Introduction
Acute liver failure (ALF), sometimes referred to as
fulminant hepatic failure, is a severe liver disease characterized
by coagulopathy and liver inflammation in patients with previously
normal liver function (1). The
onset of liver injury, often accompanied by endotoxemia and
hepatocyte death, can result in high mortality rates in animals and
humans, due to the lack of an effective therapy for ALF, other than
liver transplantation (2).
The administration of D-galactosamine (D-GalN) and
lipopolysaccharides (LPS) establishes an animal model for ALF; it
severely impairs liver function in laboratory animals, and can be
used to investigate ALF (3). LPS,
a type of endotoxin, can cause liver tissue damage through
activation of inflammatory cytokines, including interleukin
(IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and interferon-γ
(4). A report suggests that LPS
as an inflammatory mediator can stimulate the production of
reactive oxygen species (ROS) and can be used broadly to induce
research models of ALF (5). A
D-GalN/LPS-induced animal model of ALF is representative of human
liver failure and has been widely used to investigate mechanisms
and potential therapeutic drugs for clinical use in treating ALF
(2). Increasing evidence has
shown that oxidative stress and hepatocyte apoptosis are two
important pathogenic factors that contribute to D-GalN/LPS-induced
ALF (6,7).
The evaluation of mechanisms involved in
D-GalN/LPS-induced hepatic apoptosis has indicated that
mitochondria are critical targets for drug toxicity through the
formation of reactive metabolites (8,9).
In response to oxidative stress, the cellular machinery of the
antioxidative response is mediated by cytoprotective enzymes in the
liver. When it to comes to oxidation, nuclear factor-E2-related
factor 2 (Nrf2), a key sensor in antioxidative stress systems,
regulates cellular defense against oxidative damage (10), contributing to diverse cellular
functions, including proliferation, inflammation and lipid
synthesis (11). The aberrant
expression or function of Nrf2 has been linked to pathologies,
including hepatic steatosis (12)
and diabetic nephropathy (13).
Although Nrf2 is regularly sequestered by Kelch-like
ECH2-associated protein 1 (Keap1) in the cytoplasm (14), when activated, it translocates
into the nucleus and upregulates antioxidant response element (ARE)
genes, including superoxide dismutase (SOD)1/SOD2, catalase,
glutamate-cysteine ligase catalytic subunit (GCLC),
glutamate-cysteine ligase regulatory subunit (GCLM), heme
oxygenase-1 (HO-1), cytochrome P450s, multidrug-resistant proteins
and NAD(P)H dehydrogenase quinone 1 (NQO1) (15,16).
In general, the autophagy-dependent degradation of
impaired organelles, including the selective degradation of the
endoplasmic reticulum (ER; reticulophagy), mitochondria
(mitophagy), and lipid droplets (lipophagy), is a crucial cellular
pathway for maintaining cell homeostasis (17,18). However, autophagy is also regarded
as a pro-apoptotic factor and causes 'type II' programmed cell
death (19). The cellular damage
mediated by p53 has a pathological role in the progression of
hepatosteatosis. It is noteworthy that p53 can promote the
expression of damage-regulated autophagy modulator (DRAM), an
inducer of autophagy-mediated apoptosis (20).
Previous studies have revealed that human placental
hydrolysate (hPH) is a rich source of various bioactive substances,
including polydeoxyribonucleotides, RNA, DNA, peptides, amino
acids, enzymes, and trace elements, among others (21). Human placenta has been
demonstrated to possess various therapeutic properties ranging from
wound healing to immunomodulation (22). The use of aqueous extract of human
placenta in wound healing, ophthalmology, infertility, apoplexy,
and epilepsy has evolved from folk knowledge to modern scientific
practice (23). In a previous
clinical trial, it was identified that treatment of hPH ameliorated
alcoholic or nonalcoholic steatohepatitis in patients (24,25). In addition, hPH has been shown to
have a protective effect in ALF via an anti-inflammatory response
(26). However, the mechanism
underlying this anti-apoptotic action and the defense mechanism
remain to be fully elucidated. Given the various uses of human
placenta for intervention in different diseases, the present study
was undertaken to investigate the protective effects of hPH against
apoptotic hepatocyte cell death in vivo and in
vitro.
Materials and methods
Reagents and antibodies
All chemicals and solvents used were of the highest
analytical grade available. Cell culture supplies and media,
including fetal bovine serum (FBS), phosphate-buffered saline
(PBS), and penicillin-streptomycin were purchased from Thermo
Fisher Scientific, Inc. (Waltham, MA, USA). The Cell Counting Kit-8
(CCK-8; cat. no. CK04-11) was from Dojindo Molecular Laboratories,
Inc. (Kumamoto, Japan). Protease inhibitor cocktail (cat. no.
P8340) and D-GalN (cat. no. G0500) were purchased from
Sigma-Aldrich; EMD Millipore (Billerica, MA, USA).
Anti-proliferating cell nuclear antigen (PCNA; cat. no. ab15497),
anti-Tomm20 (cat. no. ab56783) and anti-Lamin B1 (cat. no. ab16048)
antibodies were purchased from Abcam (Cambridge, UK). Anti-poly
(ADP) ribose polymerase (PARP; cat. no. 9532S) antibody was
purchased from Cell Signaling Technology, Inc. (Beverly, MA, USA).
Anti-B-cell lymphoma 2 (BCL2; cat. no. SC-7382),
anti-Bcl-2-associated X protein (BAX; cat. no. SC-526), anti-DRAM
(cat. no. SC-81713), anti-SOD1 (cat. no. SC-17767), anti-SOD2 (cat.
no. SC-130345), anti-gluta-thione peroxidase (GPx; cat. no.
SC-133160), anti-Catalase (cat. no. SC-271358), anti-Keap1 (cat.
no. SC-365626), anti-HO-1 (cat. no. SC-136960), anti-p53 (cat. no.
SC-126), anti-GAPDH (cat. no. SC-20357), and anti-Nrf2 (cat. no.
SC-81342) antibodies were purchased from Santa Cruz Biotechnology,
Inc. (Santa Cruz, CA, USA). Anti-microtubule-associated protein
1A/1B-light chain 3 (LC3)I/II (cat. no. PM036),
anti-phospho-rlyated-p62 (cat. no. PM074), and anti-p62 (cat. no.
PM045) antibodies were purchased from MBL International Corporation
(Woburn, MA, USA). hPH (Laennec inj.) was manufactured by Green
Cross WellBeing Corporation (Seongnam, Korea) through the
hydrolysis of human placenta with HCl and pepsin.
Animals and experimental design
Sprague-Dawley rats (SD) rats (male, 6–8 weeks old,
weighing ~200–250 g each) were purchased from Raonbio, Inc.
(Yongin, Korea). These rats were provided with adequate food and
water ad libitum and were housed in clean cages for 1 week.
The laboratory temperature was 24±1°C and relative humidity was
40–80%. All animal experiments were performed according to the
Guide for the Care and Use of Laboratory Animals as published by
the US National Institutes of Health. The present study was
reviewed and approved by the Animal Welfare and Research Ethics
Committee at Chung-Ang University (Seoul, Korea; 2017-00003). Acute
liver injury was induced by intraperitoneal injection of LPS (15
µg/kg) together with D-GalN (700 mg/kg) dissolved in normal saline,
which can increase the sensitivity of hepatocytes. Blood was
collected from the inferior vena cava 24 h following injection of
D-GalN/LPS. The SD rats were then dissected, and liver tissues were
removed immediately for histological detection. Normal PBS was used
in control rats. hPH (1.2, 2.4, and 3.6 ml/kg) was injected
subcutaneously into each mouse 24, 48, and 72 h prior to D-GalN/LPS
injection. As a negative control, only D-GalN/LPS was injected.
Evaluation of serum alanine
aminotransferase (ALT) and aspartate aminotransferase (AST)
The collected blood samples were stored overnight at
4°C. The serum was isolated the subsequent day following
centrifugation at 15,928 × g for 10 min at 4°C. The ALT and AST
were detected using a Hitachi 7600 Series automatic biochemical
analyzer (Hitachi, Ltd., Tokyo, Japan).
Enzyme-linked immunosorbent assay (ELISA)
of cytokines
Based on a previous study, blood was collected for
measuring TNF-α (cat. no. 438207) and IL-6 (cat. no. 437107) at 24
h post-D-GalN/LPS injection. The serum was separated by
centrifugation at 15,928 × g at 4°C for 10 min. The cytokines were
measured using mouse ELISA kits (BioLegend, Inc., San Diego, CA,
USA) according to the manufacturer's protocol.
Histopathological evaluation
The liver tissues were immersed in normal 10%
neutral buffered formalin and fixed for 48 h, dehydrated in a
series of graded ethanol, embedded in paraffin wax, and cut into
5-µm sections. The paraffin-embedded sections were stained with
hematoxylin and eosin (H&E) for pathological analysis under a
light microscope. Histological changes were evaluated using a
point-counting method for the severity of hepatic injury using an
ordinal scale, as previously described (27). Briefly, the H&E-stained
sections were evaluated at ×400 magnification using the
point-counting method for the severity of hepatic injury with an
ordinal scale as follows: grade 0, minimal or no evidence of
injury; grade 1, mild injury consisting of cytoplasmic vacuolation
and focal nuclear pyknosis; grade 2, moderate to severe injury with
extensive nuclear pyknosis, cytoplasmic hypereosinophilia, and loss
of intercellular borders; and grade 3, severe necrosis with
disintegration of hepatic cords, hemorrhage, and neutrophil
infiltration.
Measurement of apoptosis via TUNEL
assay
TUNEL was performed to analyze DNA fragmentation
indicative of cellular apoptosis using the in situ cell
death detection kit (cat. no. ab206386, Abcam), according to the
manufacturer's protocol. The paraffin wax-embedded tissue sections
were treated with proteinase K, and endogenous peroxidase activity
was blocked with hydrogen peroxide. The sections were incubated at
37°C with the terminal TdT nucleotide mixture for 1 h. The
reactions were then terminated, and the slides were rinsed with
PBS. Nuclear labeling was developed with horseradish peroxidase and
diaminobenzidine. The number of apoptotic cells in each slide from
at least five different fields was analyzed using Image-Pro Plus
software (version 6.0 for Windows; Media Cybernetics, Inc.,
Rockville, MD, USA) with the assistance of a microscope (DM750;
Leica Microsystems GmbH, Wetzlar, Germany). The number of positive
cells was averaged for statistical analysis.
Liver immunohistochemistry
The paraffin sections were de-paraffinized and
rehydrated. Endogenous peroxidase was inactivated by incubation in
0.3% hydrogen peroxide in absolute methanol for 30 min. The
sections were incubated in 5% skim milk for 30 min at room
temperature. Antigen retrieval was performed by microwave (700 W)
treatment in 10 mM citrate buffer (pH 6.0) for 15 min. The sections
were incubated overnight at 4°C with anti-PCNA primary antibody
(Abcam) at a dilution of 1:500. Following washing with PBS, the
sections were incubated at room temperature for 30 min in secondary
antibody (Goat Anti-Rabbit; cat. no. ab205718; Abcam; 1:1,000). A
brown color was developed with 3 diami-nobenzidine for 2–4 min, and
the sections were then washed in distilled water. The number of
positively stained brown nuclei (denoting PCNA) in each slide from
at least five different fields was analyzed using Image-Pro Plus
software (version 6.0 for Windows) with the assistance of a
microscope (DM750, Leica Microsystems GmbH). The number of positive
cells was averaged for statistical analysis.
Cell culture and treatment
Human HepG2 cells, the HepG2 (ATCC HB 8065) cells
were purchased from the Korea Cell Line Bank (Seoul, Korea), and
were cultured in Dulbecco's modified Eagle's medium (DMEM) with 10%
(v/v) heat-inactivated FBS and 1% penicillin and incubated in a
humidified incubator with 5% CO2, 95% air at 37°C. A
dose of 5% hPH was used in the time-course investigation of the
induction of antioxidant enzymes by hPH. To investigate the
protective effects of hPH and its active mechanism on HepG2 cells
challenged by D-GaIN (Sigma; EMD Millipore), the cells were
pretreated with hPH for 2 h, stimulated with 50 mM D-GaIN at 37°C,
and harvested for the indicated experiments.
