Introduction
Various pathological conditions, including ischemia,
septic shock and diabetic comas, are accompanied by osmotic changes
which may subsequently induce cellular death and tissue damage
(1). The underlying mechanism may
be associated with the activation of survival signaling cascades
under hyperosmotic stress, which leads to the disruption of the
homeostasis between cell death and survival pathways, and the
subsequent accumulation of damaged, aggregated, misfolded and
oxidized proteins. It was demonstrated that rapid cardiomyocyte
apoptosis was simulated under hyperosmotic stress in vitro
(2), which was associated with
elevated autophagy (3,4). However, the underlying mechanism has
not been fully elucidated.
Autophagy is a highly conserved pathway in
eukaryotes, serving a critical role in cellular survival during
pathophysiological processes and hyperosmotic stress responses
(5-8). When autophagy is initiated,
autophagosome-lysosome fusion degrades cytosolic cargos
encapsulated by double membranous autophagosomes, which are tagged
by lipid-conjugated microtubule-associated protein 1 light chain 3
(LC3-II) (9). Therefore, LC3-II
is usually regarded as a marker of autophagy (10). In addition, Beclin-1, an
additional autophagy marker, serves an essential role in inducing
precursor membrane vesicle expansion through the recruitment of
additional autophagy-related (Atg) proteins at the onset of the
autophagy cascade. The subsequent selective degradation of
sequestosome-1 (SQSTM1/p62) represents the accumulation of protein
aggregates and the degree of autophagy activation (11). Notably, Atg5 is a critical factor
for regulating the maturation of autophagosomes, as Atg5-12
conjugate interacts with Atg16 like 1 to tether the complex to
phagophores and autophagosomes. Therefore, Atg5 is considered a key
element for autophagy under the majority of circumstances, though
Atg5-independent autophagy has been described previously (12).
The nuclear factor of activated T-cells 5 (NFAT5)
protein of the NFAT family, has a large C-terminal region that
harbors a hypertonicitysensitive transactivation domain.
Accumulating evidence suggests that NFAT5 is an osmosensitive
transcription factor, associated with regulating the expression of
genes responsible for cell survival under hyperosmotic conditions
(13-15). In addition, NFAT5 has been
suggested to increase DNA damage response 1 (REDD1) protein and
mammalian target of rapamycin (mTOR) complex 1 repressor
expression, and inhibit mTOR signaling (16,17); these processes subsequently
initiate the autophagic process through autophagy-associated
proteins, including BECN1 and Atg5-Atg12. Therefore, it is possible
that the NFAT5 pathway affects the autophagic pathway under
hyperosmotic stress. However, little is known about the
hyperosmotic stress-induced autophagic mechanism in terminally
differentiated cardiomyocytes.
The present study investigated the role of
hyperosmotic stress-induced autophagy in cardiomyocytes. It was
demonstrated that hyperosmotic stress increased NFAT5 expression,
which then triggered autophagy through Atg5 activity. These data
provided novel insights into the role of NFAT5 in regulating
autophagy in cardiomyocytes under hyperosmotic stress.
Materials and methods
Ethics statement
The present study was approved by the Ethics
Committee of Xuanwu Hospital Capital Medical University and
conformed to the Guide for the Care and Use of Laboratory Animals
published by the US National Institutes of Health (NIH Publication
no. 85-23, revised 1996) (18).
All animals were treated in accordance with guidelines established
by the Capital Medical University Institutional Animal Care and Use
Committee.
Cell culture and treatment
Briefly, 120 male Sprague-Dawley rats (3 months old,
~200 g) were obtained from the Laboratory Animal Center of Capital
Medical University (Beijing, China). Rats were individually housed
at 22°C with a relative humidity of 55%, acclimatized for ≥1 week,
under a 12 h light/dark cycle prior to experimentation. Water and
food were provided ad libitum. The rats were anesthetized
and heparinized. Subsequent to anaesthetizing the animals with
phenobarbital (150 mg/kg body weight), the rats were sacrificed by
excision of the heart. The rapidly excised hearts were mounted on a
Langendorff perfusion apparatus, and immediately perfused with
Ca2+-free buffer (120.4 mM NaCl, 14.7 mM KCl,
0.6 mM KH2PO4, 0.6 mM
Na2HPO4, 1.2 mM
MgSO4-7H2O, 10 mM Na-HEPES, 4.6 mM
NaHCO3, 30 mM taurine, 10 mM 2,3-butanedione monoxime
and 5.5 mM glucose, pH 7.1). Enzymatic digestion was followed by
adding 1.5 mg/ml collagenase II (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA) and CaCl2 (50 mM) to
the perfusion solution. Following digestion for 20 min, the
ventricles were quickly removed and digested in the perfusion
solution, with 10% calf serum (Chemicon International Inc.,
Temecula, CA, USA). and 12.5 mM CaCl2. The solution was
then filtered with 40 mm cell strainer (Thermo Fisher Scientific,
Inc.) to exclude undigested tissues. Following centrifugation of
the cardiomyocytes at 150 x g at 4°C for 10 min, the supernatant
was aspirated, and the cardiomyocytes were resuspended with M199
medium with selenium-insulin-transferrin (Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) in 6-well culture plates. The final cell
suspension, containing 70-80% viable myocytes, was used in
subsequent experiments.
Conditioned medium was prepared as follows:
Dulbecco's modified Eagle's medium (HyClone; GE Healthcare Life
Sciences, Logan, UT, USA; 270 mOsm/l, control medium) was modified
to reach 300, 400, 500 and 600 mOsm by adding NaCl to the medium
for 12 h at 37°C, which was confirmed with a Vapro vapor pressure
osmometer (model 5500, Wescor, Inc., Logan, UT, USA). The
time-dependent effects of osmotic stress on autophagy were
evaluated at various time point. Chloroquine (CQ; 5 µM) was used to
treat cells 1 h prior to osmotic stimulation in order to detect
autophagy flux.
