Introduction
Severe acute pancreatitis (SAP) is one of the most
common acute abdominal diseases. It is caused by pancreatic local
inflammation, necrosis and infection, and is accompanied by a
systemic inflammatory response and multiple organ dysfunction.
Although important progress has been made in the comprehensive
treatment of SAP, the mortality rate remains as high as 17%
(1,2). Thus, the study of the pathogenesis
of acute pancreatitis (AP) is helpful in its clinical
treatment.
Oxidative stress is the cause and consequence of
numerous diseases. Disequilibrium between the formation of reactive
oxygen species (ROS) and antioxidant defense systems can cause
disease directly or indirectly through the signal transduction
pathway activated by excessive ROS in organs (3). An increasing number of studies have
identified that ROS-induced oxidative stress serves an unfavorable
role not only in the pathogenesis of SAP, but also in the damage of
other organs, such as the heart, liver, lung, kidney and digestive
tract (4,5). ROS and active nitrogen produced by
oxidative stress can cause inflammation and microcirculation
disorders, and activate the pathways of cell necrosis or apoptosis,
resulting in pancreatic and other organ dysfunction (6). Pro-inflammatory factors and
oxidative stress serve a synergistic role in triggering signal
transduction in the inflammatory response of AP. Activated
mitogen-activated protein kinase and nuclear factor (NF)-κB
signaling can then lead to a cascade of amplification of
inflammation (7,8). It has been reported that ROS
produced by pancreatic follicles increased significantly in an
early SAP rat model, while the pancreatic glutathione levels were
simultaneously decreased (9).
After 6 h of pancreatic obstruction, lipid peroxidation was
significantly enhanced (10);
however, monocytes produced TNF-α until 12 h after AP, which
promoted the accumulation and activation of white blood cells in
the inflammatory site (10,11). Thus, the interaction of
inflammatory factors and oxidative stress in SAP results in
detrimental effects. Studying the mechanism of oxidative stress in
pancreatitis is, therefore, important for eliminating ROS and
treating pancreatitis.
It has been reported that the levels of interleukin
(IL)-1β, IL-6 and TNF-α in the serum of SAP patients were
significantly increased, indicating that a systemic inflammatory
response serves a critical role in the progression of SAP (12,13). The pathogenesis of SAP may be
associated with excessive activation of the NF-κB signaling
pathway, leading to a large number of inflammatory mediators
(14,15). Once the cytokines are produced,
they not only activate themselves, but also promote the production
of other cytokines, causing interlocking and amplification effects
that impair the structure and function of the pancreas (16). The inflammatory process is
important in the development of pancreatitis (17). In a healthy pancreas, the NF-κB
signaling is inactive. However, in the early stages of
pancreatitis, NF-κB signaling is activated and enhances the
inflammation process through activation of anti-apoptotic and
inflammatory genes (18). Long
periods of activated signaling in pancreatic cells lead to
pancreatic damage and fibrosis (19).
SAP is essentially a systemic inflammatory response
syndrome, and the endotoxin/Toll-like receptor 4 (TLR4)/NF-κB
signaling pathway may be important in mediating the AP inflammatory
response (20). An experimental
study demonstrated that the expression of TLR4 in SAP rats
increased in the pathogenesis of this disease, which indicates that
the expression of TLR4 directly reflects the severity of SAP
(21). TLR4 is also involved in
mediating pancreatic cell apoptosis in mice with AP (22). Taken together, TLR4 serves a vital
role in the pathogenesis of SAP, and its correlation with NF-κB
signaling is also important for studying the mechanism of SAP.
Thus, to fully understand the pathogenesis of SAP and identify a
novel target for SAP treatment, it is necessary to examine the
function of TLR4/NF-κB in SAP.
The present study focused on the effect of TLR4 and
inflammatory signaling activation on the generation of oxidative
stress in pancreatic cells. The results revealed that the
activation of inflammatory signaling increased the expression level
of TLR4. In order to prove that the increased ROS levels in
pancreatic cells were caused by TLR4 overexpression, pancreatic
acinar cells were then treated with TLR4 antagonist, and the effect
caused by inflammatory stimulation was partially reversed. The
current study indicates that the lipopolysaccharide (LPS)-induced
TLR4/NF-κB pathway is critically involved in the initiation of
inflammation, oxidative stress and decreased pancreatic cell
viability.
Materials and methods
Chemicals and materials
Fetal bovine serum (FBS) was obtained from Gibco
(Thermo Fisher Scientific, Inc., Waltham, USA). The Cell Counting
Kit-8 (CCK-8) assay kit was obtained from KeyGen Biotech Co., Ltd.
(Nanjing KeyGen Biotech Co., Ltd., Nanjing, China). Antibodies
against β-actin (sc-58673), p65 (sc-71675), phosphorylated (p)-p65
(sc-136548), B-cell lymphoma 2 (Bcl2; sc-509), Bcl2-associated X
protein (Bax; sc-20067), PMAIP1 (sc-515840) and TLR4 (sc-293072)
were purchased from Santa Cruz Biotechnology, Inc. (Dallas, TX,
USA). LPS was obtained from Sigma-Aldrich (Merck KGaA, Darmstadt,
Germany).
Primary culture of pancreatic acinar
cells and treatment
Pancreatic cells were isolated from 100 healthy
adult male (to avoid the effects of estrogen) 4-6-weeks-old
C57BL/6J mice (25-30 g, Beijing Vital River Laboratory Animal
Technology Co., Ltd., Beijing, China), according to the procedure
described in previously published study (22). The animal experiments of the
present study were approved by the Ethics Committee of Xi'an
Jiaotong University (Xi'an, China). Briefly, the pancreas was
immediately removed from the sacrificed mouse and incubated in
buffer solution (containing 130 mM NaCl, 4.7 mM KCl, 1.3 mM
CaCl2, 1.2 mM KH2PO4 and 0.2%
bovine serum albumin (Thermo Scientific Fisher, Inc.) at 37°C for
10 min. Then the cell suspension was centrifuged at 30 x g for 5
min at 4°C. Next, the acinar cell pellets were resuspended in HEPES
buffer without collagenase and centrifuged at 30 x g for 5 min at
4°C, following which the supernatant was removed. Primary
pancreatic cells were maintained in RPMI-1640 medium (Gibco; Thermo
Fisher Scientific, Inc.) with 10% FBS, antibiotics (100 U/ml
penicillin and 100 µg/ml streptomycin), 2 mM L-glutamine and
25 µg/ml gentamicin at 37°C in a humidified atmosphere with
5% CO2.
To investigate the potential protective effect of
TAK-242, an inhibitor of TLR4, primary pancreatic acinar cells were
treated with TAK-242 (1 µM) following LPS treatment (100
ng/ml) for 24 h at the temperature of 37°C. The concentration of
TAK-242 was selected according to previous published data.