Cell viability assay
The HepG2 cells, treated as described above, were
further treated with 50 mM D-GalN and incubated for another 24 h,
following which the cell viability was determined using CCK-8
(Dojindo Molecular Technologies, Inc.). To determine cell
viability, the HepG2 cells were seeded in 96-well plates at a
density of 1×104 cells/ml and grown for 24 h in a 37°C
incubator. When the cells attained 70–80% confluence, they were
either left untreated (control group) or treated with D-GalN (50
mM), 5% hPH, or 5% hPH + D-GalN (50 mM). Following incubation for
24 h, cell morphology changes were observed under Olympus inverted
microscopes (Olympus Corporation, Tokyo, Japan). The supernatants
were discarded, following which 10% CCK-8 solution in fresh DMEM
(100 µl) were added to each well. The cells were then incubated at
37°C for 1 h, and the absorbance was measured at 450 nm directly in
the wells using a SpectraMax i3x Multi-Mode detection platform
(Molecular Devices LLC, Sunnyvale, CA, USA).
Measurement of LDH release
The LDH in the culture medium was assessed with 0.2
mM NADH and 0.4 mM pyruvic acid in up to 200 µl PBS at pH 7.4. The
LDH concentration in the sample was proportional to the rate of
NADH oxidation measured by absorbance at 334 nm (OD/min) using the
SpectraMax i3x Multi-Mode detection platform. The LDH concentration
in the culture media was calculated using a commercial standard LDH
assay kit purchased from Abnova (cat. no. KA0785, Taipei,
Taiwan).
DAPI staining
Morphological changes in the apoptotic cells were
assessed by fluorescent microscopy following DAPI staining.
Briefly, the HepG2 cells were seeded at a density of
1×105 cells/ml in 6-well plates and grown for 24 h in a
37°C incubator. Following washing once with PBS, the cells were
either left untreated (control group) or treated with D-GalN (50
mM), 5% hPH, or 5% hPH + D-GalN (50 mM). Following incubation for
24 h, the cells were fixed in 4% formaldehyde for 1 h, and
permeabilized with 0.1% Triton X-100 (Biosesang, Inc., Seongnam,
Korea) for 5 min. Subsequently, DNA-specific fluorochrome DAPI
(Invitrogen; Thermo Fisher Scientific, Inc.) was applied to each
well, following which samples were incubated for 10 min in the dark
at room temperature. Finally, the cells were washed three times
with PBS and examined using a confocal microscope (Carl Zeiss AG,
Oberkochen, Germany).
Annexin V-fluorescein isothiocyanate
(FITC)/propidium iodide (PI) staining and fluorescence images
For apoptotic cell analysis, the HepG2
(1×104/well) cultures in 96-well black plates were
exposed to the indicated treatments for 12 h. The assay was
performed according to the manufacturer's protocol (BD Biosciences,
San Jose, CA, USA). Briefly, the cells were washed twice with PBS
and again with binding buffer. Annexin V-fluorescein isothiocyanate
and PI were added to each sample, and the mixture was incubated at
room temperature for 15 min. The absorbance was read directly in
the wells at 494/518 nm for FITC Annexin V and 535/617 nm for PI
using the SpectraMax i3x Multi-Mode detection platform. The
populations staining positive for Annexin V and Pl were compared
with the untreated group.
The HepG2 cells were grown to ~70% confluence;
1×105 cells were collected in a Petri dish and either
left untreated (control group) or treated with D-GalN (50 mM), 5%
hPH, or 5% hPH + D-GalN (50 mM); and then incubated for 24 h. The
cells were then washed with PBS and stained with 10 µg/ml PI for 5
min, and fluorescent images were observed under a DP70 fluorescence
microscope with DP Controller software (Olympus Corporation; ver.
3.2.276.2).
Measurement of ROS
Cytosolic and mitochondrial ROS were measured on a
96-well plate reader as previously described (28). Briefly, the HepG2 cells were
seeded at a concentration of 1×104 cells per well in
black 96-well flat bottom plates (Thermo Fisher Scientific, Inc.)
and allowed to adhere overnight. Following seeding, the cells were
washed and incubated with serum-starved medium, D-GalN (50 nM), 5%
hPH, or 5% hPH + D-GalN (50 mM) for 4 h, followed by incubation
with 2′,7′-dichlorofluorescin diacetate (DCFH-DA; 10 µM, ex/em:
518/605 nm; Invitrogen; Thermo Fisher Scientific, Inc.) or MitoSOX™
(5 µM, ex/em: 510/580 nm; Invitrogen; Thermo Fisher Scientific,
Inc.) for 20 min. The oxidative products were measured with the
SpectraMax i3x Multi-Mode detection platform.
Measurement of mitochondrial membrane
potential (ΔΨm)
The ΔΨm of intact cells was measured as previously
described, (29) with
modifications. Briefly, the cells were washed with PBS and TMRE
(200 nM, ex/em: 549/582 nm; Invitrogen; Thermo Fisher Scientific,
Inc.) was added to the cell suspension. The cells were incubated at
37°C for 30 min in the dark. The ΔΨm was measured using the
SpectraMax i3x Multi-Mode detection platform. The percentage and
mean fluorescence intensity level of the mass were calculated for
each sample.
Determination of mitochondrial mass
Mitochondrial mass was measured according to
fluorescence levels following staining with MitoTracker Green FM
(100 nM, ex/em: 490/525 nm; Invitrogen; Thermo Fisher Scientific,
Inc.) at 37°C for 30 min. Subsequently, the cells were washed once
in PBS and promptly evaluated using the SpectraMax i3x Multi-Mode
detection platform. The percentage and mean fluorescence intensity
level of the mass were calculated for each sample.
Fluorescence microscopy
The cells (1×106 cells/well) were
prepared on sterilized glass coverslips (BD Biosciences) in
triplicate and then fixed in 4% paraformaldehyde in PBS for 10 min,
permeabilized with 0.25% Triton X-100 in PBS for 10 min and
incubated with primary antibodies against Tomm20 (Abcam; 1:500),
Nrf2 (Santa Cruz Biotechnology, Inc.; 1:500), and LC3 (Abcam;
1:500) for 12 h at 4°C incubator. The cells were washed to remove
excess primary antibodies and incubated with the appropriate
fluorescently labeled secondary antibodies (1:1,000) for 1 h at
room temperature. Following mounting of slides, fluorescence images
were acquired using a confocal microscope (LSM700, Carl Zeiss
AG).
Western blot analysis
The cells were lysed in cell lysis buffer [62.5 mM
Tris-HCl (pH 6.8), 2% sodium dodecyl sulfate (SDS), 5%
β-mercaptoethanol, 2 mM phenylmethylsulfonyl fluoride, protease
inhibitors (Complete™; Roche Diagnostics GmbH, Mannheim, Germany),
1 mM Na3VO4, 50 mM NaF and 10 mM EDTA]. The
protein content of the lysates was determined using BCA Protein
Assay reagent (cat. no. 23225; Pierce; Thermo Fisher Scientific,
Inc.). A 20 µg sample of protein per lane was separated by 12%
SDS-polyacrylamide gel electrophoresis and blotted onto
polyvinylidene fluoride membranes (EMD Millipore) which were then
saturated with 5% powdered milk in Tris-buffered saline containing
0.5% Tween-20. The blots were then incubated with the appropriate
primary antibodies at a dilution of 1:1,000 or 1:2,000 for 12 h in
a 4°C incubator, and further incubated with horseradish
peroxidase-conjugated secondary antibody (1:10,000) for 2 h at
37°C. The bound antibodies were detected using enhanced
chemiluminescence (Amersham; GE Healthcare Life Sciences, Chalfont,
UK). Images of the blotted membranes were captured using the
LAS-1000 lumino-image analyzer (Fujifilm, Tokyo, Japan). The
protein levels were compared to those of the appropriate loading
control (GAPDH or non-phosphorylated proteins).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from the dorsal skin using
TRIzol (Invitrogen; Thermo Fisher Scientific, Inc.). First-strand
cDNA synthesis from the total RNA template was performed with
PrimeScript RT master mix (Takara Bio, Inc., Tokyo, Japan). The
resulting cDNA was subjected to real-time PCR (complementary cDNA,
2 µl; primer, 3 µl; 2X premix SYBR, 5 µl), using qPCR 2X PreMIX
SYBR (Enzynomics, Seoul, Korea) and a CFX-96 thermocycler (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The PCR conditions used to
amplify all genes were as follows: 10 min at 95°C and 40 cycles of
95°C for 10 sec, 60°C for 15 sec, and 72°C for 20 sec. The
expression data were calculated from the quantification cycle (Cq)
value using the ΔCq method of quantification (30). GAPDH was used for normalization.
The oligonucleotides that were used for qPCR were as follows: Human
ATG8, forward 5′-CGC ACC TTC GAA CAA AGA GT-3′ and reverse, 5′-GAC
CAT GCT GTG TCC GTT C-3′; human beclin 1 (BECN1), forward 5′-AAC
CTC AGC CGA AGA CTG AA-3′ and reverse, 5′-CCT CTA GTG CCA GCT CCT
TT-3′; human cathepsin D (CTSD), forward 5′-CAA GTT CGA TGG CAT CCT
GG-3′ and reverse 5′-CGG GTG ACA TTC AGG TAG GA-3′; human
lysosomal-associated membrane protein 1 (LAMP1), forward 5′-CTT TCA
AGG TGG AAG GTGz GC-3′ and reverse 5′-GAT AGT CTG GTA GCC TGC
GT-3′; human activating transcription factor (ATF)6, forward 5′-GTG
TCA GAG AAC CAG AGG CT-3′ and reverse 5′-GGT GCC TCC TTT GAT TTG
CA-3′; human ATF4, forward 5′-ACA CTG CTT ACG TTG CCA TG-3′ and
reverse 5′-AGA CCC ACA GAG AAC ACC TG-3′; human C/EBP homologous
protein (CHOP), forward 5′-CAT TGC CTT TCT CCT TCG GGG-3′ and
reverse 5′-CCA GAG AAG CAG GGT CAA GA-3′.
Statistical analysis
All quantitative data are presented as the mean ±
standard error of the mean for three independent experiments.
Statistical analyses were performed using the statistical package
for SPSS software version 17.0 (IBM Corp., Armonk, NY, USA).
Differences between two groups were evaluated using a paired
Student's t-test. For multiple comparisons, one-way analysis of
variance was used followed by Tukey's multiple comparisons test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Effects of hPH on mortality rates of
D-GalN/LPS-treated rats
Acute liver injury leads to high mortality rates in
rats. Therefore, the present study assessed the effects of hPH on
D-GalN (700 mg/gg)/LPS (15 µg/kg)-induced mortality. To investigate
whether hPH can protect against liver injury in rats due to
D-GalN/LPS, the survival rate of D-GalN/LPS + hPH-treated rats was
monitored for 24 h (n=12). All saline-injected rats survived as the
normal control group (n=12). No significant differences in gross
liver examination, body weight, or liver weight were found between
groups (Fig. 1A-C). As shown in
Fig. 1D, a number of rats died 7
h following D-GalN/LPS injection, and the survival rate was 33.3%
at 24 h. However, it was found that hPH pretreatment effectively
increased the survival rate of rats with liver injury induced by
D-GalN/LPS. Pretreatment with hPH (1.2 ml/kg) increased the
survival rate to 83.3%, and all hPH (2.4 and 3.6 ml/kg)-treated
rats survived. These results suggested that pretreatment with hPH
effectively protected against D-GalN/LPS-induced mortality.
