Introduction
Dyslipidemia, including hypertriglyceridemia,
hypo-high density lipoprotein (HDL)-cholesterolemia and hyper-low
density lipoprotein (LDL)-cholesterolemia, has a substantial
genetic component (1-3). Familial hypercholesterolemia is an
autosomal dominant disorder characterized by pronounced increases
in the circulating concentrations of total cholesterol and
LDL-cholesterol (1,2). One of the underlying causes of
familial hypercholesterolemia is a defect in the LDL receptor that
is responsible for the uptake of most circulating LDL-cholesterol
by the liver (2,4). In addition to mutations of the LDL
receptor gene (LDLR), familial hypercholesterolemia can be
caused by mutations in the apolipoprotein B gene (APOB),
proprotein convertase subtilisin/kexin type 9 gene (PCSK9),
cytochrome P450 family 7 subfamily A member 1 gene (CYP7A1),
and LDL receptor adaptor protein 1 gene (LDLRAP1) (2,4).
Common forms of dyslipidemia are multifactorial and polygenic
disorders, that result from an interaction between genetic
background and both lifestyle and environmental factors, such as
consumption of high-fat or high-calorie diets and physical
inactivity (1,5). The heritability of plasma
concentrations of triglycerides, HDL-cholesterol, or
LDL-cholesterol was demonstrated to be 33-43, 40-74, and 41-59%,
respectively (6-8). Given that dyslipidemia is a key risk
factor for coronary artery disease and ischemic stroke (9,10),
as well as for colorectal cancer (11,12), personalized prevention is an
important public health goal.
Genome-wide association studies (GWASs) and
gene-centric meta-analyses have implicated various genes and loci
as determinants of blood lipid levels or of predisposition to
dyslipidemia in European-ancestry populations (13-17). Genetic variants associated with
lipid profiles have been extensively investigated, with one recent
study having identified 157 such loci, including 62 variants not
previously reported (18). Recent
GWASs (19,20) or studies based on whole-exome
(21) or whole-genome (22) sequencing in European-ancestry
populations have also identified low-frequency or rare variants
associated with circulating lipid levels. A more recent exome-wide
association study (EWAS) identified 444 independent variants at 250
loci as being significantly associated with plasma levels of total
cholesterol, LDL-cholesterol, HDL-cholesterol, or triglycerides
(23). An electronic health
record-based study reported that genetic risk scores for
circulating LDL-cholesterol and triglyceride levels based on 477
single nucleotide polymorphisms (SNPs) were predictive of age at
initiation of treatment with lipid-lowering medication (24). Although various SNPs have been
demonstrated to be associated with blood lipid profiles in East
Asian (25,26) or Japanese (27) populations, genetic variants that
contribute to susceptibility to dyslipidemia in Japanese remain to
be identified definitively.
Given the substantial genetic component of the
circulating concentrations of triglycerides, HDL-cholesterol, and
LDL-cholesterol (6-8), the genetic contribution to
early-onset forms of hypertriglyceridemia,
hypo-HDL-cholesterolemia, and hyper-LDL-cholesterolemia may be
greater compared with late-onset forms (2,3).
The statistical power of genetic association studies may thus be
increased by focusing on subjects with early-onset forms of these
disorders.
The present study performed EWASs for early-onset
forms of hypertriglyceridemia, hypo-HDL-cholesterolemia, and
hyper-LDL-cholesterolemia with the use of human exome array-based
genotyping methods. The aim was to identify genetic variants that
confer susceptibility to these conditions in the Japanese
population.
Materials and methods
Study subjects
In our previous studies, the median age of subjects
with hypertriglyceridemia, hypo-HDL-cholesterolemia or
hyper-LDL-cholesterolemia was 64, 68 or 62 years, respectively
(28). Therefore, in the present
study, early-onset dyslipidemia was defined as that occurring at an
age of ≤65 years. A total of 8,073 individuals aged ≤65 years were
examined. Recruited subjects either visited outpatient clinics or
were admitted to participating hospitals in Japan (Gifu Prefectural
Tajimi Hospital, Tajimi; Gifu Prefectural General Medical Center,
Gifu; Japanese Red Cross Nagoya First Hospital, Nagoya; Northern
Mie Medical Center Inabe General Hospital, Inabe; Hirosaki
University Hospital and Hirosaki Stroke and Rehabilitation Center,
Hirosaki) because of various symptoms or for an annual health
checkup between 2002 and 2014, or were community-dwelling
individuals recruited to a population-based cohort study in Inabe
between 2010 and 2014 (29).
Venous blood was collected in the early morning
after subjects had fasted overnight. The blood samples were
centrifuged at 1,600 x g for 15 min at 4°C, and serum was separated
for subsequent analysis. Serum concentrations of triglycerides,
HDL-cholesterol, and LDL-cholesterol were measured in the clinical
laboratory of each hospital. The 2,664 subjects with
hypertriglyceridemia and 5,294 controls had serum triglyceride
concentrations of ≥1.69 mmol/l (150 mg/dl) and <1.69 mmol/l,
respectively. The 974 subjects with hypo-HDL-cholesterolemia and
7,085 controls had serum HDL-cholesterol concentrations of <1.03
mmol/l (40 mg/dl) and ≥1.03 mmol/l, respectively. The 2,911
subjects with hyper-LDL-cholesterolemia and 5,111 controls had
serum LDL-cholesterol concentrations of ≥3.62 mmol/l (140 mg/dl)
and <3.62 mmol/l, respectively. The 562 subjects with both
hypertriglyceridemia and hypo-HDL-cholesterolemia, and the 4,907
controls, overlapped between the corresponding studies, as did the
1,312 subjects with both hypertriglyceridemia and
hyper-LDL-cholesterolemia and 3,723 controls, as well as the 317
subjects with both hypo-HDL-cholesterolemia and
hyper-LDL-cholesterolemia and 4,484 controls. Individuals with
single-gene disorders, such as familial hypercholesterolemia, or
with endocrinologic or metabolic diseases that cause dyslipidemia
were excluded from the study. Those taking medications that may
cause secondary dyslipidemia were also excluded.
EWAS
Venous blood was collected into tubes containing 50
mmol/l EDTA (disodium salt), peripheral blood leukocytes were
isolated, and genomic DNA was extracted from these cells with the
use of a kit (Genomix, Talent Srl, Trieste, Italy; or SMITEST
EX-R&D, Medical & Biological Laboratories, Co., Ltd.,
Nagoya, Japan). EWASs for hypertriglyceridemia (2,664 cases and
5,294 controls), hypo-HDL-cholesterolemia (974 cases and 7,085
controls), and hyper-LDL-cholesterolemia (2,911 cases and 5,111
controls) were performed with Human Exome-12 v1.2 DNA Analysis
BeadChip or Infinium Exome-24 v1.0 BeadChip arrays (Illumina, San
Diego, CA, USA). These exome arrays include putative functional
exonic variants selected from ~12,000 individual exome and
whole-genome sequences. The exonic content consists of ~244,000
SNPs from diverse populations, including European, African,
Chinese, and Hispanic individuals (30). SNPs contained in only one of the
arrays (~2.6% of all SNPs) were excluded from analysis. Quality
control was performed, as previously described (31). Briefly, genotyping data with a
call rate of <97% were discarded, with the mean call rate for
the remaining data being 99.9%. Gender specification was checked
for all samples, and those for which gender phenotype in the
clinical records was inconsistent with genetic sex were discarded.
Duplicated samples and cryptic relatedness were checked by
calculation of identity by descent; all pairs with a value of
>0.1875 were inspected and one sample from each pair was
excluded. Heterozygosity of SNPs was calculated for all samples,
and those with extremely low or high heterozygosity (>3 standard
deviations from the mean) were discarded. SNPs in sex chromosomes
or mitochondrial DNA were excluded from analysis, as were
non-polymorphic SNPs or SNPs with a minor allele frequency of
<1.0%. SNPs whose genotype distributions deviated significantly
(P<0.01) from Hardy-Weinberg equilibrium in control individuals
were discarded. Finally, genotype data for each EWAS were examined
for population stratification by principal components analysis
(32), and population outliers
were excluded from further study. A total of 31,198, 31,133, and
31,175 SNPs passed quality control in the EWASs for
hypertriglyceridemia, hypo-HDL-cholesterolemia and
hyper-LDL-cholesterolemia, respectively, and were subjected to
further analyses.
Statistical analysis
For analysis of characteristics of the study
subjects, quantitative and categorical data were compared between
cases and controls with the unpaired Student’s t-test and Pearson’s
Chi-square test, respectively. Allele frequencies were estimated by
the gene counting method, and departure from Hardy-Weinberg
equilibrium was identified with Fisher’s exact test. The
association of allele frequencies of SNPs to hypertriglyceridemia,
hypo-HDL-cholesterolemia or hyper-LDL-cholesterolemia in the EWASs
was examined with Fisher’s exact test. To compensate for multiple
comparisons of allele frequencies with hypertriglyceridemia,
hypo-HDL-cholesterolemia, or hyper-LDL-cholesterolemia,
Bonferroni’s correction was applied for statistical significance of
association. Given that 31,198, 31,133 or 31,175 SNPs were
analyzed, a P-value of <1.60×10−6 [0.05/(31,198 or
31,175)] for hypertriglyceridemia and hyper-LDL-cholesterolemia and
a P-value of <1.61×10−6 (0.05/31,133) for
hypo-HDL-cholesterolemia was considered statistically significant
for association. The inflation factor (λ) was 1.07 for
hypertriglyceridemia, 1.05 for hypo-HDL-cholesterolemia, and 1.00
for hyper-LDL-cholesterolemia. Multivariable logistic regression
analysis was performed with hypertriglyceridemia,
hypo-HDL-cholesterolemia, or hyper-LDL-cholesterolemia as a
dependent variable and independent variables including age, sex (0,
woman; 1, man), and genotype of each SNP. Genotypes of SNPs were
assessed according to dominant [0, AA; 1, AB +
BB (A, major allele; B, minor allele)],
recessive (0, AA + AB; 1, BB), and additive
genetic models, and the P-value, odds ratio, and 95% confidence
interval were calculated. Additive models comprised additive 1 (0,
AA; 1, AB; 0, BB) and additive 2 (0,
AA; 0, AB; 1, BB) scenarios, which were
analyzed simultaneously with a single statistical model.
Associations of genotypes of identified SNPs to serum
concentrations of triglycerides, HDL-cholesterol, or
LDL-cholesterol were examined by one-way analysis of variance.
Bonferroni’s correction was also applied to other statistical
analysis as indicated. Statistical tests were performed with JMP
Genomics version 9.0 software (SAS Institute, Cary, NC, USA).
Results
Characteristics of subjects
The characteristics of the 7,958 subjects enrolled
in the hypertriglyceridemia study are listed in Table I. Age, the frequency of men, and
the prevalence of smoking, obesity, hypertension, diabetes
mellitus, chronic kidney disease, and hyperuricemia as well as body
mass index (BMI), systolic and diastolic blood pressure (BP),
fasting plasma glucose (FPG) level, blood glycosylated hemoglobin
(hemoglobin A1c) content, and serum concentrations of
creatinine and uric acid were greater, whereas the serum
concentration of HDL-cholesterol and estimated glomerular
filtration rate (eGFR) were lower, in subjects with
hypertriglyceridemia compared with controls.
 | Table ICharacteristics of subjects with
hypertriglyceridemia and control individuals. |
Table I
Characteristics of subjects with
hypertriglyceridemia and control individuals.
| Characteristic | Control |
Hypertriglyceridemia | P-value |
|---|
| Number of
subjects | 5,294 | 2,664 | |
| Age (years) | 51.3±10.2 | 53.3±8.8 | <0.0001 |
| Sex (men/women,
%) | 50.2/49.8 | 75.6/24.4 | <0.0001 |
| Smoking (%) | 35.9 | 54.0 | <0.0001 |
| Obesity (%) | 22.9 | 42.6 | <0.0001 |
| Body mass index
(kg/m2) | 22.7±3.4 | 24.8±3.6 | <0.0001 |
| Hypertension
(%) | 35.3 | 57.2 | <0.0001 |
| Systolic BP
(mmHg) | 125±23 | 132±23 | <0.0001 |
| Diastolic BP
(mmHg) | 75±14 | 80±13 | <0.0001 |
| Diabetes mellitus
(%) | 17.5 | 33.8 | <0.0001 |
| Fasting plasma
glucose (mmol/l) | 5.83±2.04 | 6.55±2.81 | <0.0001 |
| Blood hemoglobin
A1c (%) | 5.83±1.08 | 6.26±1.50 | <0.0001 |
| Serum triglycerides
(mmol/l) | 0.94±0.34 | 2.40±1.37 | <0.0001 |
| Serum
HDL-cholesterol (mmol/l) | 1.66±0.47 | 1.30±0.36 | <0.0001 |
| Serum
LDL-cholesterol (mmol/l) | 3.06±0.80 | 3.35±0.95 | 0.8593 |
| Chronic kidney
disease (%) | 12.3 | 19.9 | <0.0001 |
| Serum creatinine
(µmol/l) | 71.9±69.8 | 82.1±85.2 | <0.0001 |
| eGFR (ml
min−1 1.73 m−2) | 78.5±19.4 | 74.1±21.6 | <0.0001 |
| Hyperuricemia
(%) | 11.7 | 28.4 | <0.0001 |
| Serum uric acid
(µmol/l) | 308±90 | 362±91 | <0.0001 |
The characteristics of the 8,059 subjects enrolled
in the hypo-HDL-cholesterolemia study are listed in Table II. Age, the frequency of men, and
the prevalence of smoking, obesity, hypertension, diabetes
mellitus, chronic kidney disease, and hyperuricemia as well as BMI,
systolic and diastolic BP, FPG level, blood hemoglobin
A1c content, and serum concentrations of triglycerides,
creatinine, and uric acid were greater, whereas eGFR was lower, in
subjects with hypo-HDL-cholesterolemia compared with controls.
| Table IICharacteristics of subjects with
hypo-HDL-cholesterolemia and control individuals. |
Table II
Characteristics of subjects with
hypo-HDL-cholesterolemia and control individuals.
| Characteristic | Control |
Hypo-HDL-cholesterolemia | P-value |
|---|
| Number of
subjects | 7,085 | 974 | |
| Age (years) | 51.7±9.9 | 54.3±8.7 | <0.0001 |
| Sex (men/women,
%) | 55.1/44.9 | 85.3/14.7 | <0.0001 |
| Smoking (%) | 40.5 | 54.6 | <0.0001 |
| Obesity (%) | 27.0 | 48.2 | <0.0001 |
| Body mass index
(kg/m2) | 23.2±3.5 | 25.1±3.9 | <0.0001 |
| Hypertension
(%) | 39.6 | 64.8 | <0.0001 |
| Systolic BP
(mmHg) | 126±23 | 137±27 | <0.0001 |
| Diastolic BP
(mmHg) | 76±14 | 80±16 | <0.0001 |
| Diabetes mellitus
(%) | 19.5 | 48.2 | <0.0001 |
| Fasting plasma
glucose (mmol/l) | 5.93±2.16 | 7.16±3.26 | <0.0001 |
| Blood hemoglobin
A1c (%) | 5.89±1.17 | 6.59±1.60 | <0.0001 |
| Serum triglycerides
(mmol/l) | 1.35±0.96 | 2.12±1.62 | <0.0001 |
| Serum
HDL-cholesterol (mmol/l) | 1.63±0.42 | 0.88±0.12 | <0.0001 |
| Serum
LDL-cholesterol (mmol/l) | 3.16±0.84 | 3.13±0.99 | 0.2860 |
| Chronic kidney
disease (%) | 13.4 | 26.2 | <0.0001 |
| Serum creatinine
(µmol/l) | 72.6±66.6 | 96.0±122.7 | <0.0001 |
| eGFR (ml
min−1 1.73 m−2) | 77.5±18.5 | 73.3±29.8 | <0.0001 |
| Hyperuricemia
(%) | 15.9 | 27.2 | <0.0001 |
| Serum uric acid
(µmol/l) | 323±91 | 361±0.3 | <0.0001 |
The characteristics of the 8,022 subjects enrolled
in the hyper-LDL-cholesterolemia study are listed in Table III. Age, the prevalence of
obesity, BMI, diastolic BP, and serum concentrations of
triglycerides and uric acid were greater, whereas systolic BP and
serum concentrations of HDL-cholesterol and creatinine were lower,
in subjects with hyper-LDL-cholesterolemia compared with
controls.
| Table IIICharacteristics of subjects with
hyper-LDL-cholesterolemia and control individuals. |
Table III
Characteristics of subjects with
hyper-LDL-cholesterolemia and control individuals.
