inhibiting inflammation and oxidative
stress in human lung epithelial cells.
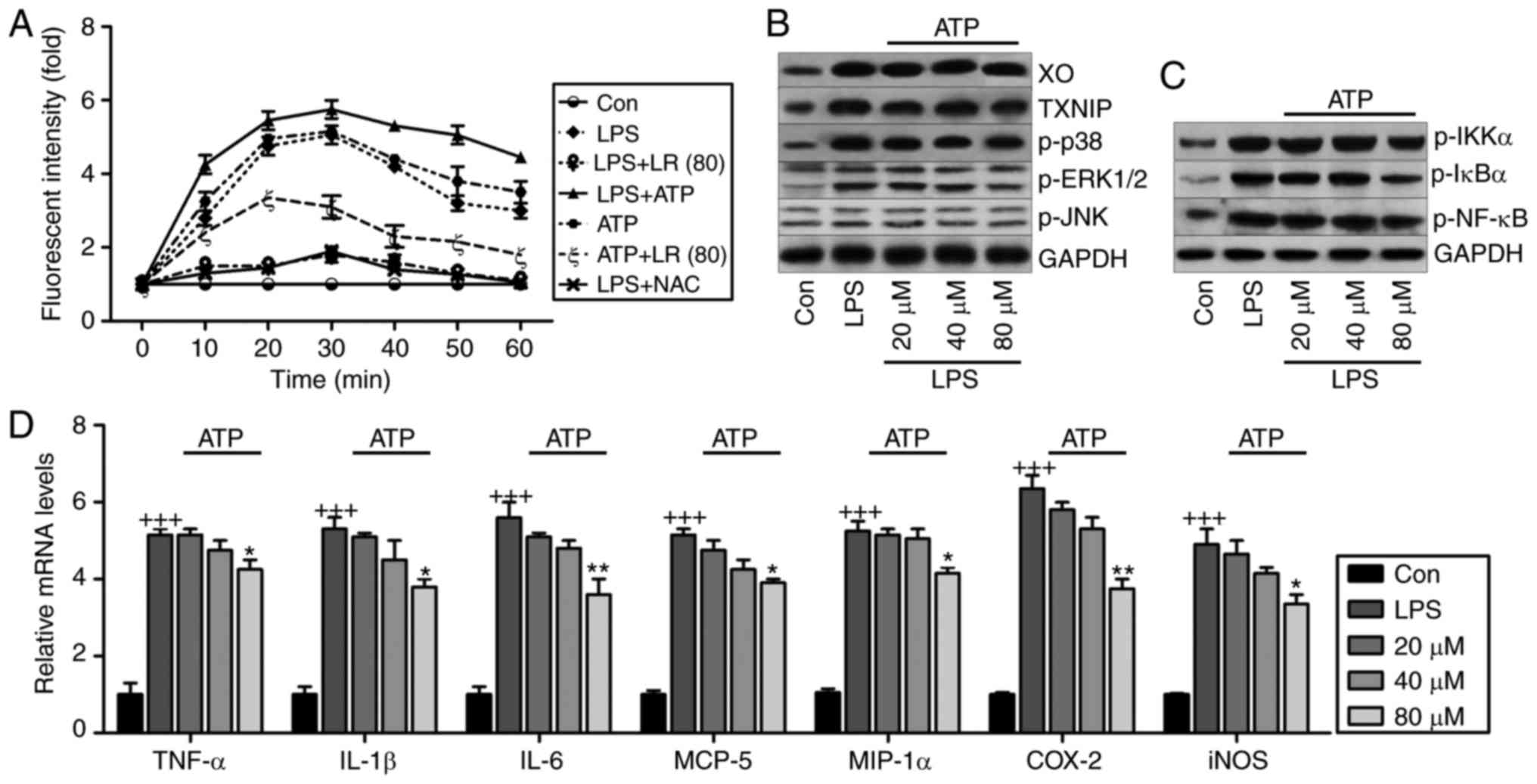 | Figure 10ROS production is involved in
LR-reduced oxidative stress and inflammation in LPS-treated cells.