Transfection
For transfection, pancreatic cells (80% confluence)
were seeded into 6-well culture plates; cells were transfected with
3 µg of the TLR4 overexpression plasmid and pCDNA3.0
(Invitrogen; Thermo Fisher Scientific, Inc.) as the control using
Lipofectamine® 3000 transfection reagent (Invitrogen;
Thermo Fisher Scientific, Inc.). The sequence of TLR4 cloned into
the pCDNA3.0 plasmid was obtained by RT-qPCR; the primers employed
for plasmid construction were: Forward, 5'-GAC GAG CTC ATG ATG CCT
CCC TGG CTC CT-3' and reverse, 5'-TAC CCG TCA GGT CCA AGT TGC CGT
TTC T-3'. After 2 days following transfection, cells were harvested
for RNA and protein isolation.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
After pancreatic cells were treated with LPS for 2 h
and transfected with plasmids, total RNA was extracted using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.). All the
procedures were conducted according to the manufacturer's protocol.
Briefly, 1 µg total RNA was reverse transcribed using an
PrimeScript™ RT reagent kit (Takara Biotechnology Co., Ltd.,
Dalian, China). The RT reaction program was as follows: 25°C for 10
min, 37°C for 120 min and 85°C for 5 min. qPCR was then conducted
using the SYBR-Green for Real-Time PCR kit and the ROCHE Light 480
detection system (Roche Diagnostics, Basel, Switzerland), and the
amplification conditions were 95°C for 10 min, followed by 50
cycles of denaturation at 95°C for 15 sec, and annealing at 62°C
for 1 min. The primers for qPCR were: TLR4 forward, 5′-AGC CAT TGC
TGC CAA CAT CA-3′ and reverse, 5′-GCC AGA GCT ACT CAG AAA C-3′.
Bcl2 forward, 5′-GGT GGG GTC ATG TGT GTG G-3′ and reverse, 5′-CGG
TTC AGG TAC TCA GTC ATC C-3′; Bax forward, 5′-CCC GAG AGG TCT TTT
TCC GAG and reverse, 5′-CCA GCC CAT GAT GGT TCT GAT-3′; PMAIP1
forward, 5′-ACC AAG CCG GAT TTG CGA TT-3′ and reverse, 5′-ACT TGC
ACT TGT TCC TCG TGG-3′ and β-actin forward, 5′-CAT GTA CGT TGC TAT
CCA GGC-3′ and reverse, 5′-CTC CTT AAT GTC ACG CAC GAT-3′. All
reactions were repeated for three times, and the relative mRNA
expression levels of target genes were normalized to β-actin.
Results were expressed as fold differences relative to the level of
β-actin using the 2-ΔΔCq method (23).
Western blot analysis
Following treatment with LPS and transfection with
plasmids, pancreatic cells were harvested for protein isolation.
Briefly, cell pellets were lysed in 200 µl of ice-cold lysis
buffer (pH 7.4; 50 mmol/l HEPES, 5 mmol/l EDTA, 100 mmol/l NaCl, 1%
Triton X-100, protease inhibitor cocktail). Protein samples (20
µg) were separated by 10% SDS-PAGE and then transferred to
polyvinylidene difluoride (PVDF) membranes. The membranes were
blocked with 5% defatted milk in Tris-buffered saline/Tween-20
(TBST) at room temperature for 1 h and then incubated with primary
antibodies overnight at 4°C. Antibody against TLR4 (1:200), p65
(1:400), p-p65 (1:200), Bcl2 (1:200), Bax (1:500) and PMAIP1
(1:100) were used. On the following day, the PVDF membranes were
washed with TBST buffer and then incubated with a mouse
peroxidase-conjugated secondary antibody (1:2,000; sc-2005, Santa
Cruz Biotechnology, Inc.) with agitation for 1 h. Finally, an
enhanced chemiluminescence solution (Thermo Scientific Fisher,
Inc.) was prepared in a dark room, and the exposure time was
determined according to the fluorescence intensity. The results
were quantified according to the intensity of the bands by GraphPad
6.0c Software, Inc. (La Jolla, CA, USA).
MTT assay
LPS was added at the concentration of 100 ng/ml
following cell attachment to the plates for 24, 48 and 72 h at the
temperature of 37°C. Then after overnight culture, baseline values
were obtained by an MTT assay (Thermo Fisher Scientific, Inc.), a
colorimetric assay to determine cell viability by measuring the
formazan reduced from MTT. After 20 min of incubation with MTT at
the temperature of 37°C, formazan was diluted with DMSO and tested
the optical density at 540 nm. MTT assay was performed at different
time points (at 24, 48 and 72 h, respectively). Experiments were
conducted in triplicate.
Cell proliferation
Pancreatic cells were suspended in complete
RPMI-1640 medium, the cell concentration was adjusted to
5x106 cells/ml, and cells were grown in 96-well cell
culture plates with 100 µl in each well. For cell
proliferation detection, cells were harvested in medium at 24 h
after transfection. Cell proliferation was detected by a CCK-8
assay kit according to the manufacturer's protocol for the duration
of 1 h. Cell viability was calculated according to the absorbance
detected at 450 nm by a microplate reader.
Cell apoptosis assay
A total of 5x105 pancreatic cells were
seeded in a 6-well cell culture dish and complete RPMI-1640 medium
was added. Following the addition of medium containing 100 ng/ml
LPS, the cells were cultured for 24 h and collected by digestion
with 0.25% trypsin. Pancreatic cells were washed twice with
pre-cooled PBS and re-suspended in 1 X binding buffer to a final
concentration of 106 cells/ml. Next, 100 µl of
cell suspension was placed in a flow tube, and 5 µl
Annexin-V-FITC (A211-01/02, Vazyme, Piscataway, NJ, USA) and 5
µl propidium iodide (A211-01/02, Vazyme) were added for a
15-min incubation in the dark. At 1 h after addition of 400
µl binding buffer to each well, the fluorescence intensity
was measured by flow cytometry.
ROS measurement
Pancreatic cells were seeded in a 6-well cell
culture dish at 60% confluence, and RPMI-1640 complete medium was
added. Subsequent to treatment with medium containing different
concentrations of LPS, cells were cultured for 24 h and collected
by digestion with 0.25% trypsin. Next, cells were washed with
pre-cooled PBS, lysed with chemical buffer and the protein
concentration was determined by the Bradford method. ROS
concentration was determined based on MDA and GPx levels (24). Following the manufacturer's
protocol of the malondialdehyde (MDA) (cat. no. A003-1) and
glutathione peroxidase (GPx) (cat. no. A005) determination kits
(Nanjing Jiancheng Bioengineering Institute, Nanjing, China), the
concentration of MDA and the activity of GPx was detected with a
spectrophotometer. The wavelength for MDA detection was 532 and 412
nm for Gpx.
Statistical analysis
Each experiment was repeated at least in triplicate.
The results are presented as the mean value ± standard deviation.
Statistical analysis for comparison of two groups was conducted
using Student's t-test, while analysis of variance was used for
multiple group comparisons. P<0.05 was considered to indicate a
statistically significant difference.