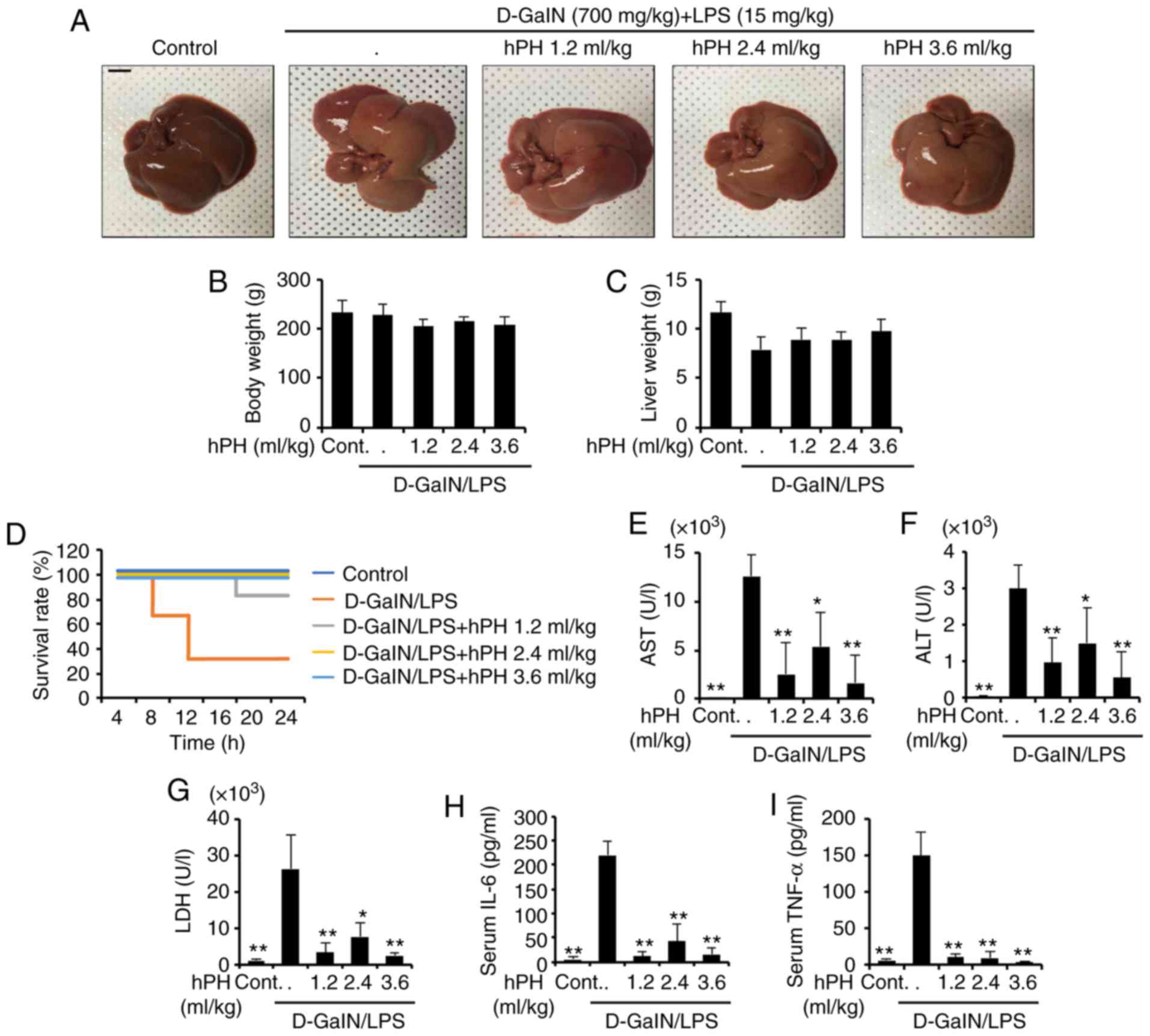 | Figure 1Protective effects of hPH treatment
on D-GalN/LPS-induced acute liver failure. hPH (1.2, 2.4, or 3.6
ml/kg) was subcutaneously administered to rats three times at
intervals of 24 h, followed by exposure to 700 mg/kg D-GalN and 15
µg/kg LPS (D-GalN/LPS). Livers from each experimental group were
examined 24 h following D-GalN/LPS challenge. (A) Gross image of
livers. Scale bar=1 cm (B) Animal weights (n=4-6) were measured
just prior to sacrifice; with no statistical difference among
groups. (C) Liver weights were measured following sacrifice, with
no statistical difference among groups (n=4-6). (D) Survival rates
of rats were observed for 24 h following D-GalN/LPS exposure
(n=12). Serum (n=4-6) was collected from animals 24 h following
exposure to D-GalN/LPS to determine levels of (E) AST, (F) ALT, (G)
LDH, (H) IL-6, and (I) TNF-α. All data are presented as the mean ±
standard error of the mean. *P<0.05 and **P<0.01,
vs. D-GalN/LPS group. hPH, human placental hydrolysate; D-GalN,
D-galactosamine; LPS, lipopolysaccharide; Cont, control; AST,
aspartate aminotransferase; ALT, alanine aminotransferase; LDH,
lactate dehydrogenase; IL-6, interleukin-6; TNF-α, tumor necrosis
factor-α. |
Effects of hPH on serum levels of ALT,
AST, and inflamma- tory cytokines in LPS/GalN-induced liver failure
in rats
The serum levels of AST, ALT, LDH, and inflammatory
cytokines are well-established markers of hepatic injury (31). In order to investigate the effects
of hPH on D-GalN/LPS-induced liver damage, hematological tests were
performed. Serum (n=4–6) was collected to detect AST, ALT, LDH,
IL-6, and TNF-α. As shown in Fig.
1E-G, LPS/D-GalN markedly increased serum levels of AST, ALT,
and LDH, whereas hPH reduced these elevations. As hepatocyte injury
resulting from acute liver injury is accompanied by an elevation in
inflammatory cyto-kines (32),
the present study examined the effect of hPH on hepatotoxicity by
measuring serum levels of IL-6 and TNF-α (Fig. 1H and I). The D-GalN/LPS-treated
rats exhibited significant increases in levels of IL-6 and TNF-α
compared with normal rats. In addition, hPH pretreatment
significantly inhibited the release of these pro-inflammatory
cytokines compared with the LPS/D-GalN group. These results
suggested that hPH effectively protected against LPS/D-GalN-induced
liver degeneration and inflammatory responses.
Effects of hPH on hepatotoxicity of
hepatocytes
The present study also investigated histological
changes following liver injury to confirm the protective effects of
hPH. The rats treated with D-GalN/LPS showed severe
histopathological degeneration evidenced by severe intrahepatic
hemorrhaging, apoptosis, necrosis, ballooned hepatocytes, and
hepatocytes expanded by fat vacuoles, whereas hPH (1.2, 2.4 and 3.6
ml/kg) treatment ameliorated these changes (Fig. 2A). It has been suggested that
D-galN/LPS-induced imbalance between apoptosis and proliferation in
hepatocytes is responsible for the impairment of liver function.
Therefore, hPH-mediated modulations of proliferation (PCNA) and
apoptosis (TUNEL) were also evaluated by immunohistochemical
analysis (Fig. 2B and C). A high
expression of PCNA and low numbers of TUNEL-positive cells were
observed in the control group. However, D-GalN/LPS markedly reduced
PCNA immunore-activity and significantly increased the number of
apoptotic cells. By contrast, pre-administration of hPH resulted in
an increase of PCNA-positive cells and reduced the number of
apoptotic cells in all three dose groups. Graphs showing results of
the staining experiments are shown in Fig. 2D-F. These observations indicated
that hPH pretreatment ameliorated hepatic injury by reducing
D-GalN/LPS-induced hepatocyte damage in rats.
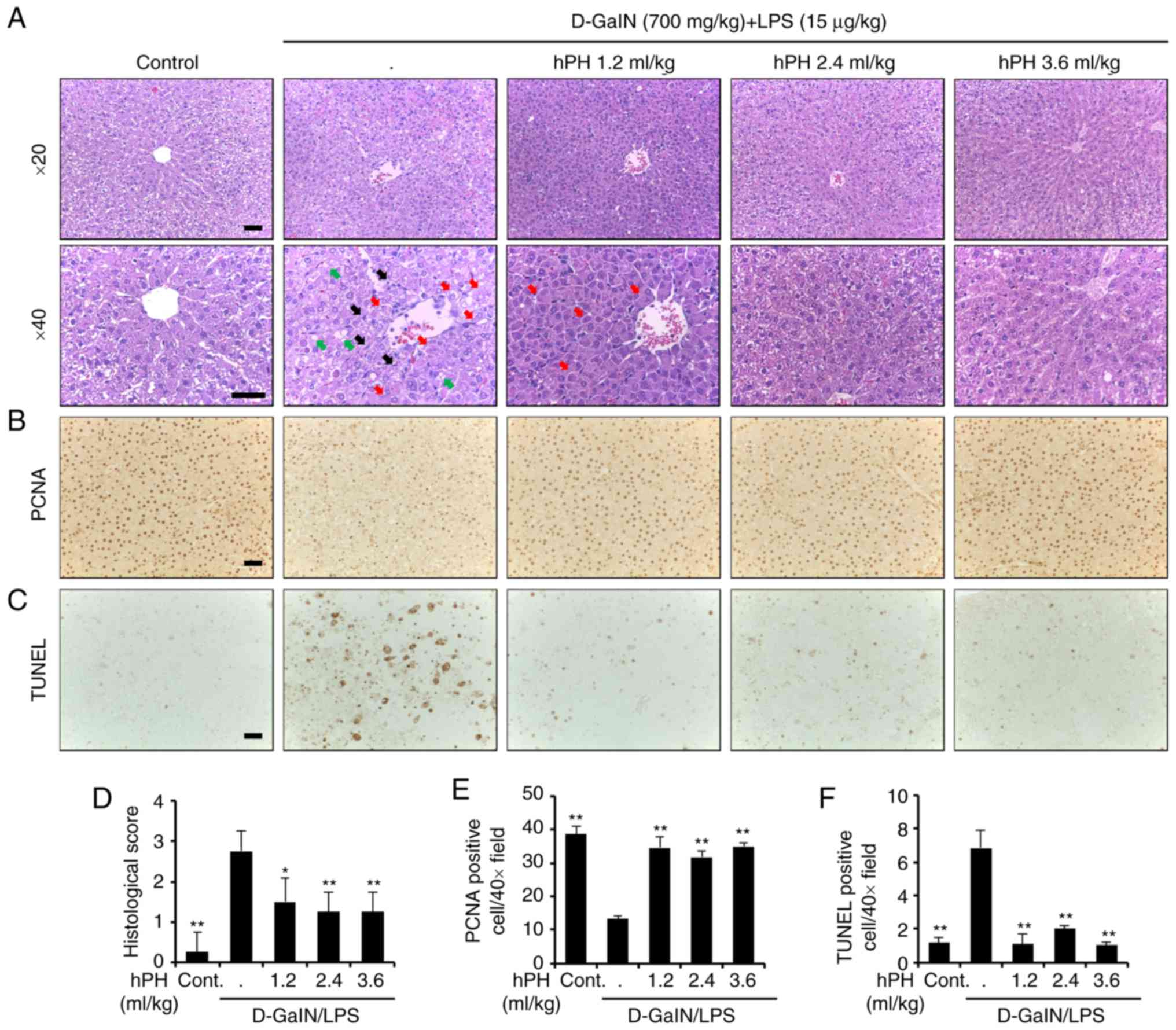 | Figure 2Effects of hPH treatment on
D-GalN/LPS-induced acute liver failure in tissue degeneration and
apoptosis. (A) Histopathological staining. Upper panel
(magnification, ×20), lower panel (magnification, ×40). Scale
bar=100 µm. The arrows indicate apoptotic hepatocytes (black), a
severely affected region of a lobule containing ballooned
hepatocytes (red), and hepatocytes expanded by fat vacuoles
(green). (B) Images showing PCNA staining. Scale bar=100 µm. (C)
Apoptotic response to D-GalN/LPS in the liver tissue of rats was
investigated using TUNEL staining. Scale bar=100 µm. (D) Stained
sections were graded for histopathology using a four-point scale
(0-3), with 0, 1, 2, and 3 representing no damage, mild damage,
moderate damage, and severe damage, respectively. (E) PCNA-positive
cells were significantly less numerous in the D-GalN/LPS-treated
group than in the control group; the addition of hPH led to
significant overexpression of PCNA in the liver compared with
D-GalN/LPS treatment alone. (F) TUNEL-positive cells was higher in
number in the group treated with D-GalN/LPS alone compared with the
control group. However, apoptotic induction by D-GalN/LPS was
further reduced in the groups treated with 1.2, 2.4, and 3.6 ml/kg
of hPH. All data are presented as the mean ± standard error of the
mean. *P<0.05 and **P<0.01, vs.
