Introduction
Subarachnoid hemorrhage (SAH) is one of the most
common cerebrovascular events, with high rates of mortality and
disability (1). SAH may occur at
different ages, ranging from children to the elderly, and the
incidence gradually increases with age. Although drugs and surgical
technologies are being continuously developed, the mortality rate
remains as high as 30–50%, and the prognosis of patients with SAH
is poor (1).
Early brain injury (EBI) refers to direct injury to
the whole brain within the initial 72 h after SAH. EBI is a
complicated event that is the result of normal brain physiological
disorder, which may lead to early brain edema, oxidative stress,
apoptosis and cerebral infarction, and may subsequently lead to
mortality or severe disability (2,3).
The term EBI has been adopted and describes an immediate injury to
the brain following SAH prior to the onset of delayed vasospasm
(4). During the EBI period, a
ruptured aneurysm causes a number of physiological disruptions,
including increased intracranial pressure (ICP), decreased cerebral
blood flow (CBF), and global cerebral ischemia. These events
initiate secondary injuries including blood-brain barrier
disruption, inflammation and oxidative cascades that all ultimately
lead to cell death (4).
Therefore, studies on EBI have substantial potential value in the
treatment and prognosis of patients with SAH. At present, a number
of studies have identified that early brain injury may be a primary
factor of poor prognosis of SAH (2,3,5).
SAH may lead to neuronal cell apoptosis, and apoptosis primarily
occurs in neurons of the hippocampus and basal cortex (6,7).
SAH also disrupts the blood-brain barrier (BBB) and increases
permeability (4,5). Cerebral edema is a direct result of
BBB destruction. It has been suggested that a number of signal
pathways serve important roles in EBI following SAH. The
phosphoinositide 3-kinase (PI3K)/protein kinase B (Akt) pathway, a
key antiapoptotic signaling pathway associated with EBI following
SAH (8–10), and the c-Jun N-terminal kinase
(JNK) associated signal pathway have been studied in EBI (7).
B-cell lymphoma 2 (Bcl-2)-associated X protein (Bax)
inhibitor-1 (BI-1), also known as TMBIM6, was first identified by
exploiting the lethal phenotype of Bax in yeast (11). BI-1 is an evolutionarily conserved
endoplasmic reticulum (ER) protein that suppresses cell death in
animal and plant cells (12).
Numerous studies have suggested a role of ER stress in neuronal
cell death (13,14).
BI-1 has an association with ER, and it may also
suppress the apoptosis induced by Bax. It has been demonstrated
that ER is involved in the apoptosis induced by unfolded protein
responses (UPRs) (15). Among the
UPRs, inositol-requiring enzymes (IRE) are associated with cell
death (14).
Serine/threonine-protein kinase/endoribonuclease IRE1 (IRE1) is an
important factor in the ER-targeted activities of Bcl-2-family
proteins (16). Glucose regulated
protein, 78 kDa (GRP78) may regulate ER function during ER stress
(17). IRE1 may activate
downstream protein kinases, particularly apoptotic signaling
kinase-1 (ASK1), which may cause JNK activation (18). IRE may also activate p38 mitogen
activated protein kinase (MAPK), which activates the
transcriptional regulator C/EBP homologous protein (CHOP) that
controls the expression of numerous apoptotic genes (19). The cytoprotective activity of BI-1
is closely associated with ER stress (20,21).
The function of BI-1 has been studied in a number of
pathological models, including non-small cell lung cancer (22), prostate cancer (23), liver regeneration (24), ischemia (21) and diabetes (25). It has been demonstrated that BI-1
protects against stroke and traumatic brain injury (20). Bax inhibitor-1 may improve
survival and neuronal differentiation of embryonic stem cells via
the differential regulation of MAPK activities (26). BI-1 overexpression in the ER is
protective in neurons (27). It
has been suggested that the expression of BI-1 may be regarded as a
novel therapy for brain-associated diseases. However, studies
concerning the association between BI-1 and SAH have rarely been
performed.
The present study investigated whether BI-1
protected the brain against EBI following SAH in rats. In addition,
the role of ER stress-mediated apoptosis in SAH rat models was
discussed.
Materials and methods
Ethics statement and animals
Seventy healthy male Sprague-Dawley rats (7-week
old), weighing 180–200 g, were purchased from the Experimental
Animal Center of Kunming Medical University (certificate no.
SCXK2005-0008). The animal experiments were approved by the Animals
Ethics Committee of Kunming Medical University and the Guide for
the Care and Use of Laboratory Animals. All rats were housed in a
humidity-controlled (50–65%) pathogen-free environment with ad
libitum access to food and water under 12/12 h light: Dark
cycle (18–22°C).
Plasmids
The rat BI-1 sequence was obtained from NCBI
(https://www.ncbi.nlm.nih.gov/nuccore/NM_019381.2),
and inserted into the pCDH plasmid (System Biosciences, LLC, Palo
Alto, CA, USA) with EcoRI and NotI enzyme sites. The short
hairpin RNA (shRNA) sequence for BI-1 was
5′-GCACCTAAAGAAGGTCTATGC-3′ (Sangon Biotech Co., Ltd., Shanghai,
China). The BI-1 oligos (forward,
5′-CCGGGCACCTAAAGAAGGTCTATGCCTCGAGGCATAGACCTTCTTTAGGTGCTTTTTG-3′
and reverse,
5′-AATTCAAAAAGCACCTAAAGAAGGTCTATGCCTCGAGGCATAGACCTTCTTTAGGTGC-3′)
(Sangon Biotech Co., Ltd.) were annealed and inserted into the
pLKO.1 plasmid (Addgene, Inc., Cambridge, MA, USA) with
EcoRI and AgeI enzyme sites. The shRNA sequence for
scrambled shRNA/pLKO.1 (Addgene, Inc.) was 5′-CCT AAG GTT AAG TCG
CCC TCG-3′. These plasmids were sequenced by Sangon Biotech Co.,
Ltd. Furthermore, the efficiency of plasmids was verified by
quantitative polymerase chain reaction (qPCR) and western blot
analysis according to the following protocol. The sequence of rat
BI-1 as follows:
5′-ATGAATATATTTGATCGGAAGATCAACTTTGATGCCCTCTTAAAATTTTCCCACATAACTCCCTCCACACAGCAGCACCTAAAGAAGGTCTATGCCAGTTTTGCACTGTGCATGTTTGTGGCAGCAGCAGGGGCCTATGTCCATGTGGTCACACGTTTCATCCAGGCTGGCCTGCTCTCTGCCCTGGGCGCCCTGGCCTTGATGATTTGCCTGATGGCCACACCTCACAGCCATGAGACGGAGCAGAAGAGGCTGGGACTGCTCGCTGGCTTCGCCTTCCTTACAGGAGTTGGCCTGGGACCTGCCCTGGAGCTGTGCATTGCCATCAACCCCAGCATCCTCCCCACGGCCTTCATGGGCACGGCCATGATCTTCACCTGCTTCAGCCTGAGTGCCCTCTACGCCAGGCGCCGGAGTTACCTCTTTTTGGGAGGTATCTTGATGTCAGCCATGAGCCTCATGTTCGTGTCCTCTCTGGGGAACCTTTTCTTTGGATCCATTTGGCTGTTCCAGGCAAACCTGTACATGGGGCTGCTGGTCATGTGCGGCTTTGTCCTCTTCGACACTCAGCTCATTATTGAAAAGGCTGAACACGGAGACAAGGATTACATCTGGCACTGCATTGACCTCTTCTTGGACTTCGTTACACTCTTCAGGAAGCTCATGCTGATCTTGGCCTTCAATGAGAAGGACAAAAAGAAAGAGAAGAAGTGA-3′.
Transfection
The following groups were designed: NC group
(transfection reagent), pCDH group, BI-1/pCDH, BI-1/pLKO.1 shRNA
and scrambled shRNA/pLKO.1. The aforementioned plasmids were
transfected with Lipo8000™ Transfection Reagent (Beyotime Institute
of Biotechnology, Haimen, China) into rat normal neuron cells (Chi
Scientific, Inc., Maynard, MA, China), respectively. A total of 48
h after transfection, all transfected cells were collected for qPCR
and western blot analysis, according to the following protocol.
SAH model and transfection
The rats were randomly divided into the following
five groups (n=8): Control; sham; SAH 24 h; SAH 48 h; and SAH 72 h
groups. The SAH model was induced in rats via puncture of the
internal carotid artery. The SAH rat model was generated according
to a protocol outlined by Prunell et al (28). Briefly, the rats were anesthetized
with sodium pentobarbital [30 mg/kg body weight, intraperitoneal
injection (i.p)]. Then, the right carotid artery branch was
exposed, and the external carotid artery (ECA) was blocked by
vascular clamp. The ECA was cut at the proximal end of the vascular
clamp, a nylon cord was inserted into the internal carotid artery
from the ECA and then moved forward 3 mm when resistance was felt.
Subsequently, the nylon cord was quickly removed. In total, the
entire procedure lasted <30 sec. The rats in the sham group only
had the right carotid artery branch exposed, without any surgery,
and rats in the control group had not undergone any surgical
procedures. Rats were euthanized with sodium pentobarbital overdose
(150 mg/kg i.p.). All of the aforementioned rats were assessed for
behavioral scores as described subsequently to determine
neurological impairment, then half of the brain samples were
collected subsequent to being perfused with PBS and fixed at room
temperature for one day in 4% paraformaldehyde for H&E
staining, immunohistochemistry, terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL) staining and
BBB injury assessment. The other half of the brain samples were
stored in liquid nitrogen for qPCR and western blot assays as
described subsequently.