(A) The lung epithelia cells of BEAS-2B were pretreated with LR (80
µM) for 24 h, followed by LPS exposure for another 1 h, and
then for addition of 10 mM N-acetylcysteine (NAC) or 5 mM ATP for
0–60 min as indicated. Then, reactive oxygen species production was
measured as the relative fluorescence intensity. The lung epithelia
cells of BEAS-2B were pretreated with different concentrations of
LR (0, 20, 40 and 80 µM) for 24 h, followed by LPS exposure
for another 1 h, and then for addition of 5 mM ATP for 60 min.
Finally, all cells were harvested for further experiments. (B) XO,
TXNIP, p-p38, p-ERK1/2 and p-JNK protein levels were measured using
western blot analysis. (C) Western blot analysis of p-IKK-α, p-IκBα
and p-NF-κB were exhibited. (D) TNF-α, IL-1β, IL-6, MCP-5, MIP-1α,
IL-10, iNOS and COX2 gene levels were evaluated using reverse
transcription-quantitative polymerase chain reaction assays. Data
are represented as mean ± standard error of the mean of three
independent experiments (n=6). +++P<0.001 vs. the Con
group in the absence of any treatments. *P<0.05, and
**P<0.01 vs. the LPS groups. LR, linarin; LPS,
lipopolysaccharide; XO, xanthine oxidase; ERK, extracellular
signal-regulated kinase; JNK, c-Jun N-terminal kinase; IKK-α, IκB
kinase-α; TNF-α, tumor necrosis factor-α; IL, interleukin; MCP-5,
monocyte chemotactic protein 5; MIP-1α, macrophage inflammatory
protein-1α; iNOS, inducible nitric oxide synthase; COX2,
cyclooxygenase 2; Con, control. |
Discussion
ALI is an important cause of significant morbidity
and mortality among patients (1,2,30).
The lung is continuously exposed to many inhaled infectious agents,
and host-derived danger signals (31). Thus, the authors studied
LPS-induced ALI in viv and in vitr and identified
inflammation and oxidative stress as significant molecular
mechanisms contributing to lung injury, findings that support
previous research (5,6,32).
Notably, LR alleviated LPS-triggered ALI via suppressing the
inflammatory response and oxidative stress by inactivating NF-κB
and TXNIP/NLRP3 signaling pathways. Therefore, LR alleviates ALI in
LPS-induced mice by suppressing inflammation and oxidation.
As previously described, activated platelet function
is needed in host defense, affecting the recruitment of lung
neutrophils and resulting in the progression of ALI. Platelets
become activated at inflammatory sites and adhere to other
platelets and leukocytes, forming NPAs or MPAs at exposed
endothelia. NPAs are formed in the lung microcirculation and rise
in blood and alveolar compartments after injury (33). Thus, circulating
leukocyte-platelet aggregates are a marker of platelet activation,
and has been reported in patients with inflammatory conditions,
such as sepsis, rheumatoid arthritis, coronary diseases, cystic
fibrosis, inflammatory bowel disease and acute respiratory distress
syndrome (34). Thromboxane A2,
an indicator of platelet activation, is rapidly hydrolyzed to the
inactive stable metabolite, TXB2 (24). CD41 is a specific platelet marker
(23,35). The authors reported that
LPS-induced increases in CD41, NPAs, MPAs and TXB2 were
downregulated by LR, suggesting that LR treatment could inhibit
platelet activation, and perhaps reverse ALI.