| Characteristic | Control |
Hyper-LDL-cholesterolemia | P-value |
|---|
| Number of
subjects | 5,111 | 2,911 | |
| Age (years) | 51.3±10.3 | 53.2±8.9 | <0.0001 |
| Sex (men/women,
%) | 59.0/41.0 | 58.2/41.8 | 0.4936 |
| Smoking (%) | 40.9 | 44.3 | 0.0034 |
| Obesity (%) | 27.7 | 32.6 | <0.0001 |
| Body mass index
(kg/m2) | 23.2±3.6 | 23.9±3.5 | <0.0001 |
| Hypertension
(%) | 42.5 | 42.6 | 0.8966 |
| Systolic BP
(mmHg) | 128±25 | 126±21 | 0.0025 |
| Diastolic BP
(mmHg) | 76±14 | 77±13 | 0.0004 |
| Diabetes mellitus
(%) | 23.2 | 22.0 | 0.2593 |
| Fasting plasma
glucose (mmol/l) | 6.11±2.49 | 6.02±2.12 | 0.0831 |
| Blood hemoglobin
A1c (%) | 5.96±1.25 | 5.99±1.26 | 0.4573 |
| Serum triglycerides
(mmol/l) | 1.40±1.19 | 1.52±0.91 | <0.0001 |
| Serum
HDL-cholesterol (mmol/l) | 1.56±0.49 | 1.51±0.42 | <0.0001 |
| Serum
LDL-cholesterol (mmol/l) | 2.70±0.56 | 3.90±0.74 | <0.0001 |
| Chronic kidney
disease (%) | 15.3 | 14.3 | 0.2118 |
| Serum creatinine
(µmol/l) | 78.3±88.7 | 71.7±58.9 | 0.0001 |
| eGFR (ml
min−1 1.73 m−2) | 77.0±21.7 | 76.8±17.8 | 0.7007 |
| Hyperuricemia
(%) | 16.3 | 18.9 | 0.0028 |
| Serum uric acid
(µmol/l) | 323±97 | 334±87 | <0.0001 |
EWASs for hypertriglyceridemia,
hypo-HDL-cholesterolemia, and hyper-LDL-cholesterolemia
The association of allele frequencies for 31,198
SNPs that passed quality control to hyper-triglyceridemia was
examined with the use of Fisher’s exact test. Following
Bonferroni’s correction, 25 SNPs were significantly associated with
hypertriglyceridemia (P<1.60×10−6; Table IV). Similar analysis of the
association of allele frequencies for 31,133 SNPs to
hypo-HDL-cholesterolemia or of those for 31,175 SNPs to
hyper-LDL-cholesterolemia revealed that 28 SNPs were significantly
associated with hypo-HDL-cholesterolemia
(P<1.61×10−6; Table
V) and 65 SNPs with hyper-LDL-cholesterolemia
(P<1.60×10−6; Table
VI).
| Table IVThe 25 SNPs significantly associated
with hypertriglyceridemia in the exome-wide association study. |
Table IV
The 25 SNPs significantly associated
with hypertriglyceridemia in the exome-wide association study.
| Gene | SNP | Nucleotide
substitutiona | Amino acid
substitution | Chromosome | Position | MAF (%) | Allele OR | Allele frequency
(P-value) |
|---|
| APOA5 | rs2075291 | G/T | G185C | 11 | 116790676 | 7.3 | 1.89 |
2.83×10−24 |
| BUD13 | rs10790162 | G/A | | 11 | 116639104 | 26.3 | 1.47 |
3.58×10−24 |
| ZPR1 | rs964184 | C/G | | 11 | 116778201 | 26.3 | 1.46 |
8.82×10−24 |
| APOA5 | rs2266788 | T/C | | 11 | 116789970 | 26.2 | 1.47 |
1.27×10−23 |
| rs7350481 | C/T | | 11 | 116715567 | 27.7 | 1.44 |
7.94×10−23 |
| rs9326246 | G/C | | 11 | 116741017 | 26.5 | 1.45 |
1.36×10−22 |
| ZPR1 | rs2075290 | T/C | | 11 | 116782580 | 26.7 | 1.43 |
1.58×10−21 |
| rs12678919 | A/G | | 8 | 19986711 | 12.6 | 0.71 |
6.07×10−11 |
| rs10503669 | C/A | | 8 | 19990179 | 12.6 | 0.71 |
8.32×10−11 |
| rs17482753 | G/T | | 8 | 19975135 | 12.6 | 0.72 |
2.04×10−10 |
| rs10096633 | C/T | | 8 | 19973410 | 12.7 | 0.72 |
2.50×10−10 |
| LPL | rs328 | C/G | S474* | 8 | 19962213 | 12.9 | 0.72 |
2.72×10−10 |
| APOA4 | rs5104 | A/G | N147S | 11 | 116821618 | 35.7 | 1.25 |
2.78×10−10 |
| rs7016880 | G/C | | 8 | 20019235 | 12.0 | 0.72 |
1.72×10−9 |
| GCKR | rs1260326 | T/C | L446P | 2 | 27508073 | 43.6 | 0.82 |
6.27×10−9 |
| GCKR | rs780093 | A/G | | 2 | 27519736 | 43.0 | 0.83 |
2.18×10−8 |
| rs1260333 | T/C | | 2 | 27525757 | 42.9 | 0.83 |
4.38×10−8 |
| rs11085421 | A/C | | 19 | 20985948 | 19.1 | 0.79 |
1.59×10−7 |
| rs1441756 | T/G | | 8 | 20010875 | 19.0 | 0.80 |
5.61×10−7 |
| rs2083637 | T/C | | 8 | 20007664 | 19.0 | 0.81 |
6.24×10−7 |
| LPL | rs301 | T/C | | 8 | 19959423 | 19.3 | 0.81 |
7.70×10−7 |
| LPL | rs13702 | A/G | | 8 | 19966981 | 19.2 | 0.81 |
9.15×10−7 |
| LPL | rs15285 | G/A | | 8 | 19967156 | 19.2 | 0.81 |
1.02×10−6 |
| rs2954033 | G/A | | 8 | 125481504 | 33.3 | 1.19 |
1.02×10−6 |
| LPL | rs326 | A/G | | 8 | 19961928 | 19.4 | 0.81 |
1.24×10−6 |
| Table VThe 28 SNPs significantly associated
with hypo-HDL-cholesterolemia in the exome-wide association
study. |
Table V
The 28 SNPs significantly associated
with hypo-HDL-cholesterolemia in the exome-wide association
study.
| Gene | SNP | Nucleotide
substitutiona | Amino acid
substitution | Chromosome | Position | MAF (%) | Allele OR | Allele frequency
(P-value) |
|---|
| LPGAT1 | rs150552771 | T/C | K200E | 1 | 211783358 | 5.0 | 16.67 |
1.09×10−12 |
| APOA5 | rs2075291 | G/T | G185C | 11 | 116790676 | 7.3 | 1.79 |
4.10×10−12 |
| COL6A5 | rs200982668 | G/A | E2501K | 3 | 130470894 | 1.3 | 0.18 |
4.51×10−11 |
| ZNF860 | rs140232911 | C/T | S161L | 3 | 31989561 | 10.4 | 4.39 |
5.21×10−11 |
| rs9261800 | C/G | | 6 | 30408822 | 2.8 | 19.65 |
6.69×10−11 |
| VPS33B | rs199921354 | C/T | R80Q | 15 | 91013841 | 1.2 | 0.17 |
1.03×10−10 |
| ADGRL3 | rs192210727 | G/T | R580I | 4 | 61909615 | 1.3 | 0.19 |
1.33×10−10 |
| TMOD4 | rs115287176 | G/A | R277W | 1 | 151170961 | 1.2 | 0.18 |
1.46×10−10 |
| COL6A3 | rs146092501 | C/T | E1386K | 2 | 237371861 | 1.2 | 0.18 |
2.10×10−10 |
| MARCH1 | rs61734696 | G/T | Q137K | 4 | 164197303 | 1.2 | 0.19 |
2.74×10−10 |
| PLCB2 | rs200787930 | C/T | E1106K | 15 | 40289298 | 1.2 | 0.19 |
2.79×10−10 |
| MOB3C | rs139537100 | C/T | R24Q | 1 | 46615006 | 1.2 | 0.19 |
2.87×10−10 |
| CXCL8 | rs188378669 | G/T | E31* | 4 | 73741568 | 1.2 | 0.19 |
3.94×10−10 |
| EHD3 | rs116417209 | G/A | V151I | 2 | 31249417 | 3.5 | 12.5 |
5.66×10−10 |
| ZNF77 | rs146879198 | G/A | R340* | 19 | 2934109 | 1.2 | 0.20 |
1.17×10−9 |
| OR4F6 | rs141569282 | G/A | A117T | 15 | 101806068 | 1.7 | 0.23 |
2.31×10−9 |
| ACAD10 | rs11066015 | G/A | | 12 | 111730205 | 27.5 | 1.37 |
2.61×10−9 |
| CACNA1D | rs35874056 | G/A | G460S | 3 | 53702798 | 2.0 | 100.00 |
2.95×10−9 |
| ALDH2 | rs671 | G/A | E504K | 12 | 111803962 | 27.6 | 1.35 |
4.23×10−9 |
| rs3764261 | G/T | | 16 | 56959412 | 19.8 | 0.69 |
7.90×10−9 |
| rs247616 | C/T | | 16 | 56955678 | 19.7 | 0.69 |
9.31×10−9 |
| BRAP | rs3782886 | A/G | | 12 | 111672685 | 29.3 | 1.34 |
1.19×10−8 |
| CETP | rs1532624 | G/T | | 16 | 56971567 | 29.6 | 0.74 |
2.63×10−8 |
| HECTD4 | rs11066280 | T/A | | 12 | 112379979 | 29.0 | 1.33 |
3.54×10−8 |
| HECTD4 | rs2074356 | C/T | | 12 | 112207597 | 25.4 | 1.32 |
1.43×10−7 |
| LILRB2 | rs73055442 | C/T | R103H | 19 | 54279838 | 1.6 | 34.09 |
1.80×10−7 |
| DCLRE1C | rs150854849 | C/T | R179Q | 10 | 14934704 | 2.4 | 59.56 |
3.16×10−7 |
| SPOPL | rs114501427 | G/A | D349N | 2 | 138568946 | 8.8 | 3.23 |
4.01×10−7 |
| Table VIThe 65 SNPs significantly associated
with hyper-LDL-cholesterolemia in the exome-wide association
study. |
Table VI
The 65 SNPs significantly associated
with hyper-LDL-cholesterolemia in the exome-wide association
study.
| Gene | SNP | Nucleotide
substitutiona | Amino acid
substitution | Chromosome | Position | MAF (%) | Allele OR | Allele frequency
(P-value) |
|---|
| APOE | rs7412 | C/T | R176C | 19 | 44908822 | 4.3 | 0.42 |
6.62×10−22 |
| COL6A3 | rs146092501 | C/T | E1386K | 2 | 237371861 | 1.2 | 2.14 |
1.42×10−11 |
| TMOD4 | rs115287176 | G/A | R277W | 1 | 151170961 | 1.2 | 2.13 |
1.83×10−11 |
| CXCL8 | rs188378669 | G/T | E31* | 4 | 73741568 | 1.2 | 2.10 |
1.95×10−11 |
| VPS33B | rs199921354 | C/T | R80Q | 15 | 91013841 | 1.2 | 2.11 |
2.32×10−11 |
| ZNF77 | rs146879198 | G/A | R340* | 19 | 2934109 | 1.2 | 2.08 |
4.91×10−11 |
| MOB3C | rs139537100 | C/T | R24Q | 1 | 46615006 | 1.2 | 2.06 |
5.58×10−11 |
| PLCB2 | rs200787930 | C/T | E1106K | 15 | 40289298 | 1.2 | 2.03 |
1.70×10−10 |
| MUC17 | rs78010183 | A/T | T1305S | 7 | 101035329 | 1.8 | 1.81 |
2.30×10−10 |
| MARCH1 | rs61734696 | G/T | Q137K | 4 | 164197303 | 1.2 | 2.01 |
3.49×10−10 |
| APOB | rs13306206 | G/A | P955S | 2 | 21019859 | 3.2 | 1.75 |
7.32×10−10 |
| COL6A5 | rs200982668 | G/A | E2501K | 3 | 130470894 | 1.3 | 1.92 |
9.85×10−10 |
| PTCH2 | rs147284320 | C/T | V503I | 1 | 44828589 | 2.0 | 1.86 |
1.23×10−9 |
| ADGRL3 | rs192210727 | G/T | R580I | 4 | 61909615 | 1.3 | 1.94 |
1.49×10−9 |
| APOC1 | rs445925 | C/T | | 19 | 44912383 | 6.6 | 0.66 |
3.62×10−9 |
| HSPA1B | rs6457452 | C/T | | 6 | 31827773 | 9.7 | 1.33 |
2.97×10−8 |
| C6orf48 | rs11968400 | C/T | | 6 | 31836952 | 9.7 | 1.33 |
3.96×10−8 |
| rs12210887 | G/T | | 6 | 31847946 | 9.7 | 1.33 |
5.26×10−8 |
| UBD | rs64036 | C/A | | 6 | 29559490 | 22.5 | 1.23 |
7.55×10−8 |
| CCHCR1 | rs147733073 | C/G | H539Q | 6 | 31145462 | 10.2 | 1.32 |
8.01×10−8 |
| VARS | rs11751198 | G/A | | 6 | 31785749 | 9.5 | 1.33 |
8.12×10−8 |
| rs2596574 | G/A | | 6 | 31366397 | 9.7 | 1.32 |
8.83×10−8 |
| LY6G6F | rs9267547 | G/A | A107T | 6 | 31707724 | 10.0 | 1.32 |
9.72×10−8 |
| NEU1 | rs13118 | T/A | | 6 | 31859509 | 9.7 | 1.32 |
1.01×10−7 |
| SLC17A3 | rs34902660 | C/A | G239V | 6 | 25850874 | 8.4 | 1.35 |
1.04×10−7 |
| FAM65B | rs150142878 | C/T | R371Q | 6 | 24847657 | 5.6 | 1.42 |
1.18×10−7 |
| TUBB | rs9500864 | G/A | | 6 | 30725455 | 20.4 | 1.23 |
1.19×10−7 |
| rs204999 | A/G | | 6 | 32142202 | 9.1 | 1.33 |
1.34×10−7 |
| rs3130663 | A/G | | 6 | 30698817 | 10.7 | 1.28 |
1.38×10−7 |
| VARS | rs5030798 | G/A | V1055I | 6 | 31779733 | 9.5 | 1.32 |
1.43×10−7 |
| LY6G6F | rs9267546 | G/A | | 6 | 31705659 | 9.8 | 1.32 |
1.44×10−7 |
| PRRC2A | rs11538264 | G/A | V1774M | 6 | 31635412 | 9.5 | 1.32 |
1.55×10−7 |
| DPCR1 | rs11970154 | G/C | G1213R | 6 | 30952101 | 13.3 | 1.28 |
1.60×10−7 |
| MSH5 | rs11754464 | C/T | | 6 | 31755958 | 9.5 | 1.32 |
1.73×10−7 |
| PCSK9 | rs151193009 | C/T | R93C | 1 | 55043912 | 1.1 | 0.42 |
1.85×10−7 |
| GABBR1 | rs29243 | C/T | | 6 | 29631325 | 10.6 | 1.30 |
1.94×10−7 |
|
KIAA0319 | rs4576240 | G/T | P133T | 6 | 24596250 | 5.5 | 1.41 |
2.10×10−7 |
| LY6G6F | rs17200983 | C/A | P34Q | 6 | 31707506 | 9.5 | 1.32 |
2.38×10−7 |
| TNXB | rs140770834 | C/G | L2271V | 6 | 32064851 | 8.8 | 1.32 |
2.42×10−7 |
| TNXB | rs11751545 | A/C | | 6 | 32073266 | 8.8 | 1.32 |
2.42×10−7 |
| DPCR1 | rs6933400 | C/T | | 6 | 30939399 | 13.2 | 1.27 |
2.43×10−7 |
| LY6G6C | rs117894946 | G/C | G75A | 6 | 31719250 | 9.5 | 1.32 |
2.66×10−7 |
| SLC44A4 | rs117127493 | G/C | E344Q | 6 | 31869232 | 8.9 | 1.32 |
3.22×10−7 |
| ZSCAN26 | rs76463649 | A/G | N15S | 6 | 28271963 | 9.6 | 1.30 |
4.26×10−7 |
| DHX16 | rs7749235 | T/C | | 6 | 30667816 | 17.8 | 1.23 |
4.67×10−7 |
|
LOC105-375015 | rs9264942 | T/C | | 6 | 31306603 | 40.1 | 1.18 |
4.82×10−7 |
| ZSCAN31 | rs6922302 | C/G | P128A | 6 | 28327533 | 9.6 | 1.30 |
5.06×10−7 |
| MDC1 | rs2269702 | A/G | | 6 | 30707358 | 17.7 | 1.23 |
5.13×10−7 |
| MDC1 | rs28986465 | C/T | P386L | 6 | 30712785 | 17.7 | 1.23 |
5.17×10−7 |
| MDC1 | rs2075015 | G/A | E371K | 6 | 30712831 | 17.7 | 1.23 |
5.17×10−7 |
| DHX16 | rs2285321 | T/C | | 6 | 30670221 | 17.8 | 1.23 |
5.18×10−7 |
| SKIV2L | rs492899 | A/G | | 6 | 31965741 | 9.1 | 1.31 |
5.33×10−7 |
| MDC1 | rs6924270 | A/G | | 6 | 30714203 | 17.8 | 1.23 |
5.81×10−7 |
| rs2508015 | C/T | | 6 | 31042423 | 11.7 | 1.27 |
6.07×10−7 |
| TUBB | rs25527 | C/T | | 6 | 30723161 | 21.0 | 1.22 |
6.08×10−7 |
| PPP1R18 | rs9468805 | G/A | | 6 | 30675932 | 17.7 | 1.23 |
7.07×10−7 |
| DHX16 | rs6937357 | T/C | | 6 | 30672547 | 17.8 | 1.23 |
7.07×10−7 |
| HECTD4 | rs11066280 | T/A | | 12 | 112379979 | 29.0 | 1.19 |
7.09×10−7 |
| TRIM40 | rs2523995 | G/A | | 6 | 30134407 | 10.6 | 1.28 |
7.88×10−7 |
| PPP1R18 | rs6457254 | C/T | | 6 | 30681357 | 17.9 | 1.22 |
1.20×10−6 |
| TUBB | rs3132584 | C/A | | 6 | 30720650 | 21.2 | 1.20 |
1.22×10−6 |
| rs3130685 | T/C | | 6 | 31238429 | 41.6 | 0.85 |
1.43×10−6 |
| GIT2 | rs2292354 | C/T | | 12 | 109930396 | 26.2 | 0.83 |
1.45×10−6 |
| PPP1R18 | rs2394392 | T/G | | 6 | 30682541 | 18.0 | 1.22 |
1.50×10−6 |
| rs2524272 | T/C | | 6 | 29714623 | 26.8 | 1.19 |
1.55×10−6 |
Multivariable logistic regression
analysis of the association of SNPs to hypertriglyceridemia,
hypo-HDL-cholesterolemia or hyper-LDL-cholesterolemia
The association of the 25 SNPs identified in the
EWAS for hypertriglyceridemia to this condition was further
examined by multivariable logistic regression analysis, following
adjustment for age and sex. All 25 SNPs were significantly
[P<0.0005 (0.05/100) in at least one genetic model] associated
with hypertriglyc-eridemia (Table
VII). Similar analysis revealed that all 28 SNPs identified in
the EWAS for hypo-HDL-cholesterolemia [P<0.0004 (0.05/112);
Table VIII] and all 65 SNPs
identified in the EWAS for hyper-LDL-cholesterolemia [P<0.0002
(0.05/260); Table IX] were
significantly associated with the respective conditions.