Results
LPS induces high-level oxidative stress
in pancreatic cells
To investigate the biological effects of
inflammatory stimuli on primary pancreatic cells, the cells were
treated with 100 ng/ml LPS to activate inflammatory signaling. The
MDA level was measured, which is an indicator of oxidative damage
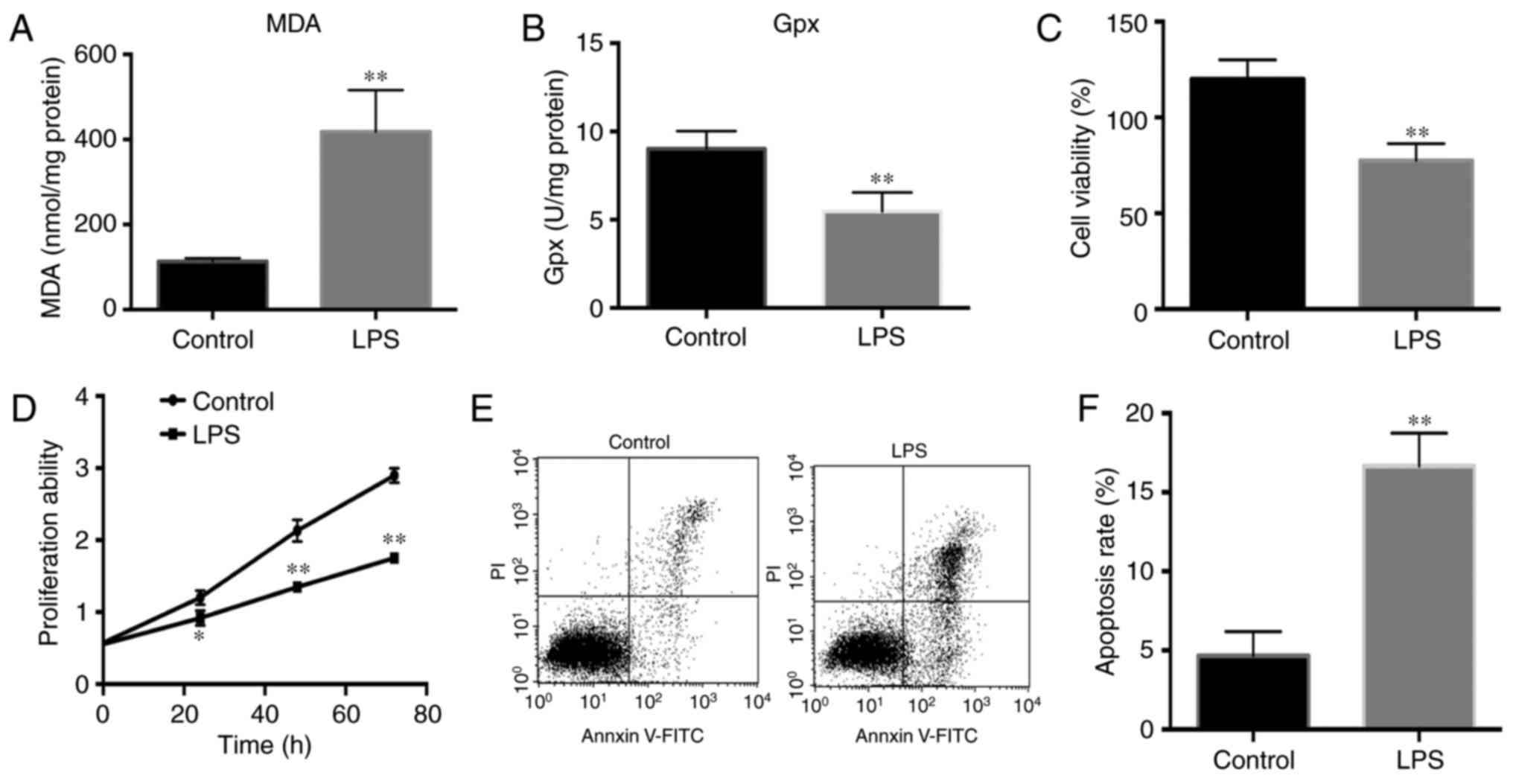
and reflects the ROS level in cells. According to the results shown
in Fig. 1A, the MDA level was
significantly increased in LPS-treated pancreatic cells, as
compared with the control cells. In addition, the GPx level was
markedly decreased in the LPS-treated group (Fig. 1B). Thus, these results indicate
that LPS increased ROS levels and thus induced oxidative stress in
pancreatic cells.
Pancreatic cell viability is decreased by
LPS treatment
To identify whether the increased ROS levels
influenced the cell viability, CCK-8 and MTT assays were applied to
detect the cell viability. Subsequent to the addition of 100 ng/ml
LPS, pancreatic cells were harvested for the detection of cell
viability. The results demonstrated that LPS significantly
decreased the pancreatic cell viability and proliferation ability
compared with the control cells (Fig.
1C and D). Furthermore, the percentage of cell apoptosis was
detected by flow cytometry. The results demonstrated that LPS
significantly increased the percentage of apoptotic cells compared
with that in the control cells (Fig.
1E and F).