Gene silencing with small interfering RNA
(siRNA)
The sequence for siRNAs were as following: Scrambled
siRNA, 5′-CUA CGC UGA GUA CUU CGA TT-3′; Beclin-1 siRNA, 5′-CTC AGG
AGA GGA GCC ATT T-3′; Atg5 siRNA, 5′-GGC CUU UCA UUC AGA AGC
UTT-3′; NFAT5 siRNA, 5′-CCA GTT CCT ACA ATG ATA A-3′. All siRNA
(200 nM) and transfection reagent (Lipofectamine® 3000;
Thermo Fisher Scientific, Inc.) were obtained from Applied
Biosystems; Thermo Fisher Scientific, Inc. Transfection with siRNAs
was conducted according to the manufacturer's protocol. After 48 h,
the efficiency of siRNA-silenced genes was confirmed by western
blot analysis as described subsequently, which was conducted to
detect the targeted proteins using specific antibodies.
Western blot analysis
Heart tissue were added into tubes with lysis (cat.
no. 1632086, Thermo Fisher Scientific, Inc.) for 1 h at 4°C and the
supernatants were collected following centrifugation (11,000 x g
for 10 min at 4°C). Subsequently, the protein concentration of
heart lysates was quantified using bovine serum albumin (cat. no.
A2058, Sigma-Aldrich; Merck KGaA) and measured via the absorption
of Coomassie Brilliant Blue in the spectrophotometer. Thereafter,
the samples were frozen at -20°C until use. Protein samples were
loaded onto SDS page (4-15%) for separation (10 µl per lane). The
separated proteins were then transferred to polyvinylidene
difluoride membranes (EMD Millipore, Billerica, MA, USA). The
membranes were blocked with 5% non-fat milk at room temperature for
1 h in TBS containing 0.1% Tween-20. Incubation with primary
antibodies [LC-3 (cat. no. 4108); Beclin-1 (cat. no. 3495); Atg5
(cat. no. 12994); SQSTM1/p62 (cat. no. 39749); ribosomal portion S6
(S6; cat. no. 2317S); phosphorylated p-S6 (cat. no. 4858S);
pro-caspase-3 (cat. no. 9664); caspase-3 (cat. no. 9662); and GAPDH
(cat. no. 2118); all from Cell Signaling Technology, Inc., Danvers,
MA, USA] and [REDD1 (cat. no. ABC245) and NFAT5 (cat. no.
SAB2107944); all from Sigma-Aldrich; Merck KGaA] was performed at
4°C overnight. All antibodies were used at a dilution of 1:1,000.
Secondary antibody incubation at 1:2,000 dilution was then
performed for 60 min at 37°C with peroxidase-conjugated Affinipure
goat anti-rabbit IgG (H+L, Cell Signaling Technology, Inc.; cat.
no. 7074) or anti-mouse IgG (H+L, Cell Signaling Technology, Inc.;
cat. no. 7076)-labeled secondary antibodies. Following washing 3
times, the membranes were subjected to enhanced chemiluminescent
detection with BeyoECL Plus (Beyotime Institute of Biotechnology,
Haimen, China). Subsequent to exposure to X-ray film, the membranes
were stripped in 5 ml stripping buffer (Bejing CoWin Biotech,
Beijing, China) for 15 min at room temperature and re-incubated
with an antibody against histone (Cell Signaling Technology, Inc.;
cat. no. 7631; 1:1,000) for normalization. Densitometric analysis
was performed using the Tanon 3500/3500R Tanon Gel Imaging System
(Tanon Science and Technology Co., Ltd., Shanghai, China).
Quantitative detection of cell
apoptosis
Apoptosis was analyzed using a fluorescein
isothiocyanate-conjugated Annexin V Apoptosis Detection kit (BD
Biosciences, San Jose, CA, USA) according to the manufacturer's
protocol. In brief, cells were harvested by trypsin treatment and
subsequently labeled with Annexin V and propidium iodide for 15 min
at 37°C. Labeled cells were analyzed with a FACScan flow cytometer
(BD Biosciences) and CellQuest software (version 5.1; BD
Biosciences).
