Introduction
Diabetes mellitus (DM) is caused by dysfunction of
the endocrine system; this metabolic dysfunction is characterized
by impaired metabolism of proteins, carbohydrates or fats, and
hyperglycemia (1). Type 2-DM is a
major public health concern, which is increasing in frequency
worldwide. In addition, it is commonly associated with various
complications and is a major chronic disease (2). It has been predicted that the
prevalence of type 2-DM will continue to rise, with >7.5% of the
global population thought to be affected by 2030 (2). Therefore, more effective methods and
therapeutic agents for treating type 2-DM and its associated
complications are urgently required (3).
A growing body of evidence has indicated that
microRNAs (miRNAs/miRs) serve important roles in the
post-transcriptional regulation of gene expression (4). Transfection of cells with
appropriate miRNAs is one way to induce the generation of
pancreatic β-cells (5). The
regulatory effects of miRNAs on cell proliferation, differentiation
and fate indicate that miRNAs serve key roles in animal development
(6-8). Embryonic stem cells could be
differentiated into fully functional islet cells, which would be
able to secrete insulin and thus, could be an alternative method to
replace damaged pancreatic islet β-cells. In recent years, it has
been indicated that the regeneration and differentiation of islet
β-cells may be induced by regulating miRNA expression (9). Similarly, the development of islet
β-cells has been revealed to be regulated by the miR-200 family and
miR-30, which control the transition from epithelial to mesenchymal
cells (9). The dynamic expression
patterns of miR-375 and miR-7 in the stem cell differentiation
process are similar to those of miR-146a and miR-34a expression in
the developing pancreas of the human fetus (10). Furthermore, V-Maf avian
musculoaponeurotic fibrosarcoma oncogene homolog B and Forkhead box
(FoxO)-A2 are targeted by miR-342, which is also associated with
the maturation and differentiation of islet β-cells (9). During the development and
differentiation of β-cells, the key transcription factors
associated with these processes are specifically targeted by miRNAs
(9,10). Therefore, miRNAs may be an
efficient, alternative strategy for the regeneration of islet
cells.
Type 2-DM has been consistently associated with
hypoadiponectinemia, which is associated with adiponectin and its
receptors. Therefore, the present study used the online software
TargetScan (targetscan.org/) to predict the
miRNAs that may regulate the expression of adiponectin receptor 1
(AdipoR1) in β-cells. It was subsequently identified that AdipoR1
may be a potential target gene of miR-6835-3p.
The present study used the MIN-6 pancreatic β-cell
line (Mus musculus; transgenic for SV40 large T antigen),
which was derived from insulinoma, and the SU.86.86 cell line
(CRL-1837™), which has pancreatic tissue origin (Homo
sapiens) and was derived from metastatic ductal carcinoma of
the liver. The aim of the present study was to evaluate the effects
of miRNA on pancreatic cell function. The two pancreatic cell
lines, SU.86.86 and MIN-6, were used to establish an appropriate
model for in vitro functional experiments.
Previous research (7) performed by our group assessed the
effects of miR-6835-3p on SU.86.86 and MIN-6 cell lines, and
revealed that AdipoR1 mRNA contained one miR-6835-3p target
sequence. The present results confirmed that AdipoR1 mRNA may be a
target of miR-6835-3p in islet β-cells. In addition, AdipoR1 may
affect the biological functions of the SU.86.86 and MIN-6 cell
lines. Therefore, the present study aimed to investigate the in
vitro effects of miR-6835-3p on the insulin secretory function
of β-cells.
Materials and methods
Cell lines
The human pancreatic ductal carcinoma cell line
SU.86.86 (CRL-1837) and the mouse pancreatic islet cell line MIN-6
were purchased from American Type Culture Collection (Manassas, VA,
USA). Cells were cultured in Dulbecco’s modified Eagle’s medium
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 15% heat-inactivated fetal bovine serum (Gibco;
Thermo Fisher Scientific, Inc.), 25 mM glucose and 5.5 mM
2-mercaptoethanol. Cells were grown at 37°C in an incubator
containing 5% CO2. Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) was used for
transfection experiments, according to the manufacturer’s protocol
(11).
Extraction of total RNA and miRNA
TRIzol® (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) was used to extract total RNA
from cells. The mirVana RNA isolation kit (Invitrogen;
Thermo Fisher Scientific, Inc.) was used to remove RNA <200 nt
from isolated total RNAs. Subsequently, cDNA was generated using
the SMART-cDNA synthesis kit (Clontech Laboratories, Inc.,
Mountainview, CA, USA), which was conducted according to the
manufacturer’s protocol. In addition, the 3′-untranslated region
(UTR) of AdipoR1 was cloned into the pmirGLO vector using the
DynaExpress miRNA Cloning kit (BioDynamics Laboratory, Inc., Tokyo,
Japan) (12).
Analysis of the reporter gene
The whole cDNA sequence that targets the 3′-UTR of
AdipoR1 mRNA was predicted and obtained from the total RNA of the
two cell lines. The reverse orientation of AdipoR1 3′-UTR was used
as a control (13). Notably,
GUGCUUUU was identified as the seed region of miR-6835-3p, which
bound to the 3′-UTR region of AdipoR1 in position 38-44. After
transfection with miR-6835-3p, the 3′-UTRs of AdipoR1, FoxO-1 and
SIRT-1 were transfected into the two cell lines. Cells were
transfected with miR-6835-3p mimics (Invitrogen; Thermo Fisher
Scientific, Inc.) (3 µg/ml) using Lipofectamine®
3000 (Invitrogen; Thermo Fisher Scientific, Inc.) for 48 h at 37°C,
and were then transfected with the 3′-UTRs of AdipoR1 (4
µg/ml), FoxO-1 and SIRT-1 based on the aforementioned
method. The Dual-Glo® Luciferase Assay system (Promega
Corporation, Madison, WI, USA) was used to determine luciferase
reporter gene activity, according to the manufacturer’s protocol.
The Site-Mutation kit (Promega Corporation) was used to construct
the mutant (mut) 3′-UTR of AdipoR1. Furthermore, FoxO-1 and SIRT-1
mRNA 3′UTRs were also obtained from the two cell lines, and
luciferase reporter assays were conducted.
Transfection with miR-6835-3p inhibitors
or mimics
For cells that were transduced with vectors and
transfected with mimics/inhibitors, transduction was performed
first. Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) was used to perform transfection experiments,
according to the manufacturer’s protocol. The miR-6835-3p mimics
(MC29537) and inhibitors (MH29537) were purchased from Thermo
Fisher Scientific, Inc., respectively. In addition, the sequence of
the negative control used was as follows:
5′-ACGUGACACGUUCGGAGAAUU-3′. The effects of miR-6835-3p inhibitors
or mimics on miR-6835-3p expression were evaluated using reverse
transcription-quantitative polymerase chain reaction (RT-qPCR). The
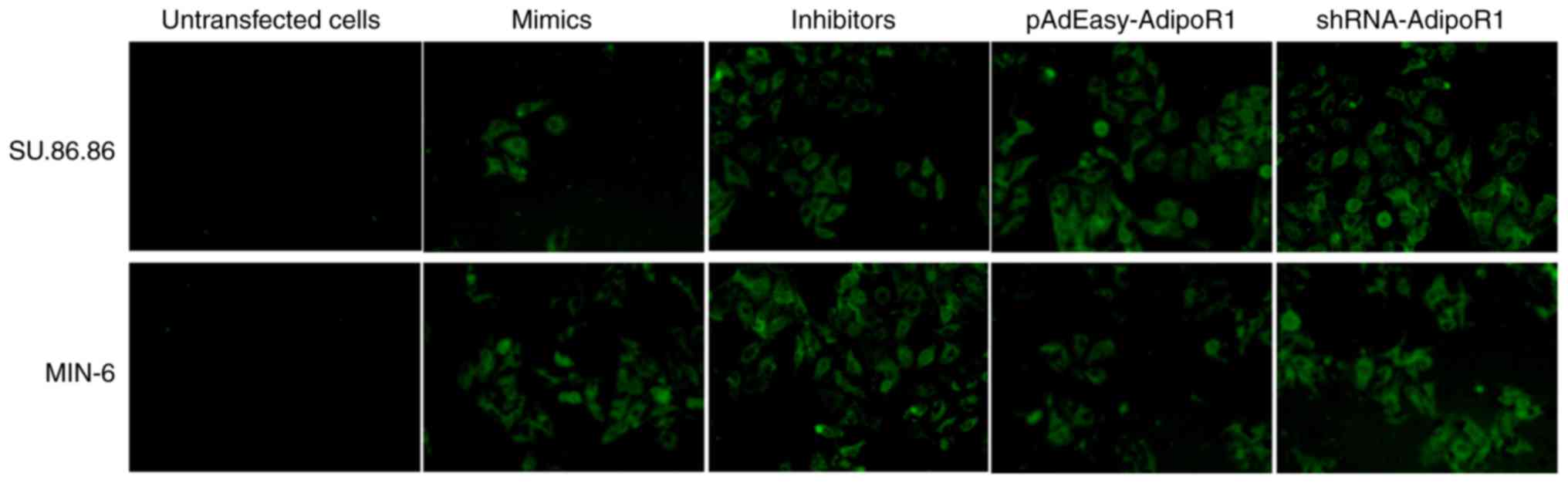
mimics/inhibitors were labeled with green fluorescent protein (GFP)
to allow for observation, in order to verify whether cell
transfection was successful (Fig.