Neutrophils and macrophages are chief inflammatory
mediators during progression of ALI (36). They infiltrate into lung tissues
and release enzymes and phagocytize pathogens. These inflammatory
cells are a fundamental source of inflammatory regulators in
vivo. In LPS-mediated inflammation, neutrophils and macrophages
are activated and recruited to the inflammatory site (37,38). Lymphocytes have also been
implicated in the progression of ALI (39,40). In the current study, the authors
confirmed that LPS increased these inflammatory cells, evidence of
ALI establishment in rodents. Importantly, LR reduced macrophages,
neutrophils and lymphocytes to alleviate ALI.
Oversecretion of pro-inflammatory cytokines,
including IL-1β, TNF-α, IL-6, MCP-5 and MIP-1α, is key to the
inflammatory response (41). Data
show that inhibiting multiple inflammatory regulators may attenuate
injury in animals (42).
Inflammatory cytokines can amplify the inflammatory response by
augmenting secretion of chemoattractant factors by airway
epithelial cells and alveolar macrophages, as well as through
expression of adhesion molecules via epithelial cells and
leukocytes (43). Activation of
the NF-κB pathway regulated by IKK-α and IκBα is essential for
regulating release of pro-inflammatory cytokines (44). In the present study, the authors
found that LR possesses anti-inflammatory ability, supported by
reduced TNF-α, IL-1β, IL-6, MCP-5 and MIP-1α, and de-phosphorylated
IKK-α, IκBα and NF-κB. IL-10, in contrast, is a potent
anti-inflammatory molecule and suppresses the action of many
pro-inflammatory molecules (45).
Consistently, LR enhanced downregulation of IL-10 induced by LPS,
confirming its anti-inflammatory capability.
Disturbance of oxidative stress is noted in
LPS-induced ALI (46,47). LPS can potentiate XO to generate
ROS, including H2O2 and
O2− (48).
LPS-treatment enhanced XO, elevated H2O2,
O2− and MDA, important products of ROS. LR
administration decreased XO, H2O2 and
O2− SOD, CAT and GPx are major scavengers of
ROS, and their activity was decreased in LPS-treated tissues.
Constitutive Nrf2 expression is important for sustaining normal
redox balance, and it is induced in response to oxidative stress,
with subsequent transcription of cytoprotective genes; it is an
essential defense against oxidative stress (49). LR induced the expression of Nrf2,
and the downstream HO-1 protein in LPS-induced lung tissues,
indicating its antioxidant ability. TXNIP binding to NLRP3 has been
implicated various diseases (50)
and NLRP3 inflammasome activation was observed in ALI (51). In the present study, the NLRP3
inflammasome was activated in LPS-induced tissues and cells, as was
its downstream signals of ASC and caspase-1, paralleling IL-1β
production and exacerbating lung injury. Furthermore, the
TXNIP/NLRP3 signaling pathway has been implicated in oxidative
stress (52,53). As previously described, activation
of MAPK, including p38, ERK1/2 and JNK, aggravates oxidative stress
and injury in lungs (29,54). Then, ROS generation activates MAPK
expression (55). In the current
study, the authors found that LR administration reduced TXNIP/NLRP3
expression and MAPK phosphorylation in LPS-treated samples.
Additionally, ATP treatment combined with LPS in vitr
enhanced XO/TXNIP/NLRP3 pathway activity and NF-κB, accelerating
cell injury. However, increased ROS induced by ATP in LPS-treated
cells were reduced by LR, which down-regulated ROS production.
Therefore, LR can suppress lung injury in mice, which might be
associated with reduced ROS, subsequently reducing inflammation and
oxidative stress.
In conclusion, LPS exacerbated inflammation,
oxidative stress and lung injury in mice by activating NF-κB,
XO/TXNIP/NLRP3 and MAPK pathways and LR scavenged ROS reduced the
inflammatory response and oxidative stress by inhibiting NF-κB,
XO/TXNIP/NLRP3 and MAPK pathways. Thus, LR may have promise for
treating ALI.
Acknowledgments
The authors thank Dr Yinghui Kong and Dr haoguang
Wang (Nanjing Medical University, Nanjing, China) for their
technical support.