In the second part of the experiments, rats were
randomly divided into the following five groups (n=6): The sham;
SAH; SAH+NC (SAH + transfection reagent); SAH+BI-1 overex-pression
(SAH+BI-1 over); and SAH+BI-1 shRNA groups. The BI-1/pCDH
overexpression and BI-1/pLKO.1 shRNA plasmid were transfected with
Entranster-in vivo DNA transfection reagent (Engreen
Biosystem NZ, Ltd., Auckland, New Zealand) by using a ventricular
puncture stereotactic apparatus and a microinjection pump. Briefly,
5 μg plasmids were dissolved in 5 μl sterile water,
and 10 μl Entranster-in vivo DNA transfection reagent
was added to 5 μl plasmid. Then, the mixture was incubated
for 15 min at room temperature. Finally, the aforementioned mixture
was injected by using a microinjection pump under a ventricular
puncture stereotactic apparatus. The SAH model was constructed 24 h
after this process. All of the rats were assessed for behavioral
scores as described subsequently to determine neurological
impairment, and all samples were collected as aforementioned.
Neurological impairment
The neurological deficits were scored blindly to
assess behavioral performance following ischemic injury according
to the method outlined by Sugawara et al (29). This method was divided into six
parts, namely: Autonomic activity; limb movement; forelimb
extension; self-climbing cage; axillary touch response; and nasal
hair touch response. Each category was scored out of 3 points, with
a total of 18 points available. The evaluation process lasted ~5
min.
Brain edema
Brain edema was examined by using the wet-dry
method: The rats were euthanized with sodium pentobarbital (150
mg/kg i.p.). The whole brain tissues were collected and weighed
immediately, the measurement of which was considered the wet
weight. Then, brain tissues were placed into the oven at 100°C for
72 h and then weighed, and this was considered dry weight. The
brain edema index (%) was calculated as follows: [(Wet weight-dry
weight)/wet weight] ×100%.
Hematoxylin and eosin (H&E)
staining
Fixed brain tissues were dehydrated with various
concentrations of xylene and ethanol (50% ethanol for 4 h, 75%
ethanol for 4 h, 85% ethanol for 3 h, 95% ethanol for 2 h, 100%
ethanol for 1 h, 100% ethanol for 1 h, 1:1 ethanol-xylene for 1 h,
xylene for 1 h and xylene for 30 min at room temperature), embedded
in paraffin, and sliced into 4 μm section. Pathological
changes were detected by H&E staining, hematoxylin staining for
5–10 min and eosin staining for 1 min at room temperature (Beijing
Solarbio Science & Technology Co., Ltd., Beijing, China). The
hippocampus in brain sections was observed under a light microscope
and the magnification was ×200 (Olympus Corporation, Tokyo,
Japan).
Immunohistochemical analysis
Sections (thickness, 4 μm) were dewaxed with
various concentrations of xylene and ethanol (xylene for 10 min,
xylene for 5 min, 100% ethanol for 5 min, 95% ethanol for 2 min,
80% ethanol for 2 min and 70% ethanol for 2 min). Sections were
incubated with biotin-labeled rabbit anti-rat IgG (dilution
1:1,000; cat. no. RS030826; ImmunoWay Biotechnology Company, Plano,
TX, USA) at 4°C overnight. Subsequent to being washed three times
with PBS, sections were incubated with horseradish peroxidase
(HRP)-conjugated streptomycin secondary antibody (1:200; cat. no.
SE068; Beijing Solarbio Science & Technology Co., Ltd.) for 2 h
at room temperature. Nuclei were stained with 0.5% hematoxylin for
5 min at room temperature. Brown staining indicated positive cells
and blue staining indicated nuclei. The brain sections were
observed under a light microscope and the magnification was ×200
(Olympus Corporation).
TdT-mediated dUTP nick-end labeling
(TUNEL) staining
A colorimetric TUNEL apoptosis assay kit was
purchased from Beyotime Institute of Biotechnology to detect
apoptosis in hippocampal neurons in the brain tissues. Experiments
were performed according to the manufacturer's protocol. Briefly,
samples were fixed at room temperature for one day in 4%
paraformaldehyde, slides were dewaxed with various concentrations
of xylene and ethanol (xylene for 10 min, xylene for 5 min, 100%
ethanol for 5 min, 95% ethanol for 2 min, 80% ethanol for 2 min and
70% ethanol for 2 min at room temperature), and incubated with 20
μg/ml proteinase K (DNase free) for 30 min at 37°C.
Subsequent to being washed three times with PBS, slides were
incubated with 3% H2O2 for 20 min at room
temperature to inactivate the endogenous peroxidase. Following
washing, slides were incubated with 50 μl TUNEL reaction
mixture for 60 min at 37°C in the dark, and then washed. Then, the
slides were incubated with 200 μl stop solution for 10 min
at room temperature. Following this, the slides were incubated with
50 μl streptavidin-HRP working solution for 30 min at room
temperature. Subsequent to washing, the slides were incubated with
400 μl DAB solution for 5–30 min at room temperature.
Apoptosis-positive cells stained brown, and cell nuclei were
stained with hematoxylin for 5 min at room temperature., which
appeared blue. The degree of apoptosis was visualized via
fluorescence light microscopy and the magnification was ×200
(Olympus Corporation). Each slice was randomly selected over three
fields.
qPCR
Total RNA from the brain tissues was isolated using
TRIzol® (Takara Biotechnology Co., Ltd., Dalian, China). A
PrimeScript™ 1st strand cDNA synthesis kit (Takara Biotechnology
Co., Ltd.) was used to synthesize cDNA. The reaction mixture
included the following: 1 μl 50 μM oligo dT primers;
1 μl dNTP mixture; and 1 μg template RNA. Then,
RNase-free dH2O was added to make up 10 μl total
volume, followed by incubation at 65°C for 5 min, and then cooling
on ice. Secondly, 10 μl reaction mixture, 4 μl 5X
PrimeScript buffer, 0.5 μl RNase inhibitor, and 1 μl
PrimeScript RTase were combined, then RNase-free dH2O
was added to make up a total reaction volume of 20 μl,
followed by incubation at 30°C for 5 min, 42°C for 30 min, and then
95°C for 5 min. qPCR was performed by using SYBR Fast qPCR Mix
(Takara Biotechnology Co., Ltd.) with an ABI 7300 PCR system. The
reaction protocol including the following: 10 μl SYBR Fast
qPCR Mix (2X); 0.8 μl PCR forward primer (10 μM); 0.8
μl PCR reverse primer (10 μM); 0.4 μl ROX
reference dye (50X); 1 μl cDNA; and dH2O up to 20
μl. The PCR thermocycler conditions were as follows: 95°C
for 30 sec, followed by 40 cycles: Denaturation at 95°C for 5 sec
and annealing at 60°C for 10 sec. Relative expression was
calculated using the 2−ΔΔCq formula (30). Data were normalized to the β-actin
gene. The following primers were used: BI-1 forward,
ACGGACTCTGGAACCATGAA; BI-1 reverse, AGCCGCCACAAACATACAA; GPP78
forward, CCACCAGGATGCAGACATTG; GPP78 reverse, AGGGCCTCCACTTCCATAGA;
IRE1 forward, AAGTTTTGGAAGAGCCAGCA; IRE1 reverse,
TGTTCTTGCCTCCAAGTGTG; JNK forward, CGGAACACCTTGTCCTGAA; JNK
reverse, TCGCCTGACTGGCTTTAAGT; ASK forward, ATCCCAGAGTCCATGTCTGC;
ASK reverse, GCAACCACATACCCGAGAGT; CHOP forward,
AGCAGAGGTCACAAGCACCT; CHOP reverse, CTGCTCCTTCTCCTTCAGC.
Western blot analysis
Proteins from the brain tissues were extracted via
lysis buffer (Beyotime Institute of Biotechnology). The total
protein concentrations were determined using a BCA protein assay
kit (Beyotime Institute of Biotechnology). Proteins (30 μg)
were separated by 10% SDS-PAGE gels and transferred to
polyvinylidene fluoride membranes. Membranes were blocked with 10%
skimmed milk in TBST (TBS containing 0.1 % Tween-20) at room
temperature for 1.5 h. Following blocking, membranes were incubated
with primary antibodies, followed by HRP Goat Anti-Rabbit IgG (H+L)
secondary antibodies (dilution 1:2,000; cat. no. AS014; ABclonal
Biotech Co., Ltd., Wuhan, China) or HRP Goat Anti-Mouse IgG (H+L)
secondary antibodies (dilution 1:2,000; cat. no. AS003; ABclonal
Biotech Co., Ltd.). The following primary antibodies were used:
BI-1 antibody (dilution 1:1,000; cat. no. sc-73483; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), CHOP antibody (dilution
1:1,000; cat. no. A11346, ABclonal, Biotech), GPR78 antibody
(dilution 1:1,000; cat. no. A12364; ABclonal Biotech), IRE1α
antibody (dilution 1:1,000; cat. no. 37073; Abcam, Cambridge, MA,
USA), JNK antibody (dilution 1:1,000; cat. no. A2462; ABclonal
Biotech), ASK1 antibody (dilution 1:1,000; cat. no. A6274; ABclonal
Biotech), caspase 3 (dilution 1:1,000; cat. no. A11319; ABclonal
Biotech), caspase 9 (dilution 1:1,000; cat. no. A7523; ABclonal
Biotech) and albumin (dilution 1:500; cat. no. A0353; ABclonal
Biotech). Subsequently, the bands were visualized by enhanced
chemiluminescence (Advansta, Inc. Menlo Park, CA, USA). The
densities of the bands were detected using Image J 2× software
(National Institutes of Health, Bethesda, MD, USA), and normalized
to β-actin.
Statistical analysis
Data are expressed as the mean ± standard deviation.