1). The images were captured using a fluorescence microscope at
×200 magnification (IX71; Olympus Corporation, Tokyo, Japan).
RT-qPCR assay
The mRNA expression levels of AdipoR1 were detected
in the two cell lines using RT-qPCR. Briefly, RT-qPCR was performed
with SYBR Green (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and
the Light-Cycler Roche 480 system (Roche Diagnostics, Basel,
Switzerland), The thermocycling conditions for qPCR were as
follows: Pre-denaturation at 95°C for 30 sec, followed by 40 cycles
of denaturation at 95°C for 5 sec and extension at 60°C for 30 sec.
The 2−ΔΔCq method was used to calculate relative
expression and quantify (12,14). The primer sequences used were as
follows: AdipoR1, forward 5′-GCAGGCACATTACACGGT-3′, reverse
5′-TCCAGTTTTTTTTTTTTTTTAGAGGTC-3′; GAPDH forward
5′-CTCATGACCACAGTCCATGCC-3′, reverse 5′-GGCATGGACTGTGGTCATGAG-3′;
miR-6835-3p, forward 5′-GACCCTCTGTCTTTTCACGAAAA-3′, reverse
5′-TTTTCGTGAAAAGACAGAGGGTC-3′; and U6, forward
5′-CTCGCTTCGGCAGCACA-3′ and reverse 5′-TGTGCTGCCGAAGCGAG-3′.
Western blot analysis
Cells were collected and total proteins were
extracted in 40 mM Tris-HCl (pH 7.4) containing 150 mM NaCl and 1%
(v/v) Triton X-100 (Sigma-Aldrich; Merck KGaA), supplemented with
protease inhibitors (Sigma-Aldrich; Merck KGaA). Protein
concentration was determined using the bicinchoninic acid protein
assay (Pierce; Thermo Fisher Scientific, Inc.). Total cell proteins
were extracted and separated by 12% SDS-PAGE; 50 µg protein
was loaded per lane. Proteins were then transferred to a
0.45-µm polyvinylidene fluoride membrane (Roche
Diagnostics), which was blocked with 1X Tris-buffered
saline-Tween-20 (0.05%) containing 5% nonfat dry milk and agitated
for 1 h at room temperature. The membranes were then incubated with
primary antibodies against AdipoR1 (1:2,000; sc-518030), Sirtuin 1
(SIRT-1) (1:2,000; sc-135791), FoxO-1 (1:2,000; sc-9808; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) and β-actin (1:3,000
dilution; 4970; Cell Signaling Technology, Inc., Danvers, MA, USA)
at 4°C overnight. The membranes were then incubated with secondary
antibodies goat anti-rabbit immunoglobulin G (IgG)-horseradish
peroxidase (HRP) (sc-2004; 1:5,000 dilution; Santa Cruz
Biotechnology, Inc.) or goat anti-mouse IgG-HRP, (sc-2005; 1:5,000
dilution; Santa Cruz Biotechnology, Inc.) for 1 h at room
temperature. Protein bands were visualized using enhanced
chemiluminescence (Amersham; GE Healthcare, Chicago, IL, USA).
Transduction of AdipoR1 and its short
hairpin (sh)RNA
For transduction of AdipoR1, PCR amplification was
performed for AdipoR1, and its open-reading frame was cloned into
the pAdEasy vector (Stratagene; Agilent Technologies, Inc., Santa
Clara, CA, USA). To generate and amplify the virus, 293A cells
(Thermo Fisher Scientific, Inc.) were employed; cells (90%
confluence) were transfected with vectors (4 µg/ml) using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) for 48 h at 37°C. Purification was subsequently
performed using the double cesium chloride gradient centrifugation
method (15). The 293A cells were
harvested, and adenovirus stocks were prepared by repeated freezing
and thawing. The infectious particles were measured in culture by a
plaque-forming unit (PFU) assay using a Cell Biolabs QuickTiter™
Adenovirus Titer Immunoassay kit (VPK-109; Cell Biolabs, Inc., San
Diego, CA, USA), which scores the number of viral plaques as a
function of dilution. The adenovirus titer reached
~3.5×109 pfu/ml. Subsequently, the viral supernatant (1
ml) was used to infect target cells (2×106 cells) for 10
days. AdipoR1 shRNA (5′-GGACAACGACUAUCUGCUACATT-3′) and the
negative control shRNA (shRNA-control; 5′-CCUACGCCACCAAUUUCGU-3′)
were obtained from Shanghai GenePharma Co., Ltd. (Shanghai, China),
and were prepared via ligation of the corresponding pairs of
oligonucleotides to the pLKO.1 vector (Sigma-Aldrich; Merck KGaA).
The pLKO.1 vectors containing AdipoR1 shRNA or shRNA-control were
introduced into target cells using Lipofectamine® 3000
(Invitrogen; Thermo Fisher Scientific, Inc.) for 48 h at 37°C. The
AdipoR1 vectors were labeled with GFP for observation, in order to
verify successful transduction/transfection into cells (Fig. 1).
Insulin secretion assay
The insulin secretion assay was conducted as
previously described (16,17).
Briefly, following a 2 h preincubation period in the modification
medium [Krebs-Ringer (Sigma-Aldrich; Merck KGaA) containing 3
mmol/l glucose], cells were treated with 20 mM glucose for 1 h.
Subsequently, the insulin levels in the cultured cell supernatant
fractions were determined using the radioimmunoassay method using a
specific insulin radioimmunoassay kit (cat. no. RI-13K; Linco
Research, Inc., St. Charles, MO, USA).
Statistical analysis
A two-tailed paired Student’s t-test was used to
analyze differences between two groups, and one-way analysis of
variance (ANOVA) was performed for multiple comparisons between
>2 groups; when the variances were equal, the least significant
difference post hoc test was used, whereas when the variances were
not uniform, Dunnett’s post hoc test was used. All data are
presented as the means ± standard deviation. SPSS software (version
18.0; SPSS, Inc., Chicago, IL, USA) was used to conduct statistical
analyses. P<0.05 was considered to indicate a statistically
significant difference.