| Table VIIAssociation of SNPs to
hypertriglyceridemia as determined by multivariable logistic
regression analysis. |
Table VII
Association of SNPs to
hypertriglyceridemia as determined by multivariable logistic
regression analysis.
| Gene | SNP | | Dominant
| Recessive
| Additive 1
| Additive 2
|
|---|
| P-value | OR | 95% CI | P-value | OR | 95% CI | P-value | OR | 95% CI | P-value | OR | 95% CI |
|---|
| APOA5 | rs2075291 | G/T | <0.0001 | 1.99 | 1.74-2.28 | <0.0001 | 3.19 | 1.79-5.66 | <0.0001 | 1.93 | 1.68-2.22 | <0.0001 | 3.50 | 1.97-6.22 |
| BUD13 | rs10790162 | G/A | <0.0001 | 1.66 | 1.51-1.83 | <0.0001 | 1.80 | 1.50-2.15 | <0.0001 | 1.58 | 1.42-1.75 | <0.0001 | 2.17 | 1.81-2.62 |
| ZPR1 | rs964184 | C/G | <0.0001 | 1.67 | 1.51-1.84 | <0.0001 | 1.74 | 1.46-2.09 | <0.0001 | 1.59 | 1.44-1.76 | <0.0001 | 2.12 | 1.76-2.56 |
| APOA5 | rs2266788 | T/C | <0.0001 | 1.67 | 1.51-1.84 | <0.0001 | 1.74 | 1.45-2.09 | <0.0001 | 1.59 | 1.44-1.76 | <0.0001 | 2.11 | 1.75-2.55 |
| rs7350481 | C/T | <0.0001 | 1.65 | 1.50-1.82 | <0.0001 | 1.68 | 1.41-2.00 | <0.0001 | 1.58 | 1.42-1.75 | <0.0001 | 2.06 | 1.72-2.47 |
| rs9326246 | G/C | <0.0001 | 1.64 | 1.49-1.81 | <0.0001 | 1.75 | 1.46-2.09 | <0.0001 | 1.56 | 1.41-1.73 | <0.0001 | 2.11 | 1.75-2054 |
| ZPR1 | rs2075290 | T/C | <0.0001 | 1.62 | 1.47-1.78 | <0.0001 | 1.70 | 1.42-2.04 | <0.0001 | 1.54 | 1.39-1.71 | <0.0001 | 2.05 | 1.70-2.46 |
| rs12678919 | A/G | <0.0001 | 0.70 | 0.62-0.79 | 0.0362 | 0.62 | 0.39-0.97 | <0.0001 | 0.71 | 0.63-0.80 | 0.0163 | 0.57 | 0.36-0.90 |
| rs10503669 | C/A | <0.0001 | 0.70 | 0.62-0.79 | 0.0434 | 0.63 | 0.40-0.99 | <0.0001 | 0.71 | 0.63-0.80 | 0.0199 | 0.58 | 0.37-0.92 |
| rs17482753 | G/T | <0.0001 | 0.71 | 0.63-0.80 | 0.0357 | 0.62 | 0.40-0.97 | <0.0001 | 0.72 | 0.64-0.82 | 0.0163 | 0.58 | 0.37-0.90 |
| rs10096633 | C/T | <0.0001 | 0.71 | 0.64-0.80 | 0.0306 | 0.61 | 0.39-0.96 | <0.0001 | 0.72 | 0.64-0.82 | 0.0138 | 0.57 | 0.37-0.89 |
| LPL | rs328 | C/G | <0.0001 | 0.72 | 0.64-0.81 | 0.0253 | 0.61 | 0.39-0.94 | <0.0001 | 0.73 | 0.64-0.82 | 0.0110 | 0.57 | 0.37-0.88 |
| APOA4 | rs5104 | A/G | <0.0001 | 1.38 | 1.25-1.52 | <0.0001 | 1.37 | 1.19-1.58 | <0.0001 | 1.32 | 1.19-1.47 | <0.0001 | 1.60 | 1.37-1.86 |
| rs7016880 | G/C | <0.0001 | 0.72 | 0.64-0.82 | 0.0548 | | | <0.0001 | 0.73 | 0.65-0.83 | 0.0327 | 0.61 | 0.39-0.96 |
| GCKR | rs1260326 | T/C | <0.0001 | 0.75 | 0.68-0.83 | <0.0001 | 0.77 | 0.68-0.88 | <0.0001 | 0.78 | 0.70-0.87 | <0.0001 | 0.67 | 0.58-0.77 |
| GCKR | rs780093 | A/G | <0.0001 | 0.77 | 0.69-0.85 | <0.0001 | 0.77 | 0.68-0.88 | <0.0001 | 0.80 | 0.72-0.89 | <0.0001 | 0.68 | 0.59-0.78 |
| rs1260333 | T/C | <0.0001 | 0.77 | 0.70-0.86 | <0.0001 | 0.77 | 0.68-0.88 | 0.0001 | 0.81 | 0.73-0.90 | <0.0001 | 0.68 | 0.59-0.78 |
| rs11085421 | A/C | <0.0001 | 0.78 | 0.70-0.86 | 0.0032 | 0.66 | 0.49-0.87 | <0.0001 | 0.80 | 0.72-0.89 | 0.0007 | 0.61 | 0.46-0.81 |
| rs1441756 | T/G | <0.0001 | 0.79 | 0.71-0.88 | 0.0125 | 0.70 | 0.53-0.93 | <0.0001 | 0.81 | 0.72-0.90 | 0.0032 | 0.66 | 0.49-0.87 |
| rs2083637 | T/C | <0.0001 | 0.79 | 0.71-0.88 | 0.0166 | 0.71 | 0.54-0.94 | <0.0001 | 0.81 | 0.72-0.90 | 0.0044 | 0.66 | 0.50-0.88 |
| LPL | rs301 | T/C | <0.0001 | 0.79 | 0.72-0.88 | 0.0107 | 0.70 | 0.53-0.92 | 0.0001 | 0.81 | 0.73-0.90 | 0.0027 | 0.66 | 0.50-0.86 |
| LPL | rs13702 | A/G | <0.0001 | 0.80 | 0.72-0.88 | 0.0130 | 0.71 | 0.54-0.93 | 0.0001 | 0.81 | 0.73-0.90 | 0.0034 | 0.66 | 0.50-0.87 |
| LPL | rs15285 | G/A | <0.0001 | 0.80 | 0.72-0.88 | 0.0149 | 0.71 | 0.54-0.94 | 0.0001 | 0.81 | 0.73-0.91 | 0.0040 | 0.67 | 0.51-0.88 |
| rs2954033 | G/A | <0.0001 | 1.26 | 1.14-1.39 | 0.0011 | 1.28 | 1.10-1.49 | 0.0001 | 1.22 | 1.10-1.35 | <0.0001 | 1.42 | 1.21-1.67 |
| LPL | rs326 | A/G | <0.0001 | 0.80 | 0.72-0.88 | 0.0233 | 0.73 | 0.56-0.96 | 0.0001 | 0.81 | 0.73-0.90 | 0.0062 | 0.69 | 0.52-0.90 |
| Table VIIIAssociation of SNPs to
hypo-HDL-cholesterolemia as determined by multivariable logistic
regression analysis. |
Table VIII
Association of SNPs to
hypo-HDL-cholesterolemia as determined by multivariable logistic
regression analysis.
| Gene | SNP | | Dominant
| Recessive
| Additive 1
| Additive 2
|
|---|
| P-value | OR | 95% CI | P-value | OR | 95% CI | P-value | OR | 95% CI | P-value | OR | 95% CI |
|---|
| LPGAT1 | rs150552771 | T/C | <0.0001 | 19.16 | 8.16-45.01 | | | | <0.0001 | 19.16 | 8.16-45.01 | | | |
| APOA5 | rs2075291 | G/T | <0.0001 | 1.85 | 1.55-2.20 | 0.0031 | 2.66 | 1.39-5.07 | <0.0001 | 1.80 | 1.50-2.16 | 0.0012 | 2.91 | 1.52-5.57 |
| COL6A5 | rs200982668 | G/A | <0.0001 | 0.18 | 0.09-0.35 | | | | <0.0001 | 0.18 | 0.09-0.35 | | | |
| ZNF860 | rs140232911 | C/T | <0.0001 | 4.88 | 3.12-7.63 | | | | <0.0001 | 4.88 | 3.12-7.63 | | | |
| rs9261800 | C/G | <0.0001 | 28.11 | 10.15-77.87 | | | | <0.0001 | 28.11 | 10.15-77.87 | | | |
| VPS33B | rs199921354 | C/T | <0.0001 | 0.17 | 0.08-0.34 | | | | <0.0001 | 0.17 | 0.08-0.34 | | | |
| ADGRL3 | rs192210727 | G/T | <0.0001 | 0.19 | 0.10-0.37 | 0.2341 | | | <0.0001 | 0.19 | 0.10-0.38 | 0.9964 | | |
| TMOD4 | rs115287176 | G/A | <0.0001 | 0.17 | 0.08-0.35 | | | | <0.0001 | 0.17 | 0.08-0.35 | | | |
| COL6A3 | rs146092501 | C/T | <0.0001 | 0.17 | 0.08-0.35 | | | | <0.0001 | 0.17 | 0.08-0.35 | | | |
| MARCH1 | rs61734696 | G/T | <0.0001 | 0.19 | 0.10-0.37 | | | | <0.0001 | 0.19 | 0.10-0.37 | | | |
| PLCB2 | rs200787930 | C/T | <0.0001 | 0.19 | 0.10-0.36 | | | | <0.0001 | 0.19 | 0.10-0.36 | | | |
| MOB3C | rs139537100 | C/T | <0.0001 | 0.19 | 0.10-0.37 | | | | <0.0001 | 0.19 | 0.10-0.37 | | | |
| CXCL8 | rs188378669 | G/T | <0.0001 | 0.19 | 0.10-0.36 | | | | <0.0001 | 0.19 | 0.10-0.36 | | | |
| EHD3 | rs116417209 | G/A | <0.0001 | 14.34 | 6.16-33.35 | | | | <0.0001 | 14.34 | 6.16-33.35 | | | |
| ZNF77 | rs146879198 | G/A | <0.0001 | 0.19 | 0.10-0.38 | | | | <0.0001 | 0.19 | 0.10-0.38 | | | |
| OR4F6 | rs141569282 | G/A | <0.0001 | 0.25 | 0.13-0.46 | | | | <0.0001 | 0.25 | 0.13-0.46 | | | |
| ACAD10 | rs11066015 | G/A | <0.0001 | 1.40 | 1.22-1.60 | <0.0001 | 1.56 | 1.26-1.93 | 0.0003 | 1.32 | 1.14-1.52 | <0.0001 | 1.77 | 1.42-2.22 |
| CACNA1D | rs35874056 | G/A | <0.0001 | 92.59 | 11.37-753.91 | | | | <0.0001 | 92.59 | 11.37-753.91 | | | |
| ALDH2 | rs671 | G/A | <0.0001 | 1.39 | 1.21-1.60 | <0.0001 | 1.55 | 1.25-1.92 | 0.0003 | 1.31 | 1.13-1.52 | <0.0001 | 1.76 | 1.40-2.20 |
| rs3764261 | G/T | <0.0001 | 0.68 | 0.58-0.79 | 0.0004 | 0.39 | 0.23-0.66 | <0.0001 | 0.72 | 0.61-0.84 | 0.0001 | 0.35 | 0.21-0.60 |
| rs247616 | C/T | <0.0001 | 0.68 | 0.58-0.79 | 0.0009 | 0.43 | 0.26-0.71 | <0.0001 | 0.71 | 0.61-0.83 | 0.0002 | 0.39 | 0.24-0.64 |
| BRAP | rs3782886 | A/G | <0.0001 | 1.39 | 1.21-1.60 | <0.0001 | 1.52 | 1.24-1.87 | 0.0003 | 1.31 | 1.13-1.52 | <0.0001 | 1.74 | 1.40-2.17 |
| CETP | rs1532624 | G/T | <0.0001 | 0.73 | 0.64-0.84 | <0.0001 | 0.50 | 0.37-0.67 | 0.0021 | 0.80 | 0.69-0.92 | <0.0001 | 0.45 | 0.33-0.61 |
| HECTD4 | rs11066280 | T/A | <0.0001 | 1.42 | 1.23-1.63 | 0.0010 | 1.43 | 1.15-1.76 | <0.0001 | 1.36 | 1.17-1.58 | <0.0001 | 1.66 | 1.32-2.08 |
| HECTD4 | rs2074356 | C/T | <0.0001 | 1.37 | 1.19-1.57 | 0.0016 | 1.45 | 1.15-1.83 | 0.0002 | 1.32 | 1.14-1.52 | <0.0001 | 1.64 | 1.29-2.09 |
| LILRB2 | rs73055442 | C/T | <0.0001 | 51.91 | 10.66-252.93 | | | | <0.0001 | 51.91 | 10.66-252.93 | | | |
| DCLRE1C | rs150854849 | C/T | <0.0001 | 78.09 | 9.17-665.03 | | | | <0.0001 | 78.09 | 9.17-665.03 | | | |
| SPOPL | rs114501427 | G/A | <0.0001 | 3.54 | 2.24-5.61 | | | | <0.0001 | 3.54 | 2.24-5.61 | | | |
| Table IXAssociation of SNPs to
hyper-LDL-cholesterolemia as determined by multivariable logistic
regression analysis. |
Table IX
Association of SNPs to
hyper-LDL-cholesterolemia as determined by multivariable logistic
regression analysis.
