Introduction
Bronchopulmonary dysplasia (BPD) is a severe and
complicated chronic respiratory disease in premature infants. In
the last 10 years, the morbidity rate of BPD has not decreased,
despite of improvements in treatment in the neonatal intensive care
unit. It has been shown that the incidence of BPD is higher in
premature infants whose gestational age is <32 weeks and who
have a lower birth weight. In a previous study, at a gestational
age of <25 weeks, the incidence of BPD was 28.1%, whereas the
incidence of BPD was only 4% at a gestational age of 29-32 weeks
(1), which seriously affected the
quality of life and long-term prognosis of patients. With the
prenatal application of glucocorticoids and the postnatal
application of pulmonary surfactants (PS), the pathological changes
and processes involved in the development of BPD vary. Compared
with classical BPD, more recently diagnosed BPD shows the major
pathological characteristics of pulmonary development arrest and
pulmonary microvascular dysplasia, with predominant manifestations
as follows: A decrease in the number and an increase in the volume
of alveoli, simplification of alveolar structure, a reduction of
alveolar septa, and abnormality of alveolar structure caused by the
abnormal morphology of pulmonary microvessels; however, airway
injury and fibrosis are mild (2).
At present, studies on the pathogenesis of BPD
mainly involve investigating oxidative stress (OS), damage of the
pulmonary epithelial barrier function and its abnormal repair
following lung injury, DNA injury, and the involvement of
epigenetics in this disease. A number of studies have demonstrated
that OS is one of the key pathogenic mechanisms of BPD (3-7).
OS is defined as the imbalance between oxidation and anti-oxidation
in the body, i.e., the imbalance between enhanced reactive
oxygen/reactive nitrogen substances and protective capacity without
antioxidants (3). OS develops
from reactive oxygen species (ROS) existing at a high level in
vivo; in a hyperoxic environment, the level of ROS may become
rapidly elevated. Several free radicals (FRs) produced by ROS,
including superoxide radicals, hydrogen peroxide and hydroxy
radicals, can interact with proteins, lipids and carbohydrates in
cells and even DNA to cause toxicity. By contrast, ROS, as a
secondary messenger, can activate a cascade reaction of proteins
and regulate the expression of nuclear factor, hypoxia-inducible
factor-1α and nuclear factor erythroid 2-related factor 2, thus
affecting the processes of cell growth and apoptosis. The OS
induced by FRs and ROS may be important contributors to several
neonatal diseases (3).
Studies have suggested that the expression levels of
partial antioxidant enzymes (AOEs), including superoxide dismutase,
catalase and glutathione peroxidase, are increased during late
pregnancy, which matches the maturity of PS. However, due to the
interruption of placental-fetal transfer and the deficiency of
endogenous production (4,5), premature infants usually lack a
normal level of AOEs, and in vivo levels of ROS and FRs are
higher than their anti-oxidative defense capacity. Additionally,
the anti-oxidative defense systems of premature infants are
immature, and thus several factors can readily increase the
occurrence of OS, including malnutrition, inflammation, mechanical
ventilation and infection. Hyperoxic exposure can further enhance
the cascade reaction of OS, affect the activity of AOEs and cause
permanent lung injury (6).
Previous studies of OS in BPD have focused on the
application of antioxidants and the epigenetic changes of
antioxidant genes (3,7). In a study of 80 very low birth
weight (VLBW) newborns with a gestational age of <32 weeks and a
birth weight of <1,250 g completed by Hsiao et al, the
levels of interleukin (IL)-6, an inflammatory indicator, and
8-hydroxydeoxyguanosine (8-OHdG), an OS marker, in serum and
bronchoalveolar lavage fluid (BALF) were markedly higher in
patients who eventually developed BPD, compared with those in
non-BPD patients. IL-6 and 8-OHdG were associated with the high
prevalence of BPD. A receiver operator characteristic curve
indicated that it was possible to predict the occurrence of BPD.
The specificity of IL-6 and 8-OHdG in BALF was up to 77.8 and
64.4%, respectively (8). In a
prospective study of 44 premature infants with respiratory distress
syndrome, Fabiano et al found that the expression level of
glutathione in BALF was lower in patients who eventually developed
BPD, compared with that in non-BPD patients, whereas the lipid
hydroperoxide level was significantly higher (9). In addition, the anti-tumor necrosis
factor drug, etanercept, markedly decreased the level of
malondialdehyde in a rat BPD model and had an effect in preventing
lung injury (10).
Several studies have indicated that antioxidants may
be used to prevent and treat BPD; however, this has yet to be shown
by a large number of clinical trials. According to the latest
study, OS and endoplasmic reticulum stress (ERS) interact; ROS and
reactive nitrogen species have mediating and regulating effects in
ERS (11). Studies have revealed
that ROS are closely associated with the protein folding capacity
of the ER. It is estimated that 25% of ROS in cells results from
the formation of the S-S bond during oxidative protein folding in
the ER. The maintenance of a redox state in the ER lumen provides a
unique microenvironment for, and contributes to, the formation of
the S-S bond; ROS produced in this process accumulate in the ER
lumen, and a change in the redox state or the increased production
of ROS directly or indirectly influences the steady state and
protein folding function of the ER (12). For example, the aggregation of
numerous ROS is an important factor causing incorrect protein
folding in the ER. ROS can directly interact with proteins to
promote the formation of S-S bonds and the breakage and degradation
of carbochain polymers, finally resulting in incorrect folding and
the inactivation of proteins to induce ERS (13). The above studies have suggested
that the OS induced by a high level of reactive oxygen radicals is
an important contributor to the incorrect protein folding observed
in the ER that is followed by ERS.
Under certain physiological or pathological
conditions, including OS, ERS can induce the aggregation of
misfolded or unfolded proteins in the ER lumen; mild ERS can be
relieved by an unfolded protein response (UPR), whereas severe ERS
may induce autophagy (14-16).
However, whether ERS is involved in the occurrence and development
of BPD remains to be fully elucidated. Based on previous reports,
hypoxia-reoxidation or ischemia-reperfusion injury can activate
autophagy and induce ERS in a lung ischemia-reperfusion model
(17-19). Zhang et al observed that
microtubule-associated protein light chain 3β (LC3B)-II can prevent
hyperoxia-induced DNA injury of pulmonary epithelial cells through
synergism in a BPD model (20,21). In the present study, a BPD rat
model was established, and ERS and autophagy in pulmonary
epithelial cells were observed. The changes in C/EBP homologous
protein (CHOP), an ERS-associated factor and LC3B, an autophagic
marker, were investigated, as were the effects of ERS on the
autophagy of lung tissues, and the roles of ERS and autophagy in
the pathogenesis of BPD.
Materials and methods
Preparation of animal models
All animal experiments were approved by the Animal
Ethics Committee of China Medical University (Shenyang, China; no.
2015PS230K), and all surgical procedures were performed under
anesthesia with chloral hydrate to minimize pain of the
experimental animals. A total of 45 specific pathogen-free
Sprague-Dawley (SD) rats (36 females and 9 males, aged 4 weeks,
220-250 g) were purchased from the Laboratory Animal Center of
China Medical University (Shenyang, China), and housed at a
temperature of 22±2°C and a relative humidity of 60-70%. All
animals were mated at a female/male ratio of 4:1, and each pregnant
SD rat was then bred independently and allowed to give birth
spontaneously at a gestational age of 22 days. At 12 h following
birth by spontaneous delivery, the newborn SD rats (with maternal
rats) were randomly divided into two groups: Model group (n=40) and
control group (n=40).
According to procedures established in our previous
study (22), the newborn rats in
the model group were placed into an oxygen chamber. The oxygen
concentration, temperature and relative humidity of the oxygen
chamber were maintained at 80-85%, 25-27°C and 60-70%,
respectively. Soda lime was used to absorb CO2 in the
oxygen chamber to maintain the CO2 concentration
<0.5%, and silica gel was used to absorb water. The oxygen
concentration was monitored with a digital oxygen analyzer every
day. In the control group, the newborn rats were allowed to breathe
in fresh air (fraction of inspired oxygen=21%). The other
conditions and control factors were the same as those in the model
group. The oxygen chamber was opened every 24 h for 30 min to
supplement food and water. The maternal rats in the model and
control groups were exchanged to prevent them succumbing to oxygen
poisoning.
Collection of lung samples
At 1, 7, 14 and 21 days following the start of the
experiment, 20 newborn SD rats were randomly selected from each
group, weighed and anesthetized by an intra-peritoneal injection of
10% chloral hydrate (3 ml/kg). Thereafter, the thoracic cavity was
opened, the left auricle sheared open, and a scalp needle inserted
into the pulmonary artery via the right ventricle. Subsequently, 10
ml of physiological saline was slowly injected to wash off the
residual blood in the lungs, and the lungs were collected. The
middle lobe of the right lung was fixed in 4% paraformaldehyde for
hematoxylin and eosin (HE) staining, immunohistochemistry and
immunofluorescence experiments. A l-mm3 section of lung
tissue was cut from the left lung and then fixed in 2.5%
glutaraldehyde for transmission electron microscopy (TEM). The
remaining lung tissues were preserved in a −80°C refrigerator for
protein and mRNA analyses.
Pathological changes of lung tissues
The tissues were observed following HE staining
under an optical microscope (H600L; Nikon Corporation, Tokyo,
Japan). The left lung tissues were fixed in 4% paraformaldehyde for
24 h, dehydrated with gradient alcohols, vitrified with xylene,
embedded with paraffin, and routinely cut into 4-µm thick
paraffin tissue sections. Thereafter, the tissue sections were
deparaffinized, hydrated, stained with HE and observed under a
light microscope. The cytoplasm and collagen fibers were colored
pink and nuclei were blue. For animals in each group, six sections
were randomly selected at various time-points, and 10 randomly
selected fields in each section were observed for recording of any
pathological changes.
Alveolar development was evaluated according to a
radial alveolar count (RAC) and the alveolar septum thickness. For
the RAC, referring to the method of Cooney and Thurlbeck, a
vertical line was plotted from the center of the respiratory
bronchiole to the closest fibrous septum (or pleura), and the
number of alveoli counted on the vertical line was designated the
RAC (23). A high RAC indicated
more mature alveolar development. The HE-stained sections were
observed under the 100X high power lens of a light microscope; 10
sections were selected from each group at various time-points, and
counting was performed three times in each section, with the
results averaged. The counts were completed by two double-blinded
independent pathological investigators. The alveolar septum
thickness was measured according to the method used in our
preliminary study (24), and the
analysis was performed using a light microscope image analysis
system. Six sections were randomly selected from each group at
various time-points, and 10 randomly selected fields were observed
in each section under the 400X high power lens of a light
microscope; the alveolar septum thickness was measured in each
field and the average result calculated. The measurements were
completed by two double-blinded independent pathological
investigators.
Ultrastructural observation of lung
tissues
Ultrastructure changes in type II alveolar
epithelial cells (AECII) were observed using transmission electron
microscopy (TEM; H-600; Hitachi, Ltd., Tokyo, Japan). The fresh
lung tissues were fixed first with 2.5% glutaraldehyde and then
with 1% osmic acid, dehydrated with acetone, soaked and embedded
with epoxy resin, polymerized for 72 h, and cut into ultrathin
sections (60 nm). Thereafter, the sections were double-stained with
uranyl acetate and lead nitrate, and then observed under TEM for
ultrastructure changes and the autophagy of AECII and organelles in
lung tissues, with images captured.