The GraphPad Prism software version 5.0a (GraphPad Software, Inc.,
La Jolla, CA, USA) was used for statistical analysis. One-way
analysis of variance, followed by Tukey's Multiple Comparison post
hoc test, was used to analyze the differences among the groups.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Neurological scoring and brain water
content
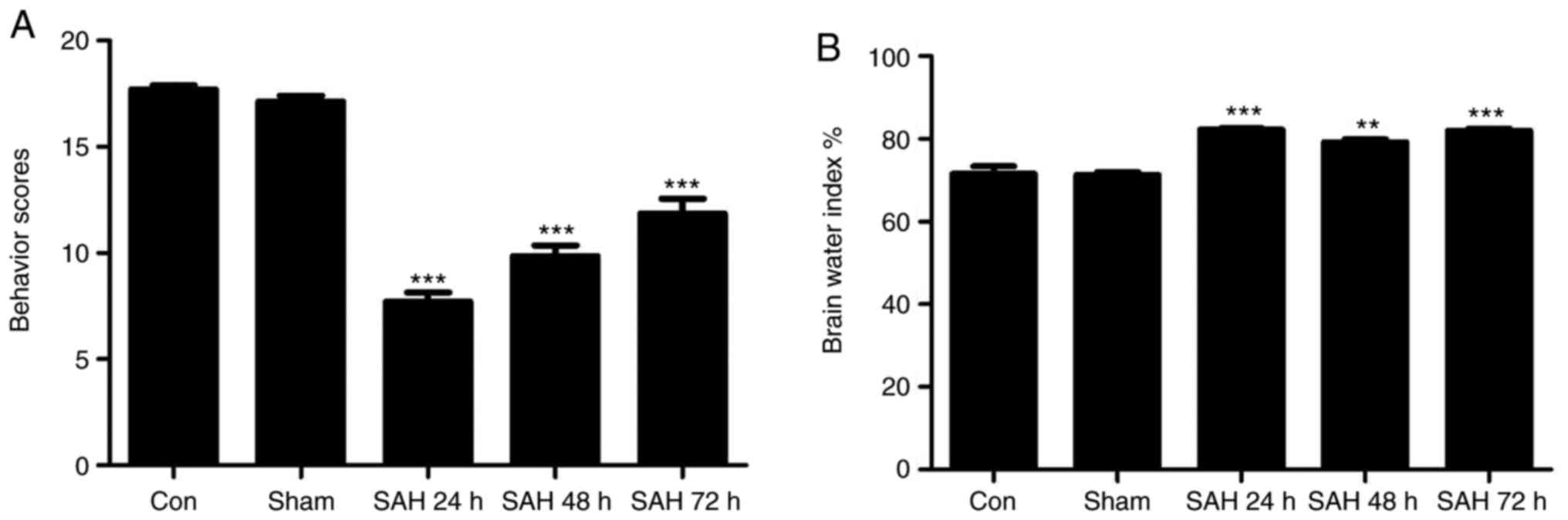
As indicated in Fig.
1A, the SAH model groups demonstrated markedly decreased
neurological scores compared with the control and sham groups, and
the SAH 24 h group exhibited lower neurological scores compared
with all the other groups. In Fig.
1B, it was indicated that the brain water indexes in the SAH
model groups were increased compared with those of the control and
sham groups. In addition, the SAH 24 h group exhibited the highest
brain water index compared with the SAH 48 h and 72 h groups.
Therefore, it was concluded that the SAH 24 h model was the most
effective.
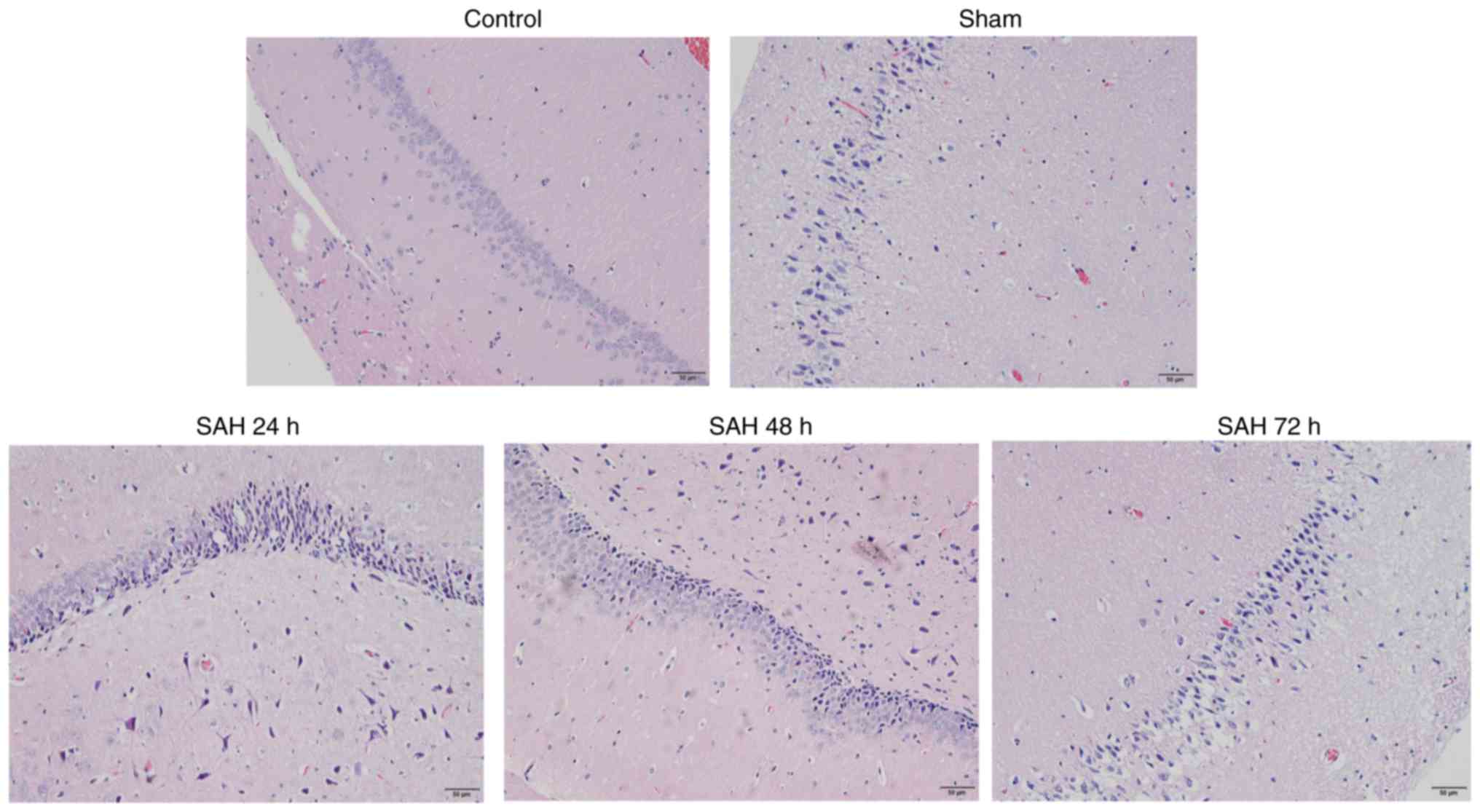
Pathological analysis of hippocampal
neurons
In the control and sham groups, the hippocampal
neurons were aligned in a well-ordered manner, with normal
morphology and without heteromorphic neurons (Fig. 2). Compared with the sham group,
the SAH 24, 48 or 72 h groups exhibited an increased number of
heteromorphic neurons in the hippocampus (Fig. 2). Additionally, the SAH 24 h group
had the highest number of heteromorphic neurons compared with the
other groups (Fig. 2). This
result indicated that SAH may induce brain injury. According to the
aforementioned results, the optimum SAH time (24 h) was selected
for subsequent SAH model experiments.
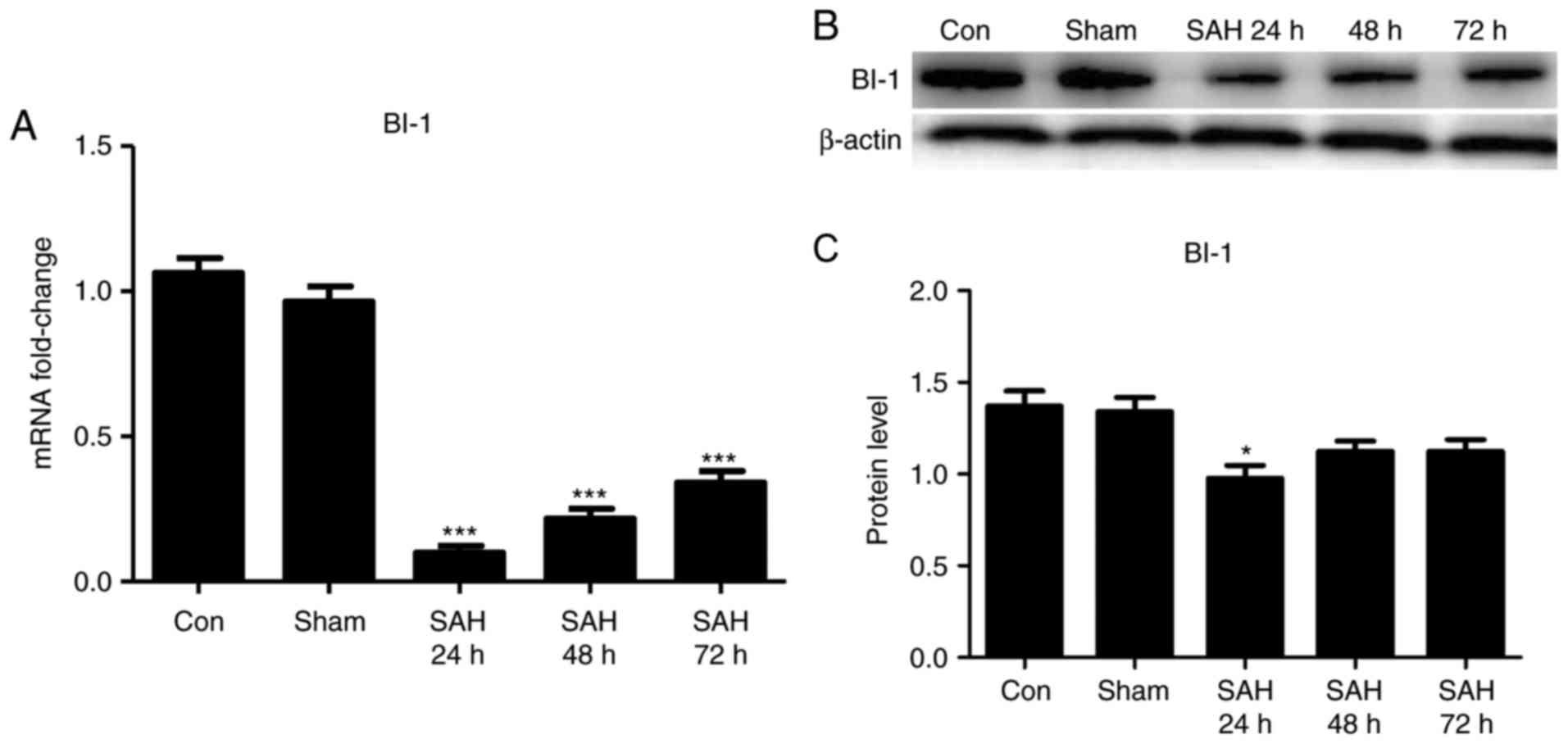
Expression levels of BI-1 in brain
tissues following SAH
To investigate the expression of BI-1 in brain
tissues following SAH, qPCR and western blot analysis were used to
detect the changes in BI-1 in all groups. It was identified that
the levels of BI-1 in the brain tissues were significantly
suppressed following SAH compared with those of the sham group, and
the level of BI-1 was the lowest at 24 h after SAH (Fig. 3). These results indicated that
BI-1 expression was associated with early brain injury following
SAH, and this expression may have an important role in brain injury
repair.