Bcl2, Bax and PMAIP1 levels in
LPS-stimulated pancreatic cells
To identify how oxidative stress induces apoptosis
in primary pancreatic cells the expression levels of Bcl2, Bax and
PMAIP1 were detected. These three genes reflect the apoptosis rate,
while they also regulate cell apoptosis. Pancreatic cells were
treated with 100 ng/ml LPS for 2 h, and then the mRNA expression
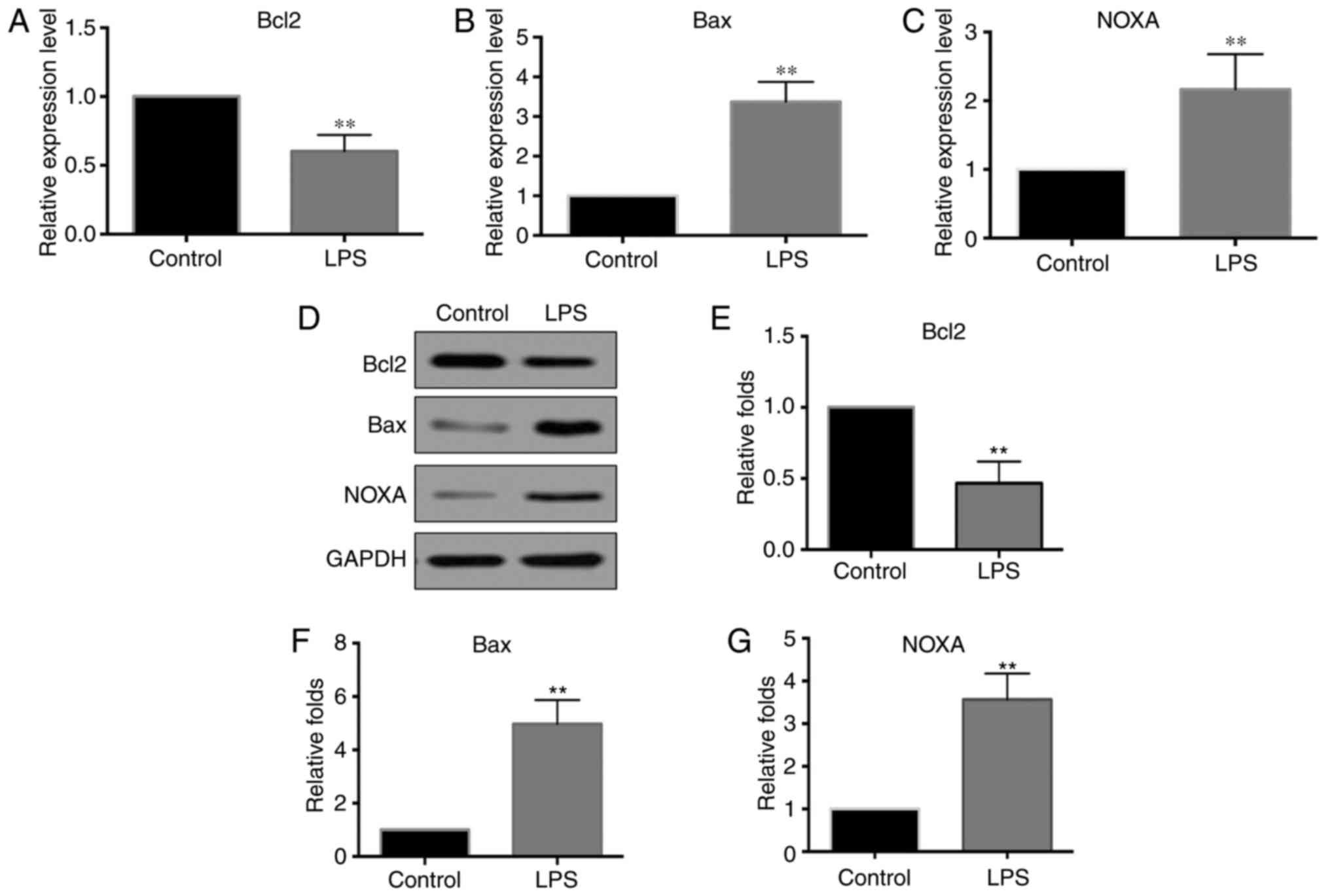
levels of Bcl2, Bax and PMAIP1 were detected by RT-qPCR. As shown
in Fig. 2A-C, the mRNA expression
level of Bcl2 was significantly decreased in LPS-treated pancreatic
cells compared with that in the controls, while the expression
levels of Bax and PMAIP1 were significantly increased in
LPS-treated pancreatic cells. The protein levels of Bcl2, Bax and
PMAIP1 were also detected at 24 h after LPS stimulation, and
changes in these levels were consistent with those observed for the
mRNA levels (Fig. 2D-G).
LPS induces TLR4 expression and
activation of NF-κB signaling in pancreatic cells
LPS is often used to activate inflammatory
signaling. In the present study, the activation of inflammatory
signaling increased the ROS level and induced cell death, Next, the
study verified the activity of inflammatory signaling by detecting
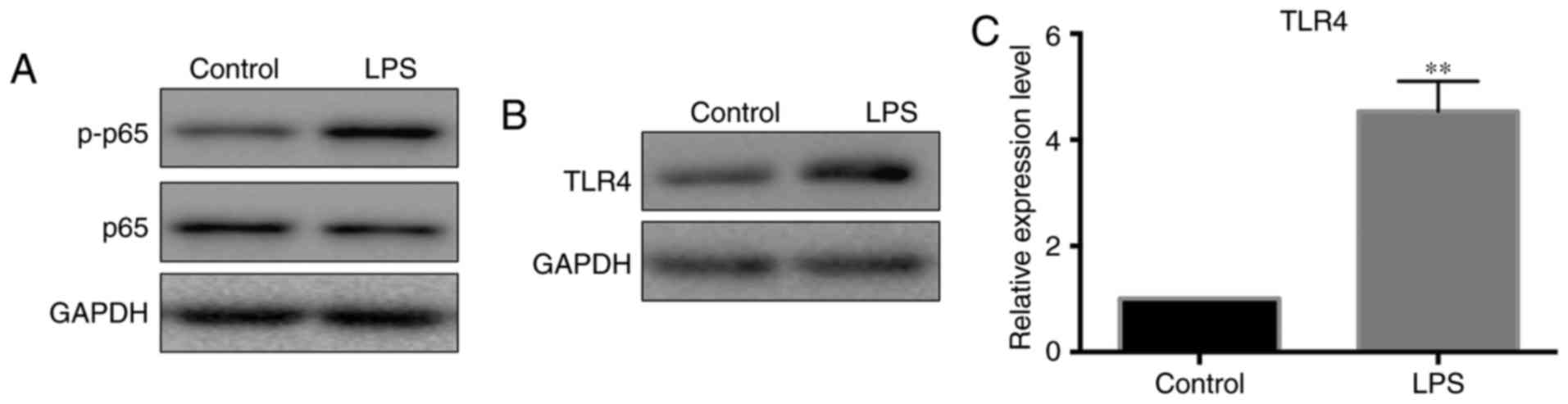
the protein levels of p-p65. According to the result shown in
Fig. 3A, p-p65 levels were
notably increased after pancreatic cells were treated with LPS.
Since TLR4 is the upstream molecule that activates NF-κB signaling,
the current study next detected the expression levels of TLR4 in
LPS-treated pancreatic cells. The results revealed that both the
mRNA and protein levels of TLR4 were increased following LPS
exposure (Fig. 3B and C).