Results
miR-6835-3p inhibits AdipoR1 expression
in SU.86.86 and MIN-6 cells
The present study used TargetScan software
(targetscan.org/) to predict the miRNA that may
regulate the expression of AdipoR1 in SU.86.86 cells. Subsequently,
AdipoR1 was identified as a potential target gene of miR-6835-3p
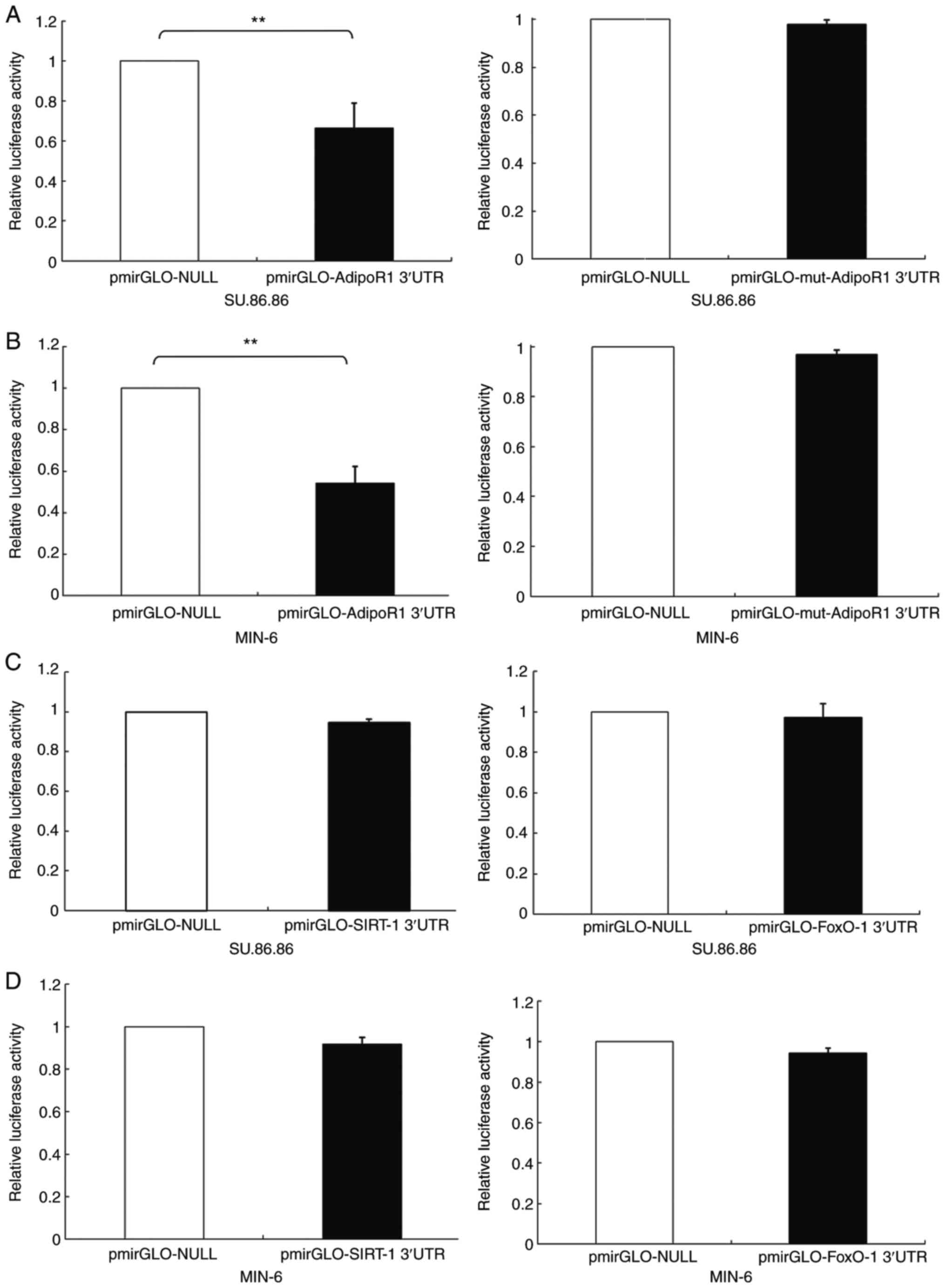
(Fig. 2). Reporter gene assays
based on luciferase activity were performed to confirm that the
AdipoR1 3′-UTR was able to bind directly to miR-6835-3p. The
results indicated that miR-6835-3p markedly downregulated 3′-UTR
AdipoR1 luciferase activity in SU.86.86 and MIN-6 cells; however,
the luciferase activity of the mut-AdipoR1 group was not influenced
by miR-6835-3p (Fig. 3A and B).
In addition, the results confirmed that miR-6835-3p did not
directly bind to FoxO-1 or SIRT-1 3′-UTRs (Fig. 3C and D). These findings indicated
that AdipoR1 mRNA was the direct target of miR-6835-3p.
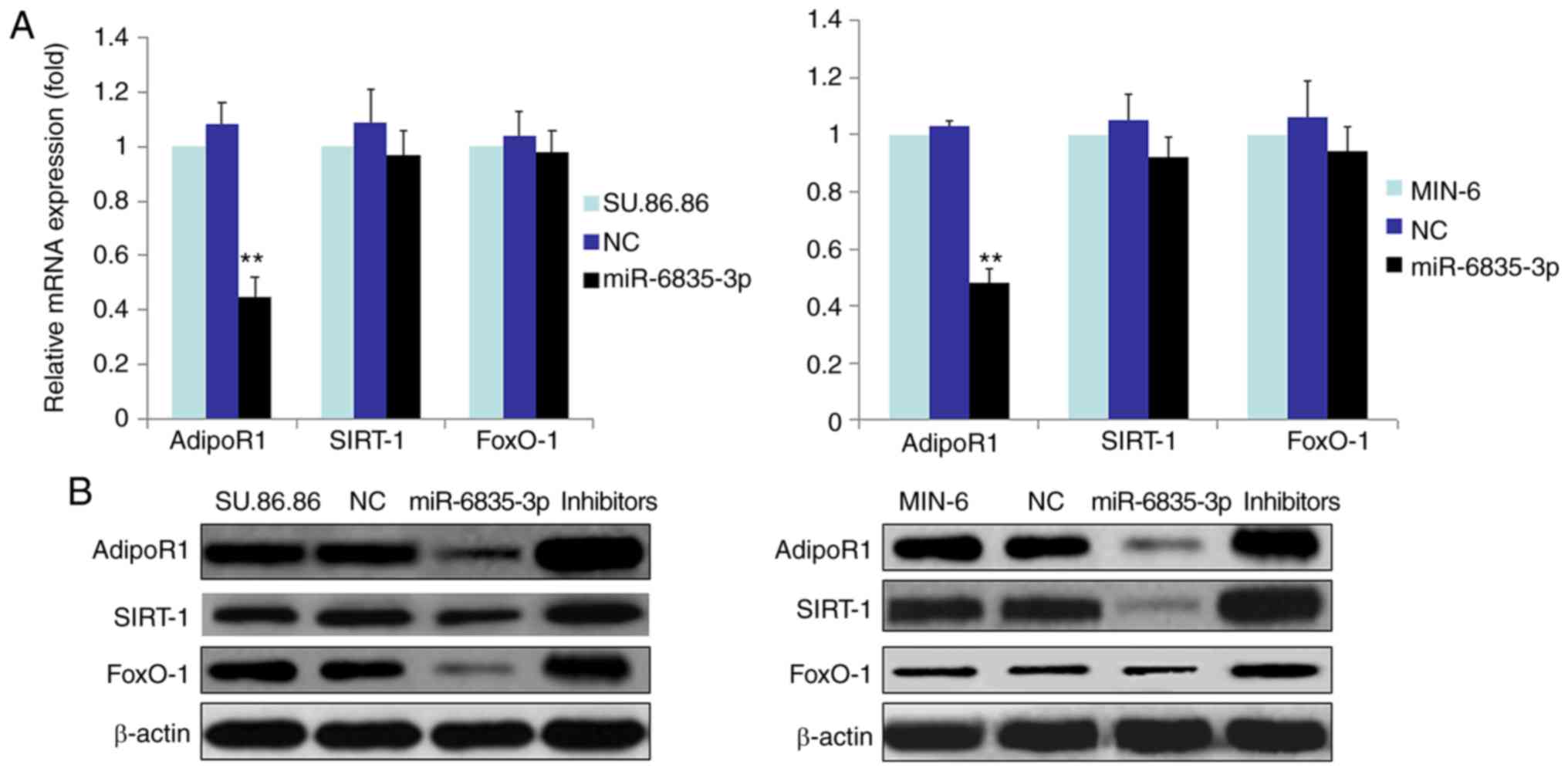
In order to determine whether miR-6835-3p affected
the mRNA and protein expression levels of FoxO-1, SIRT-1 and
AdipoR1 in SU.86.86 and MIN-6 cells, RT-qPCR and western blotting
were performed (Fig. 4). The
results demonstrated that miR-6835-3p suppressed the mRNA and
protein expression levels of AdipoR1 in SU.86.86 and MIN-6 cells
(Fig. 4A and B). Conversely,
miR-6835-3p did not affect the mRNA expression levels of FoxO-1 and
SIRT-1 (Fig. 4B); however, the
protein expression levels of FoxO-1 and SIRT-1 were decreased.