Effects of BI-1 shRNA and BI-1
overexpression on neurological scores, brain water index, H&E
staining and BBB permeability
The BI-1 levels in negative control (NC), pCDH,
BI-1/pCDH, BI-1/pLKO.1 shRNA and scrambled shRNA/pLKO.1 cells were
examined prior to use in the animal model. As demonstrated in
Fig. 4A-C, the mRNA and protein
levels of BI-1 were markedly increased in the BI-1/pCDH group
compared with that in other groups, and decreased in BI-1/pLKO.1
shRNA group compared with that in other groups. Furthermore, there
were no differences detected among the NC, pCDH and scramble
shRNA/pLKO.1 groups (Fig. 4A–C).
As no differences were detected among the NC, pCDH and scrambled
shRNA/pLKO.1 groups, only a single control group (the NC group) was
used in the subsequent animal experiments.
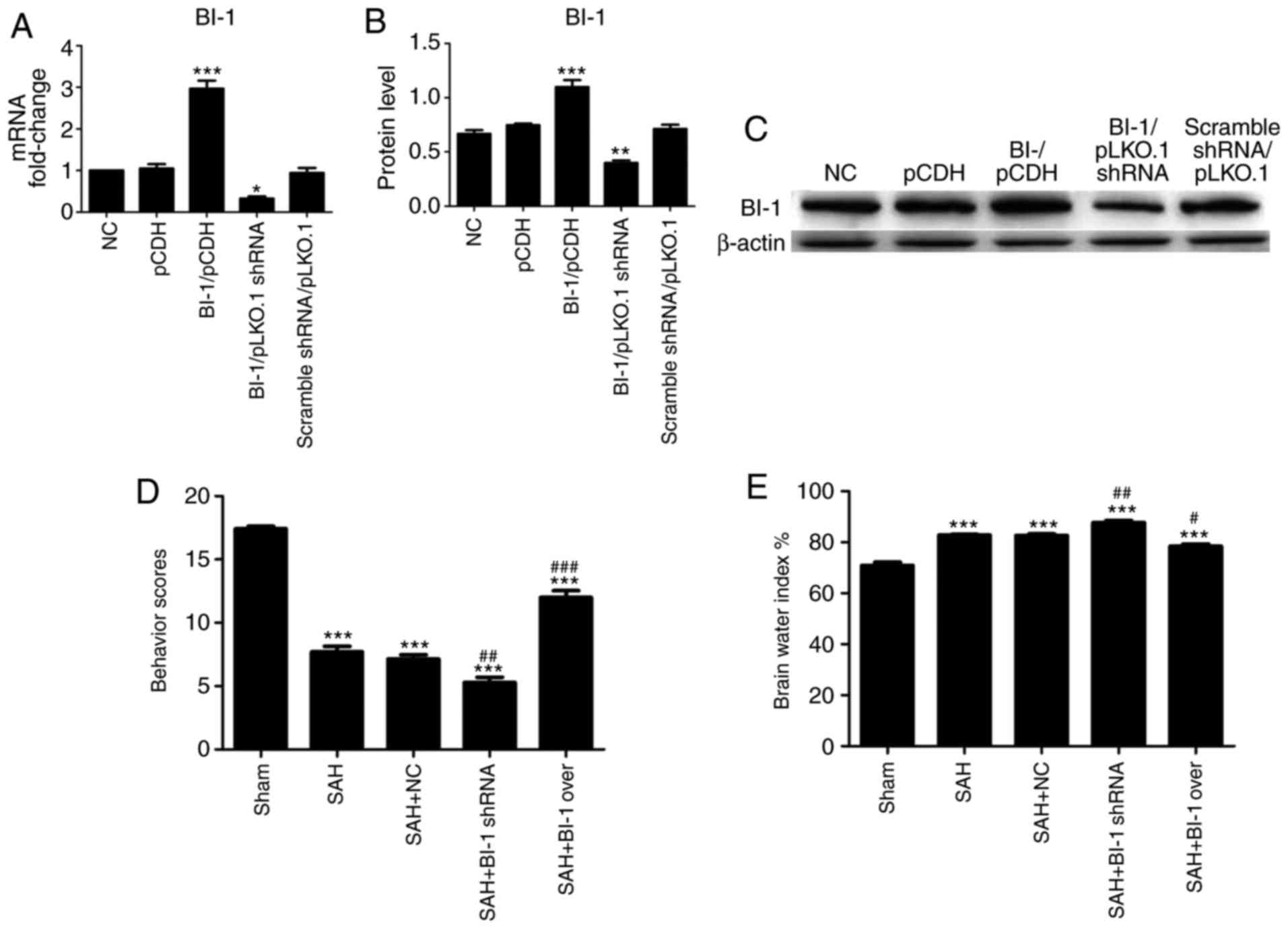 | Figure 4Effects of BI-1 on neurological
scores and brain edema in EBI following SAH. The mRNA and protein
levels of BI-1 in NC, pCDH, BI-1/pCDH, BI-1/pLKO.1 shRNA and
scrambled shRNA/pLKO.1 cells were detected by (A) quantitative
polymerase chain reaction and (B) western blot analysis. (C)
Protein levels were normalized to that of β-actin. (D) Neurological
scores were analyzed at 24, 48 and 72 h. (E) Brain water indexes
were analyzed by measuring the brain water content. All experiments
were repeated at least three times. All data are presented as the
mean ± standard deviation. *P<0.05,
**P<0.01 and ***P<0.001 vs. the NC/sham
groups. #P<0.05, ##P<0.01 and
###P<0.001 vs. the SAH group. NC, negative control;
BI-1, B-cell lymphoma 2-associated X protein-inhibitor-1; shRNA,
short hairpin RNA; SAH, subarachnoid hemorrhage; SAH+BI-1 over, SAH
+ BI-1 overexpression. |
The neurological scores were significantly decreased
in the SAH+BI-1 shRNA group and increased in the SAH+BI-1 over
group compared with those in the SAH group (Fig. 4D). The brain water indexes were
significantly increased in the SAH+BI-1 shRNA group and decreased
in the SAH+BI-1 over group compared with those in the SAH group
(Fig. 4E).
Similarly, the expression levels of albumin in the
SAH group and the SAH+BI-1 shRNA group were increased compared with
that in the sham group and were highest in the SAH+BI-1 shRNA
group. In addition, the levels of albumin in the SAH+BI-1 over
group were decreased compared with those in the SAH group but were
increased compared with those in the sham group (Fig. 5A). To additionally detect BBB
permeability, the levels of IgG in the brain tissues were also
assessed by IHC assays. In Fig.
5B, it was identified that the IgG levels in the brains were
increased following SAH compared with those in the sham group and
highest in the SAH+BI-1 shRNA group. BI-1 overexpression
significantly suppressed the IgG induced by SAH in the SAH+BI-1
over group compared with the SAH group (Fig. 5B). The quantification of
IgG-positive cells was also analyzed (Fig. 5C). The results suggested that BI-1
inhibited the BBB permeability induced by brain injury following
SAH.