TLR4 regulates ROS levels and pancreatic
cell viability
TLR4 serves a critical role in the pathogenesis of
pancreatitis and activates inflammatory signaling in via the
myeloid differentiation primary response 88-dependent and
-independent pathways (25). To
elaborate the mechanism of increased TLR4 in pancreatic cells in
response to LPS stimulation, a TLR4 overexpression plasmid was
constructed and used for cell transfection. Subsequent to
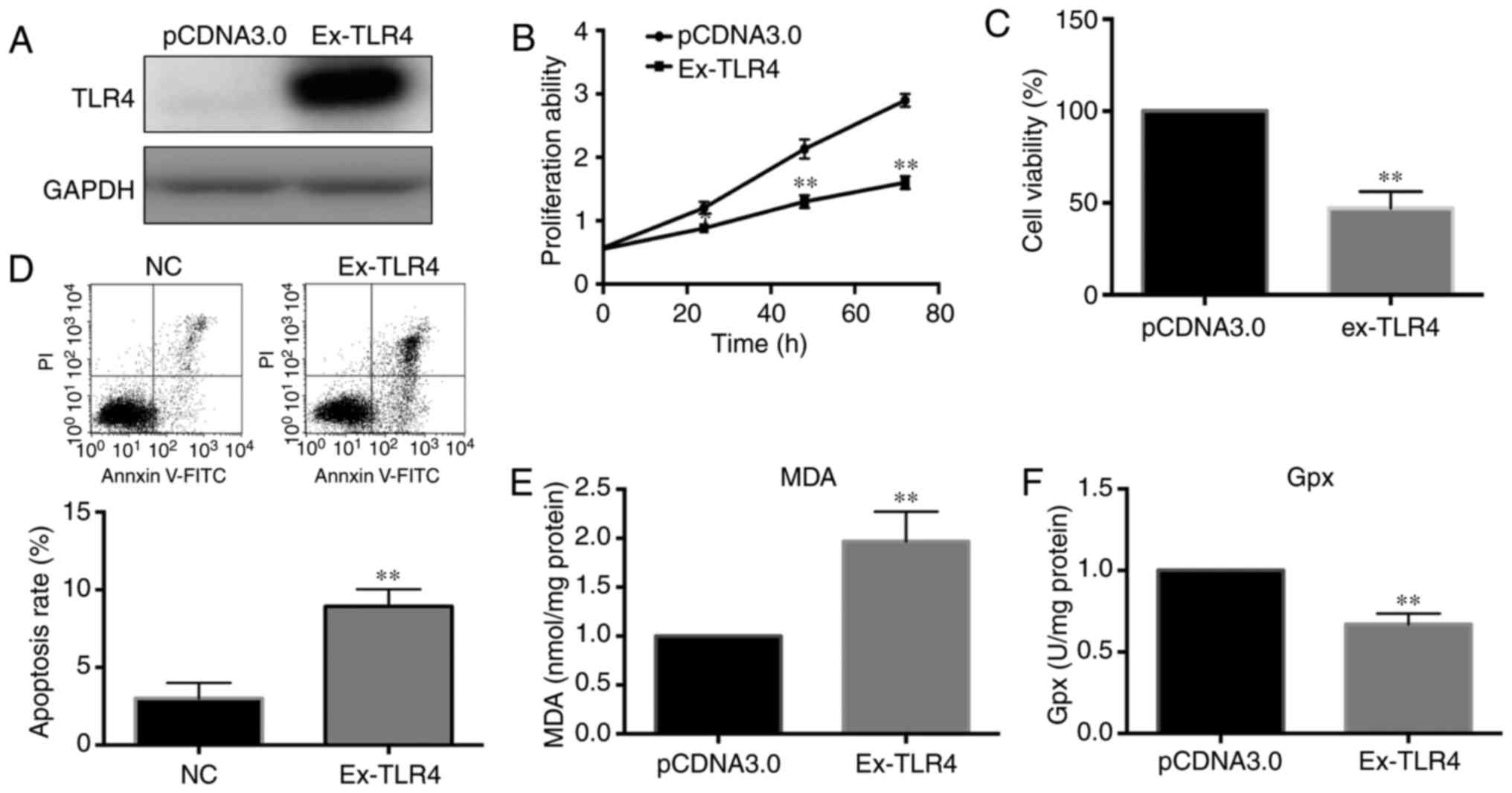
confirming the overexpression effect induced by the plasmid using
western blot analysis (Fig. 4A),
the cell viability and ROS level in pancreatic cells were detected.
CCK-8 and MTT assays demonstrated that cell viability and the
proliferation ability were significantly decreased in cells with
overexpression of TLR4 (Fig. 4B and
C). The apoptosis rate was significantly increased, which was
consistent with the change in cell viability (Fig. 4D). Finally, the ROS level we
determined following the transfection of pancreatic cells with the
TLR4 overexpression plasmid for 24 h. According to the results
shown in Fig. 4E and F, TLR4
promoted the generation of oxidative stress in pancreatic
cells.
TLR4 inhibitors reverse the LPS-induced
cellular damage to pancreatic cells and the activation of NF-κB
signaling
To confirm the contribution of NF-κB activation to
the increased ROS levels and decreased cell viability, a TLR4
inhibitor was used to inhibit the function of TLR4. The
concentration of TLR4 inhibitor used in the current study did not
affect the cell viability (data not shown). Primary pancreatic
cells were co-treated with LPS and TLR4 inhibitor. The cell
viability and ROS levels were reversed in cells co-treated with LPS
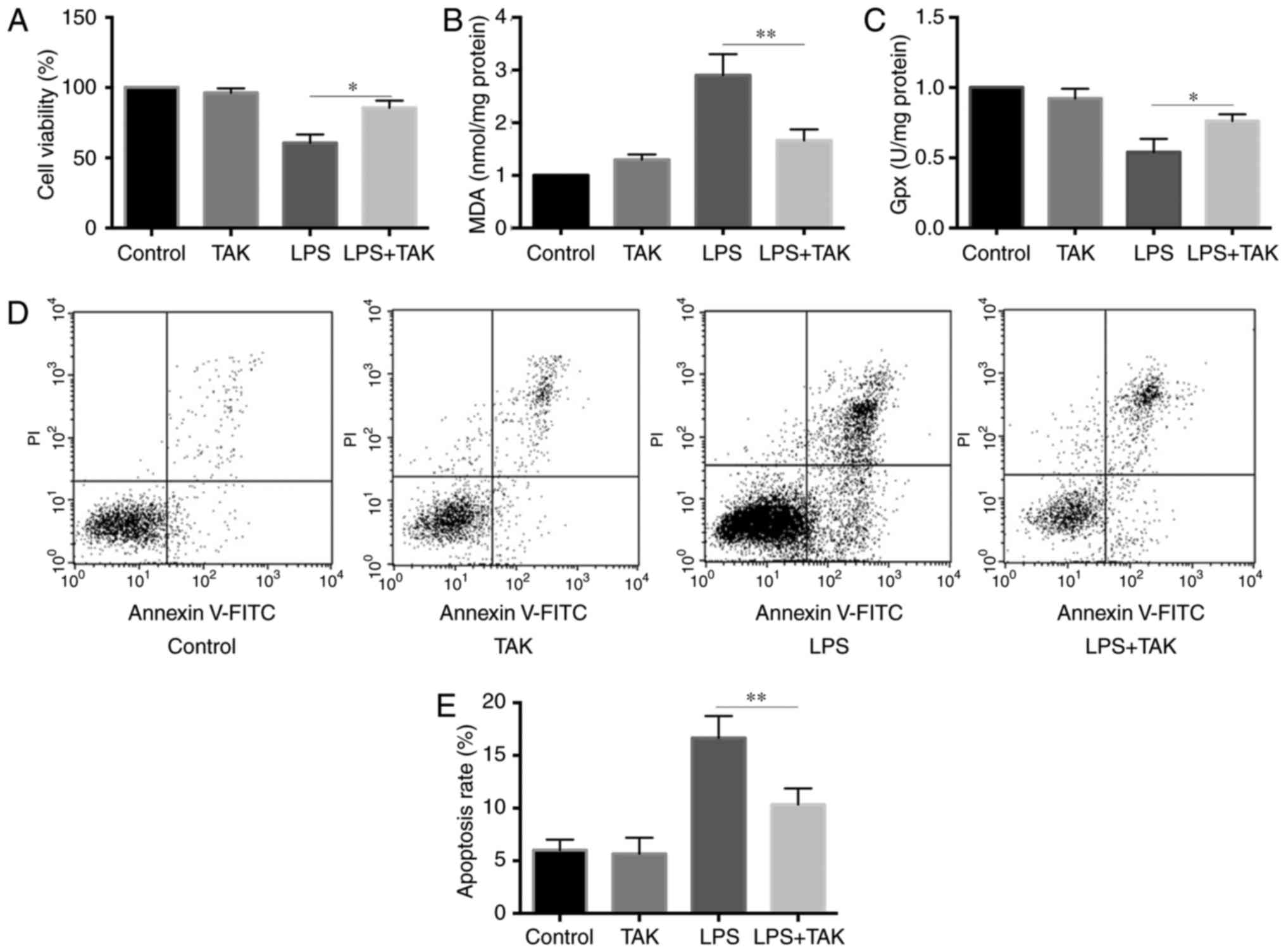
and TLR4, as compared with cells treated with LPS alone (Fig. 5A-C). Furthermore, as shown in
Fig. 5D and E, the TLR4 inhibitor
partially restored the apoptosis rate of pancreatic cells treated
by LPS for 24 h. These results suggested that TLR4 may serve a
central role in LPS-induced oxidative stress and cell
apoptosis.