Furthermore, miR-6835-3p inhibitors facilitated the expression of
AdipoR1 protein in the SU.86.86 and MIN-6 cell lines (Fig. 4B). Moreover, miR-6835-3p
inhibitors enhanced the expression of AdipoR1 protein in the
SU.86.86 and MIN-6 cell lines (Fig.
4B). Taken together, these findings indicated that miR-6835-3p
may directly modulate AdipoR1 expression by binding to the AdipoR1
3′-UTR, and the protein expression levels of FoxO-1 and SIRT-1 may
be altered in response to miR mimics.
miRNA-6835-3p regulates
glucose-stimulated insulin secretion (GSIS) via the AdipoR1
signaling pathway
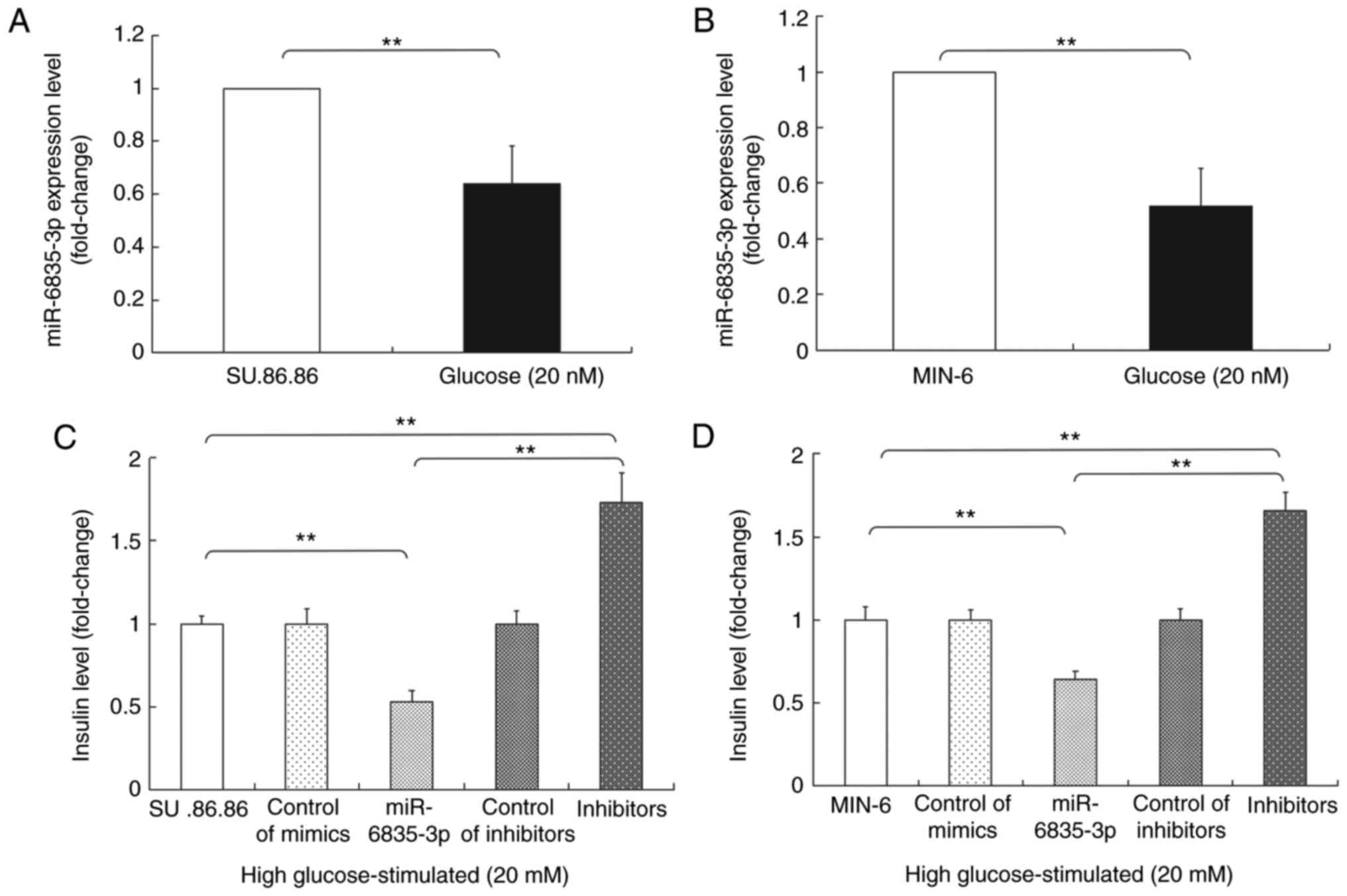
SU.86.86 and MIN-6 were cultured with glucose (20
mM), after which RT-qPCR was performed to detect the expression of
miR-6835-3p. The results revealed that the expression levels of
miR-6835-3p were lower in the high glucose-stimulated (20 mM)
SU.86.86 and MIN-6 cell groups compared with in the control group
(Fig. 5A and B), thus indicating
that the expression of miR-6835-3p changed in response to glucose
stimulation. In addition, the effects of miR-6835-3p mimics or
inhibitors on GSIS were investigated. The results indicated that
miR-6835-3p mimics suppressed GSIS; however, the inhibitors
promoted GSIS in the SU.86.86 and MIN-6 cell lines (Fig. 5C and D).