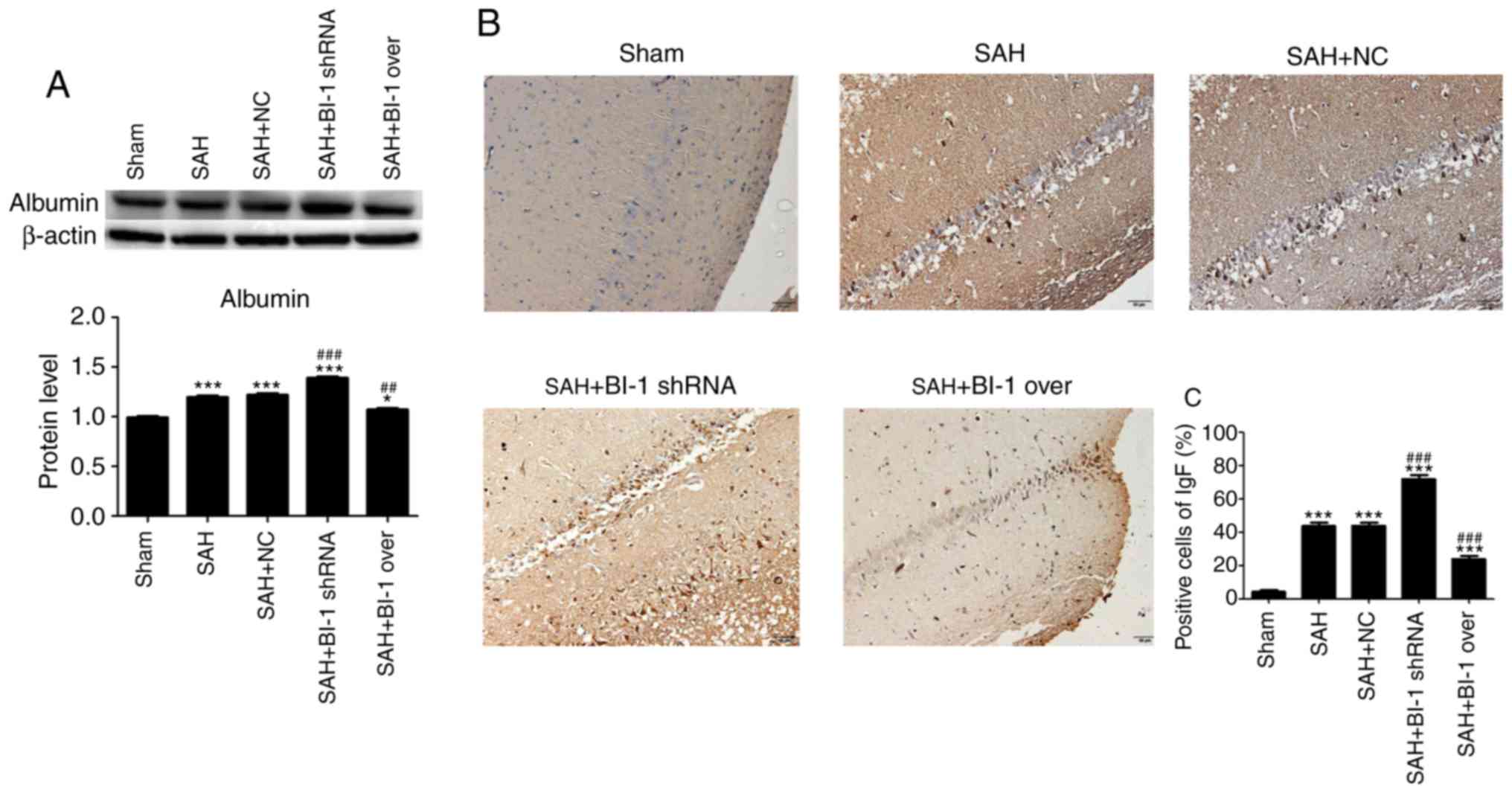 | Figure 5Effects of BI-1 on blood-brain
barrier injury during EBI following SAH. (A) The expression levels
of albumin following BI-1 overexpression and shRNA silencing were
detected by western blot analysis. (B) Immunohistochemistry of the
hippocampus in the sham, SAH, SAH+NC, SAH+BI-1 shRNA and SAH+BI-1
over groups. Scale bar=50 μm (magnification, ×200). (C)
Quantification of immunohistochemical positive cells, expressed as
the total cell percentage. All data are presented as the mean ±
standard deviation. *P<0.05 and
***P<0.001 vs. the sham group, ##P<0.01
and ###P<0.001 vs. the SAH group. BI-1, B-cell
lymphoma 2-associated X protein-inhibitor-1; shRNA, short hairpin
RNA; SAH, subarachnoid hemorrhage; SAH+BI-1 over, SAH + BI-1
overexpression; NC, negative control. |
As indicated in Fig.
6, the SAH group and the SAH+NC group contained increased
numbers of heteromorphic neurons compared with the sham group, and
the number of heteromorphic neurons in the SAH+BI-1 shRNA group was
increased compared with that in the SAH and SAN+NC groups. However,
the SAH+BI-1 over group exhibited a decreased number of
heteromorphic neurons compared with the SAH+BI-1 shRNA group, but
this number was increased compared with that in the sham group.
These results implied that BI-1 repaired and rescued brain injury
following SAH.
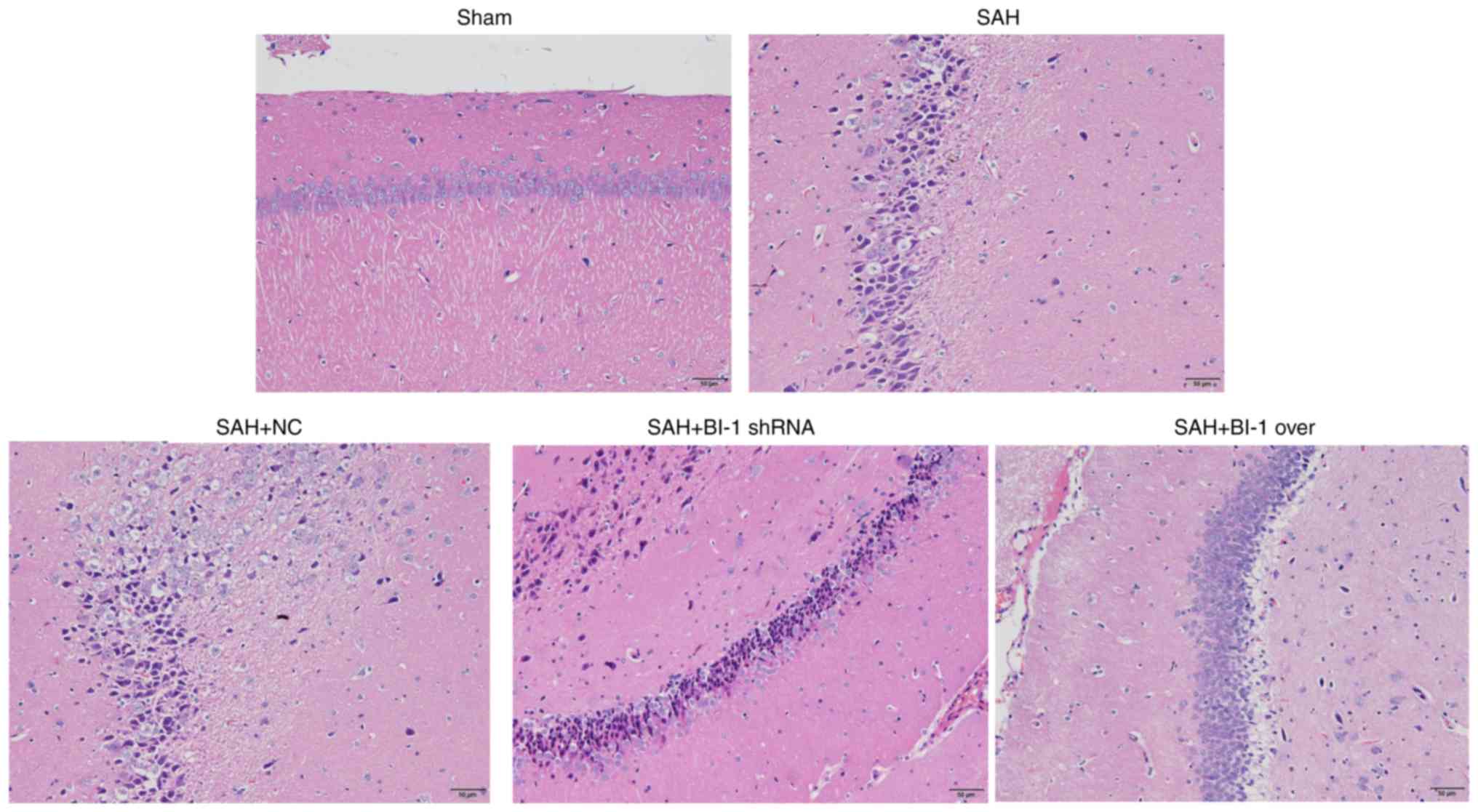 | Figure 6H&E staining of rats following
BI-1 overexpression and shRNA silencing. Normal and heteromorphic
neurons in the hippocampus of the brains were detected by H&E
staining in the sham, SAH, SAH+NC, SAH+BI-1 shRNA and SAH+BI-1 over
groups. Scale bar=50 μm (magnification, ×200). H&E,
hematoxylin and eosin; BI-1, B-cell lymphoma 2-associated X
protein-inhibitor-1; shRNA, short hairpin RNA; SAH, subarachnoid
hemorrhage; SAH+BI-1 over, SAH + BI-1 overexpression; NC, negative
control. |
Effects of BI-1 shRNA and BI-1
overexpression on ER stress-associated apoptosis
To demonstrate the effects of BI-1 on apoptosis,
TUNEL assays of brain tissues were performed. The number of
apoptosis-positive cells was markedly increased in the SAH group
and the SAH+NC group compared with the sham group, and BI-1 shRNA
significantly attenuated apoptosis in the SAH+ BI-1 shRNA group
compared with the SAH group (Fig.
7A). However, BI-1 overexpression significantly rescued the
high levels of apoptosis in the SAH+ BI-1 over group compared with
the SAH group (Fig. 7A). This
result suggested that BI-1 may inhibit the apoptosis induced by
SAH. The percentage of TUNEL-positive cells was also analysed
(Fig. 7B). The expression levels
of apoptosis-associated proteins caspase 3 and caspase 9 in these
groups via western blot analysis. These results were consistent
with TUNEL assays (Fig. 7C and
D).