TLR4 inhibitor reverses the changes in
Bcl2, Bax and PMAIP1 expression levels
As indicated earlier, the levels of Bcl2, Bax and
PMAIP1 were changed in LPS-stimulated pancreatic cells. In order to
further confirm whether the expression of these three genes was
affected by TLR4, the expression levels were tested in primary
pancreatic cells co-treated with LPS and TLR4 inhibitor. As shown
in Fig. 6A-D, the TLR4 inhibitor
partially reversed the LPS-induced changes in the expression levels
of Bcl2, Bax and PMAIP1 at both the mRNA and protein levels.
Further experiments revealed that the inhibition of TLR4 by the
selective inhibitor prevented the activation of NF-κB signaling
(Fig. 6E). The increased p-p65
level by LPS stimulation was inhibited in TLR4 inhibitor treated
cells.
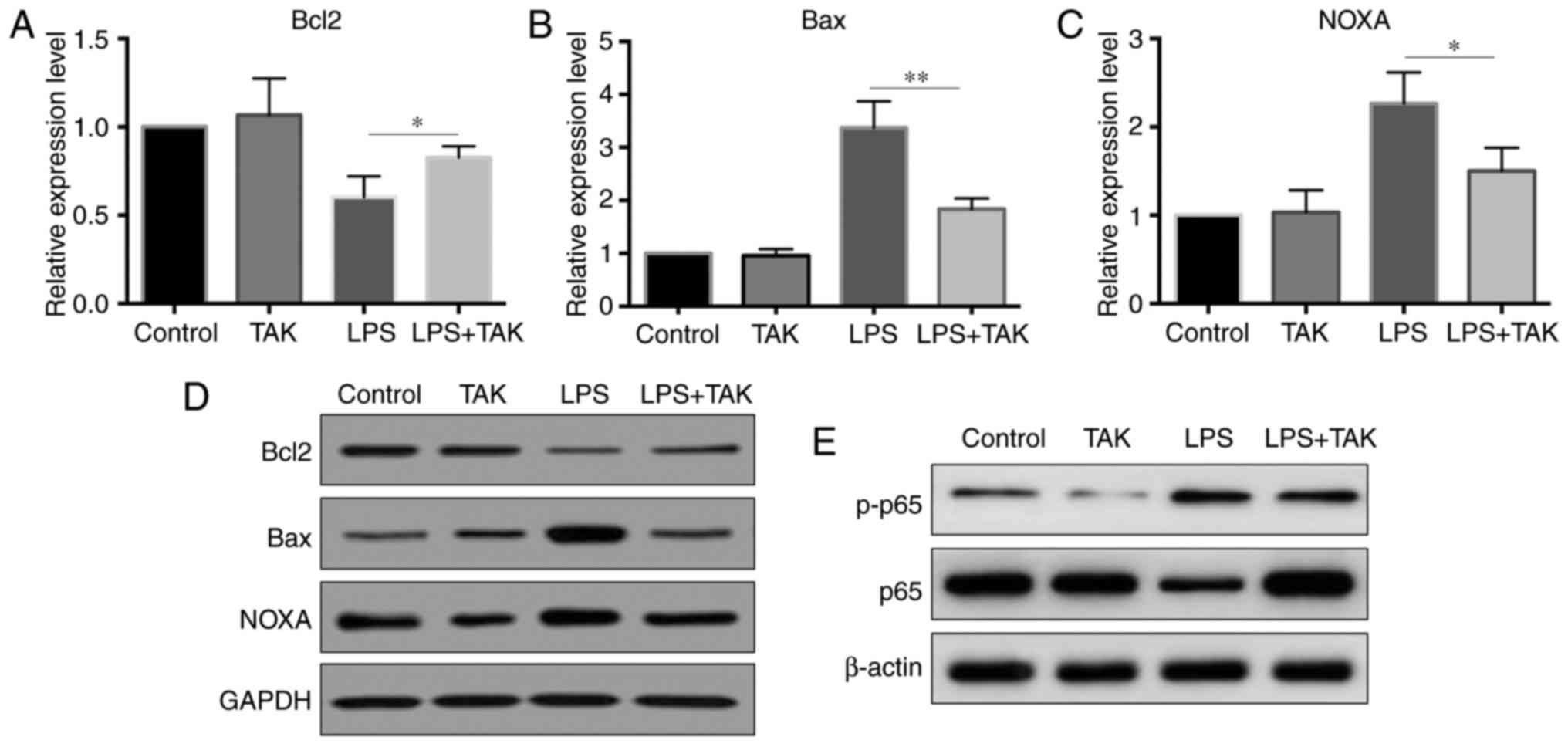 | Figure 6TLR4 inhibitor reversed the
LPS-induced changes in Bcl2, Bax and PMAIP1 expression levels.
Primary pancreatic cells were divided into four groups [control,
TAK-242 (1 µM), LPS and LPS + TAK-242] and treated for 2 h,
after which cells were harvested for RNA or protein isolation. (A)
Bcl2, (B) Bax and (C) PMAIP1 mRNA expression levels were detected
by reverse transcription-quantitative polymerase chain reaction.