D-GalN/LPS group. hPH, human placental hydrolysate; D-GalN,
D-galactosamine; LPS, lipopolysaccharide; PCNA, proliferating cell
nuclear antigen; Cont, control. |
hPH protects hepatocytes against
D-GalN-induced apoptosis
Hepatocyte apoptosis is important in the
pathogenesis of liver disease, including ALF (33). D-GalN induces hepatocyte cell
death in vivo (34) and
in vitro (35); it offers
a suitable experimental model based on its capacity to reduce the
intracellular pool of uridine monophosphate in hepatocytes,
inhibiting the synthesis of RNA and proteins, and leading to cell
apoptosis. Therefore, the present study investigated whether
D-GalN-induced hepatotoxicity occurred in HepG2 cells. Following
D-GalN stimulation, cell viability decreased markedly in a
dose-dependent manner. The results revealed a reduction of 50% at
50 mM D-GalN and of 82% at 100 mM D-GalN in the HepG2 cells
(Fig. 3A). The cytotoxic effects
of hPH on HepG2 cells have not been examined previously. The
present study used a CCK-8 assay to evaluate the dose-dependent
cytotoxic effects of hPH on HepG2 cells. The results showed that
stimulation with hPH had no significant effect on cell viability at
various concentrations (1.25–5% in serum-free DMEM) over 24 h.
Therefore, hPH was used at a concentration of 5% in subsequent
experiments (Fig. 3B). In
addition to the D-GalN-induced decrease in cell viability being
attenuated by pretreatment with hPH, the concentration of LDH
released from the hepatocytes was also reduced in the D-GalN + 5%
hPH-stimulated cells, compared with the D-GalN only-treated cells
(Fig. 3C and D). These results
indicated that hPH protected against D-GalN-induced hepato-toxicity
in HepG2 cells. Changes in cellular morphology and cell viability
were also evaluated to examine the protective effects of hPH in
D-GalN-stimulated HepG2 cells. As shown in Fig. 3E, the D-GalN-stimulated HepG2
cells exhibited a marked reduction in the number of hepatocytes.
However, hPH pretreatment effectively improved the D-GalN-mediated
cell damages.
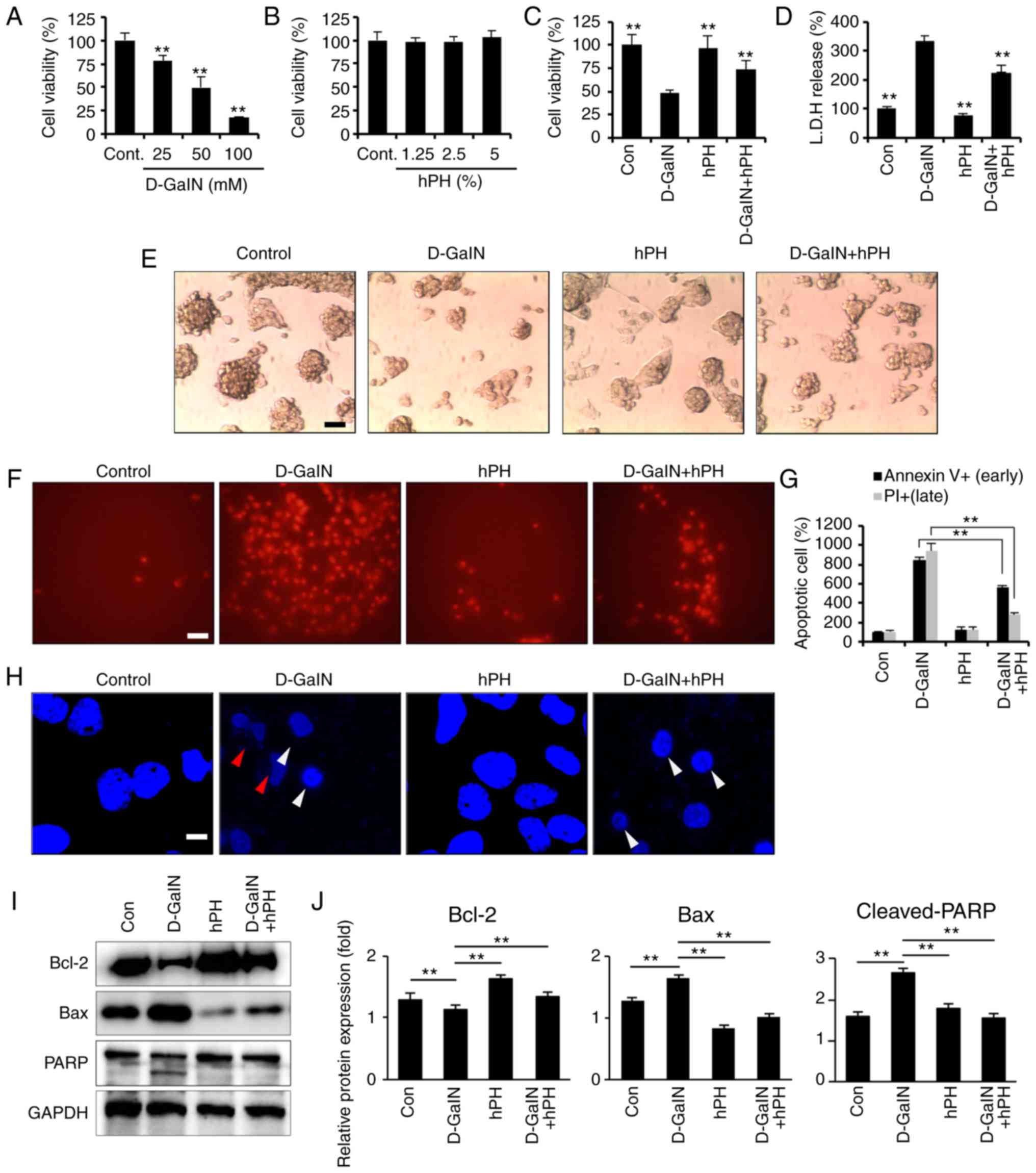 | Figure 3Effects of hPH treatment on
D-GalN-induced apoptosis in HepG2 cells. (A) HepG2 cells were
treated with 25, 50, or 100 mM D-GalN for 24 h. Treatment with 50
mM D-GalN yielded a cell viability of almost 50% compared with the
vehicle control. The concentration of 50 mM D-GalN was used to
determine the 50% inhibitory concentration. (B) HepG2 cells were
treated with increasing concentrations of hPH or vehicle control
for 24 h. HepG2 cells were treated with hPH (5%) for 2 h prior to
D-GalN stimulation (50 mM). After 24 h, the hepatoprotective
effects of hPH were assessed with the (C) Cell Counting Kit-8 assay
and (D) LDH release test for cell viability, and with an (E)
inverted phase-contrast microscope for morphological changes (scale
bar=50 µm). HepG2 cells were stimulated with D-GalN (50 mM, 24 h)
in the presence or absence of 5% hPH. (F) Cells were subjected to
fluorescence microscopy (PI stain only) and (G) Annexin V/PI
staining analyzed by a microplate (scale bar=50 µm). (H) DNA
fragmentation (red arrow) and nuclear condensation (white arrow)
were detected by DAPI staining under each condition (scale bar=5
µm). For western blot analysis, cell lysates were collected and
subjected to sodium dodecyl sulfate-PAGE, followed by immunoblot
analysis using anti-BCL2, anti-BAX and anti-PARP antibodies.
Anti-GAPDH was used as a loading control. (I) Representative images
and (J) densitometry. All data are presented as the mean ± standard
error of the mean. **P<0.01, vs. D-GalN group. hPH,
human placental hydrolysate; D-GalN, D-galactosamine; Cont,
control; LDH, lactate dehydrogenase; BCL2, B-cell lymphoma 2; BAX,
Bcl-2-associated X protein; PARP, poly (ADP) ribose polymerase; PI,
propidium iodide. |
To examine the inhibitory mechanisms of hPH in
D-GalN-induced cytotoxicity, the HepG2 cells were assessed by
Annexin V and PI staining, followed by microplate analysis
(Fig. 3F and G) or fluorescence
microscopy (Fig. 3H). D-GalN
stimulation alone facilitated the induction of apoptosis, including
levels of Annexin V+ (841%)/PI+ (948%),
compared with the controls. By contrast, pretreatment with hPH
significantly inhibited D-GalN-induced apoptosis (Annexin
V+, 801%; PI+, 280%). D-GalN-induced DNA
fragmentation was also effectively attenuated by hPH pretreatment
(Fig. 3H). The present study
further examined whether hPH regulated PARP-dependent cell death.
D-GalN induced an increase of Bax (pro-apoptotic protein) and a
decrease of Bcl-2 (anti-apoptotic protein) (Fig. 3I and J). Furthermore, the cleavage
of PARP was inhibited in the HepG2 cells by pretreatment with hPH
(Fig. 3I and J). These findings
suggested that hPH treatment significantly improved hepatocyte
degeneration through the attenuation of apoptosis in HepG2
cells.
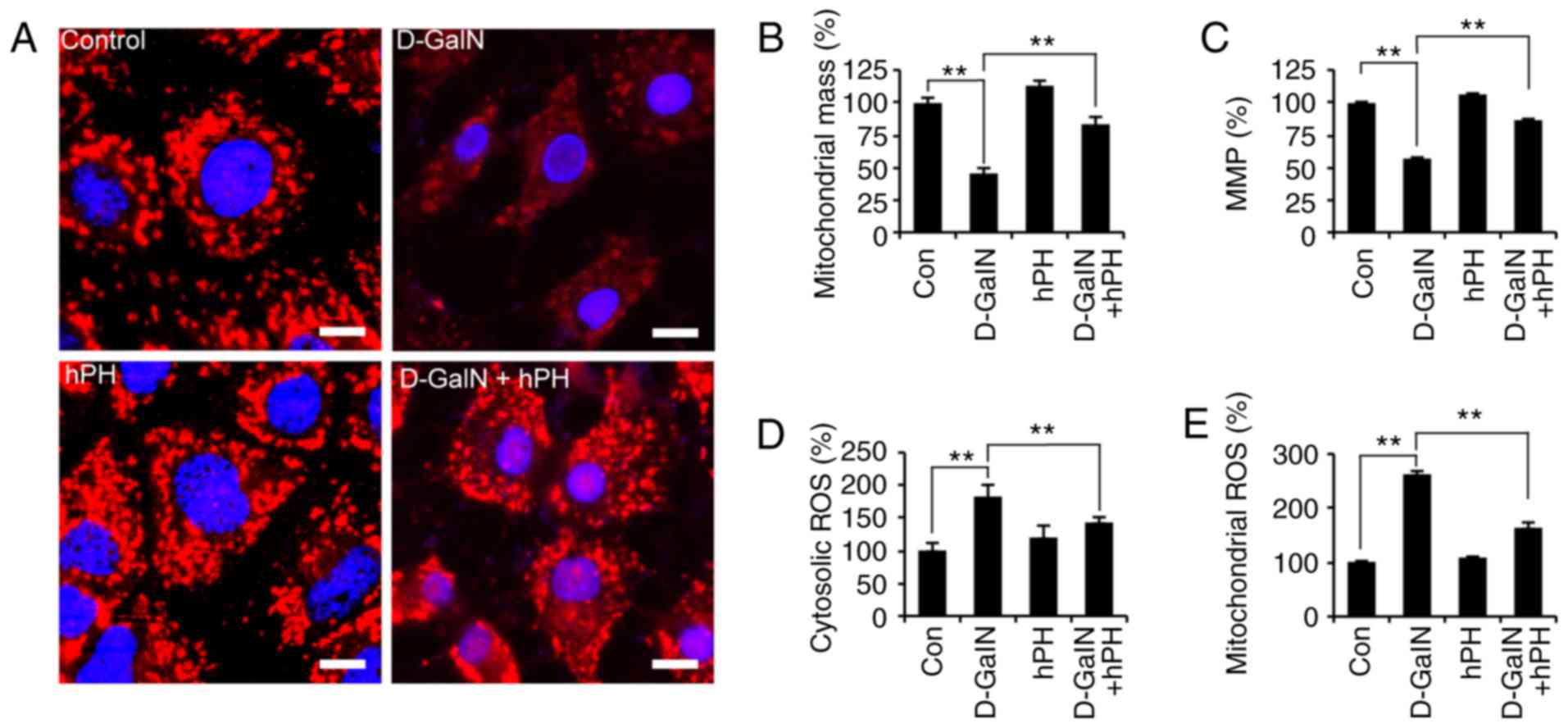
hPH regulates D-GalN-mediated
intracellular ROS generation and mitochondrial dysfunction
The rapid generation of ROS is associated with
hyperpolarization of the mitochondrial membrane potential and
apoptosis in D-GalN-treated hepa-tocytes (36). In addition, D-GalN-dependent cell
necrosis is associated with mitochondrial membrane depolarization
(37). To investigate whether the
D-GalN-mediated increase in abnormal mitochondria was rescued by
hPH treatment, the present study first evaluated mitochondrial
morphology using Tomm20 (mitochondrial outer membrane) staining
(Fig. 4A). Mitochondrial
morphology was altered at 24 h in the D-GalN-stimulated HepG2
cells. However, the increased mitochondrial damage was attenuated
significantly by hPH. In addition, mitochondrial mass decreased, by
~55%, in the D-GalN-stimulated HepG2 cells, and this was rescued by
hPH pretreatment (Fig. 4B). ΔΨm
was then evaluated using microplate analysis. The D-GalN-stimulated
HepG2 cells exhibited decreased ΔΨm; however, this decrease was
signifi-cantly attenuated in the hPH-pretreated cells and
unstimulated cells (Fig. 4C).