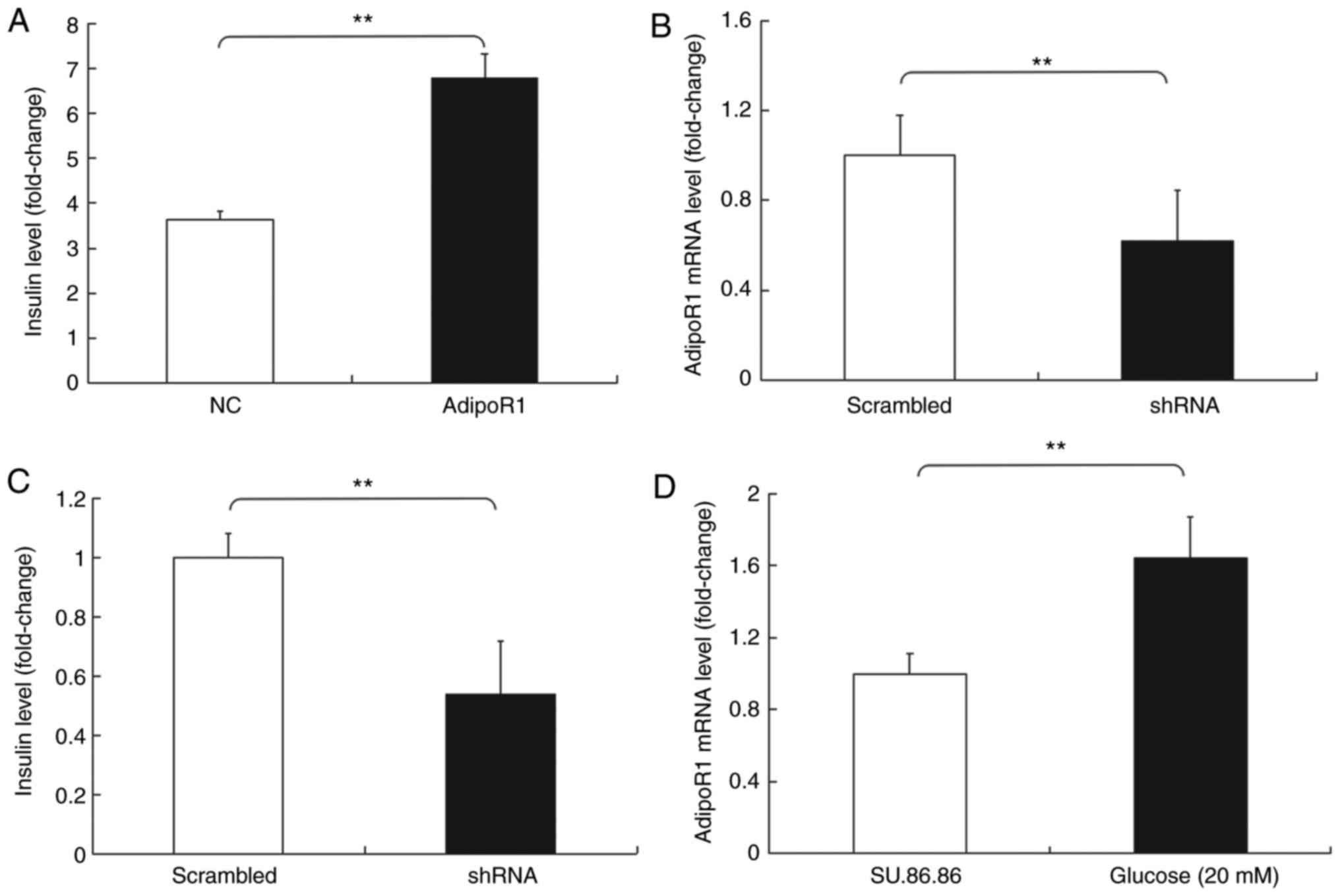
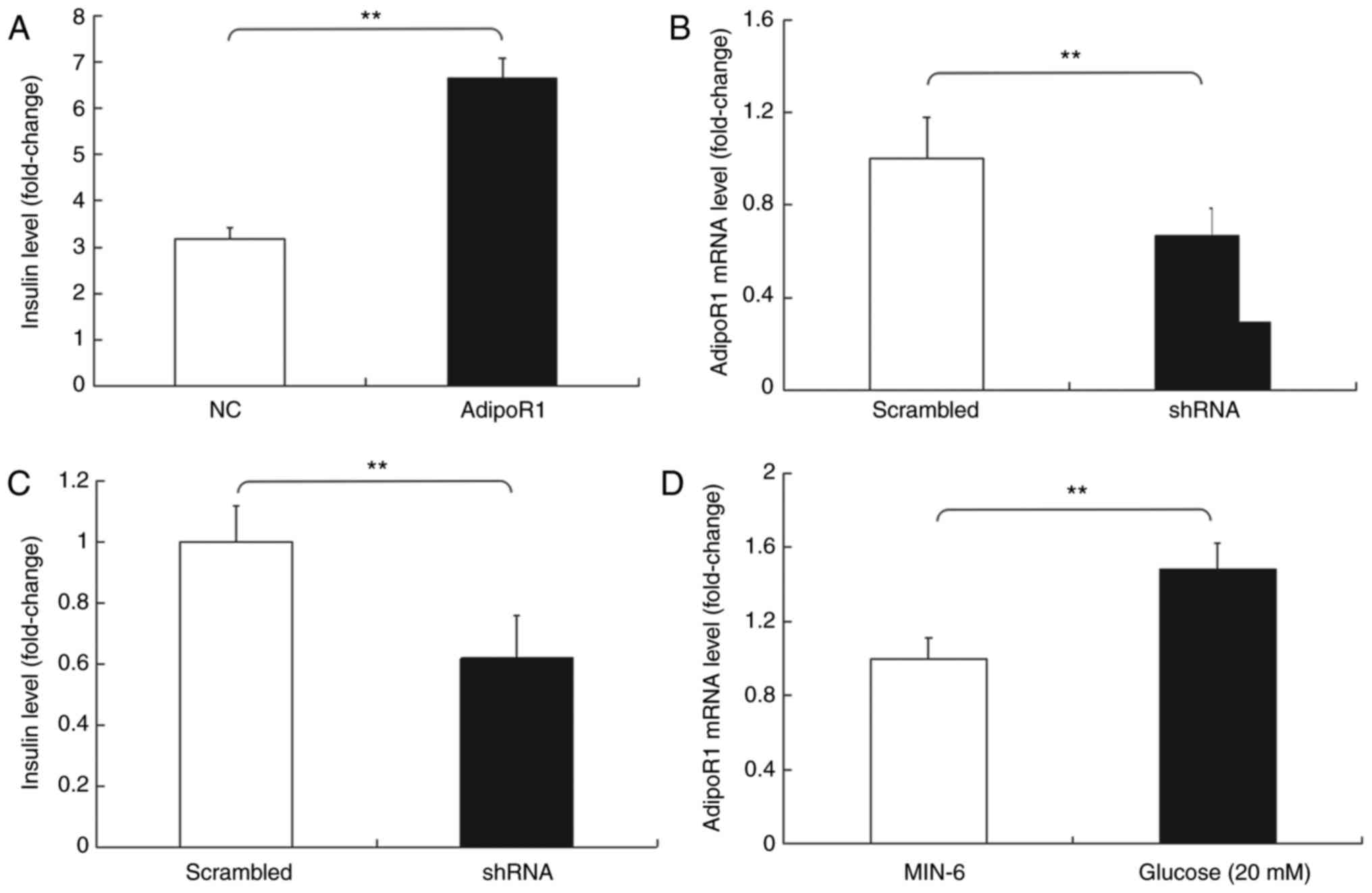
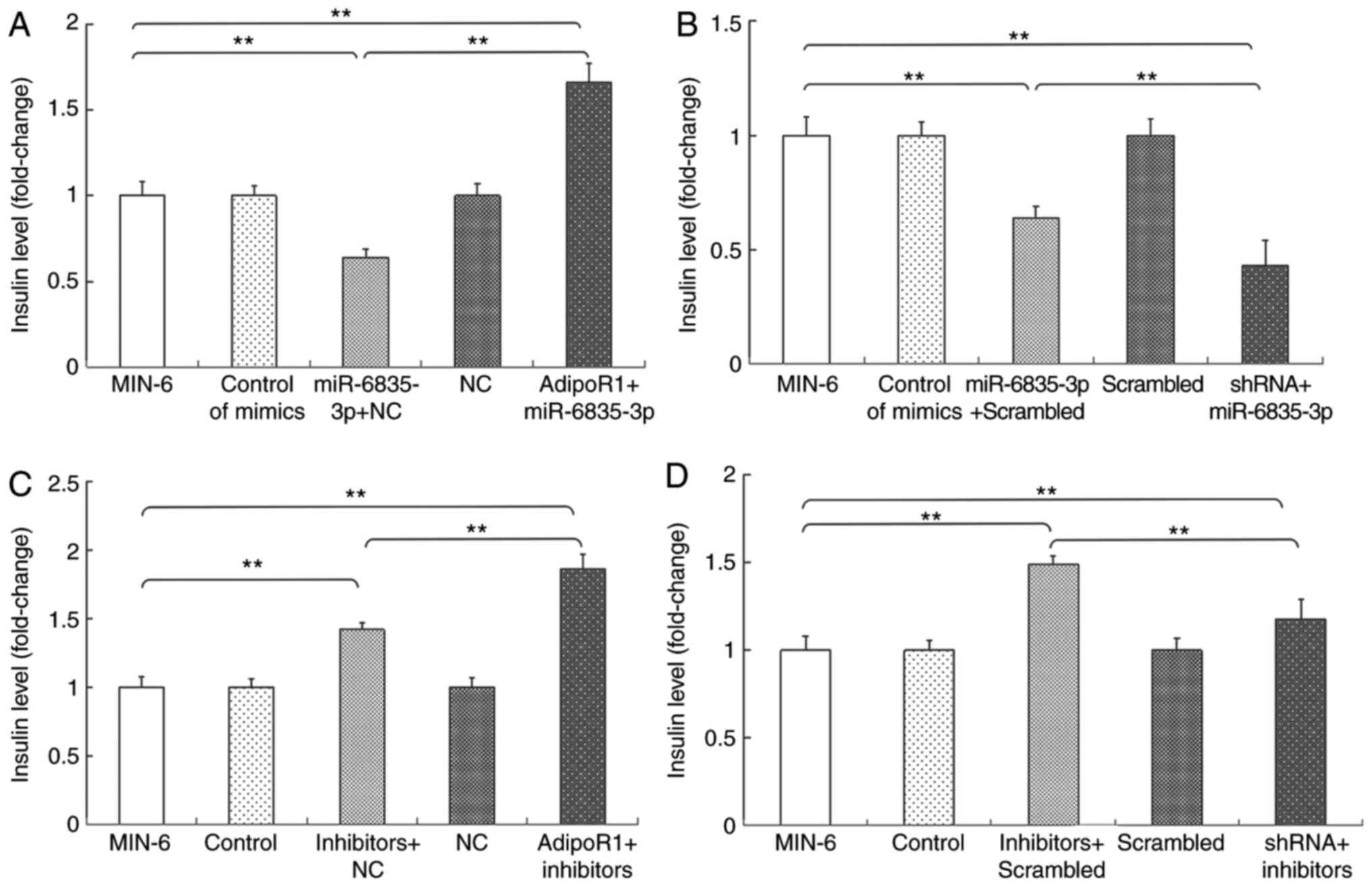
Subsequently, the effects of AdipoR1 on GSIS in the
SU.86.86 and MIN-6 cell line were determined (Figs. 6 and 7). It was demonstrated that AdipoR1
overexpression regulated GSIS in SU.86.86 and MIN-6 cells (Figs. 6A and 7A). To confirm the effects of AdipoR1 on
GSIS, the present study employed an AdipoR1 shRNA, and its effects
on AdipoR1 expression were confirmed in SU.86.86 and MIN-6 cells
(Figs. 6B and 7B). The results demonstrated that
knockdown of AdipoR1 inhibited insulin secretion stimulated by high
levels of glucose in SU.86.86 and MIN-6 cells (Figs. 6C and 7C). Furthermore, the effects of glucose
on the expression of AdipoR1 were assessed. The relative mRNA
expression levels of AdipoR1 were increased in the high
glucose-stimulated (20 mM) SU.86.86 and MIN-6 cell groups compared
with in the control groups (Figs.
6D and 7D).
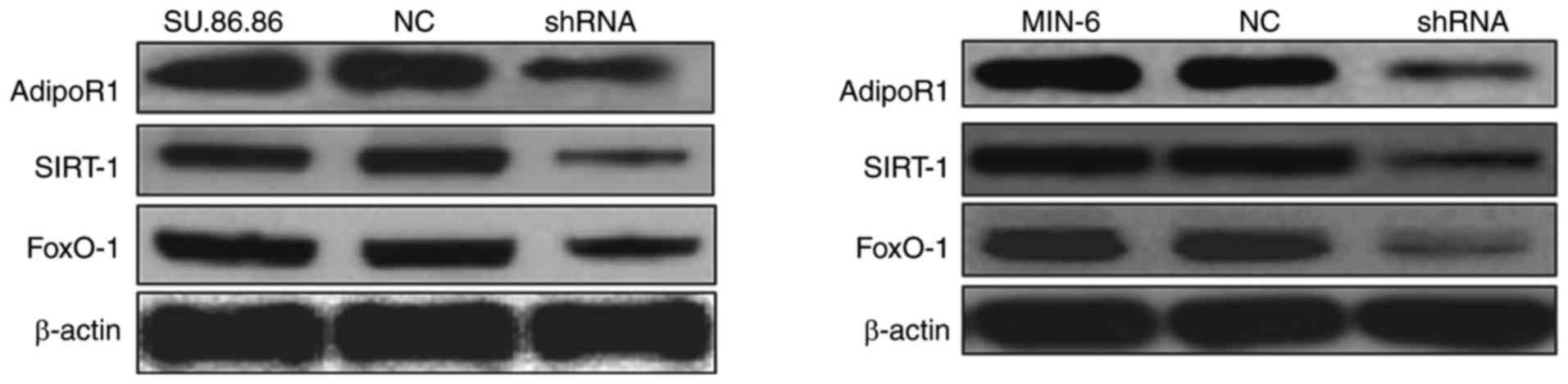
To confirm the effects of AdipoR1 on
glucose-stimulated insulin secretion, the present study transduced
SU.86.86 and MIN-6 cells with a shRNA for AdipoR1 and tested its