Immunohistochemistry
The paraffin sections of lung tissues on slides were
heated in a 60°C oven for 30 min, routinely deparaffinized,
hydrated with gradient alcohols and washed with phosphate-buffered
saline (PBS). Subsequently, 3% H2O2 was added
to eliminate endogenous peroxidases, and the slides were incubated
at 37°C for 20 min. Sodium citrate buffer was added for antigen
retrieval by microwave for 30 min, following which the slides were
cooled to room temperature and washed with PBS. Thereafter, 50
µl of goat serum (cat. no. SPN-9001,9002; Zhongshan Golden
Bridge Biotechnology Co., Ltd., Beijing, China) was added as a
blocking agent, and the slides were incubated at 37°C for 45 min.
The serum was then discarded, and 50 µl of the following
primary antibodies were added to each slide: CHOP (cat. no.
ab11419, 1:200 dilution; Abcam, Cambridge, UK), LC3B (cat. no.
3868s, 1:3,000 dilution; Cell Signaling Technology, Inc., Danvers,
MA, USA). The slides were incubated with the antibodies overnight
at 4°C, and rewarmed for 45 min. Goat anti-mouse/anti-rabbit IgG
and horseradish peroxidase-labeled streptavidin (cat. no. SPN-9001,
9002, 1:300 dilution; Zhongshan Golden Bridge Biotechnology Co.,
Ltd.) were added in sequence, and the slides were incubated at 37°C
for 20 min, following which the slides were washed with PBS, and
developed with DAB under a light microscope until brown-yellow
particles were observed in the nuclei or cytoplasm. The slides were
then washed with tap water, counterstained with hematoxylin for 5
min, blued with running water for 30 min, dehydrated with gradient
alcohols, vitrified with xylene, and mounted with neutral balsam.
PBS was used as a negative control instead of primary antibody. The
immunohistochemical kits (including goat serum, Goat
anti-mouse/anti-rabbit IgG and peroxidase-labeled streptavidin)
were purchased from ZSGB-BIO (cat. no. SPN-9001, 9002; Zhongshan
Golden Bridge Biotechnology Co., Ltd.).
Western blot analysis
Total proteins were extracted from lung tissues
using RIPA (Beyotime Institute of Biotechnology, Haimen, China) and
quantified using a BCA protein assay kit (Beyotime Institute of
Biotechnology), following with 5X loading buffer was added and
samples were boiled at 100°C for 5 min to complete protein
denaturation. A 12% SDS polyacrylamide gel was prepared. Equal
quantities of protein samples (50 µg/lane) were added to
each well, subjected to electrophoresis at 100 V for 3 h and then
transferred onto a polyvinylidene fluoride (PVDF) membrane (EMD
Millipore, Billerica, MA, USA) at 100 V for 30 min. Subsequently,
the PVDF membrane was blocked with defatted milk diluted with 5%
TBS-T [100 mM Tris base (pH 7.5), 0.9% (wt/vol) NaCl, 0.1%
{vol/vol) Tween-20] at room temperature for 1 h. The following
primary antibodies were then added: Rabbit anti-ATF4 antibody (cat.
no. ab184904, 1:500 dilution; Abcam), mouse anti-CHOP antibody
(cat. no. ab11419, 1:500 dilution; Abcam), rabbit anti-LC3B
antibody (cat. no. NB100-2220, 1:200 dilution; Novus Biologicals,
Littleton, CO, USA) and GAPDH antibody (cat. no. 10494-1-AP,
1:4,000 dilution; Wuhan Sanying Biotechnology, Wuhan, China) and
incubated overnight at 4°C. The PVDF membrane was washed three
times with TBS-T for 10 min, followed by incubation with
horseradish peroxidase-conjugated secondary antibody (goat
anti-mouse and anti-rabbit IgG-HRP; cat. nos. SA00001-1 and
SA00001-2, respectively; 1:5,000 dilution; Wuhan Sanying
Biotechnology), at room temperature for 2 h, and washing three
times with TBS-T for 10 min. Finally, Super ECL Plus (Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was added, and luminescence
analysis was performed with a chemiluminescence imaging system
(C300; Azure Biosystems, Dublin, CA, USA). The bands were
standardized according to GAPDH.
Immunofluorescence
A double immunofluorescence labeling method was
used. The paraffin lung tissue sections on slides were heated in a
60°C oven for 30 min, routinely deparaffinized and hydrated with
graded alcohols. Citrate buffer solution was added for microwave
antigen retrieval, and subsequently cooled to room temperature. The
tissue sections were blocked with goat serum (cat. no. SPN-9001;
Zhongshan Golden Bridge Biotechnology Co., Ltd.) for 45 min, and
incubated with mouse anti-rat CHOP (cat. no. ab11419, 1:50
dilution; Abcam) and rabbit anti-rat LC3B (cat. no. 3868s, 1:50
dilution; Cell Signaling Technology, Inc.), antibodies overnight at
4°C. Subsequently, fluorescent-labeled donkey anti-mouse IgG (cat.
no. ab150105, 1:200 dilution; Abcam) and donkey anti-rabbit IgG
(cat. no. ab150076, 1:200 dilution; Abcam) were added, incubated at
room temperature for 4 h, washed with PBS, stained with DAPI
(Sigma-Aldrich; EMD Millipore) for 5 min, and washed again with
PBS. The slides were observed under a confocal laser-scanning
microscope (Nikon C1 system; Nikon Corporation). Positive cells
exhibited green nuclei, and a red cytoplasm and cell membrane. For
the negative control, PBS was used instead of primary antibody.
RNA extraction
Total RNA was extracted according to the
manufacturer's protocol (Takara Bio, Inc., Otsu, Japan). The right
lung tissues were placed into an Eppendorf tube and then sheared on
ice. TRIzol (Takara Bio, Inc.) was added to each sample, which was
then repeatedly aspirated with a sample injector to form an
emulsion. Chloroform (200 µl) was then added, and the sample
was mixed evenly, incubated at room temperature for 5 min, and
centrifuged (12,000 × g) at 4°C for 15 min. The supernatant was
collected, the same volume of isopropanol added, the sample mixed
evenly, and centrifuged (12,000 × g) at 4°C for 15 min. The
supernatant was carefully removed by pipetting to leave behind a
white precipitate. Alcohol (500 µl, 70%) was added and mixed
evenly, and the sample was centrifuged (7,500 × g) at 4°C for 5
min. The supernatant was removed, 500 µl of 70% alcohol was
added, and the sample was mixed evenly and centrifuged (7,500 × g)
at 4°C for 5 min. The sample was then dried at room temperature for
15 min following removal of the supernatant, and DEPC-treated water
was added to dissolve the RNA precipitate. The OD260/OD280 was
measured with a spectrophotometer; the concentration of RNA
extracted was calculated, and the purity range was evaluated as
1.8-2.0. Finally, each RNA sample was adjusted to the same
concentration (1,000 ng/µl).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
RNA (2 µl) was pipetted from various samples
and then reverse transcribed into cDNA using a reverse
transcription system (SuperScript III; Invitrogen; Thermo Fisher
Scientific, Inc.) and a PrimeScript™ RT reagent kit (RR047A; Takara
Bio, Inc.) removal of genomic DNA (1 µl gDNA Eraser, 2
µl 5X gDNA Eraser Buffer, 2 µl RNA, 5 µl RNase
Free dH2O) with the conditions of room temperature for
15 min, and then reverse transcription reaction (add to the above
mixture: 4 µl 5X PrimeScript Buffer 2, 4 µl RNase
Free dH2O, 1 µl PrimeScript RT Enzyme Mix1 and 1
µl RT Primer Mix, total 20 µl) with the conditions of
37°C for 15 min, and 85°C for 5 sec. A 7500 Real-Time PCR system
(Applied Biosystems; Thermo Fisher Scientific, Inc.) and PCR
amplification kit (Takara Biotechnology Co., Ltd.) were used.
Adding 10 µl TB Green Premix Ex Taq (Tli RNaseH Plus) (2X),
0.4 µl forward primer, 0.4 µl reverse primer, 2
µl cDNA, 0.4 µl ROX Reference Dye II, 6.8 µl
RNase Free dH2O, total 20 µl. The reaction
conditions were as follows: 95°C for 30 sec, 95°C for 5 sec, 60°C
for 34 sec, 95°C for 15 sec, 60°C for 1 min, 95°C for 30 sec, and
60°C for 15 sec, for a total of 40 cycles. Gene-specific primers
were designed and synthesized by Takara Biotechnology Co., Ltd. A
dissolution curve was used to guarantee the specificity of PCR
products, and the relative expression levels of target genes were
normalized to GAPDH and evaluated using the 2-∆∆Cq
method (25). The primer
sequences used are listed in Table
I.
 | Table IPrimer sequences used for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction analysis.
| Primer name | Primer
sequence |
|---|
| ATF4 | F:
5′-CCTTCGACCAGTCGGGTTTG-3′ |
| R:
5′-CTGTCCCGGAAAAGGCATCC-3′ |
| CHOP | F:
5′-AGGAGAGAGAAACCGGTCCAA-3′ |
| R:
5′-GGACACTGTCTCAAAGGCGA-3′ |
| LC3B | F:
5′-ACCAGCACCCCAGCAAGA-3′ |
| R:
5′-CAGGACAGGCAGCTGCTTCT-3′ |
| GAPDH | F:
5′-GTATGACTCTACCCACGGCAAGTTC-3′ |
| R:
5′-AGCCTTCTCCATGGTGGTGAAGAC-3′ |
Statistical analysis
SPSS 21.0 software (IBM SPSS, Armonk, NY, USA) was
used for statistical analysis. The data are presented as the mean ±
standard deviation. The inter-group comparison was performed using
two independent samples t-test, and multiple comparisons by one-way
analysis of variance. Correlation analysis of the protein
expression of CHOP and LC3B-II was performed using Pearson's
correlation analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
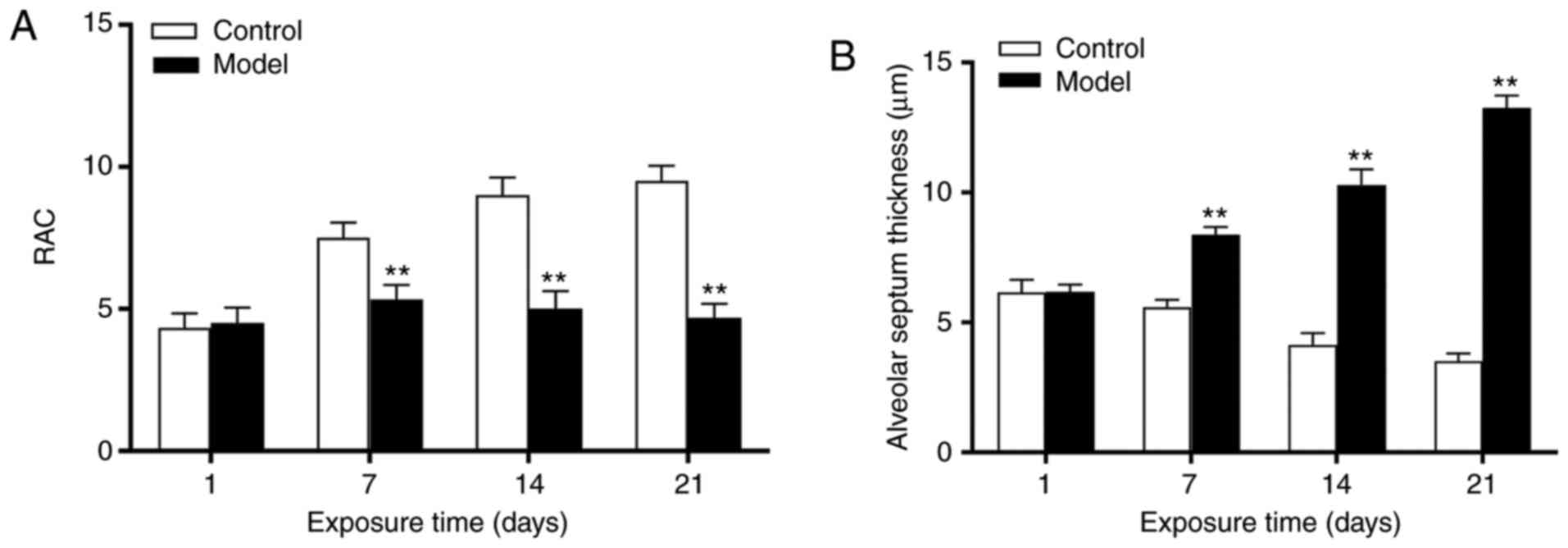
Evaluation of lung development
At day 1, there was no significant difference in the
RAC between the control and model groups (P>0.05). Between 7-21
days, the RAC continuously decreased, and a significant difference
between the two groups was noted (P<0.01; Fig. 1A). No significant difference was
observed in the alveolar septum thickness between the two groups at
1 day (P>0.05); however, this was increased at 7 days
(P<0.01), and reached a peak at 21 days (P<0.01) in the model
group (Fig. 1B).