Statistical analysis
Statistical analysis was performed using SPSS 18.0
(SPSS, Inc., Chicago, IL, USA). All results were presented as mean
± standard deviation. Comparisons of all pairs were performed using
a paired Student's t-test. Comparisons of means of multiple groups
were performed by one-way analysis of variance followed by Tukey's
post-hoc test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Hyperosmotic stress activates autophagy
in cardiomyocytes
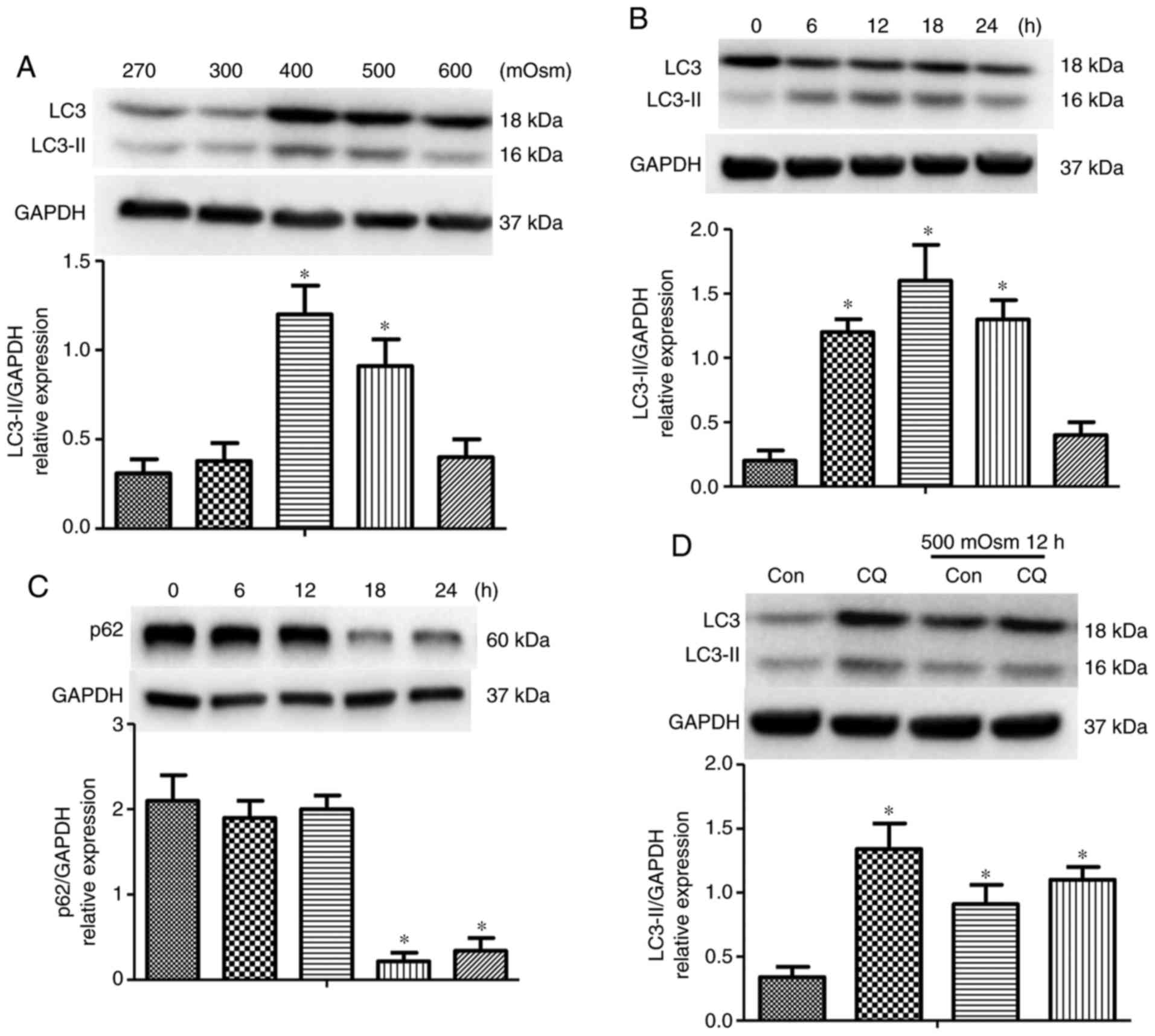
To investigate the effect of hyperosmotic stress on
autophagy, cardiomyocytes were treated with different
concentrations of NaCl as aforementioned for up to 12 h; the levels
of LC3 and LC3-II were then detected. The expression of LC3-II
protein began to accumulate at 400 mOsm and peaked at 500 mOsm
(Fig. 1A). In addition,
time-dependent changes in autophagy were determined in
cardiomyocytes with 500 mOsm stimulation. Elevation in autophagy
reached a peak at 12 h, followed by a decrease in LC3-II from 18 to
24 h subsequent to 500 mOsm stimulation (Fig. 1B). The level of SQSTM1/p62, an
autophagosome cargo protein that is concomitantly degraded with
increases in levels of autophagy (19), was then measured. The SQSTM1/p62
protein was degraded when cardiomyocytes were stimulated with 500
mOsm from 18 to 24 h, implying a continuous increase in autophagy
flux (Fig. 1C). Increases in
autophagosome level may be caused either by increased formation of
new autophagosomes, or by decreased autophagosome degradation
(20). To examine the effect of
hyperosmotic stress on autophagic flux, cells were treated with 5
µM lysosomal inhibitor CQ. Increased LC3 II levels,
indicated by the western blot analysis results, suggested that
treatment with CQ increased hyperosmotic stress-induced autophagy.
In addition, CQ application induced additional increases in
autophagosome formation in cardiomyocytes (Fig. 1D). Therefore, these results
indicated that instead of decreasing its degradation, hyperosmotic
stress induced new autophagosome formation in cardiomyocytes.