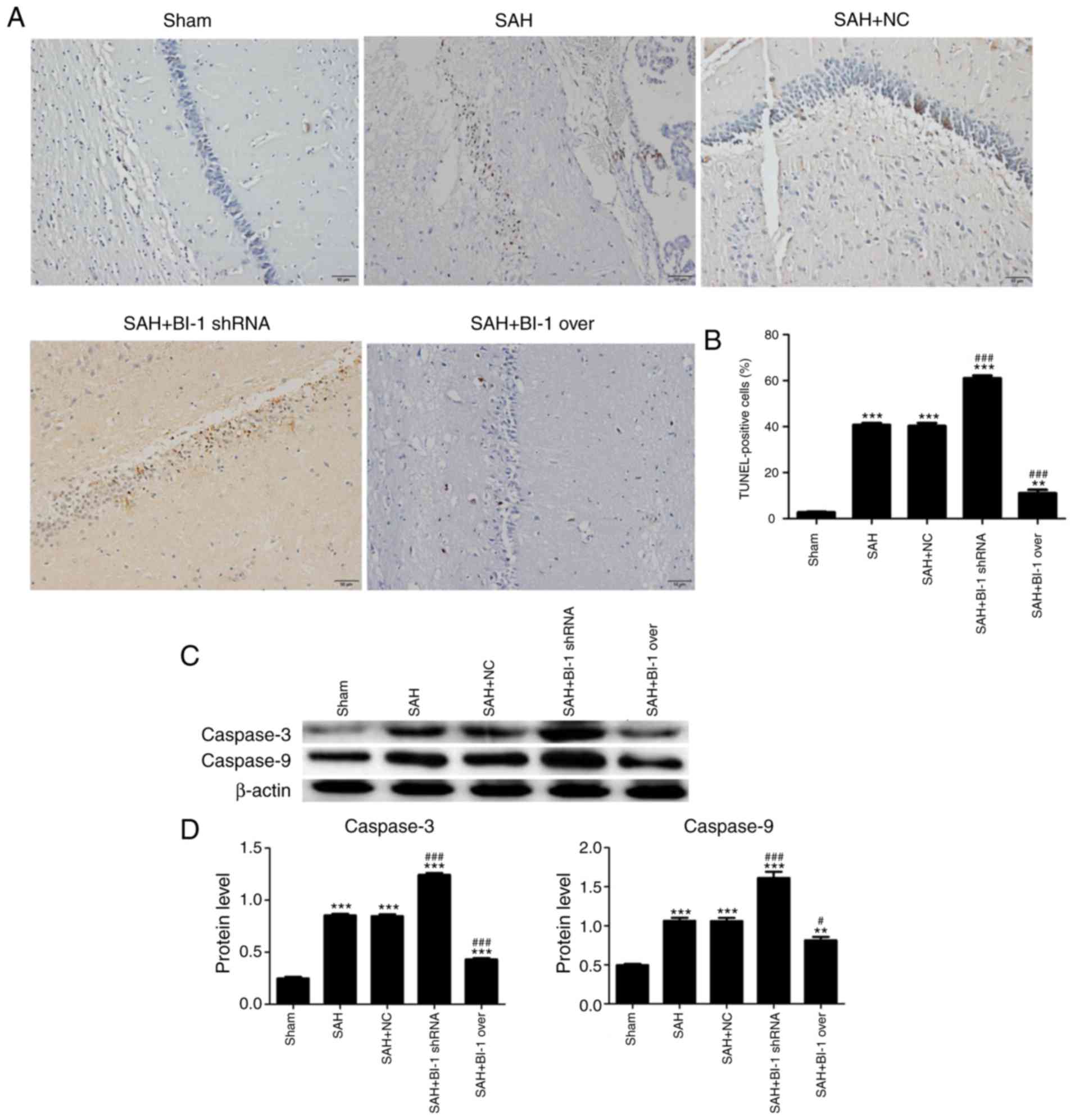 | Figure 7Effects of BI-1 on apoptosis in the
hippocampus following BI-1 overexpression and shRNA silencing. (A)
Representative TUNEL photomicrographs of the hippocampus in the
sham, SAH, SAH+NC, SAH+BI-1 shRNA and SAH+BI-1 over groups. Scale
bar=50 μm. (B) Quantification of TUNEL-positive cells in
these groups, expressed as the total cell percentage. (C) The
protein levels of caspase 3 and caspase 9 were detected by western
blot analysis. (D) Protein levels were normalized to that of
β-actin. All data are presented as the mean ± standard deviation.
**P<0.01 and ***P<0.001 vs. the sham
group. #P<0.05 and ###P<0.001 vs. the
SAH group. BI-1, B-cell lymphoma 2-associated X
protein-inhibitor-1; shRNA, short hairpin RNA; SAH, subarachnoid
hemorrhage; SAH+BI-1 over, SAH + BI-1 overexpression; NC, negative
control; TUNEL, terminal deoxynucleotidyl transferase-mediated dUTP
nick-end labeling. |
To additionally investigate the mechanism of
apoptosis, the expression levels of ER stress-associated genes
(GPR78, CHOP, IRE1, JNK and ASK1) were analyzed using qPCR and
western blot analysis. As indicated in Fig. 8, the levels of GPR78, CHOP, IRE1,
JNK and ASK1 were increased in the SAH group compared with those in
the sham group, and were also increased in the SAH+BI-1 shRNA group
compared with those in the SAH group. The expression levels of all
genes were decreased in SAH+BI-1 over group compared with those in
the SAH group; however, these levels were increased compared with
those in the sham group. No difference between the SAH and SAH +NC
groups was observed. These results indicated that BI-1 may inhibit
ER stress-associated apoptosis induced by SAH.
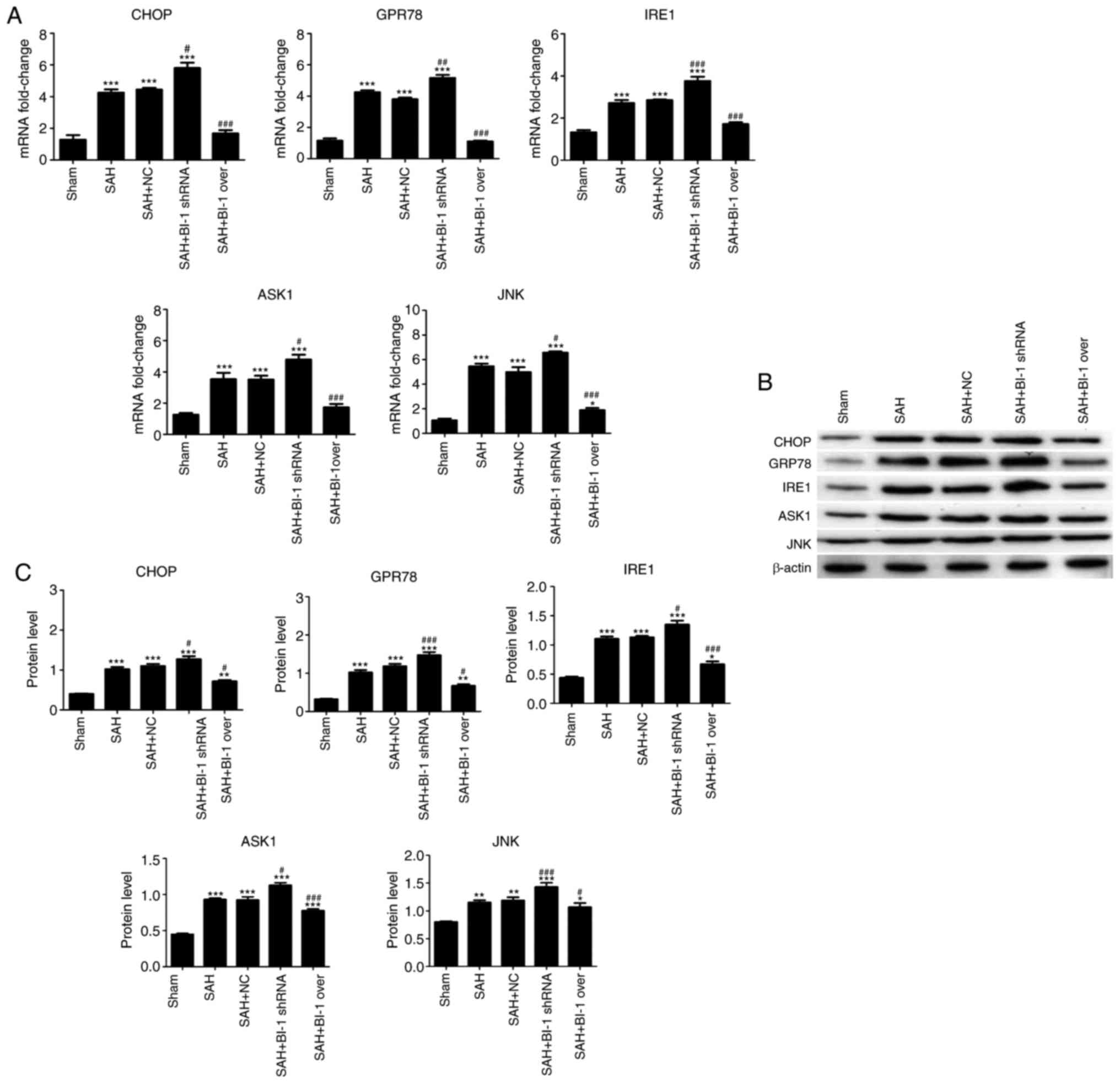 | Figure 8Potential mechanism of the effects of
BI-1 on apoptosis induced by early brain injury following SAH. (A)
The mRNA levels of GPR78, CHOP, IRE-1, JNK and ASK1 were detected
by quantitative polymerase chain reaction assays. (B) The protein
levels of GPR78, CHOP, IRE-1, JNK and ASK1 were detected by western
blot analysis. (C) Protein levels were normalized to that of
β-actin. All experiments were repeated at least three times. All
data are presented as the mean ± standard deviation.