| Gene | SNP | | Dominant
| Recessive
| Additive 1
| Additive 2
|
|---|
| P-value | OR | 95% CI | P-value | OR | 95% CI | P-value | OR | 95% CI | P-value | OR | 95% CI |
|---|
| APOE | rs7412 | C/T | <0.0001 | 0.40 | 0.33-0.48 | 0.0738 | | | <0.0001 | 0.40 | 0.33-0.49 | 0.0984 | | |
| COL6A3 | rs146092501 | C/T | <0.0001 | 2.34 | 1.87-2.93 | | | | <0.0001 | 2.34 | 1.87-2.93 | | | |
| TMOD4 | rs115287176 | G/A | <0.0001 | 2.33 | 1.86-2.91 | | | | <0.0001 | 2.33 | 1.86-2.91 | | | |
| CXCL8 | rs188378669 | G/T | <0.0001 | 2.30 | 1.85-2.87 | | | | <0.0001 | 2.30 | 1.85-2.87 | | | |
| VPS33B | rs199921354 | C/T | <0.0001 | 2.31 | 1.85-2.89 | | | | <0.0001 | 2.31 | 1.85-2.89 | | | |
| ZNF77 | rs146879198 | G/A | <0.0001 | 2.30 | 1.84-2.87 | | | | <0.0001 | 2.30 | 1.84-2.87 | | | |
| MOB3C | rs139537100 | C/T | <0.0001 | 2.27 | 1.82-2.83 | | | | <0.0001 | 2.27 | 1.82-2.83 | | | |
| PLCB2 | rs200787930 | C/T | <0.0001 | 2.22 | 1.78-2.77 | | | | <0.0001 | 2.22 | 1.78-2.77 | | | |
| MUC17 | rs78010183 | A/T | <0.0001 | 1.99 | 1.65-2.40 | | | | <0.0001 | 1.99 | 1.65-2.40 | | | |
| MARCH1 | rs61734696 | G/T | <0.0001 | 2.20 | 1.77-2.74 | | | | <0.0001 | 2.20 | 1.77-2.74 | | | |
| APOB | rs13306206 | G/A | <0.0001 | 1.69 | 1.41-2.03 | 0.0145 | 5.13 | 1.38-19.03 | <0.0001 | 1.65 | 1.37-1.98 | 0.0127 | 5.30 | 1.43-19.67 |
| COL6A5 | rs200982668 | G/A | <0.0001 | 2.12 | 1.71-2.63 | | | | <0.0001 | 2.12 | 1.71-2.63 | | | |
| PTCH2 | rs147284320 | C/T | <0.0001 | 2.02 | 1.65-2.49 | | | | <0.0001 | 2.02 | 1.65-2.49 | | | |
| ADGRL3 | rs192210727 | G/T | <0.0001 | 2.14 | 1.72-2.66 | 0.8431 | | | <0.0001 | 2.17 | 1.74-2.71 | 0.8732 | | |
| APOC1 | rs445925 | C/T | <0.0001 | 0.63 | 0.55-0.73 | 0.8258 | | | <0.0001 | 0.63 | 0.54-0.73 | 0.7212 | | |
| HSPA1B | rs6457452 | C/T | <0.0001 | 1.37 | 1.22-1.53 | 0.0284 | 1.51 | 1.04-2.20 | <0.0001 | 1.35 | 1.20-1.52 | 0.0125 | 1.61 | 1.11-2.33 |
| C6orf48 | rs11968400 | C/T | <0.0001 | 1.37 | 1.22-1.53 | 0.0425 | 1.48 | 1.01-2.15 | <0.0001 | 1.36 | 1.21-1.52 | 0.0195 | 1.57 | 1.07-2.29 |
| rs12210887 | G/T | <0.0001 | 1.37 | 1.22-1.53 | 0.0590 | | | <0.0001 | 1.36 | 1.21-1.52 | 0.0272 | 1.53 | 1.05-2.24 |
| UBD | rs64036 | C/A | <0.0001 | 1.28 | 1.17-1.40 | 0.0043 | 1.30 | 1.09-1.56 | <0.0001 | 1.26 | 1.14-1.38 | 0.0002 | 1.42 | 1.18-1.71 |
| CCHCR1 | rs147733073 | C/G | <0.0001 | 1.37 | 1.22-1.53 | 0.1210 | | | <0.0001 | 1.36 | 1.21-1.53 | 0.0592 | | |
| VARS | rs11751198 | G/A | <0.0001 | 1.36 | 1.22-1.52 | 0.0505 | | | <0.0001 | 1.35 | 1.20-1.51 | 0.0238 | 1.56 | 1.06-2.29 |
| rs2596574 | G/A | <0.0001 | 1.36 | 1.22-1.53 | 0.0807 | | | <0.0001 | 1.35 | 1.21-1.52 | 0.0377 | 1.48 | 1.02-2.14 |
| LY6G6F | rs9267547 | G/A | <0.0001 | 1.35 | 1.21-1.51 | 0.0797 | | | <0.0001 | 1.34 | 1.20-1.51 | 0.0370 | 1.47 | 1.02-2.11 |
| NEU1 | rs13118 | T/A | <0.0001 | 1.36 | 1.21-1.52 | 0.0450 | 1.47 | 1.01-2.16 | <0.0001 | 1.34 | 1.20-1.51 | 0.0217 | 1.56 | 1.07-2.29 |
| SLC17A3 | rs34902660 | C/A | <0.0001 | 1.40 | 1.24-1.57 | 0.2131 | | | <0.0001 | 1.40 | 1.24-1.57 | 0.1319 | | |
| FAM65B | rs150142878 | C/T | <0.0001 | 1.48 | 1.29-1.70 | 0.5456 | | | <0.0001 | 1.49 | 1.30-1.71 | 0.4556 | | |
| TUBB | rs9500864 | G/A | <0.0001 | 1.28 | 1.16-1.40 | 0.0025 | 1.36 | 1.11-1.66 | <0.0001 | 1.25 | 1.13-1.38 | 0.0002 | 1.47 | 1.20-1.81 |
| rs204999 | A/G | <0.0001 | 1.37 | 1.22-1.53 | 0.1767 | | | <0.0001 | 1.37 | 1.22-1.54 | 0.1002 | | |
| rs3130663 | A/G | <0.0001 | 1.35 | 1.21-1.49 | 0.1104 | | | <0.0001 | 1.34 | 1.21-1.50 | 0.0388 | 1.39 | 1.02-1.90 |
| VARS | rs5030798 | G/A | <0.0001 | 1.35 | 1.21-1.52 | 0.0609 | | | <0.0001 | 1.34 | 1.19-1.51 | 0.0296 | 1.53 | 1.04-2.24 |
| LY6G6F | rs9267546 | G/A | <0.0001 | 1.35 | 1.21-1.51 | 0.1053 | | | <0.0001 | 1.35 | 1.20-1.51 | 0.0525 | | |
| PRRC2A | rs11538264 | G/A | <0.0001 | 1.36 | 1.21-1.52 | 0.0873 | | | <0.0001 | 1.35 | 1.20-1.51 | 0.0437 | 1.48 | 1.01-2.16 |
| DPCR1 | rs11970154 | G/C | <0.0001 | 1.33 | 1.20-1.47 | 0.1235 | | | <0.0001 | 1.33 | 1.19-1.47 | 0.0445 | 1.37 | 1.01-1.86 |
| MSH5 | rs11754464 | C/T | <0.0001 | 1.35 | 1.21-1.51 | 0.0735 | | | <0.0001 | 1.34 | 1.19-1.51 | 0.0365 | 1.50 | 1.03-2.20 |
| PCSK9 | rs151193009 | C/T | <0.0001 | 0.41 | 0.29-0.59 | 0.4126 | | | <0.0001 | 0.42 | 0.29-0.59 | 0.9943 | | |
| GABBR1 | rs29243 | C/T | <0.0001 | 1.34 | 1.20-1.50 | 0.0562 | | | <0.0001 | 1.33 | 1.19-1.49 | 0.244 | 1.50 | 1.05-2.13 |
|
KIAA0319 | rs4576240 | G/T | <0.0001 | 1.47 | 1.29-1.69 | 0.7773 | | | <0.0001 | 1.49 | 1.29-1.71 | 0.6763 | | |
| LY6G6F | rs17200983 | C/A | <0.0001 | 1.35 | 1.20-1.51 | 0.0873 | | | <0.0001 | 1.34 | 1.19-1.50 | 0.0444 | 1.48 | 1.01-2.16 |
| TNXB | rs140770834 | C/G | <0.0001 | 1.36 | 1.21-1.53 | 0.1723 | | | <0.0001 | 1.36 | 1.21-1.53 | 0.1013 | | |
| TNXB | rs11751545 | A/C | <0.0001 | 1.36 | 1.21-1.53 | 0.1723 | | | <0.0001 | 1.36 | 1.21-1.53 | 0.1013 | | |
| DPCR1 | rs6933400 | C/T | <0.0001 | 1.32 | 1.20-1.47 | 0.1222 | | | <0.0001 | 1.32 | 1.19-1.47 | 0.0456 | 1.37 | 1.01-1.87 |
| LY6G6C | rs117894946 | G/C | <0.0001 | 1.35 | 1.20-1.51 | 0.0871 | | | <0.0001 | 1.34 | 1.19-1.50 | 0.0445 | 1.48 | 1.01-2.16 |
| SLC44A4 | rs117127493 | G/C | <0.0001 | 1.36 | 1.21-1.52 | 0.2033 | | | <0.0001 | 1.36 | 1.20-1.53 | 0.1218 | | |
| ZSCAN26 | rs76463649 | A/G | <0.0001 | 1.36 | 1.21-1.52 | 0.3394 | | | <0.0001 | 1.36 | 1.21-1.53 | 0.2111 | | |
| DHX16 | rs7749235 | T/C | <0.0001 | 1.28 | 1.16-1.40 | 0.0064 | 1.37 | 1.09-1.72 | <0.0001 | 1.25 | 1.13-1.38 | 0.0010 | 1.47 | 1.17-1.86 |
| LOC105- | rs9264942 | T/C | <0.0001 | 1.26 | 1.14-1.39 | 0.0004 | 1.24 | 1.10-1.39 | 0.0002 | 1.21 | 1.09-1.35 | <0.0001 | 1.38 | 1.21-1.58 |
| 375015 | | | | | | | | | | | | | | |
| ZSCAN31 | rs6922302 | C/G | <0.0001 | 1.36 | 1.21-1.52 | 0.3391 | | | <0.0001 | 1.36 | 1.21-1.53 | 0.2111 | | |
| MDC1 | rs2269702 | A/G | <0.0001 | 1.27 | 1.16-1.40 | 0.0048 | 1.39 | 1.11-1.75 | <0.0001 | 1.25 | 1.13-1.38 | 0.0007 | 1.49 | 1.18-1.88 |
| MDC1 | rs28986465 | C/T | <0.0001 | 1.27 | 1.16-1.40 | 0.0048 | 1.39 | 1.11-1.75 | <0.0001 | 1.24 | 1.13-1.38 | 0.0007 | 1.49 | 1.18-1.88 |
| MDC1 | rs2075015 | G/A | <0.0001 | 1.27 | 1.16-1.40 | 0.0048 | 1.39 | 1.11-1.75 | <0.0001 | 1.24 | 1.13-1.38 | 0.0007 | 1.49 | 1.18-1.88 |
| DHX16 | rs2285321 | T/C | <0.0001 | 1.27 | 1.16-1.40 | 0.0056 | 1.38 | 1.10-1.73 | <0.0001 | 1.25 | 1.13-1.38 | 0.0008 | 1.48 | 1.18-1.87 |
| SKIV2L | rs492899 | A/G | <0.0001 | 1.34 | 1.20-1.51 | 0.1465 | | | <0.0001 | 1.34 | 1.19-1.51 | 0.0843 | | |
| MDC1 | rs6924270 | A/G | <0.0001 | 1.27 | 1.16-1.40 | 0.0055 | 1.38 | 1.10-1.74 | <0.0001 | 1.25 | 1.13-1.38 | 0.0008 | 1.48 | 1.18-1.87 |
| rs2508015 | C/T | <0.0001 | 1.30 | 1.17-1.45 | 0.0409 | 1.39 | 1.01-1.91 | <0.0001 | 1.29 | 1.16-1.44 | 0.0166 | 1.47 | 1.07-2.02 |
| TUBB | rs25527 | C/T | <0.0001 | 1.26 | 1.14-1.38 | 0.0028 | 1.35 | 1.11-1.64 | <0.0001 | 1.23 | 1.11-1.35 | 0.0002 | 1.45 | 1.19-1.78 |
| PPP1R18 | rs9468805 | G/A | <0.0001 | 1.27 | 1.15-1.39 | 0.0048 | 1.39 | 1.11-1.75 | <0.0001 | 1.24 | 1.12-1.37 | 0.0007 | 1.49 | 1.18-1.87 |
| DHX16 | rs6937357 | T/C | <0.0001 | 1.27 | 1.15-1.39 | 0.0048 | 1.39 | 1.11-1.75 | <0.0001 | 1.24 | 1.12-1.37 | 0.0007 | 1.49 | 1.18-1.87 |
| HECTD4 | rs11066280 | T/A | <0.0001 | 1.26 | 1.15-1.38 | 0.0083 | 1.23 | 1.05-1.43 | <0.0001 | 1.24 | 1.13-1.37 | 0.0002 | 1.36 | 1.16-1.59 |
| TRIM40 | rs2523995 | G/A | <0.0001 | 1.34 | 1.20-1.49 | 0.2147 | | | <0.0001 | 1.34 | 1.20-1.50 | 0.1152 | | |
| PPP1R18 | rs6457254 | C/T | <0.0001 | 1.26 | 1.15-1.39 | 0.0063 | 1.37 | 1.09-1.72 | <0.0001 | 1.24 | 1.12-1.37 | 0.0010 | 1.47 | 1.17-1.85 |
| TUBB | rs3132584 | C/A | <0.0001 | 1.25 | 1.14-1.37 | 0.0035 | 1.34 | 1.10-1.63 | <0.0001 | 1.22 | 1.11-1.35 | 0.0003 | 1.44 | 1.18-1.76 |
| rs3130685 | T/C | 0.0005 | 0.80 | 0.71-0.91 | <0.0001 | 0.82 | 0.74-0.90 | 0.0264 | 0.86 | 0.75-0.98 | <0.0001 | 0.73 | 0.64-0.84 |
| GIT2 | rs2292354 | C/T | 0.0001 | 0.83 | 0.76-0.91 | <0.0001 | 0.68 | 0.56-0.82 | 0.0050 | 0.87 | 0.79-0.96 | <0.0001 | 0.64 | 0.52-0.78 |
| PPP1R18 | rs2394392 | T/G | <0.0001 | 1.26 | 1.15-1.39 | 0.0074 | 1.36 | 1.09-1.71 | <0.0001 | 1.24 | 1.12-1.37 | 0.0012 | 1.46 | 1.16-1.84 |
| rs2524272 | T/C | <0.0001 | 1.22 | 1.11-1.34 | 0.0010 | 1.30 | 1.11-1.52 | 0.0007 | 1.18 | 1.07-1.30 | <0.0001 | 1.40 | 1.19-1.65 |
Association of identified SNPs to serum
concentrations of triglycerides, HDL-cholesterol or
LDL-cholesterol
The association of the genotypes of identified SNPs
to serum concentrations of triglycerides, HDL-cholesterol, or
LDL-cholesterol was examined by one-way analysis of variance. The
25 SNPs identified in the EWAS for hypertriglyceridemia were all
significantly associated with serum triglyceride concentration
[P<0.0020 (0.05/25); Table X].
Among the 28 SNPs identified in the EWAS for
hypo-HDL-cholesterolemia, 27 polymorphisms were significantly
associated with the serum HDL-cholesterol level [P<0.0018
(0.05/28)], whereas rs114501427 of SPOPL was not related to
this parameter (Table XI). Among
the 65 SNPs identified in the EWAS for hyper-LDL-cholesterolemia,
37 SNPs were significantly associated with serum LDL-cholesterol
concentration [P<0.0008 (0.05/65); Table XII]. It is possible that the
lack of significant correlation between the remaining 28 SNPs and
the serum LDL-cholesterol levels may be due to effects of medical
treatment.
| Table XAssociation of SNPs identified in the
present study to serum triglyceride concentration. |
Table X
Association of SNPs identified in the
present study to serum triglyceride concentration.
| Gene | SNP | Serum triglycerides
(mmol/l) | P-value |
|---|
| APOA5 | rs2075291 | G/T | GG | GT | TT | <0.0001 |
| | | 1.39±0.98 | 1.83±1.55 | 2.56±2.27 | |
| BUD13 | rs10790162 | G/A | GG | GA | AA | <0.0001 |
| | | 1.35±1.00 | 1.54±1.18 | 1.77±1.31 | |
| ZPR1 | rs964184 | C/G | CC | CG | GG | <0.0001 |
| | | 1.35±1.00 | 1.55±1.18 | 1.75±1.30 | |
| APOA5 | rs2266788 | T/C | TT | TC | CC | <0.0001 |
| | | 1.35±1.00 | 1.54±1.17 | 1.76±1.30 | |
| rs7350481 | C/T | CC | CT | TT | <0.0001 |
| | | 1.34±0.95 | 1.56±1.24 | 1.68±1.19 | |
| rs9326246 | G/C | GG | GC | CC | <0.0001 |
| | | 1.35±1.00 | 1.54±1.18 | 1.74±1.28 | |
| ZPR1 | rs2075290 | T/C | TT | TC | CC | <0.0001 |
| | | 1.36±1.01 | 1.54±1.17 | 1.74±1.29 | |
| rs12678919 | A/G | AA | AG | GG | <0.0001 |
| | | 1.49±1.11 | 1.33±1.03 | 1.32±1.37 | |
| rs10503669 | C/A | CC | CA | AA | <0.0001 |
| | | 1.49±1.11 | 1.33±1.03 | 1.32±1.38 | |
| rs17482753 | G/T | GG | GT | TT | <0.0001 |
| | | 1.49±1.11 | 1.33±1.02 | 1.31±1.35 | |
| rs10096633 | C/T | CC | CT | TT | <0.0001 |
| | | 1.49±1.11 | 1.33±1.03 | 1.31±1.34 | |
| LPL | rs328 | C/G | CC | CG | GG | <0.0001 |
| | | 1.49±1.11 | 1.33±1.02 | 1.33±1.33 | |
| APOA4 | rs5104 | A/G | AA | AG | GG | <0.0001 |
| | | 1.37±1.00 | 1.49±1.12 | 1.57±1.28 | |
| rs7016880 | G/C | GG | GC | CC | <0.0001 |
| | | 1.49±1.11 | 1.33±1.04 | 1.30±1.35 | |
| GCKR | rs1260326 | T/C | TT | TC | CC | <0.0001 |
| | | 1.54±1.14 | 1.44±1.14 | 1.33±0.89 | |
| GCKR | rs780093 | A/G | AA | AG | GG | <0.0001 |
| | | 1.53±1.12 | 1.45±1.16 | 1.33±0.87 | |
| rs1260333 | T/C | TT | TC | CC | <0.0001 |
| | | 1.53±1.12 | 1.45±1.16 | 1.32±0.87 | |
| rs11085421 | A/C | AA | AC | CC | <0.0001 |
| | | 1.49±1.14 | 1.39±1.05 | 1.27±0.70 | |
| rs1441756 | T/G | TT | TG | GG | <0.0001 |
| | | 1.49±1.12 | 1.38±1.04 | 1.34±1.16 | |
| rs2083637 | T/C | TT | TC | CC | <0.0001 |
| | | 1.49±1.12 | 1.38±1.05 | 1.34±1.16 | |
| LPL | rs301 | T/C | TT | TC | CC | <0.0001 |
| | | 1.49±1.11 | 1.38±1.08 | 1.34±1.14 | |
| LPL | rs13702 | A/G | AA | AG | GG | <0.0001 |
| | | 1.50±1.12 | 1.38±1.04 | 1.34±1.15 | |
| LPL | rs15285 | G/A | GG | GA | AA | <0.0001 |
| | | 1.50±1.12 | 1.38±1.04 | 1.34±1.15 | |
| rs2954033 | G/A | GG | GA | AA | 0.0007 |
| | | 1.41±1.02 | 1.47±1.16 | 1.55±1.18 | |
| LPL | rs326 | A/G | AA | AG | GG | <0.0001 |
| | | 1.49±1.11 | 1.38±1.07 | 1.35±1.14 | |
| Table XIAssociation of SNPs identified in the
present study to serum HDL-cholesterol concentration. |
Table XI
Association of SNPs identified in the
present study to serum HDL-cholesterol concentration.