Atg5 is involved in hyperosmotic
stress-induced autophagy
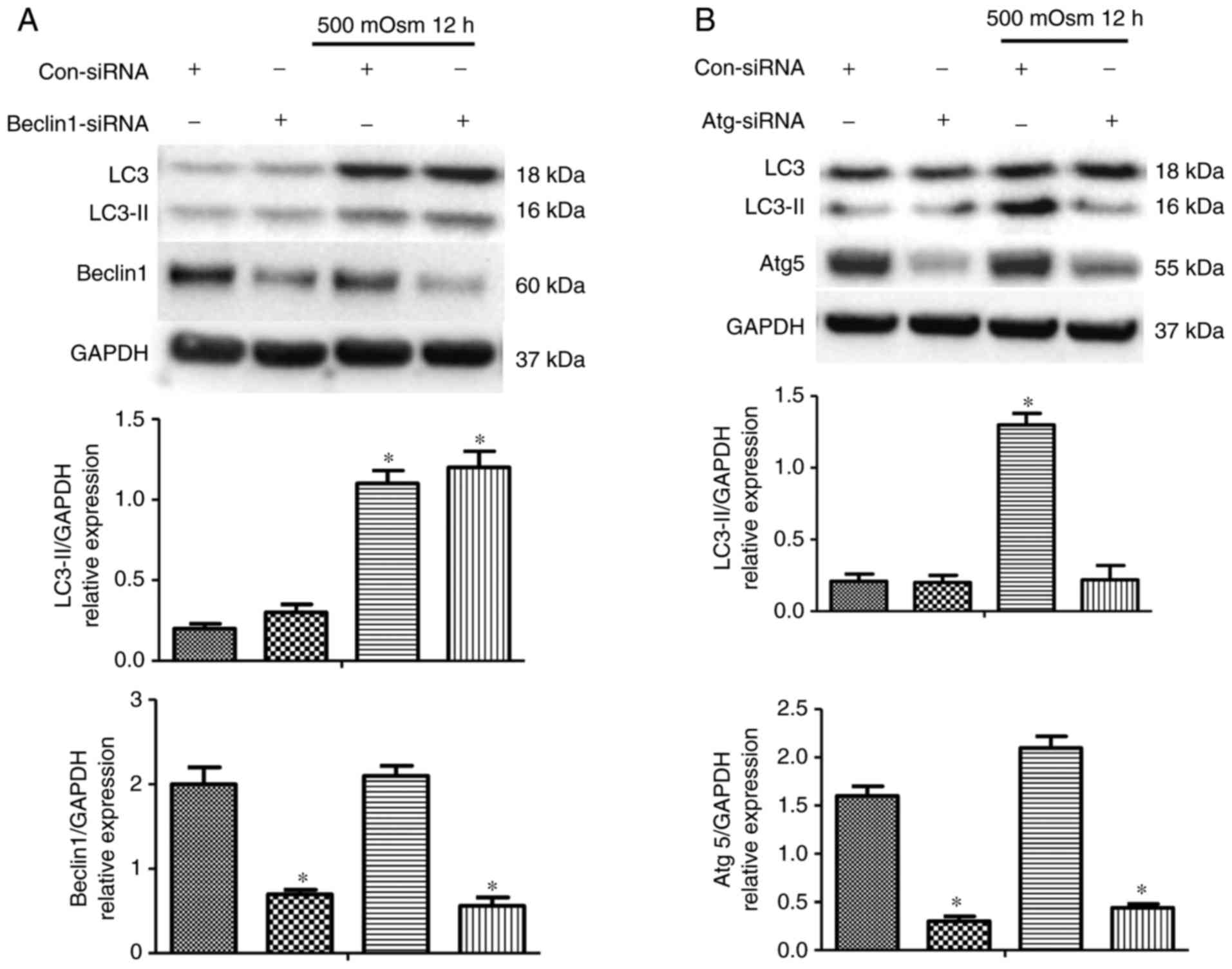
To additionally elucidate the downstream mediators
involved in hyperosmotic stress-induced autophagy, the classic
downstream components of the autophagy process were inhibited using
siRNAs. As Beclin-1 is responsible for the formation of new
autophagosomes, its involvement in hyperosmotic stress-induced
autophagy was examined through treatment with a Beclin-1-specific
siRNA, which significantly decreased Beclin-1 protein level.
Contrary to our hypothesis, the decrease in Beclin-1 did not change
the level of hyperosmotic stress-induced LC3-II accumulation
(Fig. 2A), suggesting that
Beclin-1 may not be involved in hyperosmotic stress-induced
autophagy. The role of Atg5, a known factor associated with the
processes of various stress-induced transcription factors and
protein kinases, was then examined. Using an siRNA specifically
against Atg5, it was identified that silencing Atg5 attenuated the
increase in LC3-II expression compared with Con-SiRNA group when
the cells were treated with 500 mOsm medium for 12 h (Fig. 2B). This result indicated that Atg5
was required for hyperosmotic stress-induced autophagy.
Hyperosmotic stress-induced autophagy is
regulated by NFAT5 in cardiomyocytes
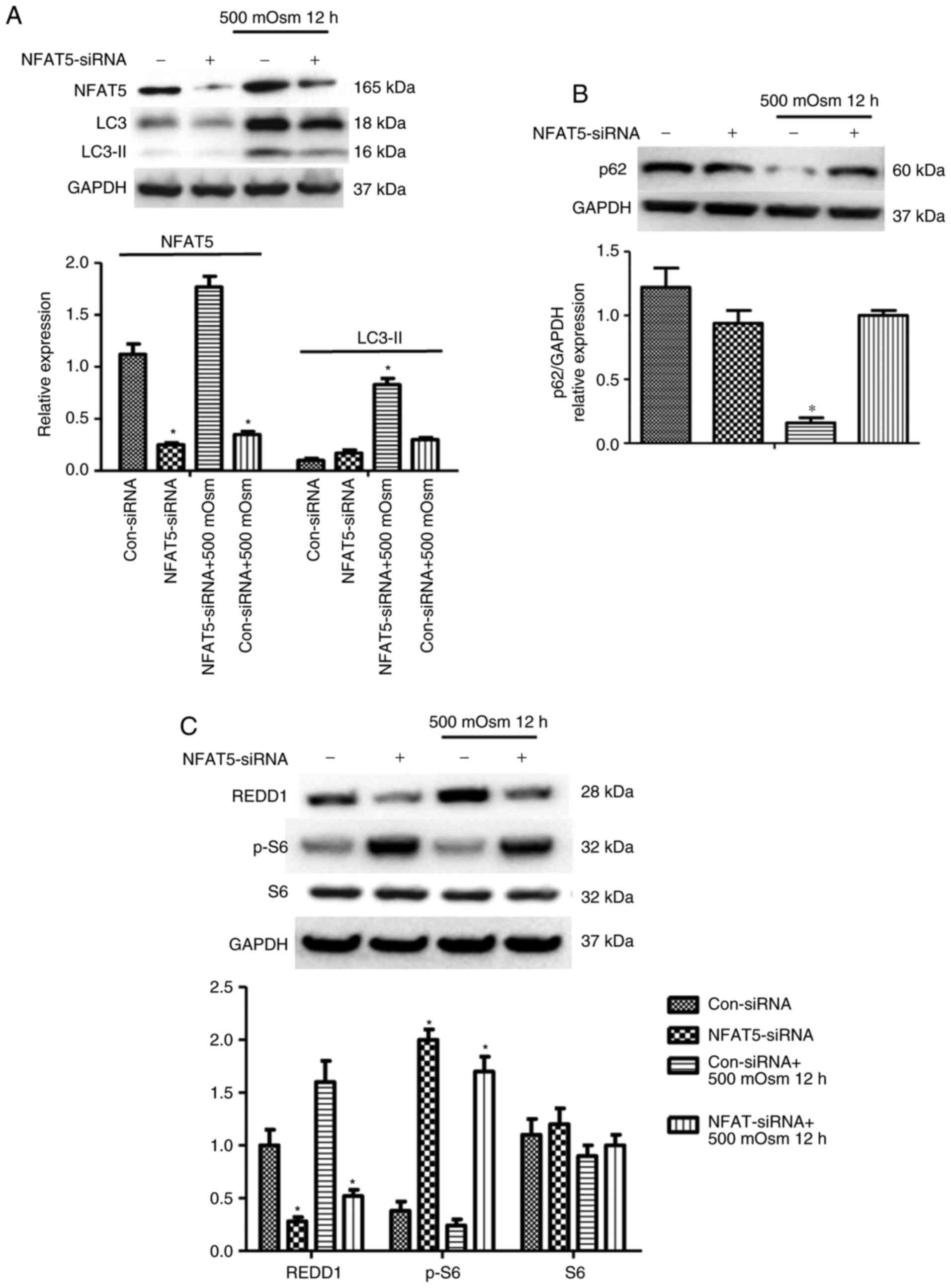
To examine the hypothesis that the interplay between
autophagy and NFAT5 pathways may affect cell survival under
hyperosmotic conditions, the role of NFAT5 in hyperosmotic