These findings suggested that hPH improved D-GalN-mediated
mitochondrial dysfunction by improving ΔΨm and inhibiting
quantitative mitochondria loss.
Subsequently, the present study determined whether
hPH was involved in the negative regulation of oxidative stress.
The HepG2 cells were stimulated with D-GalN in the presence or
absence of hPH, and ROS generation was determined using DCFH-DA or
MitoSOX (mitochondrial-specific superoxide indicator) (38) as a probe for microplate analyses
(Fig. 4D and E). D-GalN alone led
to the potent intracellular generation of ROS, whereas the
D-GalN-induced generation of ROS in HepG2 cells was attenuated
significantly by hPH pretreatment (Fig. 4D). Mitochondrial ROS increased
significantly following treatment with D-GalN (Fig. 4E), whereas D-GalN-induced
mitochondrial ROS generation was decreased by pretreatment with
hPH. These results suggested that hPH protected against
D-GalN-mediated apoptotic cell death by regulating the generation
of cytosolic and mitochondrial ROS.
hPH regulates expression of antioxidant
enzymes in HepG2 cells
As the induction of antioxidant enzymes is a
universal stress response against liver degeneration and has been
widely shown to have an anti-apoptotic effect, the expression
levels of antioxidative enzymes SOD1, SOD2, GPx, and catalase were
detected by western blot analysis (Fig. 5A). hPH induced antioxidative
enzyme expression in the HepG2 cells. These enzymes were elevated 1
h following hPH treatment and were maintained at this level until 8
h. Furthermore, to investigate the antioxidant mechanisms of hPH,
Nrf2 pathway-related gene expression on HepG2 cells was evaluated
(Fig. 5B-D). The induction of
Nrf2 upregulates antioxidant proteins, including SOD, catalase, and
HO-1, reducing ROS (15). hPH
treatment resulted in a significant decrease of Keap1 at 4–8 h.
However, hPH increased the protein levels of cytoplasmic p-p62 and
HO-1. Additionally, p62 was upregulated 4–8 h following hpH
treatment (Fig. 5B). Furthermore,
the expression of Nrf2 was decreased in the cytosol and increased
in the nucleus (active Nrf2) following hPH treatment, indicating
that translocation to the nucleus was affected by hPH treatment
(Fig. 5C and D). Together, these
findings indicated that the induction of anti-oxidative enzymes by
hPH treatment was modulated by the Keap1-p62-Nrf2 pathway.
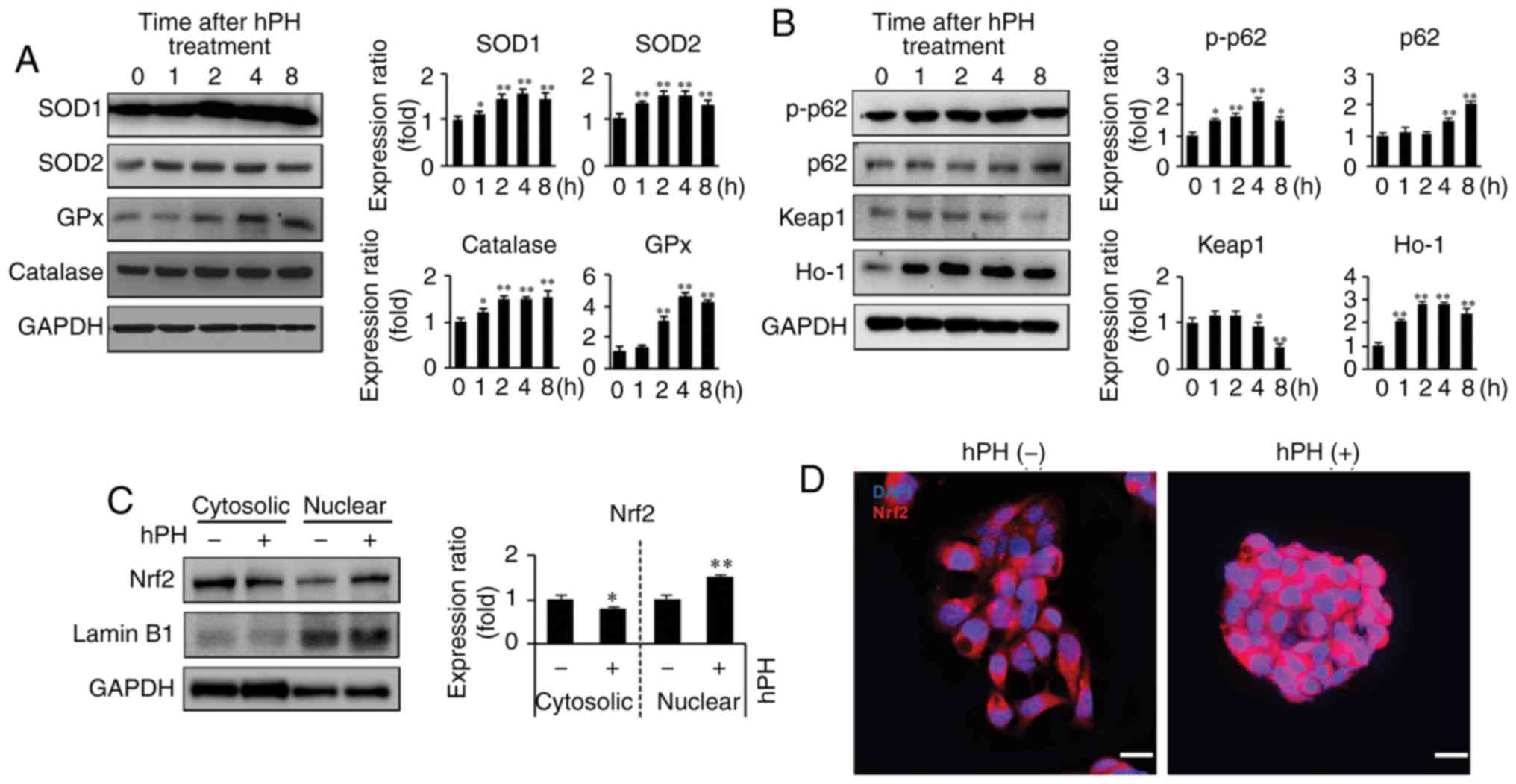 | Figure 5Upregulation of antioxidant enzymes
and regulation of Keap1-Nrf2 in hPH-treated HepG2 cells. (A)
Lysates from hPH-induced HepG2 cells were immunoblotted with
anti-SOD-1, anti-SOD-2, anti-GPx, or anti-catalase antibodies.
Representative images (left penal), densitometry results (right
panel). All data are presented as the mean ± standard error of the
mean. *P<0.05, **P<0.01 vs. 0 h time
point. (B) Following treatment with hPH, the expression levels of
p-p62, p62, Keap1, and HO-1 were determined by western blot
analysis. Representative images (left penal), densitometry results
(right panel). All data are presented as the mean ± standard error
of the mean. *P<0.05, **P<0.01, vs. 0 h
time point. (C) Nuclear localization of Nrf2 in hPH-treated HepG2
cells compared with untreated HepG2 cells. Lysates from hPH-treated
HepG2 cells were immunoblotted with anti-Nrf2, anti-Lamin B1, or
anti-GAPDH antibodies. Representative images (left penal),
densitometry results (right panel). All data are presented as the
mean ± standard error of the mean. *P<0.05,
**P<0.01, vs. 0 h time point. (D) HepG2 cells
post-hPH treatment were immunostained with anti-Nrf2 antibody.
Nuclei were identified using DAPI staining (scale bar=20 µm). hPH,
human placental hydrolysate; SOD, superoxide dismutase; GPx,
glutathione peroxidase; Keap1, Kelch-like ECH2-associated protein
1; HO-1, heme oxygenase-1; p-p62; Nrf2, nuclear factor-E2-related
factor 2. |
hPH minimizes the expression of
damage-related molecules via the regulation of autophagy
Autophagy, mitochondria, and oxidative stress are
closely intertwined in redox signalling (39). Additionally, D-GalN-induced cell
damage regulates the autophagy process in early stages. To
investigate whether the regulation of autophagy is influenced by
D-GalN stimulation and the effects of hPH, the present study
measured the level of autophagy regulation by staining cells with
anti-LC3 I/II antibody (Fig. 6A).
LC3 I/II is an established autophagy marker due to its involvement
in autophagosome membrane formation (40). In the present study, compared with
the cells treated with vehicle control or D-GalN alone, the cells
treated with hPH exhibited signifi-cantly fewer LC3 puncta per cell
(Fig. 6A). The changes in
autophagy-related genes were then quantified through assessment of
RNA and protein levels in the hPH-treated HepG2 cells. As shown in
Fig. 6B and C, hPH treatment
induced a decrease in the conversion of LC3-I to LC3-II (lipidated
form), the latter of which is a reliable marker of autopha-gosome
generation. Furthermore, the expression levels of DRAM, CHOP, and
p53 (proteins associated with autophagic cell death) indicated that
a minimized autophagy process was induced in the hPH-treated cells
(Fig. 6B) (20,41,42). Similarly, the transcription levels
of autophagy-related genes (ATG8, CTSD, BECN1, and LAMP1) and of ER
stress-related genes (ATF4, ATF6, and CHOP) were lower in the
hPH-treated cells compared with those in cells treated with D-GalN
only (Fig. 6C). These results
suggested that hPH treatment minimized the escalation of
D-GalN-induced autophagy processes in HepG2 cells.