| Gene | SNP | Serum
HDL-cholesterol (mmol/l) | P-value |
|---|
| LPGAT1 | rs150552771 | T/C | TT | TC | | <0.0001 |
| | | 1.54±0.47 | 1.11±0.63 | | |
| APOA5 | rs2075291 | G/T | GG | GT | TT | <0.0001 |
| | | 1.56±0.47 | 1.42±0.45 | 1.29±0.40 | |
|
COL6A5 | rs200982668 | G/A | GG | GA | | <0.0001 |
| | | 1.53±0.47 | 1.69±0.44 | | |
|
ZNF860 | rs140232911 | C/T | CC | CT | | 0.0003 |
| | | 1.54±0.47 | 1.37±0.59 | | |
| rs9261800 | C/G | CC | CG | | <0.0001 |
| | | 1.54±0.47 | 0.98±0.56 | | |
|
VPS33B | rs199921354 | C/T | CC | CT | | <0.0001 |
| | | 1.53±0.47 | 1.69±0.44 | | |
|
ADGRL3 | rs192210727 | G/T | GG | GT | TT | <0.0001 |
| | | 1.53±0.47 | 1.67±0.44 | 1.91±0.49 | |
| TMOD4 | rs115287176 | G/A | GG | GA | | <0.0001 |
| | | 1.53±0.47 | 1.68±0.44 | | |
|
COL6A3 | rs146092501 | C/T | CC | CT | | <0.0001 |
| | | 1.53±0.47 | 1.68±0.44 | | |
|
MARCH1 | rs61734696 | G/T | GG | GT | | <0.0001 |
| | | 1.53±0.47 | 1.69±0.44 | | |
| PLCB2 | rs200787930 | C/T | CC | CT | | <0.0001 |
| | | 1.53±0.47 | 1.69±0.44 | | |
| MOB3C | rs139537100 | C/T | CC | CT | | <0.0001 |
| | | 1.53±0.47 | 1.68±0.44 | | |
| CXCL8 | rs188378669 | G/T | GG | GT | | <0.0001 |
| | | 1.53±0.47 | 1.69±0.44 | | |
| EHD3 | rs116417209 | G/A | GG | GA | | <0.0001 |
| | | 1.54±0.47 | 1.09±0.48 | | |
| ZNF77 | rs146879198 | G/A | GG | GA | | <0.0001 |
| | | 1.53±0.47 | 1.69±0.44 | | |
| OR4F6 | rs141569282 | G/A | GG | GA | | <0.0001 |
| | | 1.48±0.46 | 1.72±0.43 | | |
|
ACAD10 | rs11066015 | G/A | GG | GA | AA | <0.0001 |
| | | 1.57±0.48 | 1.51±0.47 | 1.43±0.43 | |
|
CACNA1D | rs35874056 | G/A | GG | GA | | <0.0001 |
| | | 1.51±0.47 | 0.94±0.64 | | |
| ALDH2 | rs671 | G/A | GG | GA | AA | <0.0001 |
| | | 1.57±0.48 | 1.51±0.47 | 1.43±0.43 | |
| rs3764261 | G/T | GG | GT | TT | <0.0001 |
| | | 1.50±0.46 | 1.59±0.48 | 1.72±0.49 | |
| rs247616 | C/T | CC | CT | TT | <0.0001 |
| | | 1.50±0.46 | 1.59±0.48 | 1.71±0.50 | |
| BRAP | rs3782886 | A/G | AA | AG | GG | <0.0001 |
| | | 1.57±0.48 | 1.52±0.47 | 1.44±0.43 | |
| CETP | rs1532624 | G/T | GG | GT | TT | <0.0001 |
| | | 1.50±0.46 | 1.56±0.47 | 1.64±0.48 | |
|
HECTD4 | rs11066280 | T/A | TT | TA | AA | <0.0001 |
| | | 1.57±0.47 | 1.52±0.47 | 1.44±0.42 | |
|
HECTD4 | rs2074356 | C/T | CC | CT | TT | <0.0001 |
| | | 1.57±0.48 | 1.51±0.47 | 1.44±0.43 | |
|
LILRB2 | rs73055442 | C/T | CC | CT | | <0.0001 |
| | | 1.54±0.47 | 0.88±0.31 | | |
|
DCLRE1C | rs150854849 | C/T | CC | CT | | <0.0001 |
| | | 1.54±0.47 | 0.92±0.68 | | |
| SPOPL | rs114501427 | G/A | GG | GA | | 0.0041 |
| | | 1.54±0.47 | 1.41±0.58 | | |
| Table XIIAssociation of SNPs identified in
the present study to serum LDL-cholesterol concentration. |
Table XII
Association of SNPs identified in
the present study to serum LDL-cholesterol concentration.
| Gene | SNP | Serum LD
L-cholesterol (mmol/l) | P-value |
|---|
| APOE | rs7412 | C/T | CC | CT | TT |
<0.0001 |
| | | 3.20±0.85 | 2.74±0.80 | 2.39±1.48 | |
|
COL6A3 | rs146092501 | C/T | CC | CT | | 0.1168 |
| | | 3.16±0.86 | 3.24±0.82 | | |
| TMOD4 | rs115287176 | G/A | GG | GA | | 0.1624 |
| | | 3.16±0.86 | 3.23±0.82 | | |
| CXCL8 | rs188378669 | G/T | GG | GT | | 0.1158 |
| | | 3.16±0.86 | 3.24±0.82 | | |
|
VPS33B | rs199921354 | C/T | CC | CT | | 0.1121 |
| | | 3.16±0.86 | 3.24±0.81 | | |
| ZNF77 | rs146879198 | G/A | GG | GA | | 0.2277 |
| | | 3.16±0.86 | 3.22±0.81 | | |
| MOB3C | rs139537100 | C/T | CC | CT | | 0.2422 |
| | | 3.16±0.86 | 3.21±0.82 | | |
| PLCB2 | rs200787930 | C/T | CC | CT | | 0.1860 |
| | | 3.16±0.86 | 3.22±0.81 | | |
| MUC17 | rs78010183 | A/T | AA | AT | | 0.0096 |
| | | 3.15±0.87 | 3.26±0.78 | | |
|
MARCH1 | rs61734696 | G/T | GG | GT | | 0.3029 |
| | | 3.16±0.86 | 3.21±0.80 | | |
| APOB | rs13306206 | G/A | GG | GA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.42±0.98 | 4.20±0.86 | |
|
COL6A5 | rs200982668 | G/A | GG | GA | | 0.3549 |
| | | 3.16±0.86 | 3.20±0.80 | | |
| PTCH2 | rs147284320 | C/T | CC | CT | | 0.5075 |
| | | 3.17±0.82 | 3.20±0.82 | | |
|
ADGRL3 | rs192210727 | G/T | GG | GT | TT | 0.6309 |
| | | 3.16±0.86 | 3.20±0.81 | 2.99±0.82 | |
| APOC1 | rs445925 | C/T | CC | CT | TT |
<0.0001 |
| | | 3.19±0.86 | 2.96±0.86 | 2.99±1.21 | |
|
HSPA1B | rs6457452 | C/T | CC | CT | TT |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.38±0.88 | |
|
C6orf48 | rs11968400 | C/T | CC | CT | TT |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.37±0.88 | |
| rs12210887 | G/T | GG | GT | TT |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.37±0.89 | |
| UBD | rs64036 | C/A | CC | CA | AA |
0.0004 |
| | | 3.13±0.85 | 3.19±0.87 | 3.27±0.85 | |
|
CCHCR1 | rs147733073 | C/G | CC | CG | GG |
<0.0001 |
| | | 3.14±0.85 | 3.23±0.89 | 3.37±0.88 | |
| VARS | rs11751198 | G/A | GG | GA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.37±0.89 | |
| rs2596574 | G/A | GG | GA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.35±0.86 | |
|
LY6G6F | rs9267547 | G/A | GG | GA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.23±0.89 | 3.37±0.89 | |
| NEU1 | rs13118 | T/A | TT | TA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.37±0.89 | |
|
SLC17A3 | rs34902660 | C/A | CC | CA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.26±0.90 | 3.35±0.91 | |
|
FAM65B | rs150142878 | C/T | CC | CT | TT |
<0.0001 |
| | | 3.14±0.85 | 3.30±0.94 | 3.18±0.88 | |
| TUBB | rs9500864 | G/A | GG | GA | AA |
0.0002 |
| | | 3.13±0.85 | 3.19±0.87 | 3.28±0.90 | |
| rs204999 | A/G | AA | AG | GG |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.36±0.91 | |
| rs3130663 | A/G | AA | AG | GG | 0.0010 |
| | | 3.14±0.85 | 3.22±0.88 | 3.22±0.90 | |
| VARS | rs5030798 | G/A | GG | GA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.37±0.89 | |
|
LY6G6F | rs9267546 | G/A | GG | GA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.36±0.87 | |
|
PRRC2A | rs11538264 | G/A | GG | GA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.90 | 3.36±0.88 | |
| DPCR1 | rs11970154 | G/C | GG | GC | CC |
<0.0001 |
| | | 3.13±0.85 | 3.23±0.89 | 3.32±0.86 | |
| MSH5 | rs11754464 | C/T | CC | CT | TT |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.37±0.89 | |
| PCSK9 | rs151193009 | C/T | CC | CT | TT |
<0.0001 |
| | | 3.17±0.86 | 2.77±0.73 | 2.05 | |
|
GABBR1 | rs29243 | C/T | CC | CT | TT |
0.0001 |
| | | 3.14±0.85 | 3.23±0.89 | 3.34±0.86 | |
|
KIAA0319 | rs4576240 | G/T | GG | GT | TT |
<0.0001 |
| | | 3.14±0.85 | 3.30±0.93 | 3.17±0.89 | |
|
LY6G6F | rs17200983 | C/A | CC | CA | AA |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.36±0.88 | |
| TNXB | rs140770834 | C/G | CC | CG | GG |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.37±0.92 | |
| TNXB | rs11751545 | A/C | AA | AC | CC |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.37±0.92 | |
| DPCR1 | rs6933400 | C/T | CC | CT | TT |
<0.0001 |
| | | 3.13±0.85 | 3.23±0.89 | 3.32±0.87 | |
|
LY6G6C | rs117894946 | G/C | GG | GC | CC |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.36±0.88 | |
|
SLC44A4 | rs117127493 | G/C | GG | GC | CC |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.36±0.91 | |
|
ZSCAN26 | rs76463649 | A/G | AA | AG | GG |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.89 | 3.37±0.85 | |
| DHX16 | rs7749235 | T/C | TT | TC | CC | 0.0010 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.90 | |
|
LOC105375015 | rs9264942 | T/C | TT | TC | CC |
0.0001 |
| | | 3.13±0.84 | 3.15±0.87 | 3.25±0.88 | |
|
ZSCAN31 | rs6922302 | C/G | CC | CG | GG |
<0.0001 |
| | | 3.14±0.85 | 3.24±0.90 | 3.37±0.85 | |
| MDC1 | rs2269702 | A/G | AA | AG | GG | 0.0013 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.91 | |
| MDC1 | rs28986465 | C/T | CC | CT | TT | 0.0013 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.91 | |
| MDC1 | rs2075015 | G/A | GG | GA | AA | 0.0013 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.91 | |
| DHX16 | rs2285321 | T/C | TT | TC | CC | 0.0011 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.91 | |
|
SKIV2L | rs492899 | A/G | AA | AG | GG |
0.0001 |
| | | 3.14±0.85 | 3.23±0.89 | 3.36±0.91 | |
| MDC1 | rs6924270 | A/G | AA | AG | GG | 0.0012 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.91 | |
| rs2508015 | C/T | CC | CT | TT |
0.0005 |
| | | 3.14±0.85 | 3.22±0.88 | 3.31±0.86 | |
| TUBB | rs25527 | C/T | CC | CT | TT | 0.0008 |
| | | 3.14±0.86 | 3.19±0.86 | 3.27±0.89 | |
|
PPP1R18 | rs9468805 | G/A | GG | GA | AA | 0.0016 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.91 | |
| DHX16 | rs6937357 | T/C | TT | TC | CC | 0.0016 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.91 | |
|
HECTD4 | rs11066280 | T/A | TT | TA | AA |
0.0005 |
| | | 3.12±0.88 | 3.19±0.85 | 3.24±0.81 | |
|
TRIM40 | rs2523995 | G/A | GG | GA | AA | 0.0008 |
| | | 3.14±0.85 | 3.22±0.89 | 3.32±0.87 | |
|
PPP1R18 | rs6457254 | C/T | CC | CT | TT | 0.0010 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.91 | |
| TUBB | rs3132584 | C/A | CC | CA | AA | 0.0008 |
| | | 3.13±0.86 | 3.19±0.86 | 3.27±0.90 | |
| rs3130685 | T/C | TT | TC | CC | 0.0234 |
| | | 3.20±0.89 | 3.14±0.84 | 3.14±0.85 | |
| GIT2 | rs2292354 | C/T | CC | CT | TT | 0.0077 |
| | | 3.18±0.86 | 3.15±0.87 | 3.05±0.82 | |
|
PPP1R18 | rs2394392 | T/G | TT | TG | GG | 0.0013 |
| | | 3.14±0.85 | 3.20±0.87 | 3.28±0.91 | |
| rs2524272 | T/C | TT | TC | CC |
0.0005 |
| | | 3.13±0.86 | 3.18±0.87 | 3.27±0.87 | |
Linkage disequilibrium analysis
The linkage disequilibrium (LD) was assessed among
SNPs associated with hypertriglyceridemia,
hypo-HDL-cholesterolemia, or hyper-LDL-cholesterolemia. For the
hypertriglyceridemia study, strong LD was apparent among rs1260326
and rs780093 of GCKR and rs1260333 at chromosome 2p23
[square of the correlation coefficient (r2),
0.876-0.983]. An LD plot for the 12 SNPs located at chromosomal
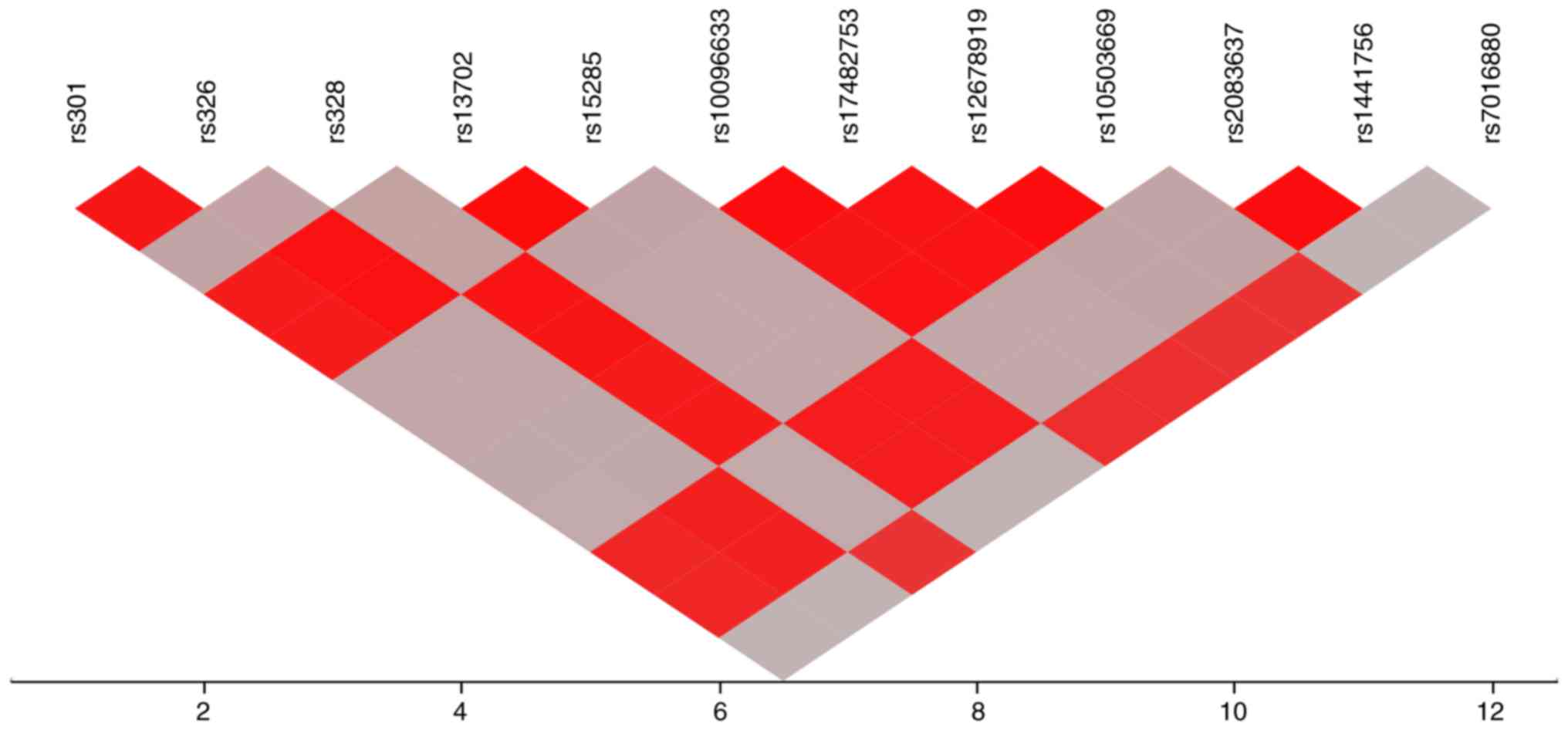
region 8p21.3 is illustrated in Fig.