Funding
No funding was received.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
XH and SG made substantial contributions to the
conception and design of the present study. XH, YW, MM, QS and HS
performed the experiments. XH wrote the paper. SG edited and
revised the manuscript critically for important intellectual
content. All authors read and approved the manuscript and agree to
be accountable for all aspects of the research in ensuring that the
accuracy or integrity of any part of the work are appropriately
investigated and resolved.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Institutional Review Board of the Huai'an First People's Hospital,
Nanjing Medical University (Nanjing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
ARDS Definition Task Force; Ranieri VM,
Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E,
Camporota L and Slutsky AS: Acute respiratory distress syndrome:
The Berlin Definition. JAMA. 307:2526–2533. 2012.PubMed/NCBI
|
|
2
|
Matthay MA and Zemans RL: The acute
respiratory distress syndrome: Pathogenesis and treatment. Annu Rev
Pathol. 6:147–163. 2011. View Article : Google Scholar :
|
|
3
|
Li B, Yang J, Huang Q, Zhang Yi, Peng C,
Zhang Y, He Y, Shi J, Li W, Hu J and Fan C: Biodistribution and
pulmonary toxicity of intratracheally instilled graphene oxide in
mice. NPG Asia Materials. 5:e442013. View Article : Google Scholar
|
|
4
|
Spragg RG, Bernard GR, Checkley W, Curtis
JR, Gajic O, Guyatt G, Hall J, Israel E, Jain M, Needham DM, et al:
Beyond mortality: Future clinical research in acute lung injury. Am
J Respir Crit Care Med. 181:1121–1127. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bouwmeester T, Bauch A, Ruffner H, Angrand
PO, Bergamini G, Croughton K, Cruciat C, Eberhard D, Gagneur J,
Ghidelli S, et al: A physical and functional map of the human
TNF-alpha/NF-kappa B signal transduction pathway. Nat Cell Biol.
6:97–105. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bhatia M and Moochhala S: Role of
inflammatory mediators in the pathophysiology of acute respiratory
distress syndrome. J Pathol. 202:145–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guzel A, Kanter M, Pergel A and Erboga M:
Anti-inflammatory and antioxidant effects of infliximab on acute
lung injury in a rat model of intestinal ischemia/reperfusion. J
Mol Histol. 43:361–369. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mao X, Yu CR, Li WH and Li WX: Induction
of apoptosis by shikonin through a ROS/JNK-mediated process in
Bcr/Abl-positive chronic myelogenous leukemia (CML) cells. Cell
Res. 18:879–888. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhou J and Chng WJ: Roles of thioredoxin
binding protein (TXNIP) in oxidative stress, apoptosis and cancer.
Mitochondrion. 13:163–169. 2013. View Article : Google Scholar
|
|
10
|
Jiao R, Liu Y, Gao H, Xiao J and So KF:
The anti-oxidant and antitumor properties of plant polysaccharides.
Am J Chin Med. 44:463–488. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang CS, Shin DM and Jo EK: The role of
NLR-related protein 3 inflammasome in host defense and inflammatory
diseases. Int Neurourol J. 16:2–12. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sun X, Jiao X, Ma Y, Liu Y, Zhang L, He Y
and Chen Y: Trimethylamine N-oxide induces inflammation and
endothelial dysfunction in human umbilical vein endothelial cells
via activating ROS-TXNIP-NLRP3 inflammasome. Biochem Biophys Res
Commun. 481:63–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rajamäki K, Lappalainen J, Oörni K,
Välimäki E, Matikainen S, Kovanen PT and Eklund KK: Cholesterol
crystals activate the NLRP3 inflammasome in human macrophages: A
novel link between cholesterol metabolism and inflammation. PLoS
One. 5:e117652010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guan J, Wu X, Arons E and Christou H: The
p38 mitogen-activated protein kinase pathway is involved in the
regulation of heme oxygenase-1 by acidic extracellular pH in aortic
smooth muscle cells. J Cell Biochem. 105:1298–1306. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim YH, Lee YS and Choi EM: Linarin
isolated from Buddleja officinali prevents hydrogen
peroxide-induced dysfunction in osteoblastic MC3T3-E1 cells. Cell
Immunol. 268:112–116. 2011. View Article : Google Scholar
|
|
16
|
Suh KS, Rhee SY, Jung WW, Kim NJ, Jang YP,
Kim HJ, Kim MK, Choi YK and Kim YS: Chrysanthemum zawadski extract
protects osteoblastic cells from highly reducing sugar-induced
oxidative damage. Int J Mol Med. 32:241–250. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Qiaoshan Y, Suhong C, Minxia S, Wenjia M,
Bo L and Guiyuan L: Preparative purification of linarin extracts
from Dendranthema indicum flowers and evaluation of its
antihypertensive effect. Evid Based Complement Alternat Med.