efficiency. AdipoR1 shRNA downregulated the protein expression
levels of AdipoR1, as well as those of SIRT1 and FoxO-1 in its
downstream signaling pathway, in the MIN-6 and SU.86.86 cell lines
(Fig. 8). These results confirmed
that the AdipoR1 shRNA regulates the protein expression levels of
AdipoR1 and its associated pathway proteins in the MIN-6 and
SU.86.86 cell lines.
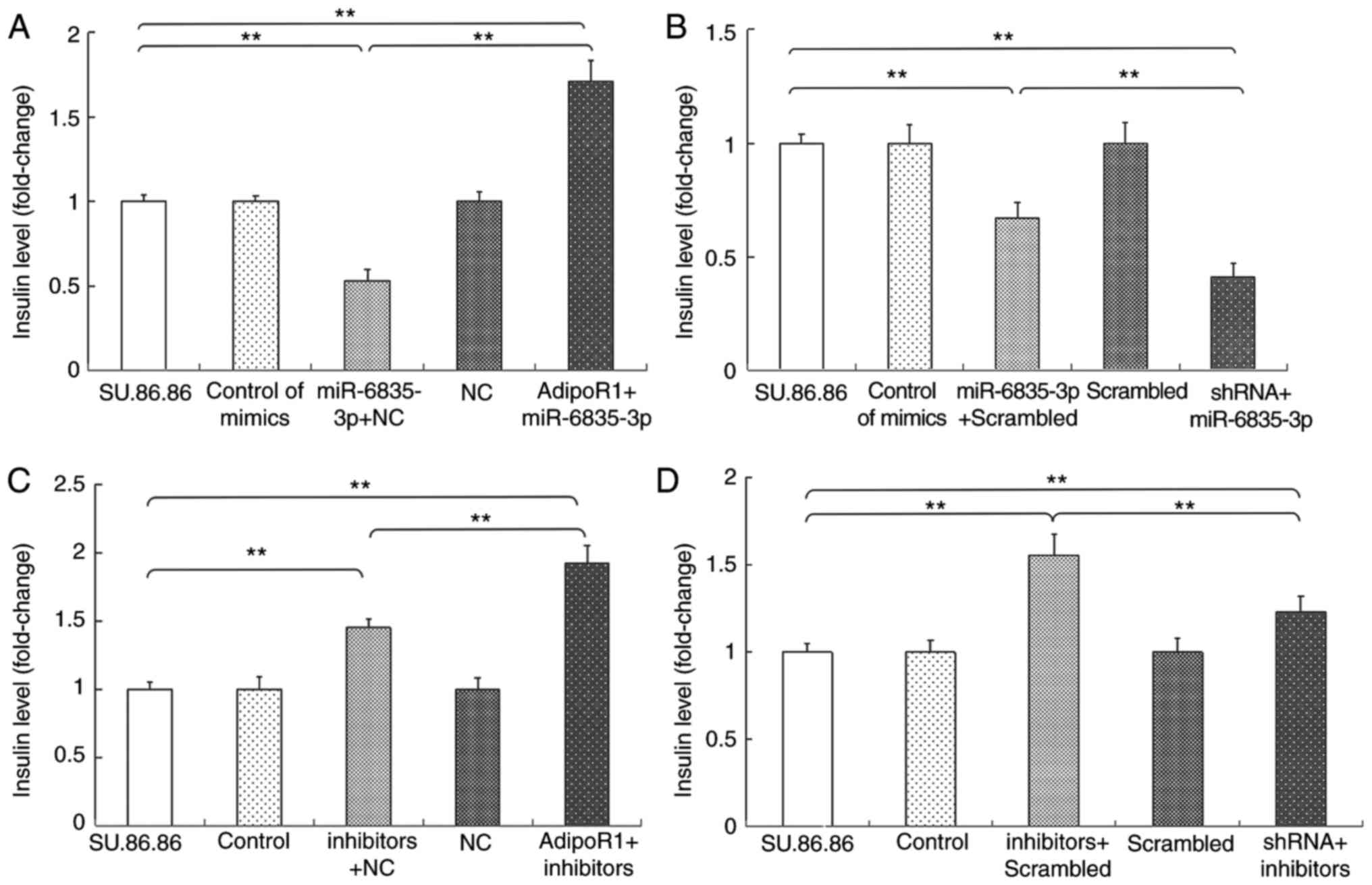
Since miR-6835-3p and AdipoR1 regulate GSIS, and
AdipoR1 mRNA is a direct target of miR-6835-3p, the present study
further investigated how the effects of miR-6835-3p on AdipoR1
expression were associated with GSIS (Figs. 9 and 10). As shown in Figs. 9A and 10A, overexpression of AdipoR1 abolished
the effects of miR-6835-3p mimics on GSIS in the SU.86.86 and MIN-6
cell lines, respectively. Conversely, treatment with miR-6835-3p
mimics and AdipoR1 shRNA synergistically inhibited GSIS (Figs. 9B and 10B). The effects of the
miR-6835-3p/AdipoR1 signaling pathway on GSIS were further
evaluated using miR-6835-3p inhibitors. The results demonstrated
that AdipoR1 overexpression significantly enhanced the effects of
miR-6835-3p inhibitors on GSIS (Figs.