(D) Protein levels of Bcl2, Bax and PMAIP1, detected by western
blot analysis. (E) Protein levels of p65 and p-p65, detected by
western blot analysis. *P<0.05 and
**P<0.01. LPS, lipopolysaccharide; TLR4, Toll-like
receptor 4; Bcl2, B-cell lymphoma 2; Bax, Bcl2-associated X
protein; p-p65, phosphorylated p65. |
Discussion
SAP is an acute inflammation process caused by the
digestion of trypsin in the pancreas and its surrounding tissues.
At present, the pathogenesis of SAP is not fully understood,
resulting in a lack of appropriate treatment strategies for this
disease. In the present study, increased expression of TLR4 in
LPS-treated pancreatic cells was observed, while increased ROS
level led to pancreatic cell damage, which was caused by TLR4. The
current study provided the specific mechanism of the TLR4/NF-κB
pathway in causing inflammation-stimulated cell death in the
pancreas, indicating a possible target for pancreatitis
treatment.
Recently, several studies have identified the
importance of TLRs in the mechanism of anti-inflammatory immunity
(26,27). TLRs are able to recognize
bacterial LPS molecules in pathogenic microorganisms and are
closely associated with clinical inflammatory diseases (27). Among these receptors, TLR4 was the
first to be identified, and its activation can lead to changes in
multiple inflammatory mediators and cytokines, which is regulated
by NF-κB signaling (28). It has
been reported that NF-κB is able to act as a messenger in the same
inflammatory response syndrome in the body. At the early onset
stage of local inflammation in an organism, inflammatory cytokines
can be activated through NF-κB signaling (29). Thus, the TLR/NF-κB pathway serves
an important role in the progression of inflammation.
Functional studies on LPS, TLR4 and NF-κB, and their
interaction in inflammatory signaling have increased the awareness
on anti-inflammatory immunity. Due to the importance of the
LPS/TLR4/NF-kB pathway in the anti-inflammatory immune response, a
number of studies have focused on its underlying mechanism. At
present, research on TLR/NF-κB has mainly focused on the
identification of TLR structure, the regulation mechanism of NF-κB
activation and the nature of TLR-oriented anti-infective drugs
(30,31). The present study focused on the
regulatory mechanism of NF-κB activation and observed that this
involved the generation of cellular ROS. In addition, the
regulatory role of TLR4 in pancreatic cells may serve as a new
target for pancreatitis treatment. However, attention must also be
paid to other pathways in anti-inflammatory immunity and their
impacts. On-going studies on these problems will broaden our
understanding of the complexities of the roles of pathogens and
anti-inflammatory immunity, and provide a theoretical and
experimental basis for the further development of drugs targeting
the LPS/TLR4/NF-kB pathway. For clinical studies, interventions in
certain aspects of the LPS/TLR4/NF-kB pathway may be effective
strategies for clinical treatment and provide a new approach for
the prevention of certain infectious diseases.
In the current study, decreased cell viability was
also observed, which may be caused by increased cell apoptosis.
Thus, the expression levels of Bcl2, Bax and PMAIP1 in LPS-treated
pancreatic cells were then examined. Bcl2 is one of the most
important anti-apoptotic genes, while Bax and PMAIP1 are two
important members of apoptotic genes. The expression levels of
these Bcl2 family genes reflect the percentage of apoptosis. One
possible mechanism through which LPS causes increased ROS level may
involve the induction of decreased cell viability, which then leads
to a change in the expression levels of Bcl2 family genes. The
expression levels of anti-apoptotic gene (Bcl2) were decreased and
the pro-apoptotic genes (Bax and PMAIP1) were increased. Another
possible mechanism is regulation of the three genes by TLR4.
According to the results of the present study, the expression
levels of Bcl2, Bax and PMAIP1 exhibited the same changes when
pancreatic cells were treated with LPS or transfected with TLR4
overexpression plasmid.
In conclusion, the data obtained in the present
study indicated that the LPS-induced TLR4/NF-κB pathway serves a
central role in the initiation of inflammation, oxidative stress
and pancreatic cell death. Furthermore, TLR4 activation was
considered necessary for LPS-induced cell damage. This mechanism
has the potential to be a novel therapeutic target in the clinical
treatment of pancreatitis.
Acknowledgements
Not applicable.
Funding
This study was supported by grants from the Science
and Technology Planning Project of Xi'an [grant no.
2017113SF/YX007(15)], the
Shaanxi Province Key Scientific and Technological Project (grant
no. 2016SF-232), and the Scientific Research Fund Project of
Shaanxi Provincial Health and Family Planning Commission (grant no.
2016D015).
Availability of data and materials
The data that support the findings of this study are
available from the corresponding author upon reasonable
request.
Authors' contributions
HP and LP designed and initiated this study. LP, LY
and LW performed the experiments of primary culture of pancreatic
acinar cells. JH and JS performed reverse
transcription-quantitative polymerase chain reaction of
lipopolysaccharide (LPS)-stimulated cells and plasmid-transfected
cells. HW and HF performed western blotting in LPS-stimulated and
plasmid-transfected pancreatic acinar cells. ZB performed cell
proliferation and viability assays. XW measured the reactive oxygen
species level in LPS-stimulated pancreatic acinar cells. HP, LP and
HF wrote the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The Ethics Committee of the Xi'an Jiaotong
University (Xi'an, China) approved the present study. Analysis was
performed in accordance with the ethical standards of the hospital
and the tenets of the Declaration of Helsinki/Declaration of
Istanbul.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
LaRusch J, Solomon S and Whitcomb DC:
Pancreatitis Overview. GeneReviews(R). Pagon RA, Adam MP, Ardinger
HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford
HC, Smith RJH, et al: University of Washington; Seattle, WA:
1993
|
|
2
|
Charbonney E and Nathens AB: Severe acute
pancreatitis: A review. Surg Infect (Larchmt). 9:573–578. 2008.
View Article : Google Scholar
|
|
3
|
Kawagishi H and Finkel T: Unraveling the
truth about antioxidants: ROS and disease: Finding the right
balance. Nat Med. 20:711–713. 2014. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Shi C, Andersson R, Zhao X and Wang X:
Potential role of reactive oxygen species in
pancreatitis-associated multiple organ dysfunction. Pancreatology.
5:492–500. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Booth DM, Mukherjee R, Sutton R and
Criddle DN: Calcium and reactive oxygen species in acute
pancreatitis: Friend or foe? Antioxid Redox Signal. 15:2683–2698.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Simon HU, Haj-Yehia A and Levi-Schaffer F:
Role of reactive oxygen species (ROS) in apoptosis induction.
Apoptosis. 5:415–418. 2000. View Article : Google Scholar
|
|
7
|
Park J, Min JS, Kim B, Chae UB, Yun JW,
Choi MS, Kong IK, Chang KT and Lee DS: Mitochondrial ROS govern the
LPS-induced pro-inflammatory response in microglia cells by
regulating MAPK and NF-κB pathways. Neurosci Lett. 584:191–196.