2014:3942762014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Han S, Sung KH, Yim D, Lee S, Lee CK, Ha
NJ and Kim K: The effect of linarin on LPS-induced cytokine
production and nitric oxide inhibition in murine macrophages cell
line RAW264.7. Arch Pharm Res. 25:170–177. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yu Q, Li X and Cao X: Linarin could
protect myocardial tissue from the injury of Ischemia-reperfusion
through activating Nrf-2. Biomed Pharmacoth. 90:1–7. 2017.
View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Kunapuli SP, Dorsam RT, Kim S and Quinton
TM: Platelet purinergic receptors. Curr Opin Pharmacol. 3:175–180.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cho MS, Bottsford-Miller J, Vasquez HG,
Stone R, Zand B, Kroll MH, Sood AK and Afshar-Kharghan V: Platelets
increase the proliferation of ovarian cancer cells. Blood.
120:4869–4872. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guo Y, Mishra A, Howland E, Zhao C, Shukla
D, Weng T and Liu L: Platelet-derived Wnt antagonist Dickkopf-1 is
implicated in ICAM-1/VCAM-1-mediated neutrophilic acute lung
inflammation. Blood. 126:2220–2229. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang L, Sammani S, Moreno-Vinasco L,
Letsiou E, Wang T, Camp SM, Bittman R, Garcia JG and Dudek SM:
FTY720(s)-phosphonate preserves sphingosine 1-phosphate receptor 1
expression and exhibits superior barrier protection to FTY720 in
acute lung injury. Criti Care Med. 42:e189–e199. 2014. View Article : Google Scholar
|
|
25
|
Chen T, Mou Y, Tan J, Wei L, Qiao Y, Wei
T, Xiang P, Peng S, Zhang Y, Huang Z and Ji H: The protective
effect of CDDO-Me on lipopolysaccharide-induced acute lung injury
in mice. Int Immunopharmaco. 25:55–64. 2015. View Article : Google Scholar
|
|
26
|
Camicia G, Pozner R and de Larrañaga G:
Neutrophil extracellular traps in sepsis. Shock. 42:286–294. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chabot F, Mitchell JA, Gutteridge JM and
Evans TW: Reactive oxygen species in acute lung injury. Eur Respir
J. 11:745–757. 1998.PubMed/NCBI
|
|
28
|
Abais JM, Xia M, Zhang Y, Boini KM and Li
PL: Redox regulation of NLRP3 inflammasomes: ROS as trigger or
effector. Antioxid Redox Sign. 22:1111–1129. 2015. View Article : Google Scholar
|
|
29
|
Chiu WH, Luo SJ, Chen CL, Cheng JH, Hsieh
CY, Wang CY, Huang WC, Su WC and Lin CF: Vinca alkaloids cause
aberrant ROS-mediated JNK activation, Mcl-1 downregulation, DNA
damage, mitochondrial dysfunction, and apoptosis in lung
adenocarcinoma cells. Biochem Pharmacol. 83:1159–1171. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim HA, Park JH, Lee S, Choi JS, Rhim T
and Lee M: Combined delivery of dexamethasone and plasmid DNA in an
animal model of LPS-induced acute lung injury. J Control Release.