Ultrastructural changes of AECII in lung
tissues
Using TEM, the AECII cells of the control group were
observed to be cuboidal in shape and with apical microvilli. Within
their cytoplasm, characteristic lamellar bodies, and ER,
mitochondria and other organelles were present with a normal
structure. In the model group, the nuclear chromatins of AECII
cells were marginally aggregated, with apical microvilli that were
sparse and detached. Lamellar bodies in the cytoplasm were damaged
and vacuolized, the mitochondria was swollen, and the ER was
markedly dilated, degranulated and pool-shaped, and was surrounded
by autophagosomes (APs). Visible cytoplasmic components were
contained in the vacuole-like structure of an AP double-layered
membrane, including mitochondria, ER and ribosomes. APs surrounded
the dilated ER (Fig. 2).
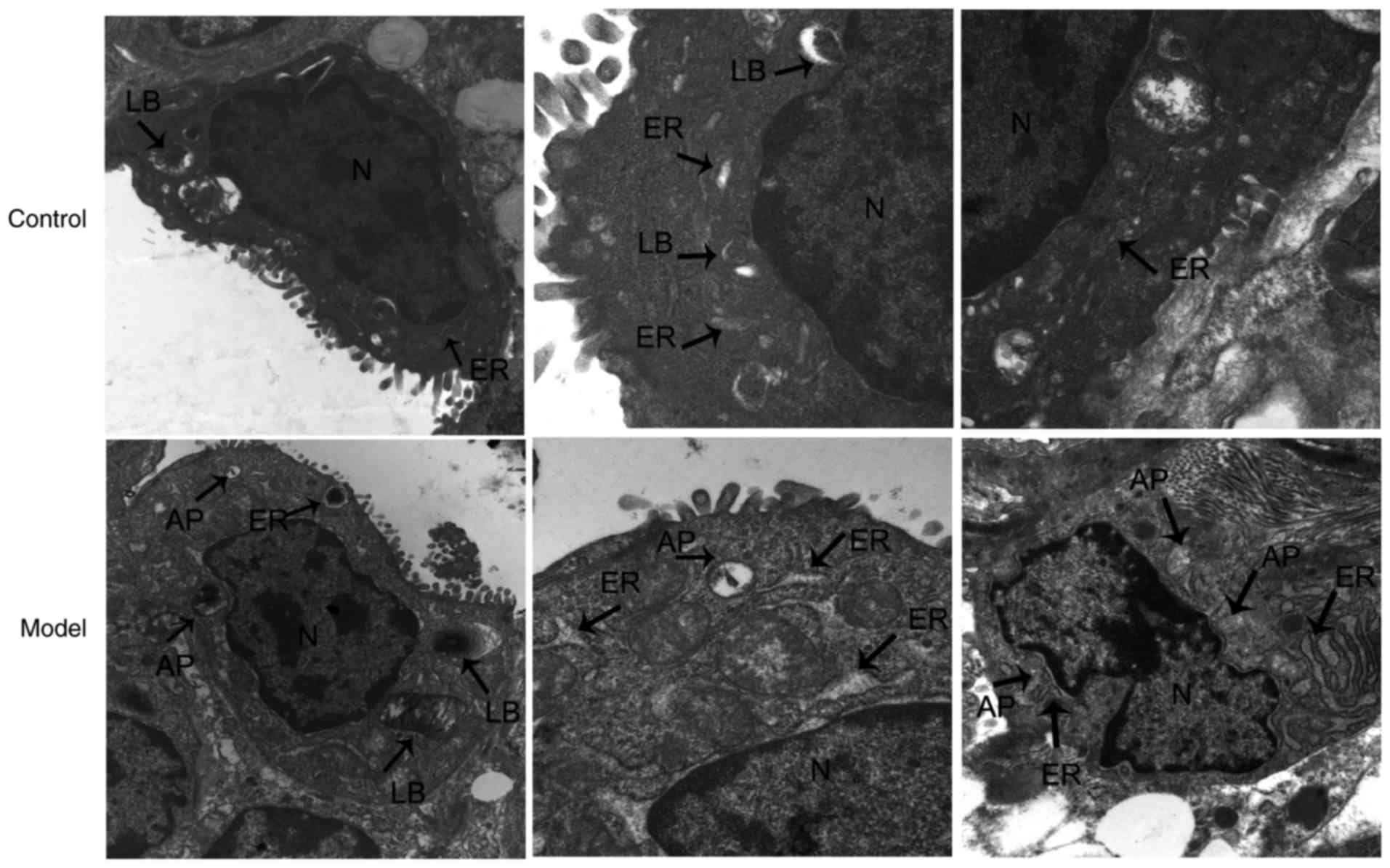 | Figure 2Transmission electron microscopy
observation of AECII ultrastructure at 14 days (magnification,
×50,000). In the control groups, AECII cells and their organelles
appeared normal. In the model groups, apical microvilli were sparse
and detached, lamellar bodies in the cytoplasm were damaged, and
the ER was markedly dilated and surrounded by autophagosomes.
AECII, type II alveolar epithelial cell; LB, lamellar body; AP,
autophagosome; ER, endoplasmic reticulum; N, AECII nucleus. |
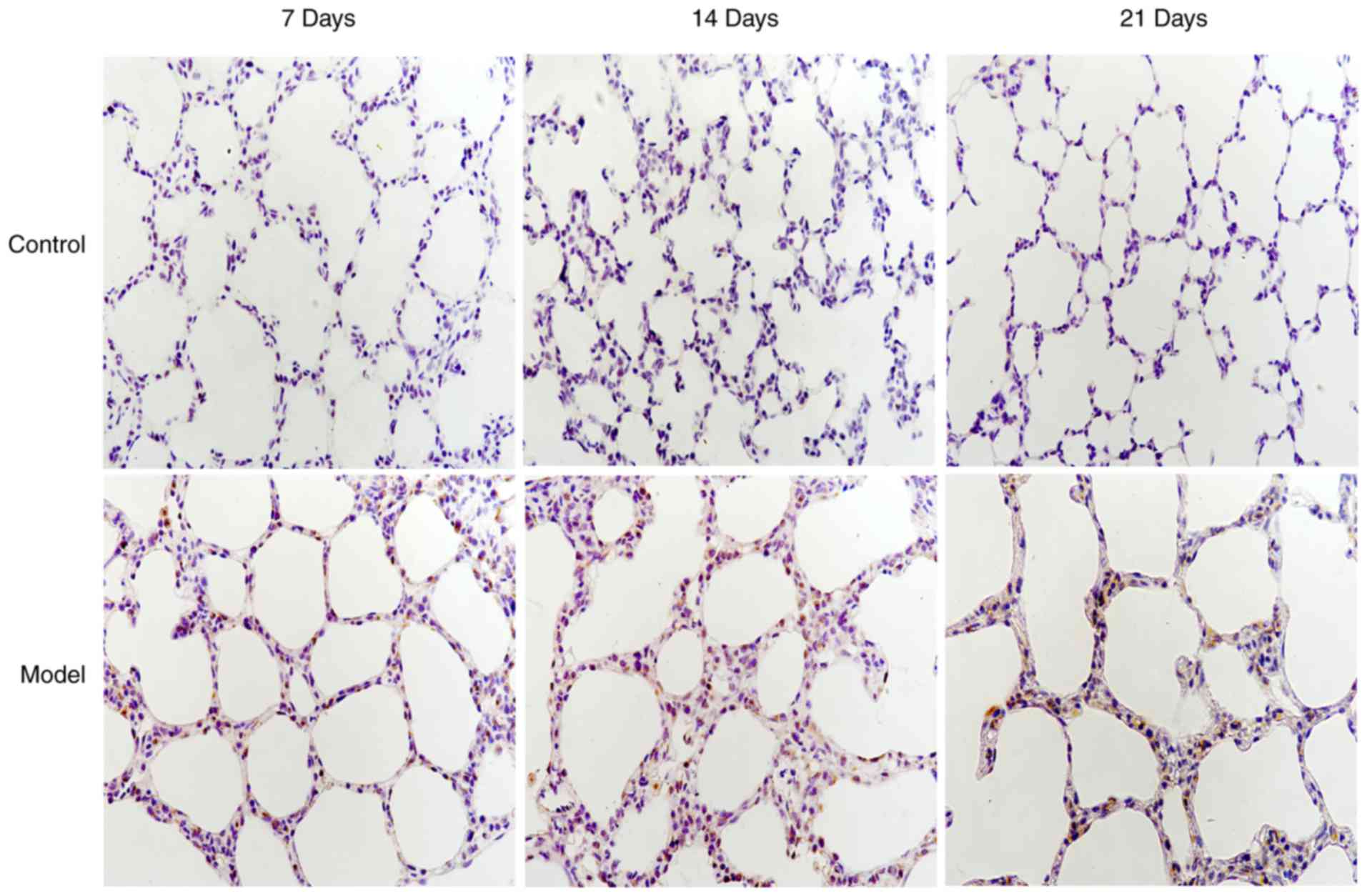
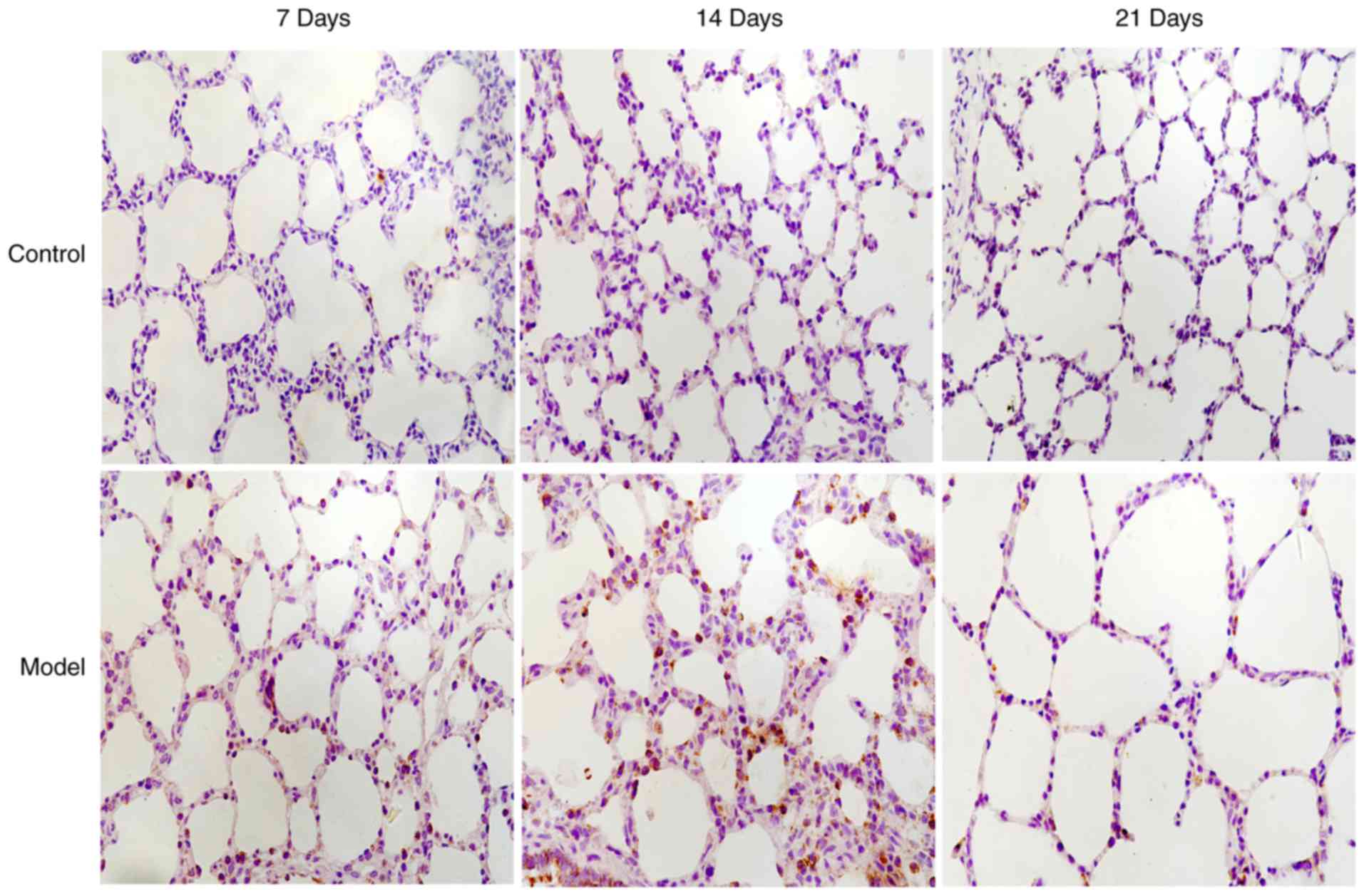
Localization of CHOP and LC3B in lung
tissues by immunohistochemistry
In the control group, LC3B protein was mainly
expressed in the cytoplasm of bronchial epithelial cells; however,
CHOP protein was not expressed. In the model group, CHOP protein
(Fig. 3) was predominantly
located in the nuclei of alveolar and bronchial epithelial cells,
and LC3B protein was aggregated in a granular and punctiform manner
in the cytoplasm (Fig. 4).