*P<0.05, **P<0.01 and
***P<0.001 vs. the control group.
#P<0.05, ##P<0.01 and
###P<0.001 vs. the SAH group. BI-1, B-cell lymphoma
2-associated X protein-inhibitor-1; shRNA, short hairpin RNA; SAH,
subarachnoid hemorrhage; SAH+BI-1 over, SAH + BI-1 overexpression;
NC, negative control; CHOP, C/EBP homologous protein; GRP78,
Glucose regulated protein, 78 kDa; IRE1, Serine/threonine-protein
kinase/endoribonuclease IRE1; ASK1, apoptotic signaling kinase-1;
JNK, c-Jun N terminal kinases. |
Discussion
The present study examined the neuroprotective and
anti-apoptotic effects of BI-1 on EBI following SAH in a rat model.
It was identified that BI-1 overexpression markedly increased
neurological scores, and decreased the brain water index and the
number of heteromorphic neurons in the hippocampal area.
Furthermore, BI-1 overexpression also restored BBB function and
suppressed the ER stress-mediated apoptosis induced by EBI
following SAH in the hippocampal area. By contrast, BI-1 shRNA
demonstrated the opposite results.
EBI is regarded as an important and potential
implementation target of treatment for SAH due to
pathophysiological variables occurring during the EBI period,
defined as the first 72 h after SAH (2,6).
During the EBI period, a number of physiological disruptions occur,
including increased ICP, decreased CBF, brain edema, BBB
disruption, inflammation and oxidative cascades that all ultimately
lead to cell death (4).
Neurological scores are associated with the pathophysiological
aspects of SAH. The present study first evaluated the neurological
scores of rats following SAH, and the lowest scores were observed
at 24 h after SAH compared with those at 48 and 72 h. Brain edema
was assessed by brain water content. The highest brain water index
was measured at 24 h after SAH compared with those of the other
groups. The H&E staining results were also consistent with
these data. The levels of BI-1 in the brain tissues at 24, 48 and
72 h after SAH were also determined. The levels of BI-1 at 24 h
were decreased compared with those of the other groups. This result
indicated that BI-1 exhibited a positive effect on brain tissues
following SAH and alleviated EBI. Therefore, 24 h after SAH was
selected the optimal time period for subsequent experiments in the
present study.
BI-1 is an evolutionarily conserved cytoprotective
protein. To additionally investigate the effect of BI-1 on EBI
following SAH, SAH models were treated with BI-1 overexpression and
shRNA plasmids. Neurological scores were significantly increased
and the brain water index was decreased in the SAH+BI-1 over group
compared with those in the SAH group. However, BI-1 shRNA
significantly decreased the neurological scores and increased the
brain water index compared with those in the SAH group. Brain edema
has been considered to have a direct effect on BBB disruption
(2). The aforementioned results
suggested that BI-1 has an important function in limiting EBI
following SAH.
In addition to brain water content, the levels of
albumin and IgG in brain tissues are also hallmarks of BBB
integrity (31,32). Under normal circumstances, the
albumin serum protein IgG cannot completely permeate the BBB.
However, following cerebral injury, albumin and IgG may permeate
the brain tissue, indicating that the BBB integrity has been
damaged and permeability is increased (33,34). Therefore, the expression of
albumin and IgG extravasation in brain tissue may be detected to
reflect the injury and permeability of the BBB. The present study
detected albumin levels using western blot analysis, and detected
IgG levels using immunohistochemical assays. The levels of albumin
were markedly increased in the SAH group compared with those in the
sham group. In addition, the levels of albumin decreased in the
SAH+BI-1 over group and increased in the SAH+BI-1 shRNA group
compared with those in the SAH group. Using IgG
immunohistochemistry, positively-stained neuronal cells in the
hippocampal area were detected in the SAH group, revealing
increased permeability of the BBB. The SAH+BI-1 over group
exhibited markedly less positive neuronal cells compared with the
SAH group, and the SAH+BI-1 shRNA group exhibited more positive
neurons compared with the SAH group. This result suggested that
BI-1 may repair and alleviate BBB damage induced by EBI following
SAH. As indicated in the results of the H&E staining, these
data were consistent with the aforementioned results.
Previous studies suggested that EBI is one of the
important factors affecting poor prognosis following SAH (35,36), and EBI is involved in a number of
processes, including apoptosis, oxidative stress and
neuroinflammation (7,37,38). It has also been suggested that the
apoptosis of neuronal cells is an important factor in EBI following
SAH, which may explain the serious outcomes of SAH (39,40). In the present study, the effect of
BI-1 on apoptosis in EBI following SAH was examined via TUNEL
assays. It was identified that the SAH group exhibited more
TUNEL-positive neuronal cells in the hippocampal area compared with
the sham group. BI-1 significantly inhibited the number of
TUNEL-positive neuronal cells in the SAH+BI-1 over group compared
with the SAH group, and the opposite results were observed in the
SAH+BI-shRNA group.
ER stress is associated with various human
neurodegenerative diseases (41).
Downstream factors of ER stress may induce neuronal apoptosis
following SAH (42). However,
certain previous studies have suggested that ER stress is induced
in cerebral ischemia and that ER stress-mediated reactions may
inhibit neuronal apoptosis (43).
ER stress appears to serve dual roles in neuronal apoptosis
(44,45). Therefore, the present study aimed
to investigate the association between ER stress and apoptosis in
EBI following SAH. The levels of ER stress signaling genes, namely
GPR78, CHOP, IRE-1, JNK and ASK1, which are associated with ER
stress, were examined via qPCR and western blot analysis. The
results suggested that ER stress-mediated apoptosis was markedly
induced in the SAH group compared with the sham groups. In
addition, BI-1 inhibited the ER stress-mediated apoptosis induced
by EBI following SAH.
In conclusion, the results from the present study
demonstrate that BI-1 exerts a neuroprotective effect on EBI
following SAH by suppressing apoptosis. The overexpression of BI-1
alleviated EBI following SAH and apoptosis, suggesting that BI-1
may represent a potential therapeutic strategy for EBI following
SAH. It was also demonstrated that ER stress mediated-apoptosis
served an important role in SAH treatment, and BI-1 may inhibit EBI
following SAH by suppressing an apoptotic pathway associated with
ER stress. Mitochondria-derived stresses are also associated with
neuropathological and neurodegenerative diseases (46). Mitochondria are central organelles
in neuronal physiology integrating several crucial functions,
including energy metabolism, cell respiration and Ca2+
homeostasis, all of which have been revealed to be dysregulated in
Alzheimer's disease and other neurodegenerative disorders, such as
Parkinson's disease (47,48). Several studies have reported that
the mitochondrial pathway is closely associated with EBI following
SAH (49,50). Therefore, the association among
BI-1, SAH and mitochondria-derived stresses will be addressed in
future studies.
Funding
The present study was supported by the Natural
Science Foundation of China (grant no. 81560227), the Scientific
Research Found Project in Yunnan Province Department of Education
(grant no. 2016ZZX046), the Joint Special Project for Applied Basic
Research of Yunnan Provincial Science and Technology
Department-Kunming Medical University [grant no. 2017FE467(-208)],
and the 'Kunhua. Aoxin' Science and Technology Project of the First
People's Hospital of Yunnan Province (grant no. 2014BS009).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JZ and JL conceived and designed the study. JL, SZ
and YZ performed the experiments. XL, WT and LJ processed data. JL
and XQ wrote the manuscript. JZ, JL, SZ, YZ and XQ reviewed and
edited the manuscript. All authors read and approved the
manuscript.
Ethics approval and consent to
participate
The animal experiments were approved by the Animals
Ethics Committee of Kunming Medical University and the Guide for
the Care and Use of Laboratory Animals.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
SAH
|
subarachnoid hemorrhage
|
|
EBI
|
early brain injury
|
|
BI-1
|
B-cell lymphoma 2-associated X
protein-inhibitor-1
|
|
ER
|
endoplasmic reticulum
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
BBB
|
blood-brain barrier
|
|
H&E
|
hematoxylin and eosin
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling
|
Acknowledgments
Not applicable.
References
|
1
|
Al-Khindi T, Macdonald RL and Schweizer
TA: Cognitive and functional outcome after aneurysmal subarachnoid
hemorrhage. Stroke. 41:e519–e536. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cahill J, Calvert JW and Zhang JH:
Mechanisms of early brain injury after subarachnoid hemorrhage. J
Cereb Blood Flow Metab. 26:1341–1353. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ayer R, Chen W, Sugawara T, Suzuki H and
Zhang JH: Role of gap junctions in early brain injury following
subarachnoid hemorrhage. Brain Res. 1315:150–158. 2010. View Article : Google Scholar :
|
|
4
|
Fujii M, Yan J, Rolland WB, Soejima Y,
Caner B and Zhang JH: Early brain injury, an evolving frontier in
subarachnoid hemorrhage research. Transl Stroke Res. 4:432–446.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ayer R and Zhang J: Connecting the early
brain injury of aneurysmal subarachnoid hemorrhage to clinical
practice. Turk Neurosurg. 20:159–166. 2010.PubMed/NCBI
|
|
6
|
Yuksel S, Tosun YB, Cahill J and Solaroglu
I: Early brain injury following aneurysmal subarachnoid hemorrhage:
Emphasis on cellular apoptosis. Turk Neurosurg. 22:529–533.
2012.PubMed/NCBI
|
|
7
|
Hasegawa Y, Suzuki H, Sozen T, Altay O and
Zhang JH: Apoptotic mechanisms for neuronal cells in early brain
injury after subarachnoid hemorrhage. Acta Neurochir Suppl.
110:43–48. 2011.
|
|
8
|
Cheng G, Chunlei W, Pei W, Zhen L and
Xiangzhen L: Simvastatin activates Akt/glycogen synthase
kinase-3beta signal and inhibits caspase-3 activation after
experimental subarachnoid hemorrhage. Vascul Pharmacol. 52:77–83.