stress-induced autophagy and its interaction with
autophagy-associated proteins was additionally investigated. Cells
were treated with 500 mOsm following transfection with an siRNA
control or siRNA against NFAT5. The expression of NFAT5 was
significantly decreased following siRNA treatment, which was
accompanied with decreased hyperosmotic stress-induced LC3-II
accumulation as determined by western blot analysis (Fig. 3A). A similar result was observed
for SQSTM1/p62 (Fig. 3B);
SQSTM1/p62 accumulation was identified in NFAT5 siRNA-treated
cells, which may be an essential response to hyperosmotic stress.
Furthermore, the absence of NFAT5 prevented REDD1 expression, and
caused an increase in the phosphorylation of S6 (Fig. 3C), a downstream mediator of mTOR
whose phosphorylation typically decreases during autophagic
process. Therefore, these results implied that NFAT5 regulated the
hyperosmotic stress-induced mTOR pathway by activating REDD1.
NFAT5 mediates the balance of apoptosis
and autophagy in response to hyperosmotic stress
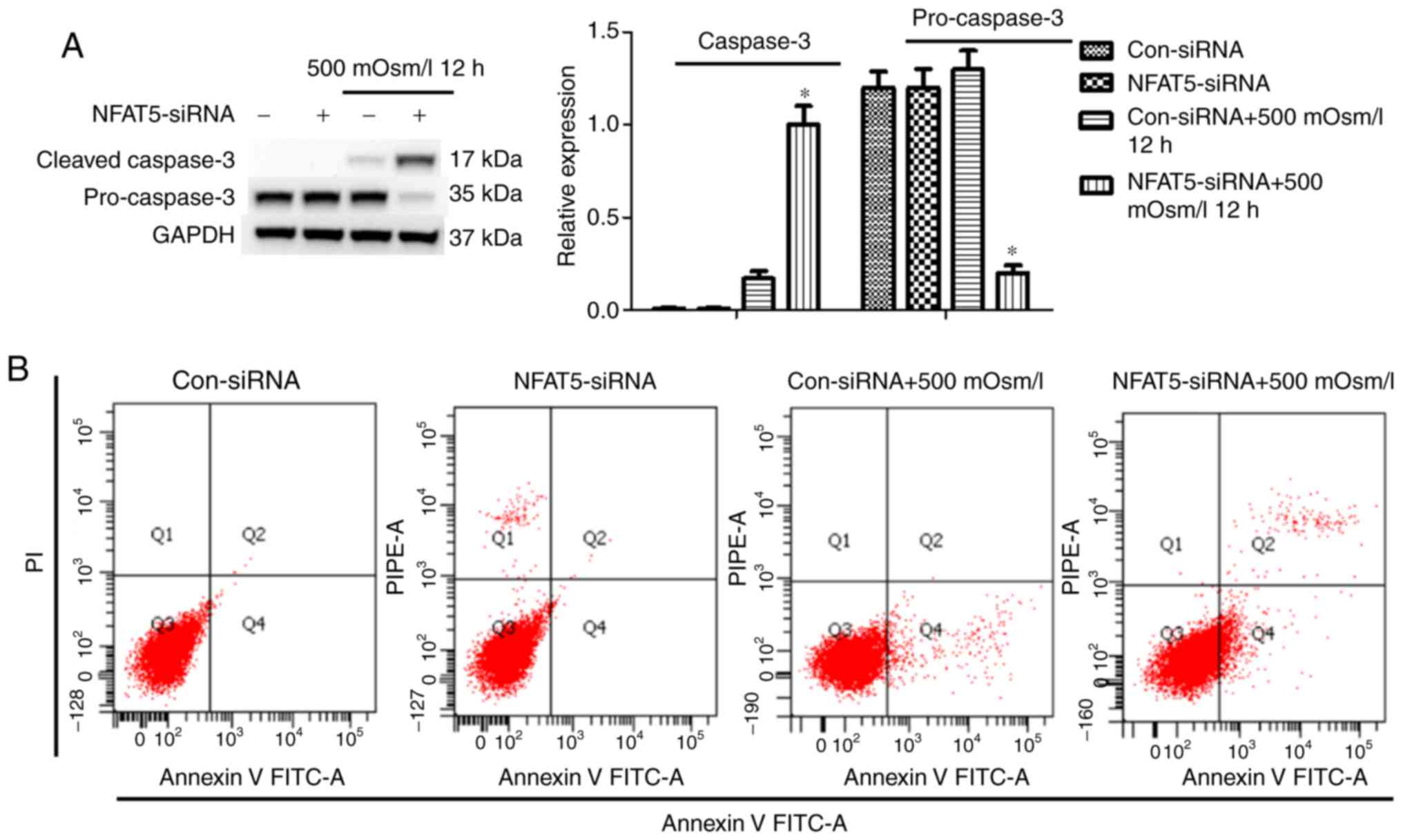
To determine the role of NFAT5 in regulating
autophagy and cell death, cell viability and cell death were
examined when treated with, or without, NFAT5 siRNA. Western blot
analysis indicated that NFAT5 downregulation exacerbated
hyperosmotic stress-induced cell death in cardiomyocytes, with
higher levels of cleaved caspase 3 and an increased number of
apoptotic cells. (Fig. 4A and B).