Expression of ATF4, CHOP and LC3B
proteins in rat lung tissues
At day 1, no statistically significant differences
were noted in the protein expression levels of ATF4, CHOP or
LC3B-II between control and model groups (P>0.05). The protein
expression levels of ATF4, CHOP and LC3B-II were significantly
higher in the model group, compared with those in the control group
at 7 days, reached a peak at 14 days (ATF4, CHOP and LC3B-II,
P<0.01) and were marginally decreased at 21 days (CHOP,
P<0.05; ATF4 and LC3B-II, P>0.05). They showed a consistent
overall trend of change (Fig.
5A–D).
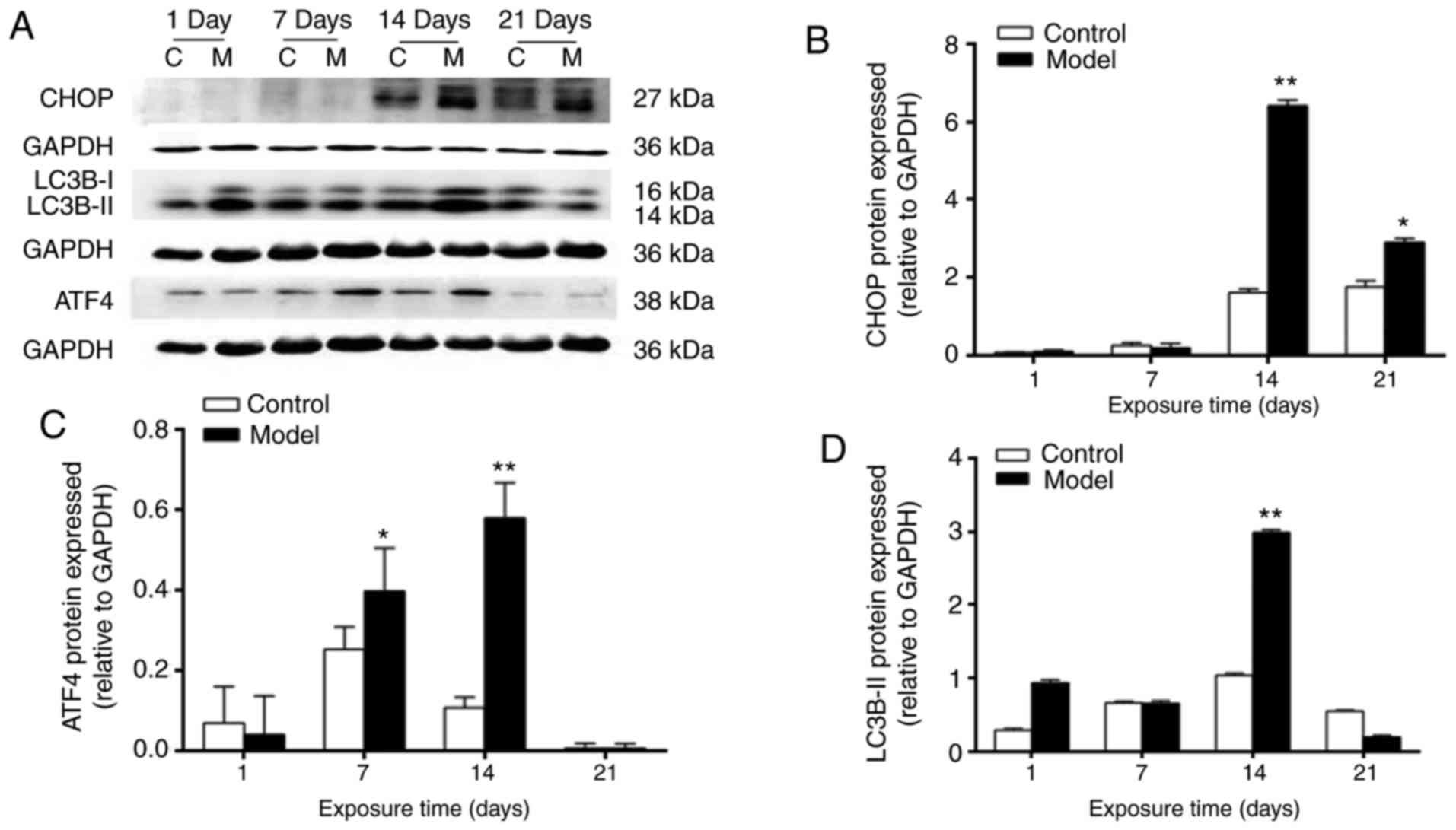 | Figure 5Expression levels of CHOP, LC3B-II,
ATF4 and GAPDH in lung tissues. (A) Western blots of CHOP, LC3B-II,
ATF4 and GAPDH in lung tissues. (B) Relative expression level of
ATF4. (C) Relative expression level of CHOP. (D) Relative
expression level of LC3B-II. *P<0.05 and
*P<0.01, compared with the control group. CHOP, C/EBP
homologous protein; LC3B, microtubule-associated protein light
chain 3β; ATF4, activating transcription factor 4; C, control; M,
model. |
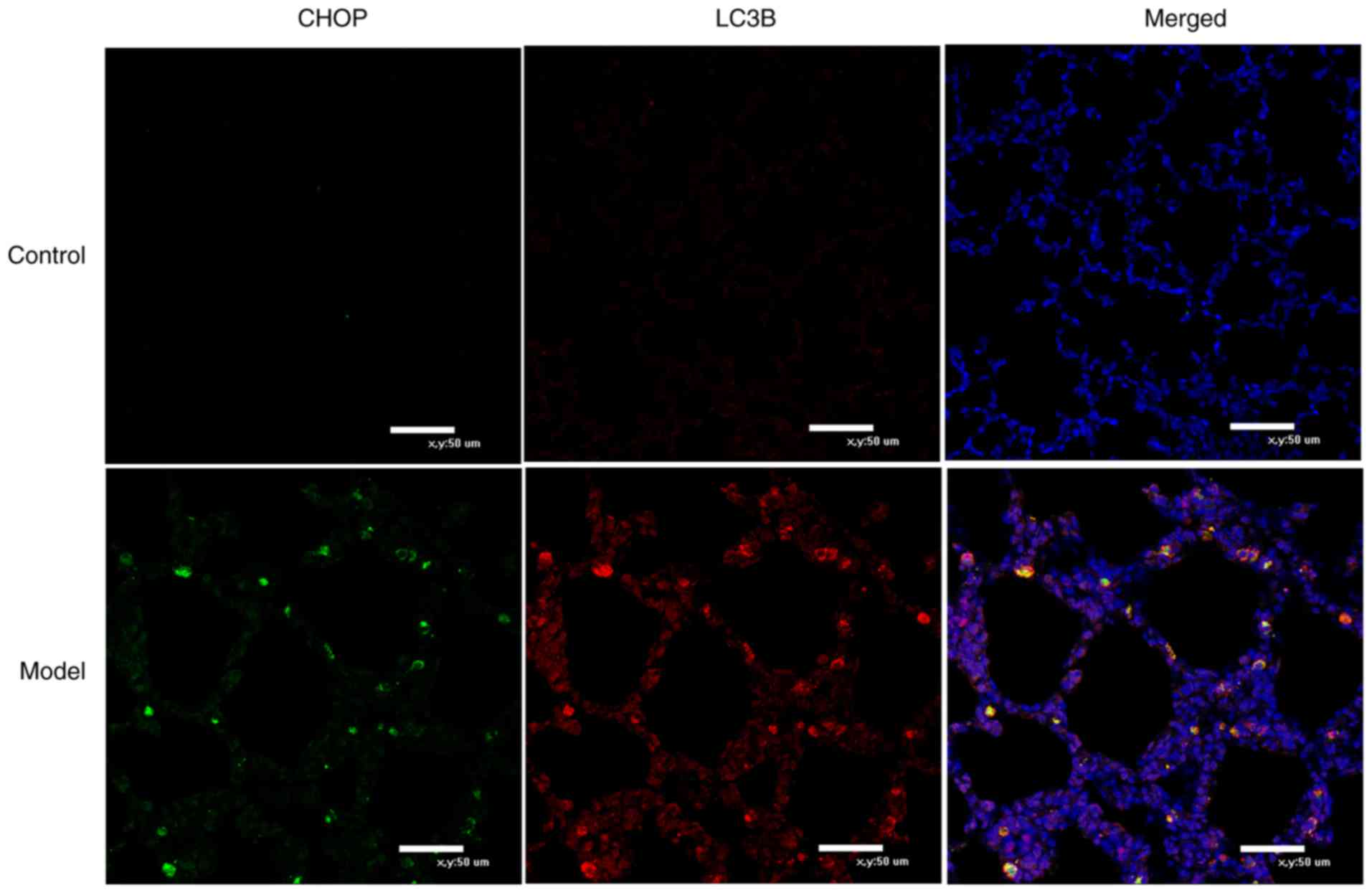
Co-localization of CHOP and LC3B in rat
lung tissues by immunofluorescence
In the control group at 14 days, LC3B, as
represented by red fluorescence, was sparsely distributed in the
alveolar walls and septa of lung tissues. Minimal green
fluorescence-labeled CHOP was evident and the two markers were not
co-expressed. In the model group, LC3B and CHOP were co-expressed
in the cytoplasm of certain cells (Fig. 6).