1. Strong LD was observed among rs301, rs326, rs13702, and
rs15285 of LPL, rs2083637 and rs1441756
(r2, 0.942-0.988), as well as among rs328 of
LPL, rs10096633, rs17482753, rs12678919, rs10503669 and
rs7016880 (r2, 0.910-0.999). An LD plot for the
eight SNPs located at chromosomal region 11q23.3 is illustrated in
Fig. 2. Significant LD was
apparent among rs10790162 of BUD13, rs7350481, rs9326246,
both rs964184 and rs2075290 of ZPR1, and rs2266788 of
APOA5 (r2, 0.687-0.990).
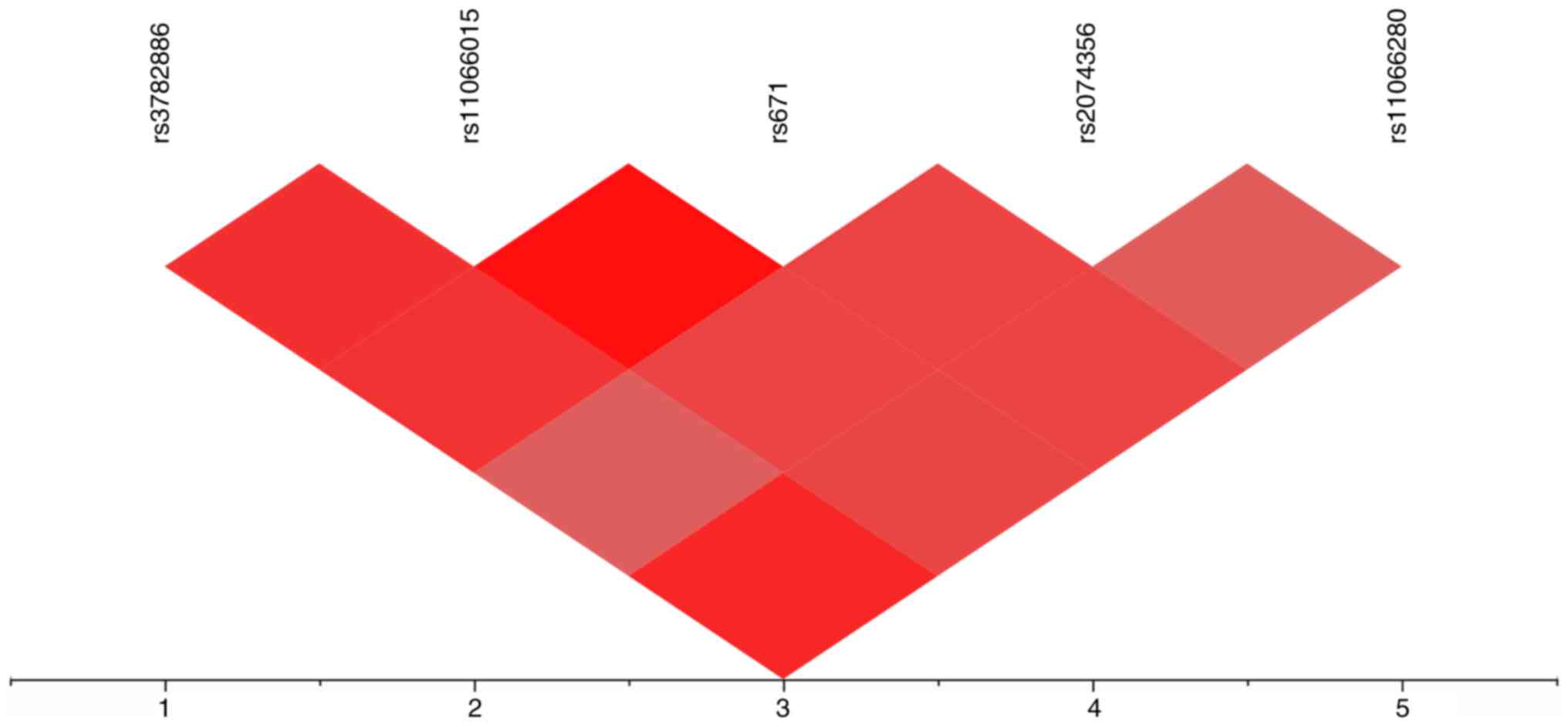
For the hypo-HDL-cholesterolemia study, significant
LD was apparent between rs139537100 of MOB3C and rs115287176
of TMOD4 (r2, 0.984); between rs116417209
of EHD3 and rs114501427 of SPOPL
(r2, 0.703); between rs140232911 of ZNF860
and rs35874056 of CACNA1D (r2, 1.00);
among rs192210727 of ADGRL3, rs188378669 of CXCL8,
and rs61734696 of MARCH1 (r2, 0.901 to
0.972); between rs199921354 of VPS33B and rs141569282 of
OR4F6 (r2, 0.994); and between rs247616
and rs3764261 at chromosome 16q13 (r2, 0.992). An
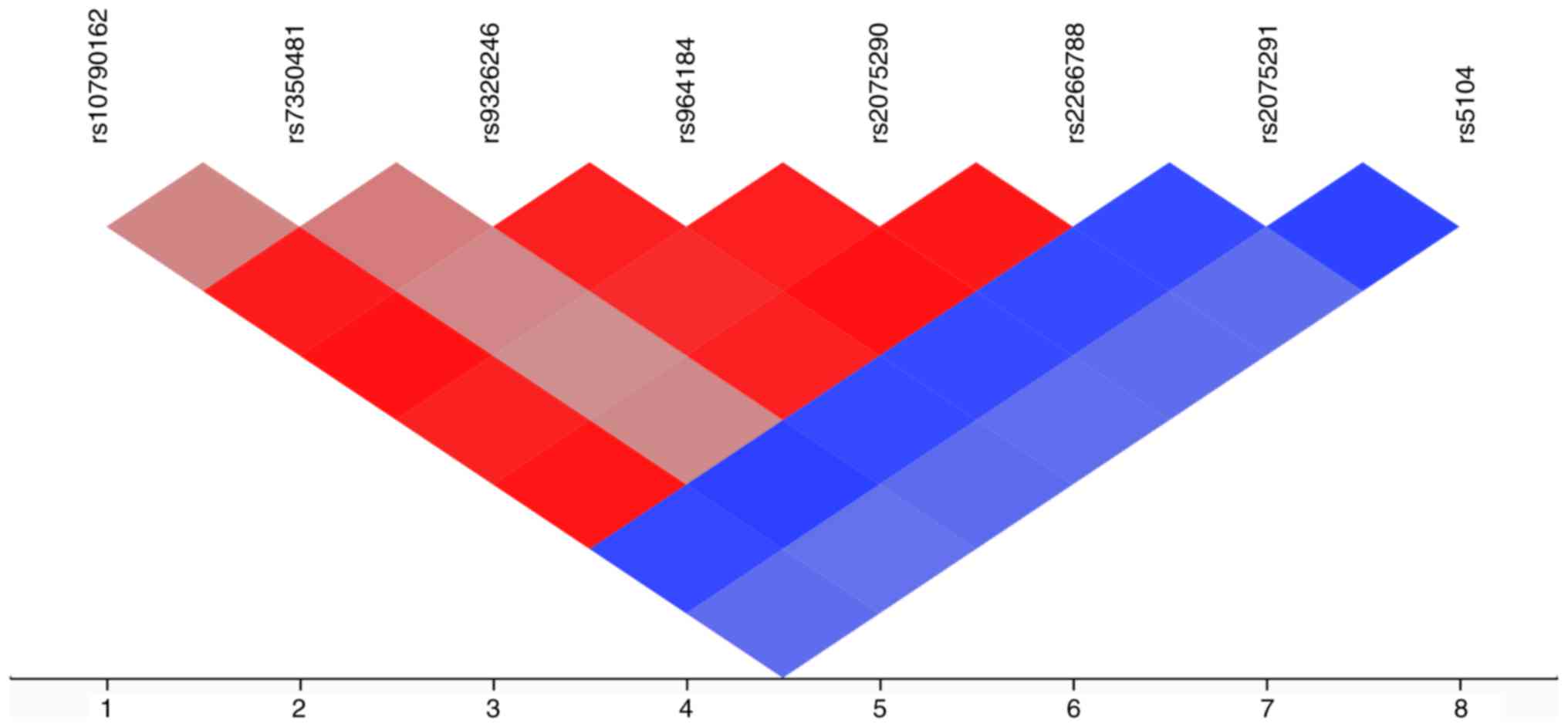
LD plot for the five SNPs located at chromosomal region 12q24.12 to
12q24.13 is illustrated in Fig.
3. Significant LD was detected among rs3782886 of BRAP,
rs11066015 of ACAD10, rs671 of ALDH2, and rs2074356
and rs11066280 of HECTD4 (r2,
0.813-0.995).
For the hyper-LDL-cholesterolemia study, significant
LD was apparent among rs147284320 of PTCH2, rs139537100 of
MOB3C and rs115287176 of TMOD4 (r2,
0.828-0.984); among rs192210727 of ADGRL3, rs188378669 of
CXCL8, and rs61734696 of MARCH1
(r2, 0.901-0.972); and between rs200787930 of
PLCB2 and rs199921354 of VPS33B
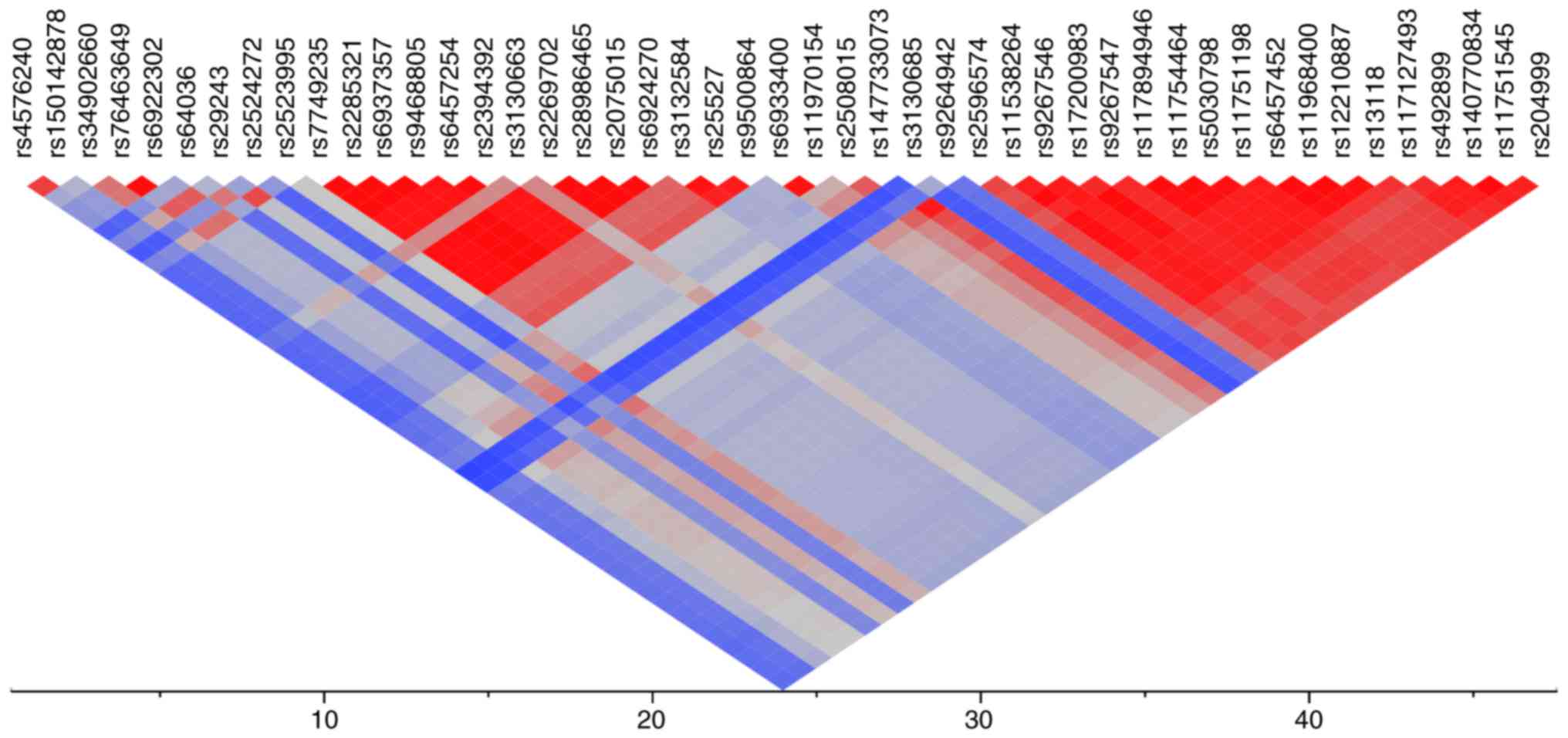
(r2, 0.994). An LD plot for the 47 SNPs located
at chromosomal region 6p22.3 to 6p21.3 is illustrated in Fig. 4. There were two major LD blocks.
The first LD block (r2, 0.571-1.000) comprised
rs7749235, rs2285321, and rs6937357 of DHX16; rs9468805,
rs6457254, and rs2394392 of PPP1R18; rs3130663 at 6p21.3;
rs2269702, rs28986465, rs2075015, and rs6924270 of MDC1; and
rs3132584, rs25527, and rs9500864 of TUBB. The second LD
block (r2, 0.762-1.000) comprised rs147733073 of
CCHCR1; rs2596574 at 6p21.3; rs11538264 of PRRC2A;
rs9267546, rs17200983, and rs9267547 of LY6G6F; rs117894946
of LY6G6C; rs11754464 of MSH5; rs5030798 and
rs11751198 of VARS; rs6457452 of HSPA1B; rs11968400
of C6orf48; rs12210887 at 6p21.3; rs13118 of NEU1;
rs117127493 of SLC44A4; rs492899 of SKIV2L;
rs140770834 and rs11751545 of TNXB; and rs204999 at 6p21.3.
Significant LD was also observed between rs4576240 of
KIAA0319 and rs150142878 of FAM65B
(r2, 0.893); among rs34902660 of SLC17A3,
rs76463649 of ZSCAN26, rs6922302 of ZSCAN31, rs29243
of GABBR1, and rs2523995 of TRIM40
(r2, 0.601-0.995); between rs64036 of UBD
and rs2524272 at 6p22.1 (r2, 0.753); and among
rs6933400 and rs11970154 of DPCR1, rs2508015 at 6p21.3, and
rs147733073 of CCHCR1 (r2,
0.596-0.987).
Relation of SNPs identified in the
present study to dyslipidemia-related phenotypes examined in
previous GWASs
The relation of genes, chromosomal loci and SNPs
identified in the present study was further analyzed against
phenotypes previously examined by GWASs available in the
Genome-Wide Repository of Associations Between SNPs and Phenotypes
(GRASP) Search v.2.0.0.0 (https://grasp.nhlbi.nih.gov/Search.aspx) developed by
the Information Technology and Applications Center, National Center
for Biotechnology Information, National Heart, Lung, and Blood
Institute, National Institutes of Health (Bethesda, MD, USA)
(33,34).