156:60–69. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wen HL, Liang ZS, Zhang R and Yang K:
Anti-inflammatory effects of triptolide improve left ventricular
function in a rat model of diabetic cardiomyopathy. Cardiovasc
Diabetol. 12:502013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ben DF, Yu XY, Ji GY, Zheng DY, Lv KY, Ma
B and Xia ZF: TLR4 mediates lung injury and inflammation in
intestinal ischemia-reperfusion. J Surg Res. 174:326–333. 2012.
View Article : Google Scholar
|
|
33
|
Alım Z, Kılınç N, İşgör MM, Şengül B and
Beydemir Ş: Some anti-inflammatory agents inhibit esterase
activities of human carbonic anhydrase isoforms I and II: An in
vitro study. Chem Biology Drug Des. 86:857–863. 2015. View Article : Google Scholar
|
|
34
|
Dopheide JF, Rubrech J, Trumpp A, Geissler
P, Zeller GC, Bock K, Dünschede F, Trinh TT, Dorweiler B, Münzel T,
et al: Leukocyte-platelet aggregates-a phenotypic characterization
of different stages of peripheral arterial disease. Platelets.
27:658–667. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hu L, Du L, Zhao Y, Li W, Ouyang Q, Zhou
D, Lu G and Lin G: Modeling Glanzmann thrombastheni using patient
specific iPSCs and restoring platelet aggregation function by CD41
over-expression. Stem Cell Res. 20:14–20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Park DW, Jiang S, Liu Y, Siegal GP, Inoki
K, Abraham E and Zmijewski JW: GSK3β-dependent inhibition of AMPK
potentiates activation of neutrophils and macrophages and enhances
severity of acute lung injury. Am J Physiol Lung Cell Mol Physiol.
307:L735–L745. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bellac CL, Dufour A, Krisinger MJ,
Loonchanta A, Starr AE, Auf dem Keller U, Lange PF, Goebeler V,
Kappelhoff R, Butler GS, et al: Macrophage matrix
metalloproteinase-12 dampens inflammation and neutrophil influx in
arthritis. Cell Rep. 9:618–632. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Nguyen KD, Fentress SJ, Qiu Y, Yun K, Cox
JS and Chawla A: Circadian gene Bmal1 regulates diurnal
oscillations of Ly6Chi inflammatory monocytes. Science.
341:1483–1488. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang J, Liu YT, Xiao L, Zhu L, Wang Q and
Yan T: Anti-inflammatory effects of apigenin in
lipopolysaccharide-induced inflammatory in acute lung injury by
suppressing COX-2 and NF-κB pathway. Inflammation. 37:2085–2090.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Natarajan V, Dudek SM, Jacobson JR,
Moreno-Vinasco L, Huang LS, Abassi T, Mathew B, Zhao Y, Wang L,
Bittman R, et al: Sphingosine-1-phosphate, FTY720, and
sphingosine-1-phosphate receptors in the pathobiology of acute lung
injury. Am J Respir Cell Mol Biol. 49:6–17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tristan M, Orozco LJ, Steed A,
Ramírez-Morera A and Stone P: Mifepristone for uterine fibroids.