Overall, these observations supported the hypothesis that
attenuated NFAT5 levels aggravated hyperosmotic stress-induced cell
death.
Discussion
In the present study, it was revealed that
hyperosmotic stress increased NFAT5 expression, which triggered
autophagy by the activation of Atg5. Conversely, silencing of NFAT5
by siRNA activated mTOR signaling and decreased REDD1 expression,
and resulted in the impairment of cardiomyocyte viability.
Collectively, these results suggested that NFAT5 served an
essential role in protecting cardiomyocytes from hyperosmotic
stress-induced cell death. Various cellular processes may be
triggered by dynamic fluctuations in extracellular osmolality
(21). The present study used a
hyperrosmotic stress condition (500 mOsm/kg) to induce a cellular
self-protective response in cardiomyocytes, which may involve
diverse compensatory mechanisms, including the accumulation of
organic osmolytes and autophagy (21-23). While autophagy serves a protective
role against hypertonic stress in certain cell types (25-27), a balance between biosynthetic and
catabolic processes, which includes the degradation of entire
organelles by autophagy, is required to maintain cell
homeostasis.
In general, autophagy is regarded as a means of
promoting the cell survival response. However, morphological
features of autophagy have also been identified in dying cells.
Although it is well known that autophagy is involved in the process
of cell death and apoptosis, the role of autophagy in
cardiomyocytes under hyperosmotic stress has not been fully
elucidated. Previous studies have indicated that hyper-osmotic
stress induces cardiac apoptosis in a tumor protein p53
(p53)-independent manner, which involves a proportion of
translocated p53 into the mitochondria during this process
(28,29). The process of p53-mediated
cytochrome C release partially involves the disruption of the
B-cell lymphoma (Bcl-2)-associated X protein-Bcl-2 and/or the poly
[adenosine 5′-diphosphate-ribose] polymerase 9-myeloid leukemia
cell-1 complexes (30,31). Nevertheless, previous studies
clearly demonstrated that autophagic cell death is regulated by a
molecular mechanism distinct from that of apoptosis, though
Beclin-1 and Bcl-2 may cooperate with Atg5 to regulate autophagy
and apoptosis (32). The
effectiveness of Atg5-induced cell death may be overidden by a
higher level of Bcl-2, supporting the hypothesis that truncated
Atg5 targets mitochondria to release cytochrome C, and perhaps
other pro-apoptotic factors (33). Therefore, Atg5 may serve a
critical role as a checkpoint responsible for switching from
apoptosis to autophagic cell death.
It is well demonstrated that the NFAT5 activity is
regulated by extracellular osmolality. Hyperosmotic stress, through
the phosphorylation of signal molecules, including Fyn (34), p38 mitogen-activated protein
kinase (p38 MAPK) and protein kinase A (35-37), increases the NFAT5 translocation
into nuclei. NFAT5, as a ubiquitously expressed transcription
factor, regulates the expression of context-dependent gene products
in a number of different cell types. Additionally, NFAT5 serves a
role in regulating cell migration and proliferation-associated gene
expression (38-40). Despite the involvement of multiple
kinases in controlling of NFAT5 activity (38,41), neither p38 MAPK nor extracellular
signal-regulated kinase 1/2-dependent prototypic stretch-activated
kinase pathways are involved in promoting NFAT5 translocation into
the nuclei.
P38 MAPK is a key regulator in directing cellular
apoptosis, cell cycle arrest, growth inhibition and differentiation
(42). P38 MAPK serves positive
and negative roles to regulate autophagy: A previous study has
indicated that MAPK14/p38a, through activating survival autophagy,
confers irinotecan resistance to p53-deficient cells (43). Conversely, it has been
demonstrated that suppression of the p38 signaling cascade in tumor
necrosis factor-α-treated L929 cells promotes necroptotic and
autophagic cell death (44).
Although the molecular mechanisms between p38 regulation and
autophagy remain largely unknown, certain molecular targets have
been suggested in the up- or downregulation of autophagy. The
phosphorylation of Atg5 at threonine 75 through p38 MAPK may
inhibit starvation-induced autophagy (45). Therefore, the data concerning p38
involvement in the control of the autophagy-apoptosis balance may
provide a novel direction for future study.
In summary, the results of the present study
demonstrated that hyperosmotic stress enhanced NFAT5 translocation,
which in turn stimulated autophagy through Atg5 activity.