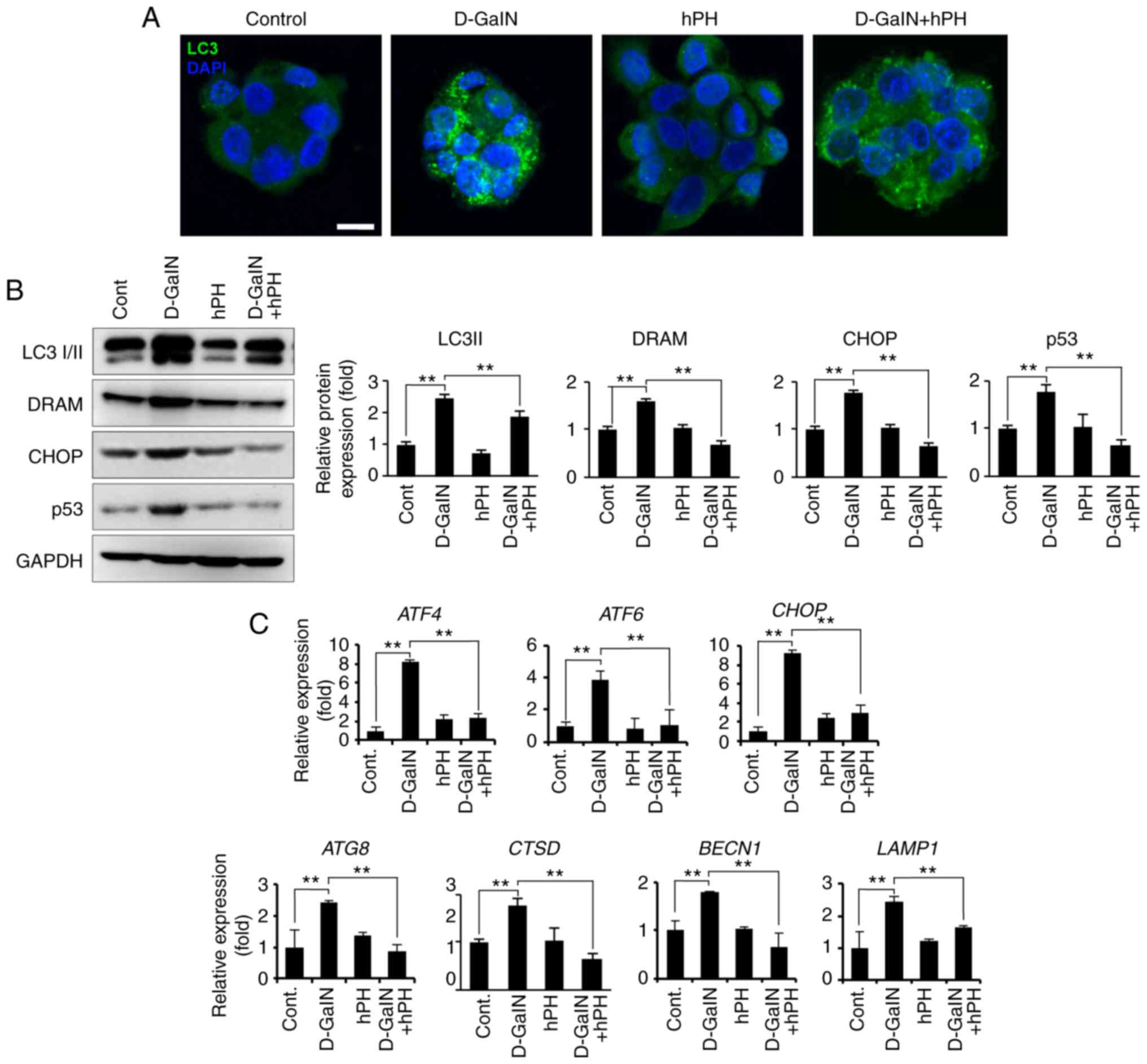 | Figure 6hPH treatment results in altered
autophagy regulation in HepG2 cells following D-GalN stimulation.
(A) HepG2 cells treated with hPH exhibit minimized autophagy flux.
To detect autophagosomes, cells were immunostained with anti-LC3
antibody. Nuclei were identified using DAPI staining (scale bar=10
µm). (B) Cell lysates were immunoblotted with anti-LC3 I/II,
anti-DRAM, anti-CHOP, anti-p53, or anti-GAPDH antibodies.
Representative western blot images (left panel) and densitometry
results (right panel). All data are presented as the mean ±
standard error of the mean. **P<0.01, vs. D-GalN
group. (C) Transcript levels of ER stress- and autophagy-related
genes were determined by reverse transcription-quantitative
polymerase chain reaction analysis. All data are presented as the
mean ± standard error of the mean. **P<0.01, vs.
D-GalN group. hPH, human placental hydrolysate; D-GalN,
D-galactosamine; Cont, control; LC3, microtubule-associated protein
1A/1B-light chain 3; DRAM, damage-regulated autophagy modulator;
CHOP, C/EBP homologous protein; ATF, activating transcription
factor; BECN1; beclin 1; LAMP1, lysosomal-associated membrane
protein 1. |
Discussion
Apoptosis and oxidative stress are interrelated
biological events that are involved in the pathogenesis of various
diseases, including ALF (43–46). Emerging evidence suggests that
D-GalN/LPS-induced oxidative stress gives rise to hepatic injuries
resulting from oxidative stress and apoptosis, which parallel those
of liver degeneration (6,7). In addition, D-GalN/LPS-induced ALF
is widely accepted as an experimental liver injury animal model,
contributing to investigation of the mechanisms underlying clinical
liver injury and the development of efficient hepatoprotective
materials (47–51). Accordingly, any approach that
relieves apoptosis and oxidative stress in vitro and in
vivo contributes to the prevention or treatment of ALF. In the
present study, it was demonstrated that pretreatment with hPH
attenuated acute liver injury associated with elevated serum levels
of ALP, AST, LDH, and pro-inflammatory cytokines (IL-6 and TNF-α).
Furthermore, it was found that apoptosis was increased following
D-GalN/LPS injection, whereas pretreatment with hPH effectively
inhibited tissue degeneration and increased cell death in
D-GalN/LPS-induced acute liver injury. Pretreatment with hPH also
reversed the extensive vacuolization, severe intrahepatic
hemorrhage, destruction of nuclei, and loss of hepatic cords.
Consistently, it has been reported that hPH possesses a variety of
biological activities, including anti-inflammatory (26,52–56) and antioxidant (26,57–59) properties. Exposure to hazardous
components from the environment can lead to pregnancy loss, uterine
dysfunction, or fetal death (60). It has been reported that oxidative
stress is mainly or partly involved in damage from these harmful
environments (61). Therefore,
the human placenta contains antioxidant defense substances to
protect embryos from oxidative stress (62–64). Human placenta extract prepared
from the placenta of healthy pregnant females is known to have
various physiological actions, including antioxidative properties
(57,65). In the present study, hPH was
prepared with human placenta, including umbilical cord, following
the provision of consent from the pregnant women. The hydrolysate
of human placenta is manufactured by a chemical process with HCl
and pepsin, followed by dialysis, heat treatment and hydrolysis.
This hydrolysate contains various amino acids, including arginine
(0.08%), lysine (0.1%), phenylalanine (0.08%), tyrosine (0.03%),
leucine (0.12%), methionine (0.03%), valine (0.04%), alanine
(0.08%), serine (0.07%), and threonine (0.06%). Based on this, hPH
is considered to contain the cleaved proteins of the amino acids
and their active ingredients.
In previous studies, it has been reported that
D-GalN leads to cytotoxicity and apoptotic cell death in HepG2
cells (66–68). D-GalN/LPS intoxication causes an
imbalance between pro-oxidants and antioxidants, involving the
excessive production of ROS and a deficiency of cellular
antioxidants, including glutathione, catalase and SOD. Oxidative
stress caused by D-GalN/LPS is a recognized phenomenon in liver
damage (47,69–71). In addition, D-GalN/LPS induces
loss of ΔΨm and production of ROS, resulting in liver damage
(36,72–74). Mitochondria are the major
subcellular organelles responsible for ROS production (75). The results of the present study
demonstrated that hPH enhanced the expression of SOD-1, SOD-2,
catalase and GPx, and increased the nuclear translocation of the
Nrf2 via a Keap1-p62-Nrf2 mechanism. The upregulation of various
important antioxidants is caused by the binding of Nrf2 to the ARE
of antioxidant target proteins, including SOD, catalase, NQO1, GCLM
and HO-1 (15,16). Nrf2 has been regarded as a novel
therapeutic target for the treatment of liver disease (76–80). In Nrf2-deficient mice, the
expression of various cytopro-tective enzymes was decreased in
hepatocytes, resulting in oxidative stress and markedly delayed
liver regeneration (81). In
addition, genetic disruption of Nrf2 triggers the progression of
necroinflammation and hepatic fibrosis in Hfe-/- mice
presenting with no liver injury (82). Pretreatment with sulforaphane
prevents hepatic damage induced by intestinal ischemia/reperfusion
in rats through an antioxidative effect via the Nrf2-ARE pathway
(79). Another study showed that
the expression of Nrf2 was upregulated to protect the liver from
inflammatory damage caused by oxidative stress during the
development of non-alcoholic fatty liver and steatohepa-titis
(78). Additionally, Farombi
et al reported that curcumin (diferuloymethane)
administration promoted the nuclear translocation and ARE-binding
of Nrf2, leading to prevention of dimethylnitrosamine-induced
hepatotoxicity (76). In
addition, β-cryptoxanthin was found to ameliorate visceral fat and
cardiometabolic health risk factors by modulating the expression of
nuclear factor-κB and Nrf2 in rats fed a high-fat diet (83).
Autophagy is an evolutionarily conserved homeostatic
process and lysosome-dependent proteolytic pathway, which is
involved in a variety of physiological and pathological processes
(84,85). According to previous data,
autophagy is involved in major liver diseases, including liver
isch-emia/reperfusion injury, hepatitis B and C, hepatocellular
carcinoma, alcoholic liver disease and non-alcoholic fatty liver
disease (86). In addition,
autophagy is induced to maintain healthy cells in the presence of
TNF-, acetaminophen-, or ethanol-induced liver injury (87–89). However, excessive autophagy can
cause autophagic cell death. Previously, autophagy has been shown
to be elevated in concanavalin A-induced acute hepatitis, leading
to the autophagic cell death of hepatoma cells in SCID/NOD mice
(90). Furthermore, autophagy
inhibition significantly improved liver graft dysfunction and the
survival rate of recipient rats with 'cold ischemia-warm
reperfusion injury' associated with liver transplantation (91), suggesting that autophagy can
aggravate liver damage. In the present study, it was found that
excessive autophagy was induced following D-GalN administration,
and that pretreatment with hPH effectively suppressed this enhanced
autophagy in D-GalN-induced HepG2 cells. It also indicated a
regulatory pathway mechanism of autophagy induction and inhibition
by D-GalN/hPH via the p53-DRAM-autophagy axis. The identification
of DRAM as a p53 target mediating the induction of autophagy
allowed for investigation of the role of autophagy in apop-tosis
(42). Liu et al reported
that p53-induced apoptosis is primarily dependent on DRAM- and
BAX-mediated autophagy in hepatosteatosis in oleic acid-treated
HepG2 cells and high-fat diet mice (92). The induction of autophagy by the
nuclear translocation of p53 involves the upregulation of DRAM and
sestrin2 in p53-sufficient, but not in p53-deficient cells
(93). The expression of CHOP is
regulated transcriptionally and post-transcriptionally in
hPH-induced HepG2 cells. CHOP, a multifunctional transcription
factor in the ER stress response, promotes autophagic apoptosis
induced by various stimuli, including tetrahydrocannabinol,
2-deoxy-D-glucose, rabbit hemorrhagic disease virus, and
apoptosis-stimulating protein of p53-2 (94-97). The data obtained in the present
study also demonstrated that hPH led to downregulation of the ER
stress-related downstream targets, ATF4 and ATF6, in D-GalN-treated
HepG2 cells. Based on the above findings, it was concluded that
autophagy, apoptosis and ER stress are closely associated with each
other, supported by the results of the present study. D-GalN
stimulation increased the expression levels of DRAM, CHOP and the
apoptosis-related protein p53 in HepG2 cells, whereas hPH
pretreatment reversed the expression of these proteins. hPH
pretreatment also resulted in LC3 lipidation, but meaningfully
minimized autophagosome expression compared with D-GalN alone.
In previous animal studies, early onset autophagy
has been shown to increase liver injury, however, this observation
has not been corroborated in patient populations. Therefore, the
epidemiological relevance of autophagy and liver damage require
investigation in the future. Taken together, the results of the
present study showed that hPH had a protective effect on hepatocyte
apoptosis through antioxidative modulation and minimization of the
autophagy process, which resulted in the inhibition of
hepatocytotoxicity. This may be useful as a target for the
treatment of acute liver injury. The identification of this pathway
can assist in understanding the molecular events leading to the
activation of oxidative stress- and autophagy-mediated cell death
in human disease, and contribute to the design of novel therapeutic
strategies for inhibiting liver disease and preventing fulminant
hepatic failure.
Funding
This study was supported by Green Cross WellBeing
Corporation, Korea (grant no. 20161015) and by the Chung-Ang
University Research Scholarship Grants in 2017.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
BJK, THK, DHB, JN, and MJC designed experiments;
DHB, JN, and MJC performed experiments; MJK, BCL, THK, DHB, JN,
MJC, CTO, JYK, and HJH analyzed data; CTO, JYK, and HJH prepared
materials; DHB, JN, and MJC prepared figures; BJK, THK, DHB, and JN
wrote the manuscript. BJK and THK had primary responsibility for
final content. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the Institutional
Animal Care and Use Committee of Chung-Ang University (2017-00003).
