1. Introduction
The response to inflammation constitutes a series of
immune events that are evoked as a result of an insult in the
tissues and aims to restore or re-establish normal structure and
function. Inflammation is characterized by an increased local
concentration of mediators, including inflammatory cells and
cytokines (1). Chronic
inflammation is a hallmark for several diseases, including cancer,
diabetes, cardiovascular and pulmonary disorders, and neurological
diseases. Furthermore, inflammation is associated with
musculoskeletal insults, among which tendon inflammation in rotator
cuff injury (RCI) is of particular concern (2). Persistent inflammation in the
shoulder tendons delays the healing responses. Therefore, the
management of inflammation and relieving of associated pain are
required for a promising therapeutic approach.
Current knowledge of the molecular mechanisms and
the primary cause of RCI associated inflammation remains limited.
It is suggested that the physiological and mechanical stress in
rotator cuff tendons results in microtrauma, which in turn leads to
inflammation (3). Apart from the
biochemical and molecular signals and pathways, epigenetic
mechanisms are also involved in the initiation, progression, onset
and regulation of inflammatory responses. Epigenetics-based
therapeutic targeting of cellular or tissue inflammation in RCI has
not been developed. Understanding the basics of epigenetic
phenomena and the regulatory signaling is likely to be useful to
elucidate the inflammatory mechanisms in RCI, the exploitation of
which can assist in designing novel therapeutic approaches for
RCI-associated inflammation.
2. DNA organization and epigenetics
Chromatin remodeling is an inevitable process that
modulates and regulates gene expression. The structural dynamics of
chromatin organization exposes the DNA strand for gene
expression/regulation machinery. Chromatin exists as a complex of
DNA, histones and non-histone proteins, which are organized into a
specific 3D architecture that undergoes reversible and dynamic
alteration during replication and gene expression. In addition,
non-mutational structural modifications in DNA, including
methylation and histone modifications, can alter gene expression
which constitutes the epigenetic regulation system. The term
‘epigenetics’ was coined by Waddington in 1968 and was referred to
as ‘the interactions between genes and their products which bring
phenotype into being’ (4).
Understanding the basic organization of the DNA in chromatin is
necessary for examining the mechanisms of epigenetics involved in
regulating gene expression.
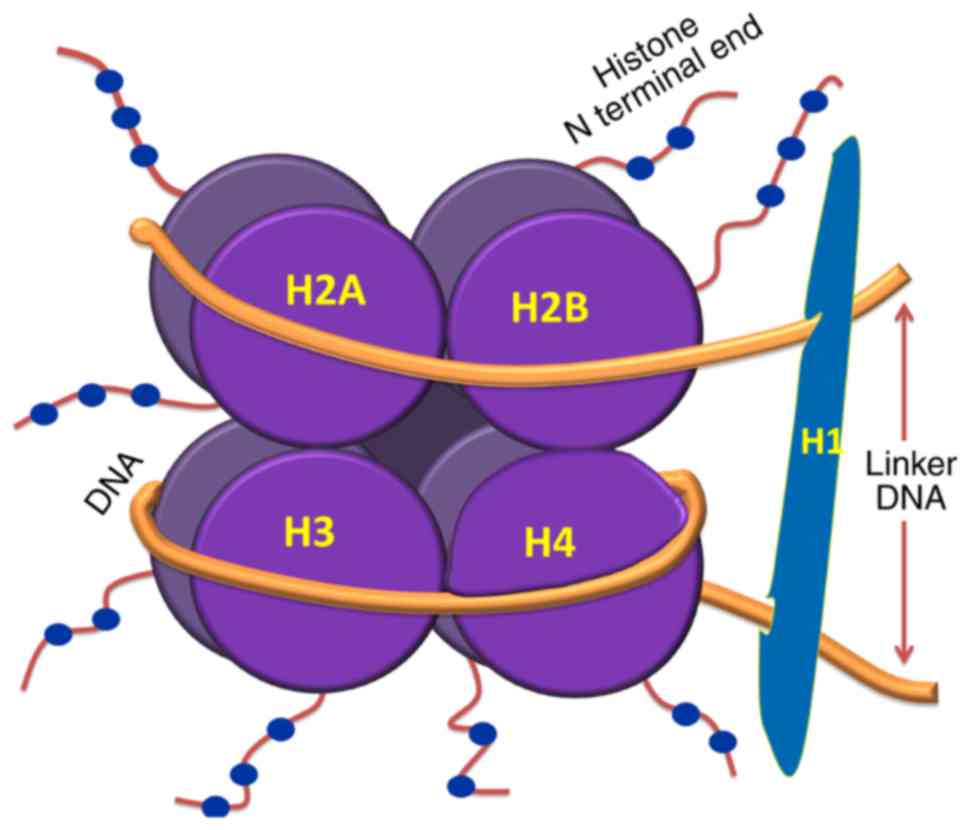
The nucleosome is the basic unit of chromatin, which
comprises ~146 base pairs (bp) of DNA wrapped around an octamer of
histone protein subunits. These subunits include two copies each of
H2A, H2B, H3 and H4. Nucleosomal organization ensures tight packing
of the histones with DNA and enhances folding and stacking of
otherwise lengthy DNA inside the nucleus. Therefore, nucleosome
organization and chromatin condensation impede gene expression,
which marks the remodeling of chromatin in the initial event of
gene activation. The N-terminal ends of histones are susceptible to
enzymatic reactions which facilitate local unwinding/rewinding of
the nucleosome and have profound effects on gene expression
(5). Linker DNA, which is between
10 and 80 bp in length, connects adjacent nucleosomes which are
folded as a compact fiber of 30 nm; these are further stacked to
form higher order structures, including chromosomes. The
nucleosomal organization of DNA is shown in Fig. 1. Linker histones (H1) are presumed
to stabilize the 30-nm fibers by binding to linker DNA. However,
this interaction between H1 and linker DNA is debated, and the
packing of DNA to its higher order structures within the nucleus
remains to be fully elucidated. In short, these epigenetic
mechanisms modulate gene function through DNA-protein interactions
without altering the genetic code.