Conversely, the absence of NFAT5 prevented REDD1 expression and
upregulated hyperosmotic stress-induced apoptosis. These data
suggested that autophagy activation is a beneficial adaptive
response to attenuate hyperosmotic stress-induced cell death.
Therefore, increasing NFAT5 activity may represent a novel
therapeutic strategy for the protection of cardiomyocytes against
hyperosmotic stress-induced damage.
Acknowledgements
The authors would like to thank the other members of
the Fusion design and Study team for their technical assistance in
the present study.
Funding
The present study was supported by the Ministry of
Education Project of Humanities and Social Sciences (grant no.
16YJAZH034).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HZ and WC performed the majority of the experiments.
HZ and YL made substantial contributions to the design of the
present study. PZ, JW completed statistical analysis and data
presentation. YQ and HZ drafted the manuscript. All authors read
and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Ethics
Committee of Xuanwu Hospital Capital Medical University, and
conformed to the Guide for the Care and Use of Laboratory Animals
published by the US National Institutes of Health (18). All animals were treated in
accordance with guidelines established by the Capital Medical
University Institutional Animal Care and Use Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
LC3
|
microtubule-associated protein 1 light
chain 3
|
|
NFAT
|
nuclear factor of activated T-cell
|
|
Atg
|
autophagy-related; SQSTM1,
sequestosome-1
|
|
PE
|
phosphatidylethanolamine
|
|
REDD1
|
DNA damage response 1
|
References
|
1
|
Wright AR and Rees SA: Cardiac cell
volume: Crystal clear or murky waters? A comparison with other cell
types. Pharmacol Ther. 80:89–121. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Galvez A, Morales MP, Eltit JM, Ocaranza
P, Carrasco L, Campos X, Sapag-Hagar M, Díaz-Araya G and Lavandero
S: A rapid and strong apoptotic process is triggered by
hyperosmotic stress in cultured rat cardiac myocytes. Cell Tissue
Res. 304:279–285. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kaur J and Debnath J: Autophagy at the
crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol.
16:461–472. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Tanaka K and Matsuda N: Proteostasis and
neurodegeneration: The roles of proteasomal degradation and
autophagy. Biochim Biophys Acta. 1843:197–204. 2014. View Article : Google Scholar
|
|
5
|
Mazure NM and Pouysségur J:
Hypoxia-induced autophagy: Cell death or cell survival? Curr Opin
Cell Biol. 22:177–180. 2010. View Article : Google Scholar
|
|
6
|
Galluzzi L, Pietrocola F, Levine B and
Kroemer G: Metabolic control of autophagy. Cell. 159:1263–1276.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Russell RC, Yuan HX and Guan KL: Autophagy
regulation by nutrient signaling. Cell Res. 24:42–57. 2014.
View Article : Google Scholar :
|
|
8
|
Levine B and Deretic V: Unveiling the
roles of autophagy in innate and adaptive immunity. Nat Rev
Immunol. 7:767–777. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Klionsky DJ, Abdalla FC, Abeliovich H,
Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M,
Agostinis P, Aguirre-Ghiso JA, et al: Guidelines for the use and
interpretation of assays for monitoring autophagy. Autophagy.
8:445–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen C, Deng M, Sun Q, Loughran P, Billiar
TR and Scott MJ: Lipopolysaccharide stimulates p62-dependent
autophagy-like aggregate clearance in hepatocytes. Biomed Res Int.
2014:2673502014.PubMed/NCBI
|
|
12
|
Nishida Y, Arakawa S, Fujitani K,
Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y
and Shimizu S: Discovery of Atg5/Atg7-independent alternative
macroau-tophagy. Nature. 461:6542009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Johnson ZI, Shapiro IM and Risbud MV:
Extracellular osmo-larity regulates matrix homeostasis in the
intervertebral disc and articular cartilage: Evolving role of
TonEBP. Matrix Biol. 40:10–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Woo SK, Lee S and Kwon MH: TonEBP
transcriptional activator in the cellular response to increased
osmolality. Pflugers Arch. 444:579–585. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Woo SK and Kwon HM: Adaptation of kidney
medulla to hypertonicity: Role of the transcription factor TonEBP.
Int Rev Cytol. 215:189–202. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ortells MC, Morancho B, Drews-Elger K,
Viollet B, Laderoute KR, López-Rodríguez C and Aramburu J:
Transcriptional regulation of gene expression during osmotic stress
responses by the mammalian target of rapamycin. Nucleic Acids Res.
40:4368–4384. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhou Y, Wang Q, Weiss HL and Evers BM:
Nuclear factor of activated T-cells 5 increases intestinal goblet
cell differentiation through an mTOR/Notch signaling pathway. Mol
Biol Cell. 25:2882–2890. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
National Research Council: Guide for the
care and use of laboratory animals. 8th edition. National Academies
Press; Washington, DC: 2011
|
|
19
|
Pursiheimo JP, Rantanen K, Heikkinen PT,
Johansen T and Jaakkola PM: Hypoxia-activated autophagy accelerates
degradation of SQSTM1/p62. Oncogene. 28:334–344. 2009. View Article : Google Scholar
|
|
20
|
Beth L and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar
|
|
21
|
Burg MB, Ferraris JD and Dmitrieva NI:
Cellular response to hyperosmotic stresses. Physiol Rev.
87:1441–1474. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hoover HE, Thuerauf DJ, Martindale JJ and
Glembotski CC: Alpha B-crystallin gene induction and
phosphorylation by MKK6-activated p38 A potential role for alpha
B-crystallin as a target of the p38 branch of the cardiac stress
response. J Biol Chem. 275:825–833. 2000. View Article : Google Scholar
|
|
23
|
Takatani T, Takahashi K, Uozumi Y, Matsuda
T, Ito T, Schaffer SW, Fujio Y and Azuma J: Taurine prevents the
ischemia-induced apoptosis in cultured neonatal rat cardio-myocytes
through Akt/caspase-9 pathway. Biochem Biophys Res Commun.