2015. View Article : Google Scholar
|
|
8
|
Cho RL, Yang CC, Lee IT, Lin CC, Chi PL,
Hsiao LD and Yang CM: Lipopolysaccharide induces ICAM-1 expression
via a c-Src/NADPH oxidase/ROS-dependent NF-κB pathway in human
pulmonary alveolar epithelial cells. Am J Physiol Lung Cell Mol
Physiol. 310:L639–L657. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lv JC, Wang G, Pan SH, Bai XW and Sun B:
Lycopene protects pancreatic acinar cells against severe acute
pancreatitis by abating the oxidative stress through JNK pathway.
Free Radic Res. 49:151–163. 2015. View Article : Google Scholar
|
|
10
|
Ramudo L, Manso MA, Sevillano S and de
Dios I: Kinetic study of TNF-alpha production and its regulatory
mechanisms in acinar cells during acute pancreatitis induced by
bile-pancreatic duct obstruction. J Pathol. 206:9–16. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Keck T, Werner J, Banafsche R, Stalmann A,
Schneider L, Gebhard MM, Herfarth C and Klar E: Oxygen radicals
promote ICAM-1 expression and microcirculatory disturbances in
experimental acute pancreatitis. Pancreatology. 3:156–163. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Song R, Yu D and Park J: Changes in gene
expression of tumor necrosis factor alpha and interleukin 6 in a
canine model of caerulein-induced pancreatitis. Can J Vet Res.
80:236–241. 2016.PubMed/NCBI
|
|
13
|
Concepcion-Martin M, Gomez-Oliva C, Juanes
A, Mora J, Vidal S, Diez X, Torras X, Sainz S, Villanueva C, Farre
A, et al: IL-6, IL-10 and TNFα do not improve early detection of
post-endoscopic retrograde cholangiopancreatography acute
pancreatitis: A prospective cohort study. Sci Rep. 6:334922016.
View Article : Google Scholar
|
|
14
|
Yang ZW, Meng XX, Zhang C and Xu P: CARD9
gene silencing with siRNA protects rats against severe acute
pancreatitis: CARD9-dependent NF-κB and P38MAPKs pathway. J Cell
Mol Med. 21:1085–1093. 2017. View Article : Google Scholar
|
|
15
|
Wang WY, Chen Y, Su X, Tang D, Ben QW, Yao
WY, Chen P and Yuan YZ: Resistin-like molecule-α causes lung injury
in rats with acute pancreatitis by activating the pi-3k/akt-NF-κB
pathway and promoting inflammatory cytokine release. Curr Mol Med.
16:677–687. 2016. View Article : Google Scholar
|
|
16
|
Jiang CY and Wang W: Resistin aggravates
the expression of proinflammatory cytokines in ceruleinstimulated
AR42J pancreatic acinar cells. Mol Med Rep. 15:502–506. 2017.
View Article : Google Scholar
|
|
17
|
DiDonato JA, Mercurio F and Karin M: NF-κB
and the link between inflammation and cancer. Immunol Rev.
246:379–400. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Gukovsky I, Gukovskaya AS, Blinman TA,
Zaninovic V and Pandol SJ: Early NF-kappaB activation is associated
with hormone-induced pancreatitis. Am J Physiol. 275:G1402–G1414.
1998.PubMed/NCBI
|
|
19
|
Huang H, Liu Y, Daniluk J, Gaiser S, Chu
J, Wang H, Li ZS, Logsdon CD and Ji B: Activation of nuclear
factor-κB in acinar cells increases the severity of pancreatitis in
mice. Gastroenterology. 144:202–210. 2013. View Article : Google Scholar
|
|
20
|
Liu Z, Liu J, Zhao K, Shi Q, Zuo T, Wang G
and Wang W: Role of daphnetin in rat severe acute pancreatitis
through the regulation of TLR4/NF-[formula: See text]B signaling
pathway activation. Am J Chin Med. 44:149–163. 2016. View Article : Google Scholar
|
|
21
|
Cen Y, Liu C, Li X, Yan Z, Kuang M, Su Y,
Pan X, Qin R, Liu X, Zheng J and Zhou H: Artesunate ameliorates
severe acute pancre-atitis (SAP) in rats by inhibiting expression
of pro-inflammatory cytokines and Toll-like receptor 4. Int
Immunopharmacol. 38:252–260. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pan LF, Yu L, Wang LM, He JT, Sun JL, Wang
XB, Bai ZH, Su LJ and Pei HH: The toll-like receptor 4 antagonist
transforming growth factor-β-activated kinase(TAK)-242 attenuates
taurocholate-induced oxidative stress through regulating
mitochondrial function in mice pancreatic acinar cells. J Surg Res.
206:298–306. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
24
|
Li X, Zhang L, Xu YW, Wang C, Zhao Y, Yu
P, Lv SW, Yan GL, Liu JQ and Luo GM: The protective effects of
6-CySeCD with GPx activity against UVB-induced injury in HaCaT
cells. Australas J Dermatol. 54:120–125. 2013. View Article : Google Scholar
|
|
25
|
Zhang X, Zhu C, Wu D and Jiang X: Possible
role of toll-like receptor 4 in acute pancreatitis. Pancreas.
39:819–824. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jimenez-Dalmaroni MJ, Gerswhin ME and
Adamopoulos IE: The critical role of toll-like receptors-from
microbial recognition to autoimmunity: A comprehensive review.
Autoimmun Rev. 15:1–8. 2016. View Article : Google Scholar
|
|
27
|
Joosten LA, Abdollahi-Roodsaz S, Dinarello
CA, O'Neill L and Netea MG: Toll-like receptors and chronic
inflammation in rheumatic diseases: New developments. Nat Rev
Rheumatol. 12:344–357. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou Y, Jin XH, Jing YX, Song Y, He XX,
Zheng LL, Wang YB, Wei ZY and Zhang GP: Porcine parvovirus
infection activates inflammatory cytokine production through
Toll-like receptor 9 and NF-κB signaling pathways in porcine kidney
cells. Vet Microbiol. 207:56–62. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lunin SM, Khrenov MO, Novoselova TV,
Parfenyuk SB, Glushkova OV, Fesenko EE and Novoselova EG:
Modulation of inflammatory response in mice with severe autoimmune
disease by thymic peptide thymulin and an inhibitor of NF-kappaB
signalling. Int Immunopharmacol. 25:260–266. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bhaskar S, Sudhakaran PR and Helen A:
Quercetin attenuates atherosclerotic inflammation and adhesion
molecule expression by modulating TLR-NF-κB signaling pathway. Cell
Immunol. 310:131–140. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu LY, Ye ZN, Zhou CH, Wang CX, Xie GB,
Zhang XS, Gao YY, Zhang ZH, Zhou ML, Zhuang Z, et al: Roles of
pannexin-1 channels in inflammatory response through the
TLRs/NF-Kappa B signaling pathway following experimental
subarachnoid hemorrhage in rats. Front Mol Neurosci. 10:1752017.
View Article : Google Scholar : PubMed/NCBI
|