9C and 10C). In addition,
GSIS was reduced in the SU.86.86 and MIN-6 cell lines treated with
miR-6835-3p inhibitors and AdipoR1 shRNA compared with in cells
transfected with miR-6835-3p inhibitors alone (Figs. 9D and 10D). Taken together, these results
confirmed that AdipoR1 was a direct target of miR-6835-3p, and
targeting AdipoR1 with miR-6835-3p inhibitors could promote
GSIS.
Discussion
Type 2-DM is associated with blindness, renal
failure and cardiovascular disease (16,18). The increasing incidence of type
2-DM is a worldwide phenomenon, which coincides with the lifestyle
changes of the last few decades (19,20). Insulin is a small peptide hormone
that is crucial for glucose homeostasis in mammals and is generated
from pancreatic islet β-cells (21-23). In healthy mammals, when blood
glucose concentration is elevated, insulin is released to promote
the absorption of glucose by fat tissues, skeletal muscles or the
liver (24,25).
miRNAs are a class of 19-22 nucleotide-long
endogenous RNAs, which can affect gene expression by binding to the
3′-UTRs of target mRNAs (26,27). It has been indicated that miRNAs
may be involved in pancreatic development and insulin secretion. In
particular, miR-375 specifically inhibits GSIS and serves a vital
role in insulin secretion (25);
miR-9 regulates GSIS of islet β-cells by targeting SIRT-1 in the
pancreas (22); miR-124a is
highly expressed in the pancreatic islets of patients with type
2-DM and inhibits insulin secretion (28); and miR-187 regulates the
expression of homeodomain interacting protein kinase 3, and is
associated with reduced GSIS (21). In addition, miR-34c reduces GSIS
by targeting vesicle-associated membrane protein 2, which induces
vital effects of insulin secretion on β-cell exocytosis (29). Although numerous studies have
reported that miRNAs target genes associated with GSIS signaling
and have revealed their physiological implications, the underlying
mechanisms associated with the modulation of GSIS have not been
fully elucidated. In the present study, the roles of miR-6835-3p in
GSIS and regulation of the AdipoR1/SIRT-1/FoxO-1 signaling pathway
were investigated.
In insulin-resistant obesity, adiponectin is
downregulated, as are adipokine factors secreted by adipose tissue.
Type 2-DM, metabolic syndrome, atherosclerosis and obesity are
consistently associated with hypoadiponectinemia (30). Adiponectin has been reported to
induce anti-atherosclerotic and anti-diabetic effects by
attenuating inflammation and oxidative stress (31,32). The biological effects induced by
adiponectin have been revealed to be associated with its receptors,
AdipoR1 and AdipoR2. In addition, AdipoR1 is able to modulate
SIRT1-FoxO1 signaling; a previous study revealed that resveratrol
increases the phosphorylation of 5′ AMP-activated protein kinase
and SIRT-1, and decreases phosphorylation of downstream effectors
FoxO-1 and FoxO-3a via increasing AdipoR1 and AdipoR2 in the renal
cortex (33). In addition,
capsaicin-activated FoxO-1 induces FoxO-1 acetylation, which is
associated with CREB binding protein and SIRT-1 (34). SIRT-1 activation may induce FoxO1
activation, and it affects insulin function through modulation of
the SIRT1-FoxO1 signaling axis (34). These findings indicated that
AdipoR1/SIRT-1/FoxO-1 signaling axis may have an important role in
insulin function. However, miRNA-mediated modulation of AdipoR1
activity in the anti-diabetes process remains unclear.
In the present study, the miRNAs that may regulate
AdipoR1 expression in the SU.86.86 and MIN-6 cell lines were
predicted using TargetScan; AdipoR1 was identified as a potential
target gene of miR-6835-3p. The present results also indicated that
miR-6835-3p markedly downregulated the luciferase activities of the
AdipoR1 3′-UTR in SU.86.86 and MIN-6 cells; however, the luciferase
activities of the mut-AdipoR1 group were not affected by
miR-6835-3p. In addition, the results of a luciferase reporter
assay confirmed that miR-6835-3p did not directly bind to the
FoxO-1 or SIRT-1 3′-UTRs. Therefore, the present study indicated
that AdipoR1 mRNA was the direct target of miR-6835-3p.
Further investigations demonstrated that miR-6835-3p
suppressed the mRNA and protein expression levels of AdipoR1 in the
SU.86.86 and MIN-6 cell lines; however, miR-6835-3p did not affect
the mRNA expression levels of FoxO-1 and SIRT-1. Conversely, the
protein expression levels of FoxO-1 and SIRT-1 were affected by
miR-6835-3p. In addition, transfection with miR-6835-3p inhibitors
facilitated the protein expression levels of AdipoR1 in SU.86.86
and MIN-6 cells. Taken together, these results indicated that
miR-6835-3p may directly modulate AdipoR1 expression by binding to
the 3′-UTR of AdipoR1 and may affect the protein expression levels
of FoxO-1 and SIRT-1 in the AdipoR1 signaling pathway.
The results demonstrated that miR-6835-3p expression
in high glucose-stimulated (20 mM) SU.86.86 and MIN-6 cells was
reduced compared with in the control group, thus indicating that
there was a change in miR-6835-3p expression in response to glucose
stimulation. In addition, transfection with miR-6835-3p mimics
suppressed GSIS; however, transfection with miR-6835-3p inhibitors
promoted GSIS in SU.86.86 and MIN-6 cells. These findings indicated
that transient transfection of cells with miR-6835-3p mimics or
inhibitors altered GSIS. Furthermore, AdipoR1 overexpression
regulated GSIS in SU.86.86 and MIN-6 cells, and AdipoR1
overexpression significantly enhanced the effects of miR-6835-3p
inhibitors on GSIS. In addition, GSIS was reduced in response to
miR-6835-3p inhibitors in the SU.86.86 and MIN-6 cell lines
following AdipoR1 knockdown with a shRNA-AdipoR1. These results
confirmed that AdipoR1 was a direct target of miR-6835-3p, and
targeting AdipoR1 with miR-6835-3p inhibitors may promote GSIS.