316:484–489. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Takatani T, Takahashi K, Uozumi Y, Shikata
E, Yamamoto Y, Ito T, Matsuda T, Schaffer SW, Fujio Y and Azuma J:
Taurine inhibits apoptosis by preventing formation of the
Apaf-1/caspase-9 apoptosome. Am J Physiol Cell Physiol.
287:C949–C953. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nunes P, Ernandez T, Roth I, Qiao X,
Strebel D, Bouley R, Charollais A, Ramadori P, Foti M, Meda P, et
al: Hypertonic stress promotes autophagy and microtubule-dependent
autopha-gosomal clusters. Autophagy. 9:550–567. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Biswas K, Khongsngi JL, Häussinger D and
Saha N: Influence of cell volume changes on autophagic proteolysis
in the perfused liver of air-breathing walking catfish (Clarias
batrachus). J Exp Zool A Ecol Genet Physiol. 311:115–124. 2009.
View Article : Google Scholar
|
|
27
|
Liu Y, Xiong Y and Bassham DC: Autophagy
is required for tolerance of drought and salt stress in plants.
Autophagy. 5:954–963. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Marchenko ND, Zaika A and Moll UM: Death
signal-induced localization of p53 protein to mitochondria a
potential role in apoptotic signaling. J Biol Chem.
275:16202–16212. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Mihara M, Erster S, Zaika A, Petrenko O,
Chittenden T, Pancoska P and Moll UM: p53 has a direct apoptogenic
role at the mitochondria. Mol Cell. 11:577–590. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chipuk JE, Kuwana T, Bouchier-Hayes L,
Droin NM, Newmeyer DD, Schuler M and Green DR: Direct activation of
Bax by p53 mediates mitochondrial membrane permeabilization and
apoptosis. Science. 303:1010–1014. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Leu JJ, Dumont P, Hafey M, Murphy ME and
George DL: Mitochondrial p53 activates Bak and causes disruption of
a Bak-Mcl1 complex. Nat Cell Biol. 6:443–450. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Zhou F, Yang Y and Xing D: Bcl-2 and
Bcl-xL play important roles in the crosstalk between autophagy and
apoptosis. FEBS J. 278:403–413. 2011. View Article : Google Scholar
|
|
33
|
Strasser A: The role of BH3-only proteins
in the immune system. Nat Rev Immunol. 5:189–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Olson JM and Hallahan AR: p38 MAP kinase:
A convergence point in cancer therapy. Trends Mol Med. 10:125–129.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Irarrazabal CE, Liu JC, Burg MB and
Ferraris JD: ATM, a DNA damage-inducible kinase, contributes to
activation by high NaCl of the transcription factor TonEBP/OREBP.
Proc Natl Acad Sci USA. 101:8809–8814. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ko BC, Lam AK, Kapus A, Fan L, Chung SK
and Chung SS: Fyn and p38 signaling are both required for maximal
hypertonic activation of the osmotic response element-binding
protein/tonicity-responsive enhancer-binding protein
(OREBP/TonEBP). J Biol Chem. 277:46085–46092. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ferraris JD, Williams CK, Persaud P, Zhang
Z, Chen Y and Burg MB: Activity of the TonEBP/OREBP transactivation
domain varies directly with extracellular NaCl concentration. Proc
Natl Acad Sci USA. 99:739–744. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Jauliac S, López-Rodriguez C, Shaw LM,
Brown LF, Rao A and Toker A: The role of NFAT transcription factors
in integrin-mediated carcinoma invasion. Nat Cell Biol. 4:540–544.
2002. View
Article : Google Scholar : PubMed/NCBI
|
|
39
|
O'Connor RS, Mills ST, Jones KA, Ho SN and
Pavlath GK: A combinatorial role for NFAT5 in both myoblast
migration and differentiation during skeletal muscle myogenesis. J
Cell Sci. 120:149–159. 2007. View Article : Google Scholar
|
|
40
|
Go WY, Liu X, Roti MA, Liu F and Ho SN:
NFAT5/TonEBP mutant mice define osmotic stress as a critical
feature of the lymphoid microenvironment. Proc Natl Acad Sci USA.
101:10673–10678. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Halterman JA, Kwon HM and Wamhoff BR:
Tonicity-independent regulation of the osmosensitive transcription
factor TonEBP (NFAT5). Am J Physiol Cell Physiol. 302:C1–C8. 2012.
View Article : Google Scholar :
|
|
42
|
Cuadrado A and Nebreda AR: Mechanisms and
functions of p38 MAPK signalling. Biochem J. 429:403–417. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Paillas S, Causse A, Marzi L, de Medina P,
Poirot M, Denis V, Vezzio-Vie N, Espert L, Arzouk H, Coquelle A, et
al: MAPK14/p38α confers irinotecan resistance to TP53-defective
cells by inducing survival autophagy. Autophagy. 8:1098–1112. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Ye YC, Yu L, Wang HJ, Tashiro S, Onodera S
and Ikejima T: TNFα-induced necroptosis and autophagy via
supression of the p38-NF-κB survival pathway in L929 cells. J
Pharmacol Sci. 117:160–169. 2011. View Article : Google Scholar
|
|
45
|
Keil E, Höcker R, Schuster M, Essmann F,
Ueffing N, Hoffman B, Liebermann DA, Pfeffer K, Schulze-Osthoff K
and Schmitz I: Phosphorylation of Atg5 by the Gadd45β-MEKK4-p38
pathway inhibits autophagy. Cell death and differentiation.
20:3212013. View Article : Google Scholar
|