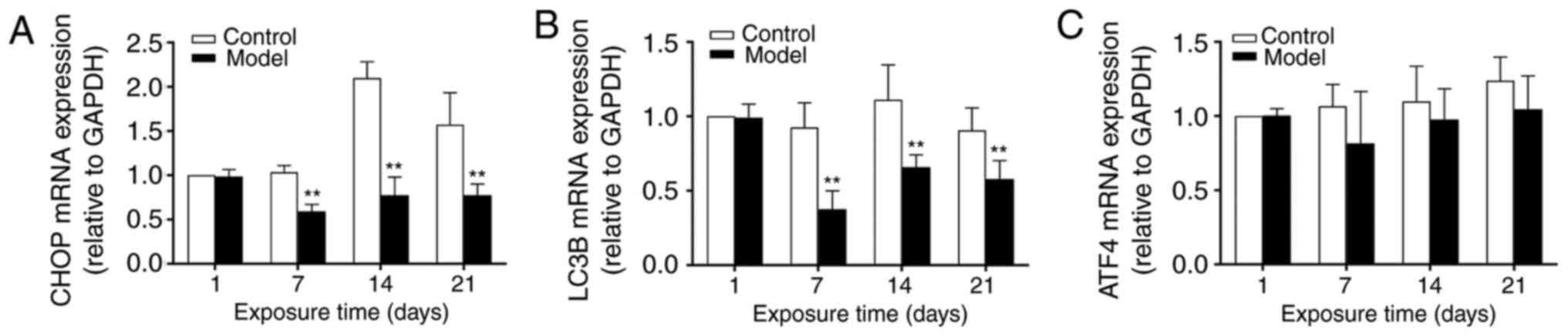
mRNA expression levels of ATF4, CHOP and
LC3B in rat lung tissues
No significant differences were observed in the mRNA
expression of ATF4 between the control and model groups
(P>0.05). In the control group, the mRNA expression level of
CHOP reached a peak at 14 days and remained high at 21 days. In the
model group, the mRNA expression of CHOP remained low with
hyperoxic exposure. At 7, 14 and 21 days, significant differences
between the control and model groups were noted (P<0.01). The
mRNA expression level of LC3B (an autophagic marker) remained high
in the control group. In the model group, it reached a trough at 7
days and was marginally increased at 14 and 21 days, but remained
significantly lower than that in the control group. At 7, 14 and 21
days, significant differences were observed between control and
model groups (P<0.01) (Fig.
7A–C).
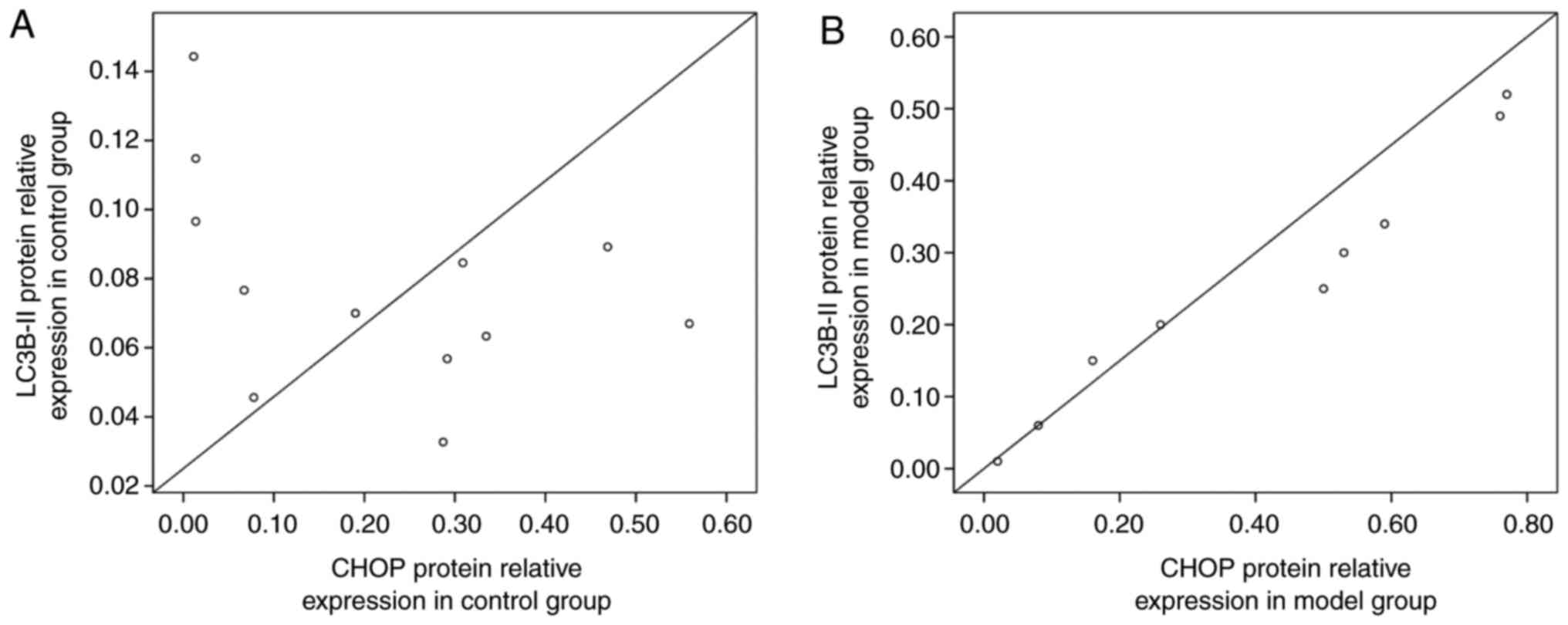
Analysis of the correlation between the
protein expression of CHOP and LC3B-II in rat lung tissues
At 14 days, the correlation between the protein
expression of CHOP and LC3B-II was analyzed. Pearson's correlation
analysis showed that there was a significant positive correlation
between the protein expression levels of CHOP and LC3B-II in the
model group (r2=0.9998,
P<0.01), and a significant positive correlation in the control
group (r2=0.9981, P<0.05)
(Fig. 8A and B).
Discussion
BPD is a common chronic respiratory disease in
premature infants. The morbidity rate of BPD associated with oxygen
therapy remains high in VLBW and in extremely low birth weight
(ELBW) infants. This is despite technological advances and
increased salvage levels in the treatment of premature infants,
improvements in the technology used in neonatal intensive care
units, the popularity of protective assisted ventilation
strategies, and increased developments in the prenatal application
of glucocorticoids and the postnatal application of PS. BPD now has
more mild pathological changes than previously and is known as 'new
BPD'. Various studies have differed in their reports of BPD
morbidity rates, which may be due to differing clinical definitions
of BPD and differences in patient demographics. The incidence rate
of BPD is inversely proportional to birth weight and gestational
age. More specifically, the severity of BPD decreases with
gestational age, and the probability of VLBW and ELBW infants
developing BPD is reported to be up to 25-35% (26). A prospective study completed by
the National Institute Child Health and Human Development on
newborn infants enrolled between October 2000 and June 2002 with a
birth weight of 501-1,249 g found that the prevalence of BPD was
25-35% (27). Similarly, American
demographic data on 4,200,000 newborn infants in 2008 found that
VLBW infants accounted for 1.46%; VLBW and ELBW infants who finally
developed BPD accounted for 20-35% (28). In addition, compared with
full-term infants, VLBW/ELBW infants have a higher mortality rate.
BPD has been reported to induce several respiratory complications
over the longer term and increase the readmission rate of patients,
which was reported to be two times higher than that of non-BPD
patients (29-33). In adolescence, the reserve
function, neural development status and growth limitation of
patients with BPD (34) cause an
increased demand for outpatient services (35). Presently, BPD treatment and its
relevant medical difficulties remain a substantial challenge for
the parents of patients and the medical industry (36); a number of studies have confirmed
that the pathological changes of hyperoxia-induced BPD models
comply with the characteristics of new BPD in premature infants,
and the establishment of BPD in rats has been demonstrated in
numerous studies (37,38).
In the present study, based on domestic and overseas
newborn rat models of hyperoxia-induced BPD, the following were
observed: Gradual thickening of alveolar walls, a decrease in the
number of alveoli, but an increase in the volume of alveoli with
structural simplifications, the interrupted formation of secondary
alveolar septa, a decreased RAC, and a markedly increased alveolar
septum thickness. The above findings indicated the arrest of
alveolar development, which is coincident with the characteristics
of pulmonary maldevelopment in new BPD. These results are
consistent with the results of our previous studies (37,38).
Numerous studies have demonstrated that OS is a key
link in the occurrence of BPD (39-41). For example, Abdel Ghany et
al (39) reported in a study
of 200 neonates that, compared with full-term infants, levels of
antioxidant vitamins A and E and hydrogen peroxidase, and the
overall anti-oxidative level were markedly decreased in premature
infants. Additionally, the level of malondialdehyde, a lipid
peroxidation marker, was markedly increased, whereas that of
vitamin A promoted cell differentiation and the synthesis of
surfactants (42,43). Other studies have revealed that a
higher dose of vitamin A administered enterally in premature
infants may decrease the incidence rates of BPD, intraventricular
hemorrhage and necrotizing enterocolitis, and improve prognosis
(39). The resuscitation of
premature infants with a hypoxic strategy can reduce the demand for
respiratory support, the expression of OS markers and the morbidity
rate of BPD (44). The levels of
total hydrogen peroxide, oxidative protein products and
non-protein-bound iron in the umbilical cord blood have shown a
significant positive correlation with the prevalence of BPD
(45). A hyperoxic environment
leads to the marked aggregation of ROS and thus triggers OS, with
OS and ROS in a mutually causal relationship.
Wang et al found in their study of HepG2
cells that surfactin induced the production of ROS to thus induce
ERS (46). In a study of
atherosclerosis, Toma et al observed that the intracellular
ROS level was decreased if human endothelial cells cultured with
saccharified low density lipoprotein were treated using the ERS
inhibitors salubrinal and polychlorobenzoic acid, whereas the
intracellular level of ERS was lowered if the cells were treated
using the antioxidant, N-acetylcysteine (47).
A number of the aforementioned studies have
suggested an association between ERS and ROS (46-49). For example, ERS can also induce
the production of ROS. Increased ATP is consumed when the ER folds
proteins; however, ATP consumption triggers incorrect protein
folding and elevates the oxidative phosphorylation level of
mitochondria, thus increasing the production of ATP and ROS. The ER
is a Ca2+ bank and unfolded proteins, which accumulate
in the ER, can induce a Ca2+ imbalance, with
Ca2+ released into the ER lumen. The Ca2+
released into the cytoplasm can then produce ROS via an inositol
triphosphate receptor (48,49), thus causing OS. It is crucial for
a cell defense response to maintain the dynamic balance between the
ER and cytoplasm, in which the transmission of ER signals has a
dominant role.