In the hypertriglyceridemia study, GCKR,
chromosomal region 2p23, LPL, region 8p21.3, region 8q24.2,
BUD13, region 11q23.3, ZPR1, APOA5, and
APOA4 have been previously reported to be associated with
circulating triglyceride concentrations, whereas chromosome 19p12
had not been related to triglyceride levels or other
dyslipidemia-related traits (Table
XIII).
| Table XIIIRelation of genes, chromosomal loci,
and SNPs associated with hypertriglyceridemia in the present study
to previously examined dyslipidemia-related phenotypes. |
Table XIII
Relation of genes, chromosomal loci,
and SNPs associated with hypertriglyceridemia in the present study
to previously examined dyslipidemia-related phenotypes.
| Gene/chr
locus | SNP | Chr | Position | Previously
examined phenotypes |
|---|
| GCKR | rs1260326 | 2 | 27508073 | Triglycerides
(20686565, 21423719, 23063622, 22629316, 19060906, 18193043) |
| rs780093 | 2 | 27519736 | |
| 2p23 | rs1260333 | 2 | 27525757 | Triglycerides
(20686565, 19060906, 20864672, 19802338) |
| LPL | rs301 | 8 | 19959423 | Triglycerides
(20686565) |
| rs326 | | 19961928 | |
| rs328 | | 19962213 | |
| rs13702 | | 19966981 | |
| rs15285 | | 19967156 | |
| 8p21.3 | rs10096633 | 8 | 19973410 | Triglycerides
(20686565, 19060906, 19060911, 21943158, 20139978, 23063622,
23505323, 19060910, 19802338) |
| 8p21.3 | rs17482753 | 8 | 19975135 | Triglycerides
(20686565, 23063622, 19060906, 18193043, 21943158, 18179892,
19913121, 17463246) |
| 8p21.3 | rs12678919 | 8 | 19986711 | Triglycerides
(20686565, 19060906, 21909109, 21943158, 20139978, 23063622,
23505323) |
| 8p21.3 | rs10503669 | 8 | 19990179 | Triglycerides
(20686565, 23063622, 21909109, 19060906, 18193043, 21943158,
19913121) |
| 8p21.3 | rs2083637 | 8 | 20007664 | Triglycerides
(20686565, 19060906, 19060911, 21943158, 19802338, 23063622) |
| 8p21.3 | rs1441756 | 8 | 20010875 | Triglycerides
(20686565, 19060906) |
| 8p21.3 | rs7016880 | 8 | 20019235 | Triglycerides
(20686565, 19060906) |
| 8q24.2 | rs2954033 | 8 | 125481504 | Triglycerides
(22479202, 20686565, 23063622, 19060906, 21386085) |
| BUD13 | rs10790162 | 11 | 116639104 | Triglycerides
(20686565, 23202125, 23063622, 20139978, 19060906, 20864672,
18193046) |
| 11q23.3 | rs7350481 | 11 | 116715567 | Triglycerides
(20686565, 20139978, 19060906) |
| 11q23.3 | rs9326246 | 11 | 116741017 | Triglycerides
(23202125, 20686565, 19060906) |
| ZPR1 | rs964184 | 11 | 116778201 | Triglycerides
(20686565, 23063622, 21909109, 19060906, 20864672, 22629316,
20139978, 21943158, 23505323, 23726366, 19913121) |
| rs2075290 | 11 | 116782580 | |
| APOA5 | rs2266788 | 11 | 116789970 | Triglycerides
(20686565, 23063622, 22629316, 19060906, 21943158, 19913121,
18193043, 19802338, 23505323, 23236364) |
| rs2075291 | 11 | 116790676 | |
| APOA4 | rs5104 | 11 | 116821618 | Triglycerides
(20686565, 23063622, 19060906, 23505323, 19197348, 19913121) |
| 19p12 | rs11085421 | 19 | 20985948 | None |
In the study of hypo-HDL-cholesterolemia,
APOA5, BRAP, ALDH2, HECTD4, chromosome
16q13, CETP, and LILRB2 were previously reported to
be associated with circulating HDL-cholesterol levels, whereas
CXCL8, ACAD10, and PLCB2 were associated with
circulating levels of total cholesterol, LDL-cholesterol, and
triglycerides, respectively (Table
XIV). MARCH1, chromosome 6p22.1, and VPS33B were
also associated with adiponectin concentrations, type 1 diabetes
mellitus, and type 2 diabetes mellitus, respectively. The remaining
13 genes (MOB3C, TMOD4, LPGAT1, EHD3,
SPOPL, COL6A3, ZNF860, CACNA1D,
COL6A5, ADGRL3, DCLRE1C, OR4F6,
ZNF77) had not been related to circulating HDL-cholesterol
or other dyslipidemia-related phenotypes.
| Table XIVRelation of genes, chromosomal loci
and SNPs associated with hypo-HDL-cholesterolemia in the present
study to previously examined dyslipidemia-related phenotypes. |
Table XIV
Relation of genes, chromosomal loci
and SNPs associated with hypo-HDL-cholesterolemia in the present
study to previously examined dyslipidemia-related phenotypes.
| Gene/chr
locus | SNP | Chr | Position | Previously
examined phenotypes |
|---|
| MOB3C | rs139537100 | 1 | 46615006 | None |
| TMOD4 | rs115287176 | 1 | 151170961 | None |
|
LPGAT1 | rs150552771 | 1 | 211783358 | None |
| EHD3 | rs116417209 | 2 | 31249417 | None |
| SPOPL | rs114501427 | 2 | 138568946 | None |
|
COL6A3 | rs146092501 | 2 | 237371861 | None |
|
ZNF860 | rs140232911 | 3 | 31989561 | None |
|
CACNA1D | rs35874056 | 3 | 53702798 | None |
|
COL6A5 | rs200982668 | 3 | 130470894 | None |
|
ADGRL3 | rs192210727 | 4 | 61909615 | None |
| CXCL8 | rs188378669 | 4 | 73741568 | Total cholesterol
(23063622) |
|
MARCH1 | rs61734696 | 4 | 164197303 | Adiponectin
concentrations (20887962) |
| 6p22.1 | rs9261800 | 6 | 30408822 | Type 1 diabetes
(17554300) |
|
DCLRE1C | rs150854849 | 10 | 14934704 | None |
| APOA5 | rs2075291 | 11 | 116790676 | HDL-cholesterol
(23063622, 20686565, 22629316, 21386085) |
| BRAP | rs3782886 | 12 | 111672685 | HDL-cholesterol
(21572416) |
|
ACAD10 | rs11066015 | 12 | 111730205 | LDL-cholesterol
(20686565) |
| ALDH2 | rs671 | 12 | 111803962 | HDL-cholesterol
(21572416) |
|
HECTD4 | rs2074356 | 12 | 112207597 | HDL-cholesterol
(21572416, 21909109, 22751097) |
| rs11066280 | 12 | 112379979 | |
| PLCB2 | rs200787930 | 15 | 40289298 | Triglycerides
(23063622) |
|
VPS33B | rs199921354 | 15 | 91013841 | Type 2 diabetes
(22885922) |
| OR4F6 | rs141569282 | 15 | 101806068 | None |
| 16q13 | rs247616 | 16 | 56955678 | HDL-cholesterol
(20686565, 19060906, 20339536, 20838585, 19060911, 20031538) |
| 16q13 | rs3764261 | 16 | 56959412 | HDL-cholesterol
(20686565, 23063622, 19060911, 21943158, 21347282, 18193043,
20694148, 21589926, 19802338, 19913121) |
| CETP | rs1532624 | 16 | 56971567 | HDL-cholesterol
(20686565, 23063622, 22629316, 21347282, 21589926, 19060911,
20031564, 19060906, 18193044, 21943158) |
| ZNF77 | rs146879198 | 19 | 2934109 | None |
|
LILRB2 | rs73055442 | 19 | 54279838 | HDL-cholesterol
(20686565) |
In the study of hyper-LDL-cholesterolemia,
PCSK9, APOB, chromosome 6p21.3, SKIV2L,
HECTD4, APOE, and APOC1 were previously
reported to be associated with circulating LDL-cholesterol levels,
whereas CXCL8, 6p21.3, CCHCR1, PRRC2A,
LY6G6F, MSH5, C6orf48, SLC44A4,
TNXB, and PLCB2 were associated with circulating
concentrations of total cholesterol or triglycerides (Table XV). MARCH1,
SLC17A3, ZSCAN31, GABBR1, 6p22.1,
TRIM40, PPP1R18, 6p21.3, MDC1, TUBB,
DPCR1, LY6G6C, VARS, HSPA1B, and
VPS33B were associated with diabetes mellitus-associated
phenotypes. The remaining 16 genes (PTCH2, MOB3C,
TMOD4, COL6A3, COL6A5, ADGRL3,
KIAA0319, FAM65B, ZSCAN26, UBD,
DHX16, LOC105375015, NEU1, MUC17,
GIT2, ZNF77) had not been related to circulating
LDL-cholesterol or other dyslipidemia-related phenotypes.
| Table XVRelation of genes, chromosomal loci
and SNPs associated with hyper-LDL-cholesterolemia in the present
study to previously examined dyslipidemia-related phenotypes. |
Table XV
Relation of genes, chromosomal loci
and SNPs associated with hyper-LDL-cholesterolemia in the present
study to previously examined dyslipidemia-related phenotypes.
| Gene/chr
locus | SNP | Chr | Position | Previously
examined phenotypes |
|---|
| PTCH2 | rs147284320 | 1 | 44828589 | None |
| MOB3C | rs139537100 | 1 | 46615006 | None |
| PCSK9 | rs151193009 | 1 | 55043912 | LDL-cholesterol
(23063622, 21347282, 18193044, 20686565, 22629316) |
| TMOD4 | rs115287176 | 1 | 151170961 | None |
| APOB | rs13306206 | 2 | 21019859 | LDL-cholesterol
(20686565, 23202125, 23063622) |
|
COL6A3 | rs146092501 | 2 | 237371861 | None |
|
COL6A5 | rs200982668 | 3 | 130470894 | None |
|
ADGRL3 | rs192210727 | 4 | 61909615 | None |
| CXCL8 | rs188378669 | 4 | 73741568 | Total cholesterol
(23063622) |
|
MARCH1 | rs61734696 | 4 | 164197303 | Adiponectin
concentrations (20887962) |
|
KIAA0319 | rs4576240 | 6 | 24596250 | None |
|
FAM65B | rs150142878 | 6 | 24847657 | None |
|
SLC17A3 | rs34902660 | 6 | 25850874 | Serum urate
(23263486), type 1 diabetes (17554300) |
|
ZSCAN26 | rs76463649 | 6 | 28271963 | None |
|
ZSCAN31 | rs6922302 | 6 | 28327533 | Type 1 diabetes
(17554300) |
| UBD | rs64036 | 6 | 29559490 | None |
|
GABBR1 | rs29243 | 6 | 29631325 | Type 1 diabetes
(17554300) |
| 6p22.1 | rs2524272 | 6 | 29714623 | Type 1 diabetes
(17554300) |
|
TRIM40 | rs2523995 | 6 | 30134407 | Type 1 diabetes
(17554300, 17632545) |
| DHX16 | rs7749235 | 6 | 30667816 | None |
| rs2285321 | 6 | 30670221 | |
| rs6937357 | 6 | 30672547 | |
|
PPP1R18 | rs9468805 | 6 | 30675932 | Type 1 diabetes
(17632545) |
| rs6457254 | 6 | 30681357 | |
| rs2394392 | 6 | 30682541 | |
| 6p21.3 | rs3130663 | 6 | 30698817 | Type 1 diabetes
(17632545, 17554300) |
| MDC1 | rs2269702 | 6 | 30707358 | Fat mass
(19584900), type 1 diabetes (17554300), body mass index
(19584900) |
| rs28986465 | 6 | 30712785 | |
| rs2075015 | 6 | 30712831 | |
| rs6924270 | 6 | 30714203 | |
| TUBB | rs3132584 | 6 | 30720650 | Type 1 diabetes
(17554300) |
| rs25527 | 6 | 30723161 | |
| rs9500864 | 6 | 30725455 | |
| DPCR1 | rs6933400 | 6 | 30939399 | Type 1 diabetes
(17554300, 17632545) |
| rs11970154 | 6 | 30952101 | |
| 6p21.3 | rs2508015 | 6 | 31042423 | Type 1 diabetes
(17632545), triglycerides (20686565) |
|
CCHCR1 | rs147733073 | 6 | 31145462 | Triglycerides
(20686565) |
| 6p21.3 | rs3130685 | 6 | 31238429 | Triglycerides
(20686565), type 1 diabetes (17554300) |
|
LOC105375015 | rs9264942 | 6 | 31306603 | None |
| 6p21.3 | rs2596574 | 6 | 31366397 | LDL-cholesterol
(23063622), total cholesterol (23063622), type 1 diabetes
(17554300, 17632545) |
|
PRRC2A | rs11538264 | 6 | 31635412 | Type 1 diabetes
(17554300, 17632545), triglycerides (20686565), total cholesterol
(20686565) |
|
LY6G6F | rs9267546 | 6 | 31705659 | Triglycerides
(20686565) |
| rs17200983 | 6 | 31707506 | |
| rs9267547 | 6 | 31707724 | |
|
LY6G6C | rs117894946 | 6 | 31719250 | Type 1 diabetes
(17554300, 17632545) |
| MSH5 | rs11754464 | 6 | 31755958 | Type 1 diabetes
(17554300, 17632545), triglycerides (20686565), total cholesterol
(20686565) |
| VARS | rs5030798 | 6 | 31779733 | Type 1 diabetes
(17632545) |
| rs11751198 | 6 | 31785749 | |
|
HSPA1B | rs6457452 | 6 | 31827773 | Type 1 diabetes
(17554300, 17632545) |
|
C6orf48 | rs11968400 | 6 | 31836952 | Triglycerides
(20686565), total cholesterol (20686565), |
| | | | type 1 diabetes
(17632545) |
| 6p21.3 | rs12210887 | 6 | 31847946 | Type 1 diabetes
(17554300, 17632545,), triglycerides (20686565), total cholesterol
(20686565) |
| NEU1 | rs13118 | 6 | 31859509 | None |
|
SLC44A4 | rs117127493 | 6 | 31869232 | Type 1 diabetes
(17632545), triglycerides (20686565), total cholesterol
(20686565) |
|
SKIV2L | rs492899 | 6 | 31965741 | LDL-cholesterol
(20686565), type 1 diabetes (17554300, 17632545), triglycerides
(20686565), total cholesterol (20686565) |
| TNXB | rs140770834 | 6 | 32064851 | Type 1 diabetes
(17554300, 17632545), triglycerides (20686565), total cholesterol
(20686565) |
| rs11751545 | 6 | 32073266 | |
| 6p21.3 | rs204999 | 6 | 32142202 | Type 1 diabetes
(17632545) |
| MUC17 | rs78010183 | 7 | 101035329 | None |
| GIT2 | rs2292354 | 12 | 109930396 | None |
|
HECTD4 | rs11066280 | 12 | 112379979 | LDL-cholesterol
(21572416, 20686565), HDL-cholesterol (21572416, 21909109,
22751097) |
| PLCB2 | rs200787930 | 15 | 40289298 | Triglycerides
(23063622) |
|
VPS33B | rs199921354 | 15 | 91013841 | Type 2 diabetes
(22885922) |
| ZNF77 | rs146879198 | 19 | 2934109 | None |
| APOE | rs7412 | 19 | 44908822 | LDL-cholesterol
(23100282, 23063622, 20686565, 22629316, 19060911) |
| APOC1 | rs445925 | 19 | 44912383 | LDL-cholesterol
(20686565, 21347282, 18193044, 23063622, 18193043, 20864672,
23100282) |
Network analysis of genes identified in
the present study
Network analysis of the 10 genes (MOB3C,
TMOD4, LPGAT1, EHD3, COL6A3,
ZNF860, CACNA1D, COL6A5, DCLRE1C,
ZNF77) or three genes (KIAA0319, FAM65B,
UBD) associated in the present study with
hypo-HDL-cholesterolemia or hyper-LDL-cholesterolemia,
respectively, was performed to predict functional gene-gene
interactions, with the use of the GeneMANIA Cytoscape plugin
(http://apps.cytoscape.org/apps/genemania; Donnelly
Centre for Cellular and Biomolecular Research, University of
Toronto, Toronto, Canada) (35-37) and Cytoscape v3.4.0 software
(http://www.cytoscape.org; The
Cytoscape Consortium, San Diego, CA, USA) (38). Given that LOC105375015
protein has not been characterized, it was not examined. A set of
50 dyslipidemia-related genes were selected from the DisGeNET
database (http://www.disgenet.org/web/DisGeNET; Integrative
Biomedical Informatics Group, Research Programme on Biomedical
Informatics, Barcelona Biomedical Research Park, Barcelona, Spain)
(39,40) according to the rank order (high to
low) of scores for association with dyslipidemia. The network
analysis revealed that the 10 or three genes associated in the
present study with hypo-HDL-cholesterolemia or
hyper-LDL-cholesterolemia, respectively, have potential direct or
indirect interactions with the 50 genes previously demonstrated to
be associated with dyslipidemia (Fig.
5).
 | Figure 5Network analysis of the genes
identified in the present study with known dyslipidemia-related
genes. The 10 genes (MOB3C, TMOD4, LPGAT1,
EHD3, COL6A3, ZNF860, CACNA1D,
COL6A5, DCLRE1C, ZNF77) and three genes
(KIAA0319, FAM65B, UBD) associated in the
present study with hypo-HDL-cholesterolemia and
hyper-LDL-cholesterolemia, respectively, are indicated in red
circles. The 50 dyslipidemia-related genes selected from the
DisGeNET database (http://www.disgenet.org/web/DisGeNET) are indicated
in green circles. Interactions between red circles or between red
and green circles are indicated by bold lines. Molecules
represented by gray circles are putative mediators of interactions
between the genes. |
Discussion
Dyslipidemia is an important public health problem
because of its high prevalence and role as a risk factor for more
serious conditions, including coronary artery disease, ischemic
stroke, and colorectal cancer (9-12).
The identification of genetic variants that confer susceptibility
to dyslipidemia is thus clinically important for prevention of
these conditions. The present study has performed novel EWASs for
hypertriglyceridemia, hypo-HDL-cholesterolemia, and
hyper-LDL-cholesterolemia in subjects with early-onset forms of
these disorders, which may have a greater genetic component
compared with late-onset forms.
Among 25 SNPs in six genes and five chromosomal
loci significantly associated with early-onset
hypertriglyceridemia, the present study newly identified rs11085421
at 19p12 as a susceptibility locus for this condition. This SNP was
significantly associated with serum triglyceride concentration,
with the minor C allele being protective against
hypertriglyceridemia.
Among 28 SNPs in 24 genes and two chromosomal loci
significantly associated with hypo-HDL-cholesterolemia, the current
study newly identified 13 genes (MOB3C, TMOD4,
LPGAT1, EHD3, SPOPL, COL6A3,
ZNF860, CACNA1D, COL6A5, ADGRL3,
DCLRE1C, OR4F6, ZNF77) as potential
susceptibility loci for this condition. SNPs of 12 of these genes
were significantly associated with serum HDL-cholesterol levels,
with minor alleles of LPGAT1, EHD3, ZNF860,
CACNA1D, and DCLRE1C representing risk factors for
hypo-HDL-cholesterolemia and minor alleles of MOB3C,
TMOD4, COL6A3, COL6A5, ADGRL3,
OR4F6, and ZNF77 being protective against this
condition. Given that rs114501427 of SPOPL was not
significantly related to serum HDL-cholesterol concentration, this
gene was excluded as a novel susceptibility locus. Furthermore,
given that significant LD was apparent among SNPs of ADGRL3,
CXCL8, and MARCH1, as well as between SNPs of
OR4F6 and VPS33B, ADGRL3 and OR4F6 were
also removed from the final list of novel significant loci. In
addition, significant LD was detected between SNPs of MOB3C
and TMOD4 as well as between those of ZNF860 and
CACNA1D. We thus identified eight novel loci
(MOB3C-TMOD4, LPGAT1, EHD3, COL6A3,
ZNF860-CACNA1D, COL6A5, DCLRE1C, ZNF77)
that confer susceptibility to early-onset
hypo-HDL-cholesterolemia.