Cochrane Database Syst Rev. 8:CD0076872012.
|
|
42
|
Yousef AA, Amr YM and Suliman GA: The
diagnostic value of serum leptin monitoring and its correlation
with tumor necrosis factor-alpha in critically ill patients: A
prospective observational study. Crit Care. 14:R332010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Eickmeier O, Seki H, Haworth O, Hilberath
JN, Gao F, Uddin M, Croze RH, Carlo T, Pfeffer MA and Levy BD:
Aspirin-triggered resolvin D1 reduces mucosal inflammation and
promotes resolution in a murine model of acute lung injury. Mucosal
Immunol. 6:256–266. 2013. View Article : Google Scholar
|
|
44
|
Ganeff C, Remouchamps C, Boutaffala L,
Benezech C, Galopin G, Vandepaer S, Bouillenne F, Ormenese S,
Chariot A, Schneider P, et al: Induction of the alternative NF-κB
pathway by lymphotoxin αβ (LTαβ) relies on internalization of LTβ
receptor. Mol Cell Biol. 31:4319–4334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Akdis CA and Blaser K: Mechanisms of
interleukin-10-mediated immune suppression. Immunology.
103:131–136. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Lee JH, Jo YH, Kim K, Lee JH, Rim KP, Kwon
WY, Suh GJ and Rhee JE: Effect of N-acetylcysteine (NAC) on acute
lung injury and acute kidney injury in hemorrhagic shock.
Resuscitation. 84:121–127. 2013. View Article : Google Scholar
|
|
47
|
Zhao W, Gan X, Su G, Wanling G, Li S, Hei
Z, Yang C and Wang H: The interaction between oxidative stress and
mast cell activation plays a role in acute lung injuries induced by
intestinal ischemia-reperfusion. J Surg Res. 187:542–552. 2014.
View Article : Google Scholar
|
|
48
|
Miura D, Miura Y and Yagasaki K:
Resveratrol inhibits hepatoma cell invasion by suppressing gene
expression of hepatocyte growth factor via its reactive oxygen
species-scavenging property. Clin Exp Metastasis. 21:445–451. 2004.
View Article : Google Scholar
|
|
49
|
Kovac S, Angelova PR, Holmström KM, Zhang
Y, Dinkova-Kostova AT and Abramov AY: Nrf2 regulates ROS production
by mitochondria and NADPH oxidase. Biochim Biophys Acta.
1850:794–801. 2015. View Article : Google Scholar :
|
|
50
|
Abais JM, Xia M, Li G, Chen Y, Conley SM,
Gehr TW, Boini KM and Li PL: Nod-like receptor protein 3 (NLRP3)
inflammasome activation and podocyte injury via
thioredoxin-interacting protein (TXNIP) during
hyperhomocysteinemia. J Biol Chem. 289:27159–27168. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhang B, Wang B, Cao S, Wang Y and Wu D:
Silybin attenuates LPS-induced lung injury in mice by inhibiting
NF-κB signaling and NLRP3 activation. Int J Mol Med. 39:1111–1118.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Li Y, Li J, Li S, Li Y, Wang X, Liu B, Fu
Q and Ma S: Curcumin attenuates glutamate neurotoxicity in the
hippocampus by suppression of ER stress-associated TXNIP/NLRP3
inflammasome activation in a manner dependent on AMPK. Toxicol Appl
Pharm. 286:53–63. 2015. View Article : Google Scholar
|
|
53
|
Liu W, Gu J, Qi J, Zeng XN, Ji J, Chen ZZ
and Sun XL: Lentinan exerts synergistic apoptotic effects with
paclitaxel in A549 cells via activating ROS-TXNIP-NLRP3
inflammasome. J Cell Mol Med. 19:1949–1955. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Fielhaber JA, Carroll SF, Dydensborg AB,
Shourian M, Triantafillopoulos A, Harel S, Hussain SN, Bouchard M,
Qureshi ST and Kristof AS: Inhibition of mammalian target of
rapamycin augments lipopolysaccharide-induced lung injury and
apoptosis. J Immunol. 188:4535–4542. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Chen J, Guo R, Yan H, Tian L, You Q, Li S,
Huang R and Wu K: Naringin inhibits ROS-activated MAPK pathway in
high glucose-induced injuries in H9c2 cardiac cells. Basic Clin
Pharmacol Toxicol. 114:293–304. 2014. View Article : Google Scholar
|