In a rat ischemia-reperfusion brain injury model,
the protective effect of hydrogen-rich water was found to be
associated with a decrease in oxidative products and an increase in
AOEs. In addition, with an increase in the activity of GRP78, a
molecular chaperone (50), and a
decrease in the activity of CHOP, hydrogen-rich water significantly
improved the survival rate and neurological function of rats
(51). Tabas and Ron and
Marciniak et al identified that the ERS-associated factor,
CHOP, was also involved in regulating OS (52,53). Song et al noted that,
following knockout of the CHOP gene, the OS of islet β cells
declined, whereas the antioxidant protein-encoding capacity and
antioxidant function improved (54). Han et al found in a
chromatin immunoprecipitation experiment that CHOP and ATF4 were
able to bind the promoter region of protein synthesis-promoting
genes by interaction, and induced the increased synthesis of
proteins; however, if occurring prior to the recovery of an ER
internal steady state, the increased synthesis of proteins
triggered OS and led to an increase in the aggregation of ROS
(55). The above findings further
suggest that ERS is closely associated with OS.
In the present study, hyperoxic treatment resulted
in the production of ROS during the establishment of a BPD newborn
rat model; ROS may induce cell death, influence the alveolation
process, and cause lung injury and abnormal injury repair (56). AECII cells are the major cells
attacked by ROS in hyperoxia-induced lung injury (41); they are rich in ER, and thus have
the conditions and foundation for the occurrence of ERS.
Previously, it was found that, in addition to necrosis, apoptosis
and transdifferentiation, the ultrastructure of AECII cells changed
during BPD lung injury. For example, microvilli were detached and
sparse, nuclear chromatin became marginally aggregated, lamellar
bodies became damaged and vacuolized, and mitochondria became
swollen. In the present study, it was also observed that the ER,
which had a laminar structure under physiological conditions,
became markedly dilated, pool-shaped and degranulated; surrounding
APs were sparsely distributed. This suggested that ERS is somewhat
associated with autophagy in OS. ERS and autophagy are two
important pathways of cell protein degradation. The latest data
show that ERS adapts to endogenous and exogenous pressures by
strengthening its protein folding capacity and activating
protective mechanisms, including autophagy and an antioxidant
response.
In addition to calcium storage, the ER is an
important organelle for protein synthesis, whereby newly
synthesized proteins are folded into mature proteins in the ER
lumen, and then transferred to the cytoplasm or out of cells. Under
several physiological or pathological conditions, including
hypoxia, viral infection and ischemia, the intracellular steady
state of the ER is damaged and dysfunction occurs. This leads to a
metabolic imbalance of carbohydrates and lipids, and a disturbance
in calcium balance in the ER, in addition to a change in the
environment of the ER lumen for redox reactions. Such a change of
the internal environment leads to the incorrect folding and
maturation of proteins, thus causing the aggregation of incorrectly
folded/unfolded proteins in the ER lumen, leading to a failure in
the effective output of proteins and eventually inducing ERS
(57,58). The imbalance between oxidation and
anti-oxidative defense capacity keeps cells in an OS state, whereas
the ROS produced by OS can attach to the organelle membrane to
induce a peroxidative injury, an important factor in causing
incorrect protein folding in the ER and thus inducing ERS. ERS is
involved in proinflammatory signaling, the apoptosis of AECs, and
the transdifferentiation of epithelial to mesenchymal cells.
CHOP, being downstream of ATF4, is an ERS-specific
transcriptional factor. The expression levels of ATF4 and CHOP were
significantly increased in the presence of ERS, and the formation
of ERS was inhibited when CHOP was knocked down in the cells
(59). In the ERS pathway,
activated CHOP mediates LPS-induced lung injury (60); the over-expression of CHOP in
renal cells and podocytes causes an increase in the production of
ROS, indicating that CHOP is involved in OS (61). In a previous study, Lozon et
al found that, when MLE-12 cells were exposed to 95%
O2 for 24 h, the protein expression of CHOP increased.
In CHOP−/− mice, pneumonedema and increased permeability
occurred under a hyperoxic environment, which suggested that CHOP
may have a protective role in an oxidizing environment (62). Choo-Wing et al showed that
hyperoxic stimulation in a BPD mouse model led to a marked increase
in the protein and mRNA expression levels of CHOP, in vitro
and in vivo. The knockout of CHOP by siRNA was found to
reduce mouse MLE-12 cell death and relieve hyperoxia-induced
alveolation disorders. Coincidently, CHOP levels were increased in
the lung tissues of patients with BPD (63). Lu et al demonstrated that
the protein level of CHOP was significantly increased during
hyperoxic exposure (64).
In the present study, it was found that the protein
levels of ATF4 and CHOP in the model group were higher compared
with those in the control group at 7 days, which was consistent
with the study results of Choo-Wing et al (63) and Lu et al (64) The protein expression level of CHOP
reached a peak at 14 days and was marginally decreased at 21 days
in the present study, whereas it was continuously increased in the
study of Lu et al. One possible cause for this difference
may be that premature rats were delivered by cesarean section at
day 21 of gestation in the model used by Lu et al, whereas
the newborn rats were delivered spontaneously at 22 days of
gestation in the present study. Such a difference in the premature
reduction of the protein expression level of CHOP may be attributed
to the varying anti-oxidative defense capacity of individuals. No
significant difference was found in mRNA expression level of ATF4
between the control and model groups. The mRNA expression level of
CHOP was significantly higher in the control group, compared with
that in the model group. This differs from the results of the above
study on the protein expression level of CHOP, and the high protein
expression of CHOP induced by ERS can promote apoptosis. Several
studies have shown that minimal CHOP protein is expressed in normal
tissues and cells (65).
Therefore, the differential protein and mRNA expression of CHOP may
be regulated not at the gene level, but at a post-transcriptional
level, by, for example, via shortening of protein half-life, enzyme
degradation or mRNA regulation.
In the present study, it was revealed by TEM that,
compared with the control group, the morphology and organelle
structure of AECII cells were seriously damaged; the ER was dilated
and surrounded by marginally aggregated APs with a double-layer
membrane, Autophagy of AECII cells in the model group was also
noted at day 14. This intuitively indicates that ERS and autophagy
co-exist in AECII cells. Studies have shown that autophagy has a
specific function in AECII cells, namely, it is involved in the
synthesis of pulmonary surfactants. CHOP is a recognized mediator
of apoptosis; however, a previous study suggested that CHOP
promoted a survival-promoting autophagic process via synergistic
reaction with ATF4 (66). LC3B-II
is a key protein for the maturation of APs; following the
activation of autophagy, the autophagic marker LC3B-I forms LC3B-II
by esterified coupling with phosphatidyl-ethanolamine. LC3B-II then
translocates to the membrane of APs, with the level of LC3B-II
reflecting the number of APs to a certain degree (67). Zhang et al identified that,
with the prolongation of hyperoxic exposure time, the level of
LC3B-II was markedly increased. The AECII-to-AECI
transdifferentiation capacity is partially restored by suppressing
the level of LC3B-II (21).
The present study revealed that the protein
expression level of LC3B-II in the model group was significantly
higher, compared with that in the control group at 7 days, and
reached a peak at 14 days, which coincided with the study results
of Zhang et al, who did not investigate a 21-day group. In
the present study, following 21 days of hyperoxic exposure, the
level of autophagy was decreased or suppressed. In addition,
observations at a gene level showed that the mRNA expression level
of LC3B was markedly lower in the model group, compared with that
in the control group at ≥7 days. This suggested that
autophagy/autophagic flow, or post-transcriptional regulation may
have been suppressed. It is generally considered that the formation
and degradation of APs are in a dynamic balance, and when such a
balance is damaged, if the degradation of APs is limited or
autophagy is suppressed, it affects the production of APs to a
certain extent. Autophagy, as an outlet of ERS, is one of the
important pathways for protein degradation in cells, and can
increase the threshold for the promotion of apoptosis in the dual
effects of ERS and reduce the pressure load of cells. The
suppression of autophagic flow can increase the aggregation of
proteins and worsen ERS (68-70).
Previous studies have suggested that there is an
association between ERS and autophagy. These two systems may be
mutually independent in function, but have a dependent association
in regulation; changing the activity of one system may influence
the other. According to previous studies, continuous ERS may
increase the activity of autophagy (71), whereas autophagy may alleviate ERS
by eliminating unfolded proteins; in contrast, interrupting
autophagy may worsen ERS (72,73). However, there have been few
studies on the association between autophagy and ERS in BPD. A
series of electron microscopic results have provided evidence for
the role of the ER membrane in the formation and maturation of APS.
The immunoelectron microscopy study by Dunn showed that APs
acquired lysosomal markers from the ER during their formation and
gradual maturation (74). Ueno
et al found that, following treatment with leupeptin, an ER
membrane marker was present on the vesicle membrane of APs isolated
from the rat liver (75). Bernale
et al observed a dilated ER with the formation of AP-like
structures, and that intensive and selective enclosure was from the
membrane components of the UPR (71). Axe et al noted that AP
formation from membrane compartments was enriched in
phosphatidylinositol 3-phosphate and dynamically connected to the
ER, which showed a close association between the formation of APs
and the ER (76). Hart et
al suggested that at least part of the membrane of APs was
derived from the ER membrane (77).
Using TEM and immunofluorescence techniques, the
present study observed the co-presence of ERS and autophagy in
AECII cells, and the aggregation of APs surrounding dilated ER. In
the model group, CHOP and LC3B were simultaneously increased at 7
days, and reached a peak at 14 days, indicating a simultaneous
increase of ERS and levels of autophagy during hyperoxic exposure.
Peaks formed at the same time, and an almost consistent change in
trend suggested that CHOP and LC3B may influence and promote each
other, and be mutually dependent.
A previous chromatin immunoprecipitation experiment
demonstrated that ATF4 and CHOP directly induce the transcriptional
expression of autophagy (Atg)-related genes Atg10, Atg5, p62 and
Atg7 by binding with the GTGCAACC region of cis-acting
elements in the promoters of these genes. These genes have a
non-negligible role in the formation, prolongation and functional
maintenance of autophagosomes (78). Based on previous reports, ATF4 and
CHOP have a targeted effect on LC3B-II and Atg5 via the PKR-like
endoplasmic reticulum kinase-eIF2a pathway (79,80).
The high protein expression level of CHOP is
associated with the esterification capacity of LC3B, and can
promote the transformation of LC3B-I to LC3B-II (78). Another study demonstrated that
autophagy regulated the transcription of CHOP, and that CHOP
promoted survival via autophagy (66) to offset excessive UPR signals,
which further suggests a possible co-dependent association between
CHOP and autophagy. However, the present study found that, at 21
days, the level of CHOP in the model group was higher, compared
with that in the control group, whereas the level of LC3B was
lower. This suggested that the autophagosome elimination pathway
may be inhibited in this phase, causing the suppression of
autophagic flow and the inability of cells to degrade proteins via
an autophagy pathway. As a result, the expression level of CHOP was
continuously high, but remained lower, compared with that at day
14. This further indicated that CHOP and LC3B may have a mutually
dependent relationship, which is consistent with the views of Xu
et al (81) as well as the
views of other studies (82-84). Their views were specifically as
follows: There is an interaction between ERS and autophagy;
continuous ERS can increase autophagic activity; autophagy may
alleviate ERS by eliminating unfolded proteins, and, in contrast,
interrupting autophagy may increase ERS. The double
immunofluorescence staining in the present study showed that CHOP
and LC3B were co-expressed in the model group at day 14. Analysis
at this time-point showed a positive correlation between CHOP and
LC3B in the control and model groups. This further supported the
above views.