Among 65 SNPs in 44 genes and two chromosomal loci
significantly associated with hyper-LDL-cholesterolemia, the
present study newly identified 16 genes (PTCH2,
MOB3C, TMOD4, COL6A3, COL6A5,
ADGRL3, KIAA0319, FAM65B, ZSCAN26,
UBD, DHX16, LOC105375015, NEU1,
MUC17, GIT2, ZNF77) as potential
susceptibility loci for this condition. SNPs of six of these genes
(KIAA0319, FAM65B, ZSCAN26, UBD,
LOC105375015, NEU1) were significantly associated
with serum LDL-cholesterol levels, with minor alleles of these
genes representing risk factors for hyper-LDL-cholesterolemia.
Given that SNPs of 10 genes (PTCH2, MOB3C,
TMOD4, COL6A3, COL6A5, ADGRL3,
DHX16, MUC17, GIT2, ZNF77) were not
significantly related to the serum LDL-cholesterol concentration,
these genes were removed from the list of novel susceptibility
loci, even though this discrepancy may be attributable to the
effects of lipid-lowering medication. Given that rs13118 of
NEU1 was in significant LD with rs492899 of SKIV2L in
a large LD block and that rs76463649 of ZSCAN26 was in LD
with SNPs of SLC17A3, ZSCAN31, GABBR1, and
TRIM40, both NEU1 and ZSCAN26 were also
excluded from the final results. In addition, significant LD was
detected between SNPs of KIAA0319 and FAM65B. We thus
identified three novel loci (KIAA0319-FAM65B, UBD,
LOC105375015) that confer susceptibility to early-onset
hyper-LDL-cholesterolemia.
A recent study has reported that network analysis
of functional gene-gene interactions may be informative with regard
both to clarification of biological processes underlying coronary
artery disease and to the identification of therapeutic targets for
this condition (41). A network
analysis was therefore performed in the present study in order to
predict biological processes related to the identified genes and
the interactions of these genes with those previously known to be
associated with dyslipidemia. The results from the network analysis
revealed that the 10 or three genes associated in the present study
with hypo-HDL-cholesterolemia or hyper-LDL-cholesterolemia
respectively, had direct or indirect interactions with the 50
dyslipidemia-related genes selected from the DisGeNET database
(39,40). The underlying molecular mechanisms
of these interactions, however, remain to be elucidated.
Our group has previously reported that two, three
and nine SNPs were associated with hypertriglyceridemia
(P<1.71×10−4), hypo-HDL-cholesterolemia
(P<1.44×10−4), and hyper-LDL-cholesterolemia
(P<1.10×10−4), respectively, as determined by
multivariable logistic regression analysis with adjustment for age
and sex, following an initial EWAS screening of allele frequencies
among subjects with early- or late-onset forms of these conditions
(28). The associations of the
two SNPs [rs10790162 (P=3.58×10−24) and rs7350481
(P=7.94×10−23)] with hypertriglyceridemia were
replicated (P<0.05) in the present study. The associations of
two of the three SNPs [rs147317864 (P=0.0139) and rs12229654 (P=
0.0001)] with hypo-HDL-cholesterolemia were replicated in the
present study. The associations of eight of the nine SNPs
[rs7771335 (P=1.00×10−7), rs2071653
(P=1.61×10−5), rs2853969 (P= 6.16×10−8),
rs2269704 (P=2.92×10−7), rs2269703 (P=3.67×10
−7), rs495089 (P= 0.0 0 01), rs2269702
(P=5.13×10−7) and rs1233399 (P=0.0009)] with
hyper-LDL-cholesterolemia were replicated in the present study.
Although the association of most SNPs with dyslipidemia identified
in our previous study (28) was
replicated, genetic variants associated with hypertriglyceridemia,
hypo-HDL-cholesterolemia, or hyper-LDL-cholesterolemia appear to
differ, at least in part, between early- and late-onset forms of
the diseases.
There are several limitations to the present study.
Firstly, given that the results were not replicated, their
validation will be necessary in independent study populations or in
other ethnic groups. The identified SNPs were not tested in a
clinical setting to validate the assessment of genetic risk for
dyslipidemia in patients. In addition, it is possible that SNPs
identified in the present study may be in LD with other genetic
variants in the same gene or in nearby genes that are actually
responsible for the development of hypertriglyceridemia,
hypo-HDL-cholesterolemia, or hyper-LDL-cholesterolemia. Finally,
the functional relevance of identified SNPs to the pathogenesis of
hypertriglyceridemia, hypo-HDL-cholesterolemia, or
hyper-LDL-cholesterolemia remains to be elucidated.
In conclusion, the present study has identified
chromosome 19p12, eight loci (MOB3C-TMOD4, LPGAT1,
EHD3, COL6A3, ZNF860-CACNA1D, COL6A5,
DCLRE1C, ZNF77) and three loci
(KIAA0319-FAM65B, UBD, LOC105375015) that
confer susceptibility to early-onset hypertriglyceridemia,
hypo-HDL-cholesterolemia and hyper-LDL-cholesterolemia,
respectively. Determination of genotypes for the SNPs at these loci
may prove informative for assessment of genetic risk for
hypertriglyceridemia, hypo-HDL-cholesterolemia and
hyper-LDL-cholesterolemia in the Japanese population.
Acknowledgments
Not applicable.
Funding
This work was supported by CREST, Japan Science and
Technology Agency (grant no. JPMJCR1302; to YYam, JS and IT).
Availability of data and materials
All data underlying the findings described in the
article are available on request from the corresponding author.
Authors’ contributions
YYam contributed to conception and design of the
study; to acquisition, analysis, and interpretation of the data;
and to drafting of the manuscript. KK, MO, HH, and TF each
contributed to acquisition of the data and to revision of the
manuscript. YYas, IT, and JS contributed to analysis and
interpretation of the data as well as to revision of the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The study protocol complied with the Declaration of
Helsinki and was approved by the Committees on the Ethics of Human
Research of Mie University Graduate School of Medicine, Hirosaki
University Graduate School of Medicine, and participating hospitals
(Gifu Prefectural Tajimi Hospital, Gifu Prefectural General Medical
Center, Japanese Red Cross Nagoya First Hospital, Northern Mie
Medical Center Inabe General Hospital, and Hirosaki Stroke and
Rehabilitation Center). Written informed consent for participation
in the study was obtained from all subjects.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Durrington P: Dyslipidaemia. Lancet.
362:717–731. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Paththinige CS, Sirisena ND and
Dissanayake V: Genetic determinants of inherited susceptibility to
hypercholesterolemia-a comprehensive literature review. Lipids
Health Dis. 16:1032017. View Article : Google Scholar
|
|
3
|
Dron JS and Hegele RA: Genetics of
triglycerides and the risk of atherosclerosis. Curr Atheroscler
Rep. 19:312017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Soutar AK and Naoumova RP: Mechanism of
disease: Genetic causes of familial hypercholesterolemia. Nat Clin
Pract Cardiovasc Med. 4:214–225. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Heller DA, DeFaire U, Pedersen N, Dahlén G
and McClearn GE: Genetic and environmental influences on serum
lipid levels in twins. N Engl J Med. 328:1150–1156. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abney M, McPeek MS and Ober C: Broad and
narrow herita-bilities of quantitative traits in a founder
population. Am J Hum Genet. 68:1302–1307. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
van Dongen J, Willemsen G, Chen WM, de
Geus EJ and Boomsma DI: Heritability of metabolic syndrome traits
in a large population-based sample. J Lipid Res. 54:2914–2923.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Woo JG, Morrison JA, Stroop DM, Aronson
Friedman L and Martin LJ: Genetic architecture of lipid traits
changes over time and differs by race: Princeton Lipid Follow-up
Study. J Lipid Res. 55:1515–1524. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Benjamin EJ, Virani SS, Callaway CW,
Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling
FN, Deo R, et al: Heart disease and stroke statistics-2018 update:
A report from the American Heart Association. Circulation.
137:e67–e492. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Law MR, Wald NJ and Rudnicka AR:
Quantifying effect of statins on low density lipoprotein
cholesterol, ischaemic heart disease, and stroke: Systematic review
and meta-analysis. Br Med J. 326:14232003. View Article : Google Scholar
|
|
11
|
Agnoli C, Grioni S, Sieri S, Sacerdote C,
Vineis P, Tumino R, Giurdanella MC, Pala V, Mattiello A, Chiodini
P, et al: Colorectal cancer risk and dyslipidemia: A case-cohort
study nested in an Italian multicentre cohort. Cancer Epidemiol.
38:144–151. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yao X and Tian Z: Dyslipidemia and
colorectal cancer risk: A meta-analysis of prospective studies.
Cancer Causes Control. 26:257–268. 2015. View Article : Google Scholar
|
|
13
|
Kathiresan S, Melander O, Guiducci C,
Surti A, Burtt NP, Rieder MJ, Cooper GM, Roos C, Voight BF,
Havulinna AS, et al: Six new loci associated with blood low-density
lipoprotein cholesterol, high-density lipoprotein cholesterol or
triglycerides in humans. Nat Genet. 40:189–197. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kathiresan S, Willer CJ, Peloso GM,
Demissie S, Musunuru K, Schadt EE, Kaplan L, Bennett D, Li Y,
Tanaka T, et al: Common variants at 30 loci contribute to polygenic
dyslipidemia. Nat Genet. 41:56–65. 2009. View Article : Google Scholar
|
|
15
|
Aulchenko YS, Ripatti S, Lindqvist I,
Boomsma D, Heid IM, Pramstaller PP, Penninx BW, Janssens AC, Wilson
JF, Spector T, et al: Loci influencing lipid levels and coronary
heart disease risk in 16 European population cohorts. Nat Genet.
41:47–55. 2009. View
Article : Google Scholar :
|
|
16
|
Teslovich TM, Musunuru K, Smith AV,
Edmondson AC, Stylianou IM, Koseki M, Pirruccello JP, Ripatti S,
Chasman DI, Willer CJ, et al: Biological, clinical and population
relevance of 95 loci for blood lipids. Nature. 466:707–713. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Asselbergs FW, Guo Y, van Iperen EP,
Sivapalaratnam S, Tragante V, Lanktree MB, Lange LA, Almoguera B,
Appelman YE, Barnard J, et al: Large-scale gene-centric
meta-analysis across 32 studies identifies multiple lipid loci. Am
J Hum Genet. 91:823–838. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Willer CJ, Schmidt EM, Sengupta S, Peloso
GM, Gustafsson S, Kanoni S, Ganna A, Chen J, Buchkovich ML, Mora S,
et al: Discovery and refinement of loci associated with lipid
levels. Nat Genet. 45:1274–1283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Peloso GM, Auer PL, Bis JC, Voorman A,
Morrison AC, Stitziel NO, Brody JA, Khetarpal SA, Crosby JR,
Fornage M, et al: Association of low-frequency and rare
coding-sequence variants with blood lipids and coronary heart
disease in 56,000 whites and blacks. Am J Hum Genet. 94:223–232.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Surakka I, Horikoshi M, Mägi R, Sarin AP,
Mahajan A, Lagou V, Marullo L, Ferreira T, Miraglio B, Timonen S,
et al: The impact of low-frequency and rare variants on lipid
levels. Nat Genet. 47:589–597. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Lange LA, Hu Y, Zhang H, Xue C, Schmidt
EM, Tang ZZ, Bizon C, Lange EM, Smith JD, Turner EH, et al:
Whole-exome sequencing identifies rare and low-frequency coding
variants associated with LDL cholesterol. Am J Hum Genet.
94:233–245. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Helgadottir A, Gretarsdottir S,
Thorleifsson G, Hjartarson E, Sigurdsson A, Magnusdottir A,
Jonasdottir A, Kristjansson H, Sulem P, Oddsson A, et al: Variants
with large effects on blood lipids and the role of cholesterol and
triglycerides in coronary disease. Nat Genet. 48:634–639. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu DJ, Peloso GM, Yu H, Butterworth AS,
Wang X, Mahajan A, Saleheen D, Emdin C, Alam D, Alves AC, et al:
Exome-wide association study of plasma lipids in >300,000
individuals. Nat Genet. 49:1758–1766. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hoffmann TJ, Theusch E, Haldar T,
Ranatunga DK, Jorgenson E, Medina MW, Kvale MN, Kwok PY, Schaefer
C, Krauss RM, et al: A large electronic-health-record-based
genome-wide study of serum lipids. Nat Genet. 50:401–413. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Peloso Lu X, Liu GM, Wu DJ, Zhang Y, Zhou
H, Li W, Tang J, Dorajoo CS, Li RH, et al: Exome chip meta-analysis
identifies novel loci and East Asian-specific coding variants that
contribute to lipid levels and coronary artery disease. Nat Genet.
49:1722–1730. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Spracklen CN, Chen P, Kim YJ, Wang X, Cai
H, Li S, Long J, Wu Y, Wang YX, Takeuchi F, et al: Association
analyses of East Asian individuals and trans-ancestry analyses with
European individuals reveal new loci associated with cholesterol
and triglyceride levels. Hum Mol Genet. 26:1770–1784. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kurano M, Tsukamoto K, Kamitsuji S,
Kamatani N, Hara M, Ishikawa T, Kim BJ, Moon S, Jin Kim Y and
Teramoto T: Genome-wide association study of serum lipids confirms
previously reported associations as well as new associations of
common SNPs within PCSK7 gene with triglyceride. J Hum Genet.
61:427–433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamada Y, Sakuma J, Takeuchi I, Yasukochi
Y, Kato K, Oguri M, Fujimaki T, Horibe H, Muramatsu M, Sawabe M, et
al: Identification of eight genetic variants as novel determinants
of dyslipidemia in Japanese by exome-wide association studies.
Oncotarget. 8:38950–38961. 2017.PubMed/NCBI
|
|
29
|
Yamada Y, Matsui K, Takeuchi I, Oguri M
and Fujimaki T: Association of genetic variants with hypertension
in a longitudinal population-based genetic epidemiological study.
Int J Mol Med. 35:1189–1198. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Grove ML, Yu B, Cochran BJ, Haritunians T,
Bis JC, Taylor KD, Hansen M, Borecki IB, Cupples LA, Fornage M, et
al: Best practices and joint calling of the HumanExome BeadChip:
The CHARGE Consortium. PLoS One. 8:e680952013. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Anderson CA, Pettersson FH, Clarke GM,
Cardon LR, Morris AP and Zondervan KT: Data quality control in
genetic case-control association studies. Nat Protoc. 5:1564–1573.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Price AL, Patterson NJ, Plenge RM,
Weinblatt ME, Shadick NA and Reich D: Principal components analysis
corrects for stratification in genome-wide association studies. Nat
Genet. 38:904–909. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Leslie R, O’Donnell CJ and Johnson AD:
GRASP: Analysis of genotype-phenotype results from 1,390
genome-wide association studies and corresponding open access
database. Bioinformatics. 30:i185-i1942014. View Article : Google Scholar
|
|
34
|
Eicher JD, Landowski C, Stackhouse B,
Sloan A, Chen W, Jensen N, Lien JP, Leslie R and Johnson AD: GRASP
v2.0: An update on the genome-wide repository of associations
between SNPs and phenotypes. Nucleic Acids Res. 43(Database Issue):
D799–D804. 2015. View Article : Google Scholar :
|
|
35
|
Warde-Farley D, Donaldson SL, Comes O,
Zuberi K, Badrawi R, Chao P, Franz M, Grouios C, Kazi F, Lopes CT,
et al: The GeneMANIA prediction server: Biological network
integration for gene prioritization and predicting gene function.
Nucleic Acids Res. 38:W214–W220. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Montojo J, Zuberi K, Rodriguez H, Kazi F,
Wright G, Donaldson SL, Morris Q and Bader GD: GeneMANIA cytoscape
plugin: Fast gene function predictions on the desktop.
Bioinformatics. 26:2927–2928. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Montojo J, Zuberi K, Rodriguez H, Bader GD
and Morris Q: GeneMANIA: Fast gene network construction and
function prediction for Cytoscape. F1000Res. 3:1532014.PubMed/NCBI
|
|
38
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Piñero J, Queralt-Rosinach N, Bravo À,
Deu-Pons J, Bauer-Mehren A, Baron M, Sanz F and Furlong LI:
DisGeNET: A discovery platform for the dynamical exploration of
human diseases and their genes. Database (Oxford) 2015. bav0282015.
View Article : Google Scholar
|
|
40
|
Piñero J, Bravo À, Queralt-Rosinach N,
Gutiérrez-Sacristán A, Deu-Pons J, Centeno E, García-García J, Sanz
F and Furlong LI: DisGeNET: A comprehensive platform integrating
information on human disease-associated genes and variants. Nucleic
Acids Res. 45:D833–D839. 2017. View Article : Google Scholar :
|
|
41
|
Lempiäinen H, Brænne I, Michoel T,
Tragante V, Vilne B, Webb TR, Kyriakou T, Eichner J, Zeng L,
Willenborg C, et al: Network analysis of coronary artery disease
risk genes elucidates disease mechanisms and druggable targets. Sci
Rep. 8:34342018. View Article : Google Scholar : PubMed/NCBI
|