Placental tissues were used following the provision of informed
consent from the pregnant women.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Bernal W, Auzinger G, Dhawan A and Wendon
J: Acute liver failure. Lancet. 376:190–201. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Han DW: Intestinal endotoxemia as a
pathogenetic mechanism in liver failure. World J Gastroenterol.
8:961–965. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sathivel A, Balavinayagamani, Hanumantha
Rao BR and Devaki T: Sulfated polysaccharide isolated from Ulva
lactuca attenuates d-galactosamine induced DNA fragmentation and
necrosis during liver damage in rats. Pharm Biol. 52:498–505. 2014.
View Article : Google Scholar
|
|
4
|
Masaki T, Chiba S, Tatsukawa H, Yasuda T,
Noguchi H, Seike M and Yoshimatsu H: Adiponectin protects
LPS-induced liver injury through modulation of TNF-α in KK-Ay obese
mice. Hepatology. 40:177–184. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wu Z, Han M, Chen T, Yan W and Ning Q:
Acute liver failure: Mechanisms of immune-mediated liver injury.
Liver Int. 30:782–794. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Sheriff SA, Shaik Ibrahim S, Devaki T,
Chakraborty S, Agarwal S and Pérez-Sánchez H: Lycopene prevents
mitochondrial dysfunction during
d-galactosamine/lipopolysaccharide-induced fulminant hepatic
failure in albino rats. J Proteome Res. 16:3190–3199. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dong L, Yin L, Quan H, Chu Y and Lu J:
Hepatoprotective effects of
kaempferol-3-O-α-l-arabinopyranosyl-7-O-α-l-rhamn opyranoside on
d-Galactosamine and lipopolysaccharide caused hepatic failure in
mice. Molecules. 22:E17552017. View Article : Google Scholar
|
|
8
|
Lee SB, Kang JW, Kim SJ, Ahn J, Kim J and
Lee SM: Afzelin ameliorates D-galactosamine and
lipopolysaccharide-induced fulminant hepatic failure by modulating
mitochondrial quality control and dynamics. Br J Pharmacol.
174:195–209. 2017. View Article : Google Scholar
|
|
9
|
Decker CW, Casian JG, Nguyen KT, Horton
LA, Rao MP, Silkwood KH and Han D: The critical role of
mitochondria in drug-induced liver injury. Molecules, Systems and
Signaling in Liver Injury. Springer; pp. 159–181. 2017, View Article : Google Scholar
|
|
10
|
Nguyen T, Nioi P and Pickett CB: The
Nrf2-antioxidant response element signaling pathway and its
activation by oxidative stress. J Biol Chem. 284:13291–13295. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bryan HK, Olayanju A, Goldring CE and Park
BK: The Nrf2 cell defence pathway: Keap1-dependent and-independent
mechanisms of regulation. Biochem Pharmacol. 85:705–717. 2013.
View Article : Google Scholar
|
|
12
|
Zhou R, Lin J and Wu D: Sulforaphane
induces Nrf2 and protects against CYP2E1-dependent binge
alcohol-induced liver steatosis. Biochim Biophys Acta.
1840:209–218. 2014. View Article : Google Scholar
|
|
13
|
Jiang T, Huang Z, Lin Y, Zhang Z, Fang D
and Zhang DD: The protective role of Nrf2 in streptozotocin-induced
diabetic nephropathy. Diabetes. 59:850–860. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kobayashi A, Kang MI, Okawa H, Ohtsuji M,
Zenke Y, Chiba T, Igarashi K and Yamamoto M: Oxidative stress
sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to
regulate proteasomal degradation of Nrf2. Mol Cell Biol.
24:7130–7139. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bataille A and Manautou J: Nrf2: A
potential target for new therapeutics in liver disease. Clin
Pharmacol Ther. 92:340–348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jian Z, Li K, Song P, Zhu G, Zhu L, Cui T,
Liu B, Tang L, Wang X, Wang G, et al: Impaired activation of the
Nrf2-ARE signaling pathway undermines H response: A possible
mechanism for melanocyte degeneration in 2O2-induced oxidative
stress vitiligo. J Invest Dermatol. 134:2221–2230. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gozuacik D and Kimchi A: Autophagy and
cell death. Curr Top Dev Biol. 78:217–245. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Crighton D, Wilkinson S, O'Prey J, Syed N,
Smith P, Harrison PR, Gasco M, Garrone O, Crook T and Ryan KM:
DRAM, a p53-induced modulator of autophagy, is critical for
apoptosis. Cell. 126:121–134. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tonello G, Daglio M, Zaccarelli N,
Sottofattori E, Mazzei M and Balbi A: Characterization and
quantitation of the active poly-nucleotide fraction (PDRN) from
human placenta, a tissue repair stimulating agent. J Pharm Biomed
Anal. 14:1555–1560. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Sur TK, Biswas TK, Ali L and Mukherjee B:
Anti-inflammatory and anti-platelet aggregation activity of human
placental extract. Acta Pharmacol Sin. 24:187–192. 2003.PubMed/NCBI
|
|
23
|
Chakraborty PD and Bhattacharyya D:
Isolation of fibronectin type III like peptide from human placental
extract used as wound healer. J Chromatogr B Analyt Technol Biomed
Life Sci. 818:67–73. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choi JY, Lee K, Lee SM, Yoo SH, Hwang SG,
Choi JY, Lee SW, Hwang JS, Kim KK, Kang HC, et al: Efficacy and
safety of human placental extract for alcoholic and nonalcoholic
steato-hepatitis: An open-label, randomized, comparative study.
Biol Pharm Bull. 37:1853–1859. 2014. View Article : Google Scholar
|
|
25
|
Shimokobe H, Sumida Y, Tanaka S, Mori K,
Kitamura Y, Fukumoto K, Kakutani A, Ohno T, Kanemasa K, Imai S, et
al: Human placental extract treatment for non-alcoholic
steato-hepatitis non-responsive to lifestyle intervention: A pilot
study. Hepatol Res. 45:1034–1040. 2015. View Article : Google Scholar
|
|
26
|
Park S, Phark S, Lee M, Lim J and Sul D:
Anti-oxidative and anti-inflammatory activities of placental
extracts in benzo[a] pyrene-exposed rats. Placenta. 31:873–879.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Camargo CA Jr, Madden JF, Gao W, Selvan RS
and Clavien P: Interleukin-6 protects liver against warm
ischemia/reperfusion injury and promotes hepatocyte proliferation
in the rodent. Hepatology. 26:1513–1520. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Okoh VO, Felty Q, Parkash J, Poppiti R and
Roy D: Reactive oxygen species via redox signaling to PI3K/AKT
pathway contribute to the malignant growth of 4-hydroxy
estradiol-transformed mammary epithelial cells. PLoS One.
8:e542062013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jasek E, Lis GJ, Jasińska M, Jurkowska H
and Litwin JA: Effect of histone deacetylase inhibitors
trichostatin A and valproic acid on etoposide-induced apoptosis in
leukemia cells. Anticancer Res. 32:2791–2799. 2012.PubMed/NCBI
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
31
|
Cassidy W and Reynolds T: Serum lactic
dehydrogenase in the differential diagnosis of acute hepatocellular
injury. J Clin Gastroenterol. 19:118–121. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Rolando N, Wade J, Davalos M, Wendon J,
Philpott-Howard J and Williams R: The systemic inflammatory
response syndrome in acute liver failure. Hepatology. 32:734–739.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guicciardi M and Gores GJ: Apoptosis: A
mechanism of acute and chronic liver injury. Gut. 54:1024–1033.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stachlewitz RF, Seabra V, Bradford B,
Bradham CA, Rusyn I, Germolec D and Thurman RG: Glycine and uridine
prevent d-galactosamine hepatotoxicity in the rat: Role of kupffer
cells. Hepatology. 29:737–745. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Thabrew MI, Hughes RD and McFarlane IG:
Screening of hepa-toprotective plant components using a HepG2 cell
cytotoxicity assay. J Pharm Pharmacol. 49:1132–1135. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
González R, Ferrín G, Hidalgo AB, Ranchal
I, López-Cillero P, Santos-Gónzalez M, López-Lluch G, Briceño J,
Gómez MA, Poyato A, et al: N-acetylcysteine, coenzyme Q10 and
superoxide dismutase mimetic prevent mitochondrial cell dysfunction
and cell death induced by d-galactosamine in primary culture of
human hepatocytes. Chem Biol Interact. 181:95–106. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zhao Y, Li S, Childs EE, Kuharsky DK and
Yin XM: Activation of pro-death Bcl-2 family proteins and
mitochondria apoptosis pathway in tumor necrosis factor-α-induced
liver injury. J Biol Chem. 276:27432–27440. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kuznetsov AV, Kehrer I, Kozlov AV, Haller
M, Redl H, Hermann M, Grimm M and Troppmair J: Mitochondrial ROS
production under cellular stress: Comparison of different detection
methods. Anal Bioanal Chem. 400:2383–2390. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee J, Giordano S and Zhang J: Autophagy,
mitochondria and oxidative stress: Cross-talk and redox signalling.
Biochem J. 441:523–540. 2012. View Article : Google Scholar :
|
|
40
|
Yang Z and Klionsky DJ: Mammalian
autophagy: Core molecular machinery and signaling regulation. Curr
Opin Cell Biol. 22:124–131. 2010. View Article : Google Scholar :
|
|
41
|
Liu GY, Jiang XX, Zhu X, He WY, Kuang YL,
Ren K, Lin Y and Gou X: ROS activates JNK-mediated autophagy to
counteract apoptosis in mouse mesenchymal stem cells in vitro. Acta
Pharmacol Sin. 36:1473–1479. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Maiuri MC, Zalckvar E, Kimchi A and
Kroemer G: Self-eating and self-killing: Crosstalk between
autophagy and apoptosis. Nat Rev Mol Cell Biol. 8:741–752. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Cichoż-Lach H and Michalak A: Oxidative
stress as a crucial factor in liver diseases. World J
Gastroenterol. 20:8082–8091. 2014. View Article : Google Scholar
|
|
44
|
Jenner P: Oxidative stress in Parkinson's
disease. Ann Neurol. 53(Suppl 3): S26–S38. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Kujoth G, Hiona A, Pugh T, Someya S,
Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA,
et al: Mitochondrial DNA mutations, oxidative stress, and apoptosis
in mammalian aging. Science. 309:481–484. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Radi E, Formichi P, Battisti C and
Federico A: Apoptosis and oxidative stress in neurodegenerative
diseases. J Alzheimers Dis. 42(Suppl 3): S125–S152. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Osakabe N, Yasuda A, Natsume M, Sanbongi
C, Kato Y, Osawa T and Yoshikawa T: Rosmarinic acid, a major
polyphenolic component of Perilla frutescens, reduces
lipopolysaccharide (LPS)-induced liver injury in D-galactosamine
(D-GalN)-sensitized mice. Free Radic Biol Med. 33:798–806. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nowak M, Gaines GC, Rosenberg J, Minter R,
Bahjat FR, Rectenwald J, MacKay SL, Edwards CK III and Moldawer LL:
LPS-induced liver injury in D-galactosamine-sensitized mice
requires secreted TNF-alpha and the TNF-p55 receptor. Am J Physiol
Regul Integr Comp Physiol. 278:R1202–R1209. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Wu YL, Lian LH, Wan Y and Nan JX:
Baicalein inhibits nuclear factor-κB and apoptosis via c-FLIP and
MAPK in D-GalN/LPS induced acute liver failure in murine models.
Chem Biol Interact. 188:526–534. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Chen L, Ren F, Zhang H, Wen T, Piao Z,
Zhou L, Zheng S, Zhang J, Chen Y, Han Y, et al: Inhibition of
glycogen synthase kinase 3β ameliorates D-GalN/LPS-induced liver
injury by reducing endoplasmic reticulum stress-triggered
apoptosis. PloS One. 7:e452022012. View Article : Google Scholar
|
|
51
|
Wang H, Xu DX, Lv JW, Ning H and Wei W:
Melatonin attenuates lipopolysaccharide (LPS)-induced apoptotic
liver damage in D-galactosamine-sensitized mice. Toxicology.
237:49–57. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kawakatsu M, Urata Y, Goto S, Ono Y and Li
TS: Placental extract protects bone marrow-derived stem/progenitor
cells against radiation injury through anti-inflammatory activity.