Taken together, the results of the present study
suggested that hyperoxic stimulation activated ERS and autophagy.
The AECII cells of newborn rats under hyperoxic exposure exhibited
dilated, pool-shaped ER and APs containing ER and other organelles.
In addition, CHOP and LC3B were overex-pressed and correlated with
each other, with both increasing and reaching a peak at the same
time. This suggested that ERS and autophagic activation may be
associated with and regulate the occurrence and development of BPD.
This may be a novel and potential method to treat hyperoxia-induced
BPD effectively in clinic, by adjusting the balance of ERS and
autophagy. However, the specific mechanisms involved in ERS and
autophagy under hyperoxic conditions were not investigated in the
present study; these mechanisms require further investigation.
Acknowledgments
The authors would like to thank Liu Dongyan and Miao
Jianing for their excellent technical assistance.
Abbreviations:
|
BPD
|
bronchopulmonary dysphasia
|
|
HE
|
hematoxylin and eosin
|
|
RAC
|
radial alveolar count
|
|
TEM
|
transmission electron microscopy
|
|
ER
|
endoplasmic reticulum
|
|
ROS
|
reactive oxygen species
|
|
AECII
|
type II alveolar epithelial cells
|
|
OS
|
oxidative stress
|
|
FRs
|
free radicals
|
|
AOEs
|
antioxidant enzymes
|
|
PS
|
pulmonary surfactants
|
|
VLBW
|
very low birth weight
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
UPR
|
unfolded protein response
|
|
APs
|
autophagosomes
|
Funding
This study was supported by the National Natural
Science Foundation of China (grant no. 81571479) and the Key
laboratory of basic research projects in Liaoning province
Department of Education (grant no. LZ2015070).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
ML was involved in drafting the manuscript or
revising it critically for important intellectual content. BP
collected the rat lung tissue and made contributions to the
bronchopulmonary dysphasia rat model. YS made contributions to
analysis and interpretation of data. JF made contributions to the
conception and design of the project. XX gave final approval of the
version to be published.
Ethics approval and consent to
participate
All animal experiments were approved by the Animal
Ethics Committee of China Medical University (no. 2015PS230K), and
all surgical procedures were performed under anesthesia with
chloral hydrate to minimize pain of the experimental animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Isayama T, Lee SK, Mori R, Kusuda S,
Fujimura M, Ye XY and Shah PS; Canadian Neonatal Network, Neonatal
Research Network of Japan: Comparison of mortality and morbidity of
very low birth weight infants between Canada and Japan. Pediatrics.
130:e957–e965. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Silva DM, Nardiello C, Pozarska A and
Morty RE: Recent advances in the mechanisms of lung alveolarization
and the pathogenesis of bronchopulmonary dysplasia. Am J Physiol
Lung Cell Mol Physiol. 309:L1239–L1272. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Dani C and Poggi C: The role of genetic
polymorphisms in antioxidant enzymes and potential antioxidant
therapies in neonatal lung disease. Antioxid Redox Signal.
21:1863–1880. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gitto E, Pellegrino S, D'Arrigo S, Barberi
I and Reiter RJ: Oxidative stress in resuscitation and ventilation
of newborns. Eur Respir J. 34:1461–1469. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Perrone S, Negro S, Tataranno ML and
Buonocore G: Oxidative stress and antioxidant strategies in
newborns. J Matern Fetal Neonatal Med Suppl. 3:63–65. 2010.
View Article : Google Scholar
|
|
6
|
Perrone S, Tataranno ML and Buonocore G:
Oxidative stress and bronchopulmonary dysplasia. J Clin Neonatol.
1:109–114. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wang X, Li W, Liu W, Cai B, Cheng T, Gao
C, Mo L, Yang H and Chang L: GSTM1 and GSTT1 gene polymorphisms as
major risk factors for bronchopulmonary dysplasia in a Chinese Han
population. Gene. 533:48–51. 2014. View Article : Google Scholar
|
|
8
|
Hsiao CC, Chang JC, Tsao LY, Yang RC, Chen
HN, Lee CH, Lin CY and Tsai YG: Correlates of elevated
interleukin-6 and 8-Hydroxy-2′-Deoxyguanosine levels in tracheal
aspirates from very low birth weight infants who develop
bronchopulmonary dysplasia. Pediatr Neonatol. 58:63–69. 2017.
View Article : Google Scholar
|
|
9
|
Fabiano A, Gavilanes AW, Zimmermann LJ,
Kramer BW, Paolillo P, Livolti G, Picone S, Bressan K and Gazzolo
D: The development of lung biochemical monitoring can play a key
role in the early prediction of bronchopulmonary dysplasia. Acta
Paediatr. 105:535–541. 2016. View Article : Google Scholar
|
|
10
|
Kaya G, Saldir M, Polat A, Fidanci MK,
Erdem A, Erdem G, Kurt YG, Cetinkaya M, Cekmez F, Onguru O and Tunc
T: Evaluation of etanercept treatment in newborn rat model with
hyperoxic lung injury. Fetal Pediatr Pathol. 35:327–338. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Görlach A, Klappa P and Kietzmann T: The
endoplasmic reticulum: Folding, calcium homeostasis, signaling, and
redox control. Antioxid Redox Signal. 8:1391–1418. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tu BP and Weissman JS: Oxidative protein
folding in eukaryotes: Mechanisms and consequences. J Cell Biol.
164:341–346. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sevier CS and Kaiser CA: Ero1 and redox
homeostasis in the endoplasmic reticulum. Biochim Biophys Acta.
1783:549–556. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Younce CW, Wang K and Kolattukudy PE:
Hyperglycaemia-induced cardiomyocyte death is mediated via MCP-1
production and induction of a novel zinc-finger protein MCPIP.
Cardiovasc Res. 87:665–674. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rothermel BA and Hill JA: Myocyte
autophagy in heart disease: Friend or foe? Autophagy. 3:632–634.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Takemura G, Miyata S, Kawase Y, Okada H,
Maruyama R and Fujiwara H: Autophagic degeneration and death of
cardiomyocytes in heart failure. Autophagy. 2:212–214. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jian X, Xiao-yan Z, Bin H, Yu-feng Z, Bo
K, Zhi-nong W and Xin N: MiR-204 regulate cardiomyocyte autophagy
induced by hypoxia-reoxygenation through LC3-II. Int J Cardiol.
148:110–112. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang C, Li YZ, Wang XR, Lu ZR, Shi DZ and
Liu XH: Panax quinquefolium saponins reduce myocardial
hypoxia-reoxygenation injury by inhibiting excessive endoplasic
reticulum stress. Shock. 37:228–233. 2012. View Article : Google Scholar
|
|
19
|
Fan T, Huang Z, Chen L, Wang W, Zhang B,
Xu Y, Pan S, Mao Z, Hu H and Geng Q: Associations between
autophagy, the ubiquitin-proteasome system and endoplasmic
reticulum stress in hypoxia-deoxygenation or ischemia-reperfusion.
Eur J Pharmacol. 791:157–167. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang L, Zhao S, Yuan LJ, Wu HM, Jiang H,
Zhao SM, Luo G and Xue XD: Autophagy regulates hyperoxia-induced
intracellular accumulation of surfactant protein C in alveolar type
II cells. Mol Cell Biochem. 408:181–189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang L, Zhao S, Yuan L, Wu H, Jiang H and
Luo G: Hyperoxia-mediated LC3B activation contributes to the
impaired transdifferentiation of type II alveolar epithelial cells
(AECIIs) to type I cells (AECIs). Clin Exp Pharmacol Physiol.
43:834–843. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
You K, Xu X, Fu J, Xu S, Yue X, Yu Z and
Xue X: Hyperoxia disrupts pulmonaryepithelial barrier in newborn
rats via the deterioration of occludin and ZO-1. Respir Res.
13:362012. View Article : Google Scholar
|
|
23
|
Cooney TP and Thurlbeck WM: The radial
alveolar count method of Emery and Mithal: A reappraisal
1-postnatal lung growth. Thorax. 37:572–579. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hou A, Fu J, Yang H, Zhu Y, Pan Y, Xu S
and Xue X: Hyperoxia stimulates the transdifferentiation of type II
alveolar epithelial cells in newborn rats. Am J Physiol Lung Cell
Mol Physiol. 308:L861–L872. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−delta delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
26
|
Abman SH and Groothius JR: Pathophysiology
and treatment of bronchopulmonary dysplasia. Current issues.
Pediatr Clin North Am. 41:277–315. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Walsh MC, Yao Q, Gettner P, Hale E,
Collins M, Hensman A, Everette R, Peters N, Miller N, Muran G, et
al: Impact of a physiologic definition on bronchopulmonary
dysplasia rates. Pediatrics. 114:1305–1311. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Martin JA, Hamilton BE, Sutton PD, Ventura
SJ, Mathews TJ and Osterman MJ: Births: Final data for 2008. Natl
Vital Stat Rep. 59(1): 3–71. 2010.
|
|
29
|
Chien YH, Tsao PN, Chou HC, Tang JR and
Tsou KI: Rehospitalization of extremely-low-birth-weight infants in
first 2 years of life. Early Hum Dev. 66:33–40. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Doyle LW, Ford G and Davis N: Health and
hospitalisations after discharge in extremely low birth weight
infants. Semin Neonatol. 8:137–145. 2003. View Article : Google Scholar
|
|
31
|
Lamarche-Vadel A, Blondel B, Truffer P,
Burguet A, Cambonie G, Selton D, Arnaud C, Lardennois C, du
Mazaubrun C, N'Guyen S, et al: Rehospitalization in infants younger
than 29 weeks' gestation in the EPIPAGE cohort. Acta Paediatr.
93:1340–1345. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Luu TM, Lefebvre F, Riley P and
Infante-Rivard C: Continuing utilisation of specialised health
services in extremely preterm infants. Arch Dis Child Fetal
Neonatal Ed. 95:F320–F325. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Smith VC, Zupancic JA, McCormick MC, Croen
LA, Greene J, Escobar GJ and Richardson DK: Rehospitalization in
the first year of life among infants with bronchopulmonary
dysplasia. J Pediatr. 144:799–803. 2004.PubMed/NCBI
|
|
34
|
Broström EB, Thunqvist P, Adenfelt G,
Borling E and Katz-Salamon M: Obstructive lung disease in children
with mild to severe BPD. Respir Med. 104:362–370. 2010. View Article : Google Scholar
|
|
35
|
Hintz SR, Kendrick DE, Vohr BR, Poole WK
and Higgins RD; National institute of child health and human
development (NICHD) neonatal research network: Community supports
after surviving extremely low-birthweight, extremely preterm birth:
Special outpatient services in early childhood. Arch Pediatr
Adolesc Med. 162:748–755. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Groothuis JR and Makari D: Definition and
outpatient management of the very low-birth-weight infant with
bronchopulmonary dysplasia. Adv Ther. 29:297–311. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yang H, Fu J, Xue X, Yao L, Qiao L, Hou A,
Jin L and Xing Y: Epithelial-mesenchymal transitions in
bronchopulmonary dysplasia of newborn rats. Pediatr Pulmonol.
49:1112–1123. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Hou A, Fu J, Shi Y, Qiao L, Li J, Xing Y
and Xue X: Decreased ZONAB expression promotes excessive
transdifferentiation of alveolar epithelial cells in
hyperoxia-induced bronchopulmonary dysplasia. Int J Mol Med.
41:2339–2349. 2018.PubMed/NCBI
|
|
39
|
Abdel Ghany EA, Alsharany W, Ali AA,
Younass ER and Hussein JS: Anti-oxidant profiles and markers of
oxidative stress in preterm neonates. Paediatr Int Child Health.