In conclusion, miR-6835-3p exerted effects on
insulin secretion in the SU.86.86 and MIN-6 cell lines, which were
mediated by regulating the expression of AdipoR1 and the associated
signaling pathway proteins. AdipoR1 was observed to be a direct
target of miR-6835-3p, thus suggesting that miR-6835-3p may be a
crucial regulator of GSIS through inhibition of the
AdipoR1/SIRT-1/FoxO-1 signaling pathway. The results of the present
study suggested that inhibitors of miR-6835-3p may be a potential
therapeutic strategy for promoting GSIS by targeting AdipoR1, and
therefore may be considered an effective treatment for type
2-DM.
Acknowledgments
Not applicable.
Funding
The present study was supported by grants from the
National Natural Science Foundation of China (grant nos. 30600524
and 81341067).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors’ contributions
HW conducted the experiments. LJ, ZL and WW
participated in the experiments. CH designed the experiments and
wrote the paper.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Asif M: The prevention and control the
type-2 diabetes by changing lifestyle and dietary pattern. J Educ
Health Promot. 3:1–8. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Riobo Servan P: Obesity and diabetes. Nutr
Hosp. 28:138–143. 2013.PubMed/NCBI
|
|
3
|
Xiao J, Chen T and Cao H: Flavonoid
glycosylation and biological benefits. Biotechnol Adv. May
22–2014.Epub ahead of print. View Article : Google Scholar
|
|
4
|
Bartel DP: MicroRNAs: Target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lahmy R, Soleimani M, Sanati MH, Behmanesh
M, Kouhkan F and Mobarra N: Pancreatic islet differentiation of
human embryonic stem cells by microRNA overexpression. J Tissue Eng
Regen Med. 10:527–534. 2016. View Article : Google Scholar
|
|
6
|
Stefani G and Slack FJ: Small non-coding
RNAs in animal development. Nat Rev Mol Cell Biol. 9:219–230. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi Y and Jin Y: MicroRNA in cell
differentiation and development. Sci China C Life Sci. 52:205–211.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Carrington JC and Ambros V: Role of
microRNAs in plant and animal development. Science. 301:336–338.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaviani M, Azarpira N, Karimi MH and
Al-Abdullah I: The role of microRNAs in islet β-cell development.
Cell Biol Int. 40:1248–1255. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Dalgaard LT and Eliasson L: An
‘alpha-beta’ of pancreatic islet microribonucleotides. Int J
Biochem Cell Biol. 88:208–219. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shewade YM, Umrani M and Bhonde RR:
Large-scale isolation of islets by tissue culture of adult mouse
pancreas. Transplant Proc. 31:1721–1723. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
O’Connell RM, Taganov KD, Boldin MP, Cheng
G and Baltimore D: MicroRNA-155 is induced during the macrophage
inflammatory response. Proc Natl Acad Sci USA. 104:1604–1609. 2007.
View Article : Google Scholar
|
|
13
|
Esquela-Kerscher A and Slack FJ:
Oncomirs-microRNAs with a role in cancer. Nat Rev Cancer.
6:259–269. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
15
|
Nikolai VK and Steve JU: Association of
RNA polymerase complexes of the parasitic protozoan cryptosporidium
parvum with virus-like particles: Heterogeneous system. J Virol.
74:5788–5795. 2000. View Article : Google Scholar
|
|
16
|
Sebastiani G, Po A, Miele E, Ventriglia G,
Ceccarelli E, Bugliani M, Marselli L, Marchetti P, Gulino A,
Ferretti E and Dotta F: MicroRNA-124a is hyperexpressed in type 2
diabetic human pancreatic islets and negatively regulates insulin
secretion. Acta Diabetol. 52:523–530. 2015. View Article : Google Scholar
|
|
17
|
da Silva Xavier G, Loder MK, McDonald A,
Tarasov AI, Carzaniga R, Kronenberger K, Barg S and Rutter GA:
TCF7L2 regulates late events in insulin secretion from pancreatic
islet beta-cells. Diabetes. 58:894–905. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Weir GC and Bonner-Weir S: Sleeping islets
and the relationship between β-cell mass and function. Diabetes.
60:2018–2019. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kahn SE: The relative contributions of
insulin resistance and beta-cell dysfunction to the pathophysiology
of type 2 diabetes. Diabetologia. 46:3–19. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Maris M, Ferreira GB, D’Hertog W, Cnop M,
Waelkens E, Overbergh L and Mathieu C: High glucose induces
dysfunction in insulin secretory cells by different pathways: A
proteomic approach. J Proteome Res. 9:6274–6287. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Locke JM, da Silva Xavier G, Dawe HR,
Rutter GA and Harries LW: Increased expression of miR-187 in human
islets from individuals with type 2 diabetes is associated with
reduced glucose-stimulated insulin secretion. Diabetologia.
57:122–128. 2014. View Article : Google Scholar
|
|
22
|
Ramachandran D, Roy U, Garg S, Ghosh S,
Pathak S and Kolthur-Seetharam U: Sirt1 and mir-9 expression is
regulated during glucose-stimulated insulin secretion in pancreatic
β-islets. FEBS J. 278:1167–1174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Osmai M, Osmai Y, Bang-Berthelsen CH,
Pallesen EM, Vestergaard AL, Novotny GW, Pociot F and
Mandrup-Poulsen T: MicroRNAS as regulators of beta-cell function
and dysfunction. Diabetes Metab Res Rev. 32:334–349. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thirone AC, Huang C and Klip A:
Tissue-specific roles of IRS proteins in insulin signaling and
glucose transport. Trends Endocrinol Metab. 17:72–78. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Poy MN, Eliasson L, Krutzfeldt J, Kuwajima
S, Ma X, Macdonald PE, Pfeffer S, Tuschl T, Rajewsky N, Rorsman P
and Stoffel M: A pancreatic islet-specific microRNA regulates
insulin secretion. Nature. 432:226–230. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ambros V: The functions of animal
microRNAs. Nature. 431:350–355. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bartel DP: MicroRNAs: Genomics,
biogenesis, mechanism, and function. Cell. 116:281–297. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baroukh N, Ravier MA, Loder MK, Hill EV,
Bounacer A, Scharfmann R, Rutter GA and Van Obberghen E:
MicroRNA-124a regulates Foxa2 expression and intracellular
signaling in pancreatic beta-cell lines. J Biol Chem.
282:19575–19588. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hu S, Wang H, Chen K, Cheng P, Gao S, Liu
J, Li X and Sun X: MicroRNA-34c downregulation
ameliorates-amyloid-β-induced synaptic failure and memory deficits
by targeting VAMP2. J Alzheimers Dis. 48:673–686. 2015. View Article : Google Scholar
|
|
30
|
Fisman EZ and Tenenbaum A: Adiponectin: A
manifold therapeutic target for metabolic syndrome, diabetes, and
coronary disease. Cardiovasc Diabetol. 13:1032014. View Article : Google Scholar
|
|
31
|
Yamauchi T, Kamon J, Ito Y, Tsuchida A,
Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et
al: Cloning of adiponectin receptors that mediate antidiabetic
metabolic effects. Nature. 423:762–769. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yamauchi T, Iwabu M, Okada-Iwabu M and
Kadowaki T: Adiponectin receptors: A review of their structure,
function and how they work. Best Pract Res Clin Endocrinol Metab.
28:15–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Park HS, Lim JH, Kim MY, Kim Y, Hong YA,
Choi SR, Chung S, Kim HW, Choi BS, Kim YS, et al: Resveratrol
increases AdipoR1 and AdipoR2 expression in type 2 diabetic
nephropathy. J Transl Med. 14:1762016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pramanik KC, Fofaria NM, Gupta P and
Srivastava SK: CBP-mediated FOXO-1 acetylation inhibits pancreatic
tumor growth by targeting SirtT. Mol Cancer Ther. 13:687–698. 2014.
View Article : Google Scholar : PubMed/NCBI
|