J Radiat Res. 54:268–276. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Park JY, Lee J, Jeong M, Min S, Kim SY,
Lee H, Lim Y and Park HJ: Effect of Hominis Placenta on cutaneous
wound healing in normal and diabetic mice. Nutri Res Pract.
8:404–409. 2014. View Article : Google Scholar
|
|
54
|
Lee KH, Kim TH, Lee WC, Kim SH, Lee SY and
Lee SM: Anti-inflammatory and analgesic effects of human placenta
extract. Nat Prod Res. 25:1090–1100. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lee KW, Ji HM, Kim DW, Choi SM, Kim S and
Yang EJ: Effects of Hominis placenta on LPS-induced cell toxicity
in BV2 microglial cells. J Ethnopharmacol. 147:286–292. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Akagi H, Imamura Y, Makita Y, Nakamura H,
Hasegawa N, Fujiwara SI and Wang PL: Evaluation of collagen type-1
production and anti-inflammatory activities of human placental
extracts in human gingival fibroblasts. J Hard Tissue Biol.
25:277–281. 2016. View Article : Google Scholar
|
|
57
|
Watanabe S, Togashi S, Takahashi N and
Fukui T: L-tryptophan as an antioxidant in human placenta extract.
J Nutri Sci Vitaminol (Tokyo). 48:36–39. 2002. View Article : Google Scholar
|
|
58
|
Togashi SI, Takahashi N, Iwama M, Watanabe
S, Tamagawa K and Fukui T: Antioxidative collagen-derived peptides
in human-placenta extract. Placenta. 23:497–502. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Rozanova S, Cherkashina Y, Repina S,
Rozanova K and Nardid O: Protective effect of placenta extracts
against nitrite-induced oxidative stress in human erythrocytes.
Cell Mol Biol Lett. 17:240–248. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Halliwell B and Gutteridge JM: Free
Radicals in Biology and Medicine. Oxford University Press; USA:
2015, View Article : Google Scholar
|
|
61
|
Wells PG and Winn LM: Biochemical
toxicology of chemical teratogenesis. Crit Rev Biochem Mol Biol.
31:1–40. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Avissar N, Whitin JC, Allen PZ, Wagner DD,
Liegey P and Cohen HJ: Plasma selenium-dependent glutathione
peroxidase. Cell of origin and secretion. J Biol Chem.
264:15850–15855. 1989.PubMed/NCBI
|
|
63
|
Thomas EL, Learn DB, Jefferson MM and
Weatherred W: Superoxide-dependent oxidation of extracellular
reducing agents by isolated neutrophils. J Biol Chem.
263:2178–2186. 1988.PubMed/NCBI
|
|
64
|
Kankofer M: Antioxidative defence
mechanisms against reactive oxygen species in bovine retained and
not-retained placenta: Activity of glutathione peroxidase,
glutathione transferase, catalase and superoxide dismutase.
Placenta. 22:466–472. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Mochizuki H and Kada T: Restorative
effects of human placenta extract in X-ray-irradiated mice. J
Radiat Res. 23:403–410. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
González R, Collado JA, Nell S, Briceño J,
Tamayo MJ, Fraga E, Bernardos A, López-Cillero P, Pascussi JM,
Rufián S, et al: Cytoprotective properties of α-tocopherol are
related to gene regulation in cultured D-galactosamine-treated
human hepato-cytes. Free Radic Biol Med. 43:1439–1452. 2007.
View Article : Google Scholar
|
|
67
|
Siendones E, Fouad D, Abou-Elella AMKE,
Quintero A, Barrera P and Muntané J: Role of nitric oxide in
d-galactos-amine-induced cell death and its protection by PGE 1 in
cultured hepatocytes. Nitric Oxide. 8:133–143. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Mahmoud MF, Hamdan DI, Wink M and
El-Shazly AM: Hepatoprotective effect of limonin, a natural
limonoid from the seed of Citrus aurantium var. bigaradia, on
D-galactosamine- induced liver injury in rats. Naunyn-Schmiedebergs
Arch Pharmacol. 387:251–261. 2014. View Article : Google Scholar
|
|
69
|
Wang Y, Li Y, Xie J, Zhang Y, Wang J, Sun
X and Zhang H: Protective effects of probiotic Lactobacillus casei
Zhang against endotoxin-and d-galactosamine-induced liver injury in
rats via anti-oxidative and anti-inflammatory capacities. Int
Immunopharmacol. 15:30–37. 2013. View Article : Google Scholar
|
|
70
|
Xia X, Su C, Fu J, Zhang P, Jiang X, Xu D,
Hu L, Song E and Song Y: Role of α-lipoic acid in LPS/d-GalN
induced fulminant hepatic failure in mice: Studies on oxidative
stress, inflammation and apoptosis. Int Immunopharmacol.
22:293–302. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Shin JW, Wang JH, Park HJ, Choi MK, Kim HG
and Son CG: Herbal formula CGX ameliorates
LPS/D-galactosamine-induced hepatitis. Food Chem Toxicol.
49:1329–1334. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Lee HJ, Oh YK, Rhee M, Lim JY, Hwang JY,
Park YS, Kwon Y, Choi KH, Jo I, Park SI, et al: The role of
STAT1/IRF-1 on synergistic ROS production and loss of mitochondrial
transmembrane potential during hepatic cell death induced by
LPS/d-GalN. J Mol Biol. 369:967–984. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Liu LM, Zhang JX, Luo J, Guo HX, Deng H,
Chen JY and Sun SL: A role of cell apoptosis in lipopolysaccharide
(LPS)-induced nonlethal liver injury in D-galactosamine
(D-GalN)-sensitized rats. Dig Dis Sci. 53:1316–1324. 2008.
View Article : Google Scholar
|
|
74
|
Soriano ME, Nicolosi L and Bernardi P:
Desensitization of the permeability transition pore by cyclosporin
A prevents activation of the mitochondrial apoptotic pathway and
liver damage by tumor necrosis factor-alpha. J Biol Chem.
279:36803–36808. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Indo HP, Davidson M, Yen HC, Suenaga S,
Tomita K, Nishii T, Higuchi M, Koga Y, Ozawa T and Majima HJ:
Evidence of ROS generation by mitochondria in cells with impaired
electron transport chain and mitochondrial DNA damage.
Mitochondrion. 7:106–118. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Farombi EO, Shrotriya S, Na HK, Kim SH and
Surh YJ: Curcumin attenuates dimethylnitrosamine-induced liver
injury in rats through Nrf2-mediated induction of heme oxygenase-1.
Food Chem Toxicol. 46:1279–1287. 2008. View Article : Google Scholar
|
|
77
|
Klaassen CD and Reisman SA: Nrf2 the
rescue: Effects of the antioxidative/electrophilic response on the
liver. Toxicol Appl Pharmacol. 244:57–65. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Xu W, Shao L, Zhou C, Wang H and Guo J:
Upregulation of Nrf2 expression in non-alcoholic fatty liver and
steatohepatitis. Hepatogastroenterology. 58:2077–2080. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Zhao HD, Zhang F, Shen G, Li YB, Li YH,
Jing HR, Ma LF, Yao JH and Tian XF: Sulforaphane protects liver
injury induced by intestinal ischemia reperfusion through Nrf2-ARE
pathway. World J Gastroenterol. 16:3002–3010. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Pan CW, Pan ZZ, Hu JJ, Chen WL, Zhou GY,
Lin W, Jin LX and Xu CL: Mangiferin alleviates lipopolysaccharide
and D-galactosamine-induced acute liver injury by activating the
Nrf2 pathway and inhibiting NLRP3 inflammasome activation. Eur J
Pharmacol. 770:85–91. 2016. View Article : Google Scholar
|
|
81
|
Beyer TA, Xu W, Teupser D, auf dem Keller
U, Bugnon P, Hildt E, Thiery J, Kan YW and Werner S: Impaired liver
regeneration in Nrf2 knockout mice: Role of ROS-mediated
insulin/IGF-1 resistance. EMBO J. 27:212–223. 2008. View Article : Google Scholar
|
|
82
|
Duarte TL, Caldas C, Santos AG,
Silva-Gomes S, Santos-Gonçalves A, Martins MJ, Porto G and Lopes
JM: Genetic disruption of NRF2 promotes the development of
necroinflam-mation and liver fibrosis in a mouse model of
HFE-hereditary hemochromatosis. Redox Biol. 11:157–169. 2017.
View Article : Google Scholar
|
|
83
|
Sahin K, Orhan C, Akdemir F, Tuzcu M,
Sahin N, Yılmaz I and Juturu V: β-Cryptoxanthin ameliorates
metabolic risk factors by regulating NF-κB and Nrf2 pathways in
insulin resistance induced by high-fat diet in rodents. Food Chem
Toxicol. 107:270–279. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Uchiyama Y, Shibata M, Koike M, Yoshimura
K and Sasaki M: Autophagy-physiology and pathophysiology. Histochem
Cell Biol. 129:407–420. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Rautou PE, Mansouri A, Lebrec D, Durand F,
Valla D and Moreau R: Autophagy in liver diseases. J Hepatol.
53:1123–1134. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Amir M, Zhao E, Fontana L, Rosenberg H,
Tanaka K, Gao G and Czaja MJ: Inhibition of hepatocyte autophagy
increases tumor necrosis factor-dependent liver injury by promoting
caspase-8 activation. Cell Death Differ. 20:878–887. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Donohue TM Jr: Autophagy and
ethanol-induced liver injury. World J Gastroenterol. 15:1178–1185.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lin Z, Wu F, Lin S, Pan X, Jin L, Lu T,
Shi L, Wang Y, Xu A and Li X: Adiponectin protects against
acetaminophen-induced mitochondrial dysfunction and acute liver
injury by promoting autophagy in mice. J Hepatol. 61:825–831. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Chang CP and Lei HY: Autophagy induction
in T cell-independent acute hepatitis induced by concanavalin A in
SCID/NOD mice. Int J Immunopathol Pharmacol. 21:817–826. 2008.
View Article : Google Scholar
|
|
91
|
Gotoh K, Lu Z, Morita M, Shibata M, Koike
M, Waguri S, Dono K, Doki Y, Kominami E, Sugioka A, et al:
Participation of autophagy in the initiation of graft dysfunction
after rat liver transplantation. Autophagy. 5:351–360. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Liu K, Lou J, Wen T, Yin J, Xu B, Ding W,
Wang A, Liu D, Zhang C, Chen D and Li N: Depending on the stage of
hepa-tosteatosis, p53 causes apoptosis primarily through either
DRAM-induced autophagy or BAX. Liver Int. 33:1566–1574.
2013.PubMed/NCBI
|
|
93
|
Maiuri MC, Malik SA, Morselli E, Kepp O,
Criollo A, Mouchel PL, Carnuccio R and Kroemer G: Stimulation of
autophagy by the p53 target gene Sestrin2. Cell Cycle. 8:1571–1576.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Salazar M, Carracedo A, Salanueva ÍJ,
Hernández-Tiedra S, Lorente M, Egia A, Vázquez P, Blázquez C,
Torres S, García S, et al: Cannabinoid action induces
autophagy-mediated cell death through stimulation of ER stress in
human glioma cells. J Clin Invest. 119:1359–1372. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Xi H, Kurtoglu M, Liu H, Wangpaichitr M,
You M, Liu X, Savaraj N and Lampidis TJ: 2-Deoxy-D-glucose
activates autophagy via endoplasmic reticulum stress rather than
ATP depletion. Cancer Chemother Pharmacol. 67:899–910. 2011.
View Article : Google Scholar :
|
|
96
|
Liu K, Shi Y, Guo X, Wang S, Ouyang Y, Hao
M, Liu D, Qiao L, Li N, Zheng J and Chen D: CHOP mediates
ASPP2-induced autophagic apoptosis in hepatoma cells by releasing
Beclin-1 from Bcl-2 and inducing nuclear translocation of Bcl-2.
Cell Death Dis. 5:e13232014. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Tuñón MJ, San-Miguel B, Crespo I, Laliena
A, Vallejo D, Álvarez M, Prieto J and González-Gallego J: Melatonin
treatment reduces endoplasmic reticulum stress and modulates the
unfolded protein response in rabbits with lethal fulminant
hepatitis of viral origin. J Pineal Res. 55:221–228. 2013.
View Article : Google Scholar : PubMed/NCBI
|