36:134–140. 2016. View Article : Google Scholar
|
|
40
|
Jobe AH and Bancalari E: Bronchopulmonary
dysplasia. Am J Respir Crit Care Med. 163:1723–1729. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Saugstad OD: Oxygen and oxidative stress
in bronchopulmonary dysplasia. J Perinat Med. 38:571–577. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Biesalski HK and Nohr D: Importance of
vitamin-A for lung function and development. Mol Aspects Med.
24:431–440. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Metzler MD and Snyder JM: Retinoic acid
differentially regulates expression of surfactant associated
proteins in human fetal lung. Endocrinology. 133:1990–1998. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Vento M, Moro M, Escrig R, Arruza L,
Villar G, Izquierdo I, Roberts LJ II, Arduini A, Escobar JJ, Sastre
J and Asensi MA: Preterm resuscitation with low oxygen causes less
oxidative stress, inflammation and chronic lung disease.
Pediatrics. 124:e439–e449. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Perrone S, Tataranno ML, Negro S, Longini
M, Marzocchi B, Proietti F, Iacoponi F, Capitani S and Buonocore G:
Early identification of the risk for free radical-related diseases
in preterm newborns. Early Hum Dev. 86:241–244. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang CL, Liu C, Niu LL, Wang LR, Hou LH
and Cao XH: Surfactin-induced apoptosis through
ROS-ERS-Ca2+-ERK pathways in HepG2 cells. Cell Biochem
Biophys. 67:1433–1439. 2013. View Article : Google Scholar :
|
|
47
|
Toma L, Sanda GM, Deleanu M, Stancu CS and
Sima AV: Glycated LDL increase VCAM-1 expression and secretion in
endothelial cells and promote monocyte adhesion through mechanisms
involving endoplasmic reticulum stress. Mol Cell Biochem.
417:169–179. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Malhotra JD and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress: A vicious cycle or a
double-edged sword? Antioxid Redox Signal. 9:2277–2293. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Cioffi DL: Redox regulation of endothelial
canonical transient receptor potential channels. Antioxid Redox
Signal. 15:1567–1582. 2011. View Article : Google Scholar :
|
|
50
|
Flodby P, Li C, Liu Y, Wang H, Marconett
CN, Laird-Offringa IA, Minoo P, Lee AS and Zhou B: GRP78 regulates
ER homeostasis and distal epithelial cell survival during lung
development. Am J Respir Cel Mol Biol. 55:135–149. 2016. View Article : Google Scholar
|
|
51
|
Gao Y, Gui Q, Jin L, Yu P, Wu L, Cao L,
Wang Q and Duan M: Hydrogen-rich saline attenuates hippocampus
endoplasmic reticulum stress after cardiac arrest in rats. Neurosci
Lett. 640:29–36. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Tabas I and Ron D: Integrating the
mechanisms of apoptosis induced by endoplasmic reticulum stress.
Nat Cell Biol. 13:184–190. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Marciniak SJ, Yun CY, Oyadomari S, Novoa
I, Zhang Y, Jungreis R, Nagata K, Harding HP and Ron D: CHOP
induces death by promoting protein synthesis and oxidation in the
stressed endoplasmic reticulum. Genes Dev. 18:3066–3077. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Song B, Scheuner D, Ron D, Pennathur S and
Kaufman RJ: Chop deletion reduces oxidative stress, improves beta
cell function, and promotes cell survival in multiple mouse models
of diabetes. J Clin Invest. 118:3378–3389. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Han J, Back SH, Hur J, Lin YH,
Gildersleeve R, Shan J, Yuan CL, Krokowski D, Wang S, Hatzoglou M,
et al: ER-stress-induced transcriptional regulation increases
protein synthesis leading to cell death. Nat Cell Biol. 15:481–490.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Zhang X, Shan P, Sasidhar M, Chupp GL,
Flavell RA, Choi AM and Lee PJ: Reactive oxygen species and
extracellular signal-regulated kinase 1/2 mitogen-activated protein
kinase mediate hyperoxia-induced cell death in lung epithelium. Am
J Respir Cell Mol Biol. 28:305–315. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Cao SS and Kaufman RJ: Targeting
endoplasmic reticulum stress in metabolic disease. Expert Opin Ther
Targets. 17:437–448. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Roussel BD, Kruppa AJ, Miranda E, Crowther
DC, Lomas DA and Marciniak SJ: Endoplasmic reticulum dysfunction in
neurological disease. Lancet Neurol. 12:105–118. 2013. View Article : Google Scholar
|
|
59
|
Kroemer G and Blomgren K: Mitochondrial
cell death control in familial Parkinson disease. PLoS Biol.
5:e2062007. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Endo M, Oyadomari S, Suga M, Mori M and
Gotoh T: The ER stress pathway involving CHOP is activated in the
lungs of LPS-treated mice. J Biochem. 138:501–507. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Bek MF, Bayer M, Müller B, Greiber S, Lang
D, Schwab A, August C, Springer E, Rohrbach R, Huber TB, et al:
Expression and function of C/EBP homology protein (GADD153) in
podocytes. Am J Pathol. 168:20–32. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Lozon TI, Eastman AJ, Matute-Bello G, Chen
P, Hallstrand TS and Altemeier WA: PKR-dependent CHOP induction
limits hyperoxiainduced lung injury. Am J Physiol Lung Cell Mol
Physiol. 300:L422–L429. 2011. View Article : Google Scholar
|
|
63
|
Choo-Wing R, Syed MA, Harijith A, Bowen B,
Pryhuber G, Janér C, Andersson S, Homer RJ and Bhandari V:
Hyperoxia and interferon-γ-induced injury in developing lungs occur
via cyclo-oxygenase-2 and theendoplasmic reticulum stress-dependent
pathway. Am J Respir Cell Mol Biol. 48:749–757. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Lu HY, Zhang J, Wang QX, Tang W and Zhang
LJ: Activation of the endoplasmic reticulum stress pathway
involving CHOP in the lungs of rats with hyperoxia-induced
bronchopulmonary dysplasia. Mol Med Rep. 12:4494–4500. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bozi LH, Jannig PR, Rolim N, Voltarelli
VA, Dourado PM, Wisløff U and Brum PC: Aerobic exercise training
rescues cardiac protein quality control and bluntsendoplasmic
reticulum stress in heart failure rats. J Cell Mol Med.
20:2208–2212. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
B'Chir W, Chaveroux C, Carraro V, Averous
J, Maurin AC, Jousse C, Muranishi Y, Parry L, Fafournoux P and
Bruhat A: Dual role for CHOP in the crosstalk between autophagy and
apoptosis to determine cell fate in response to amino acid
deprivation. Cell Signal. 26:1385–1391. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kouno T, Mizuguchi M, Tanida I, Ueno T,
Kanematsu T, Mori Y, Shinoda H, Hirata M, Kominami E and Kawano K:
Solution structure of microtubule-associated protein light chain 3
and identification of its functional subdomains. J Biol Chem.
280:24610–24617. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Schönthal AH: Endoplasmic reticulum stress
and autophagy as targets for cancer therapy. Cancer Lett.
275:163–169. 2009. View Article : Google Scholar
|
|
69
|
Høyer-Hansen M and Jäättelä M: Connecting
endoplasmic reticulum stress to autophagy by unfolded protein
response and calcium. Cell Death Differ. 14:1576–1582. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Mathew R, Karantza-Wadsworth V and White
E: Role of autophagy in cancer. Nat Rev Cancer. 7:961–967. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Bernales S, McDonald KL and Walter P:
Autophagy counterbalances endoplasmic reticulum expansion during
the unfolded protein response. PLoS Biol. 4:e4232006. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Cheng Y and Yang JM: Survival and death of
endoplasmic-reticulum-stressed cells: Role of autophagy. World J
Biol Chem. 2:226–231. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Schleicher SM, Moretti L, Varki V and Lu
B: Progress in the unraveling of the endoplasmic reticulum
stress/autophagy pathway and cancer: Implications for future
therapeutic approaches. Drug Resist Updat. 13:79–86. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dunn WA Jr: Studies on the mechanisms of
autophagy: Formation of the autophagic vacuole. J Cell Biol.
110:1923–1933. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Ueno T, Muno D and Kominami E: Membrane
markers of endoplasmic reticulum preserved in autophagic vacuolar
membranes isolated from leupeptin-administered rat liver. J Biol
Chem. 266:18995–18999. 1991.PubMed/NCBI
|
|
76
|
Axe EL, Walker SA, Manifava M, Chandra P,
Roderick HL, Habermann A, Griffiths G and Ktistakis NT:
Autophagosome formation from membrane compartments enriched in
phosphatidylinositol 3-phosphate and dynamically connected to the
endoplasmic reticulum. J Cell Biol. 182:685–701. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Hart LS, Cunningham JT, Datta T, Dey S,
Tameire F, Lehman SL, Qiu B, Zhang H, Cerniglia G, Bi M, et al: ER
stress-mediated autophagy promotes Myc-dependent transformation and
tumor growth. J Clin Invest. 122:4621–4634. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
B'chir W, Maurin AC, Carraro V, Averous J,
Jousse C, Muranishi Y, Parry L, Stepien G, Fafournoux P and Bruhat
A: The eIF2α/ATF4 pathway is essential for stress-induced autophagy
gene expression. Nucleic Acids Res. 41:7683–7699. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Rouschop KM, van den Beucken T, Dubois L,
Niessen H, Bussink J, Savelkouls K, Keulers T, Mujcic H, Landuyt W
and Voncken JW: The unfolded protein response protects human tumor
cells during hypoxia through regulation of the autophagy genes
MAP1LC3B and ATG5. J Clin Invest. 120:127–141. 2010. View Article : Google Scholar :
|
|
80
|
Avivar-Valderas A, Salas E,
Bobrovnikova-Marjon E, Diehl JA, Nagi C, Debnath J and
Aguirre-Ghiso JA: PERK integrates autophagy and oxidative stress
responses to promote survival during extracellular matrix
detachment. Mol Cell Biol. 31:3616–3629. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Xu Y, Yu H, Qin H, Kang J, Yu C, Zhong J,
Su J, Li H and Sun L: Inhibition of autophagy enhances cisplatin
cytotoxicity through endoplasmic reticulum stress in human cervical
cancer cells. Cancer Lett. 314:232–243. 2012. View Article : Google Scholar
|
|
82
|
Kouroku Y, Fujita E, Tanida I, Ueno T,
Isoai A, Kumagai H, Ogawa S, Kaufman RJ, Kominami E and Momoi T: ER
stress (PERK/eIF2alpha phosphorylation) mediates the
polyglutamine-induced LC3 conversion, an essential step for
autophagy formation. Cell Death Differ. 14:230–239. 2007.
View Article : Google Scholar
|
|
83
|
Nijholt DA, de Graaf TR, van Haastert ES,
Oliveira AO, Berkers CR, Zwart R, Ovaa H, Baas F, Hoozemans JJ and
Scheper W: Endoplasmic reticulum stress activates autophagy but not
the proteasome in neuronal cells: Implications for Alzheimer's
disease. Cell Death Differ. 18:1071–1081. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Adolph TE, Tomczak MF, Niederreiter L, Ko
HJ, Böck J, Martinez-Naves E, Glickman JN, Tschurtschenthaler M,
Hartwig J, Hosomi S, et al: Paneth cells as a site of origin for
intestinal inflammation. Nature. 503:272–276. 2013. View Article : Google Scholar : PubMed/NCBI
|