2010. View Article : Google Scholar
|
|
9
|
Kusaka G, Ishikawa M, Nanda A, Granger DN
and Zhang JH: Signaling pathways for early brain injury after
subarachnoid hemorrhage. J Cereb Blood Flow Metab. 24:916–925.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Endo H, Nito C, Kamada H, Yu F and Chan
PH: Akt/GSK3beta survival signaling is involved in acute brain
injury after subarach-noid hemorrhage in rats. Stroke.
37:2140–2146. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu Q and Reed JC: Bax inhibitor-1, a
mammalian apoptosis suppressor identified by functional screening
in yeast. Mol Cell. 1:337–346. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chae HJ, Kim HR, Xu C, Bailly-Maitre B,
Krajewska M, Krajewski S, Banares S, Cui J, Digicaylioglu M, Ke N,
et al: BI-1 regulates an apoptosis pathway linked to endoplasmic
reticulum stress. Mol Cell. 15:355–366. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bredesen DE, Rao RV and Mehlen P: Cell
death in the nervous system. Nature. 443:796–802. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim I, Xu W and Reed JC: Cell death and
endoplasmic reticulum stress: Disease relevance and therapeutic
opportunities. Nat Rev Drug Discov. 7:1013–1030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Patil C and Walter P: Intracellular
signaling from the endoplasmic reticulum to the nucleus: The
unfolded protein response in yeast and mammals. Curr Opin Cell
Biol. 13:349–355. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hetz C, Bernasconi P, Fisher J, Lee AH,
Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A,
Glimcher LH and Korsmeyer SJ: Proapoptotic BAX and BAK modulate the
unfolded protein response by a direct interaction with IRE1alpha.
Science. 312:572–576. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Szegezdi E, Logue SE, Gorman AM and Samali
A: Mediators of endoplasmic reticulum stress-induced apoptosis.
EMBO Rep. 7:880–885. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nishitoh H, Matsuzawa A, Tobiume K,
Saegusa K, Takeda K, Inoue K, Hori S, Kakizuka A and Ichijo H: ASK1
is essential for endoplasmic reticulum stress-induced neuronal cell
death triggered by expanded polyglutamine repeats. Genes Dev.
16:1345–1355. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lei K and Davis RJ: JNK phosphorylation of
Bim-related members of the Bcl2 family induces Bax-dependent
apoptosis. Proc Natl Acad Sci USA. 100:2432–2437. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Krajewska M, Xu L, Xu W, Krajewski S,
Kress CL, Cui J, Yang L, Irie F, Yamaguchi Y, Lipton SA and Reed
JC: Endoplasmic reticulum protein BI-1 modulates unfolded protein
response signaling and protects against stroke and traumatic brain
injury. Brain Res. 1370:227–237. 2011. View Article : Google Scholar :
|
|
21
|
Bailly-Maitre B, Fondevila C, Kaldas F,
Droin N, Luciano F, Ricci JE, Croxton R, Krajewska M, Zapata JM,
Kupiec-Weglinski JW, et al: Cytoprotective gene bi-1 is required
for intrinsic protection from endoplasmic reticulum stress and
ischemia-reperfusion injury. Proc Natl Acad Sci USA. 103:2809–2814.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lu B, Li Y, Li H, Zhang Y, Xu J, Ren L, Fu
S and Zhou Y: Bax inhibitor-1 is overexpressed in non-small cell
lung cancer and promotes its progression and metastasis. Int J Clin
Exp Pathol. 8:1411–1418. 2015.PubMed/NCBI
|
|
23
|
Grzmil M, Thelen P, Hemmerlein B, Schweyer
S, Voigt S, Mury D and Burfeind P: Bax inhibitor-1 is overexpressed
in prostate cancer and its specific down-regulation by RNA
interference leads to cell death in human prostate carcinoma cells.
Am J Pathol. 163:543–552. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Bailly-Maitre B, Bard-Chapeau E, Luciano
F, Droin N, Bruey JM, Faustin B, Kress C, Zapata JM and Reed JC:
Mice lacking bi-1 gene show accelerated liver regeneration. Cancer
Res. 67:1442–1450. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bailly-Maitre B, Belgardt BF, Jordan SD,
Coornaert B, von Freyend MJ, Kleinridders A, Mauer J, Cuddy M,
Kress CL, Willmes D, et al: Hepatic Bax inhibitor-1 inhibits
IRE1alpha and protects from obesity-associated insulin resistance
and glucose intolerance. J Biol Chem. 285:6198–6207. 2010.
View Article : Google Scholar
|
|
26
|
Jeon K, Lim H, Kim JH, Han D, Lee ER, Yang
GM, Song MK, Kim JH and Cho SG: Bax inhibitor-1 enhances survival
and neuronal differentiation of embryonic stem cells via
differential regulation of mitogen-activated protein kinases
activities. Biochim Biophys Acta. 1823:2190–2200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dohm CP, Siedenberg S, Liman J, Esposito
A, Wouters FS, Reed JC, Bähr M and Kermer P: Bax inhibitor-1
protects neurons from oxygen-glucose deprivation. J Mol Neurosci.
29:1–8. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Prunell GF, Mathiesen T and Svendgaard NA:
A new experimental model in rats for study of the pathophysiology
of subarachnoid hemorrhage. Neuroreport. 13:2553–2556. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sugawara T, Ayer R, Jadhav V and Zhang JH:
A new grading system evaluating bleeding scale in filament
perforation subarachnoid hemorrhage rat model. J Neurosci Methods.
167:327–334. 2008. View Article : Google Scholar
|
|
30
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
31
|
Alafuzoff I, Adolfsson R, Bucht G and
Winblad B: Albumin and immunoglobulin in plasma and cerebrospinal
fluid, and blood-cerebrospinal fluid barrier function in patients
with dementia of Alzheimer type and multi-infarct dementia. J
Neurol Sci. 60:465–472. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Sorjonen DC: Total protein, albumin quota,
and electrophoretic patterns in cerebrospinal fluid of dogs with
central nervous system disorders. Am J Vet Res. 48:301–305.
1987.PubMed/NCBI
|
|
33
|
Kumar A, Mittal R, Khanna HD and Basu S:
Free radical injury and blood-brain barrier permeability in
hypoxic-ischemic encephalopathy. Pediatrics. 122:e722–e727. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Schmidt-Kastner R, Szymas J and Hossmann
KA: Immunohistochemical study of glial reaction and serum-protein
extravasation in relation to neuronal damage in rat hippocampus
after ischemia. Neuroscience. 38:527–540. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Suzuki H: What is early brain injury?
Transl Stroke Res. 6:1–3. 2015. View Article : Google Scholar
|
|
36
|
Sehba FA, Hou J, Pluta RM and Zhang JH:
The importance of early brain injury after subarachnoid hemorrhage.
Prog Neurobiol. 97:14–37. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hosaka K and Hoh BL: Inflammation and
cerebral aneurysms. Transl Stroke Res. 5:190–198. 2014. View Article : Google Scholar
|
|
38
|
Chen S, Yang Q, Chen G and Zhang JH: An
update on inflammation in the acute phase of intracerebral
hemorrhage. Transl Stroke Res. 6:4–8. 2015. View Article : Google Scholar
|
|
39
|
Sabri M, Kawashima A, Ai J and Macdonald
RL: Neuronal and astrocytic apoptosis after subarachnoid
hemorrhage: A possible cause for poor prognosis. Brain Res.
1238:163–171. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Matz PG, Fujimura M and Chan PH:
Subarachnoid hemolysate produces DNA fragmentation in a pattern
similar to apoptosis in mouse brain. Brain Res. 858:312–319. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Lindholm D, Wootz H and Korhonen L: ER
stress and neurodegenerative diseases. Cell Death Differ.
13:385–392. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
He Z, Ostrowski RP, Sun X, Ma Q, Huang B,
Zhan Y and Zhang JH: CHOP silencing reduces acute brain injury in
the rat model of subarachnoid hemorrhage. Stroke. 43:484–490. 2012.
View Article : Google Scholar :
|
|
43
|
Roussel BD, Kruppa AJ, Miranda E, Crowther
DC, Lomas DA and Marciniak SJ: Endoplasmic reticulum dysfunction in
neurological disease. Lancet Neurol. 12:105–118. 2013. View Article : Google Scholar
|
|
44
|
Placido AI, Pereira CM, Duarte AI,
Candeias E, Correia SC, Carvalho C, Cardoso S, Oliveira CR and
Moreira PI: Modulation of endoplasmic reticulum stress: An
opportunity to prevent neurodegeneration? CNS Neurol Disord Drug
Targets. 14:518–533. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Stefani IC, Wright D, Polizzi KM and
Kontoravdi C: The role of ER stress-induced apoptosis in
neurodegeneration. Curr Alzheimer Res. 9:373–387. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
McManus MJ, Murphy MP and Franklin JL:
Mitochondria-derived reactive oxygen species mediate
caspase-dependent and-independent neuronal deaths. Mol Cell
Neurosci. 63:13–23. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Winklhofer KF and Haass C: Mitochondrial
dysfunction in Parkinson's disease. Biochim Biophys Acta.
1802:29–44. 2010. View Article : Google Scholar
|
|
48
|
Itoh K, Nakamura K, Iijima M and Sesaki H:
Mitochondrial dynamics in neurodegeneration. Trends Cell Biol.
23:64–71. 2013. View Article : Google Scholar :
|
|
49
|
Chen J, Wang L, Wu C, Hu Q, Gu C, Yan F,
Li J, Yan W and Chen G: Melatonin-enhanced autophagy protects
against neural apoptosis via a mitochondrial pathway in early brain
injury following a subarachnoid hemorrhage. J Pineal Res. 56:12–19.
2014. View Article : Google Scholar
|
|
50
|
Liang Y, Che X, Zhao Q, Darwazeh R, Zhang
H, Jiang D, Zhao J, Xiang X, Qin W, Liu L and He Z:
Thioredoxin-interacting protein mediates mitochondrion-dependent
apoptosis in early brain injury after subarachnoid hemorrhage. Mol
Cell Biochem. 2018. View Article : Google Scholar
|