As compatible chromatin (heterochromatin) makes DNA
inaccessible for gene expression machinery, the events that induce
structural alterations in chromatin are crucial for the initiation
of gene expression. However, actively expressing genes
(euchromatin) are more susceptible for the action of specific
enzymes and regulatory proteins involved in gene expression
(6,7). The transient modifications in
chromatin structure are facilitated by remodeling complexes which
shuffle nucleosomes to randomly expose the wound DNA for a limited
time (7). Chromatin remodeling
complexes can be ATP-dependent, which can move nucleosome positions
to induce a conformational change to enable the DNA accessible on
the histone surface. This complex mediates ATPase activity for
energy and is predominantly associated with members of the SWI/SNF
family. SWI/SNF subfamilies with helicase domains have also been
established as epigenetic modulators. Remodeling complexes act by
disrupting chromatin structure and by inducing covalent
modifications to the nucleosomes through acetylation and
methylation, with methylation occurring particularly at the histone
N-termini. These remodeling complexes are selective to the genes to
be expressed and the transcription factors to be recruited during
gene expression (8).
Mammalian epigenetic systems include DNA
methylation, histone modifications and RNA interference. These
systems can act individually, autonomously or cooperatively and can
persist throughout the cell cycle, including during
mitosis/meiosis. Disruption of these mechanisms may lead to the
loss of cellular integrity, alterations in phenotype and disease
progression, and impairments in normal development (9). Large-scale genome sequence variation
(insertions, deletions or chromosomal rearrangements) ranging
between 1 kbp and several mbps, collectively termed copy number
variants (CNVs), is also considered to be similar to epigenetic
phenomenon (10,11).
3. DNA methylation
DNA methylation is crucial for the normal
development mediated through genomic imprinting, X chromosome
inactivation, transcriptional repression and transposition. The
failure to impart appropriate methylation tags to DNA contributes
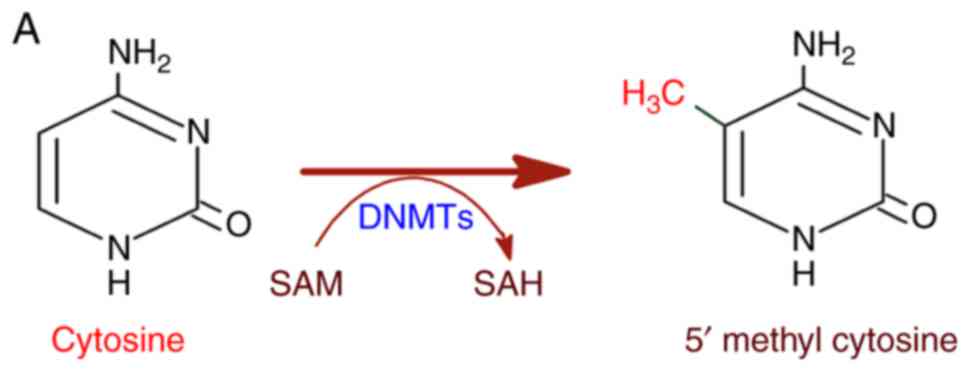
to genetic diseases and cancer (12). DNA methylation is catalyzed by DNA
methyl transferases (DNMTs) with S-adenosyl methionine (SAM) as a
methyl group donor (13,14). DNMTs transfer the methyl group
from SAM to cytosine residues on DNA, specifically at the CpG
dinucleotide sequence yielding a 5-methylcytosine residue (Fig. 2A). In this regard, methylation in
the promoter regions of the genes implies the extent of gene
repression (15). The functional
outcomes of DNMTs are appreciated in X chromosome inactivation and
genomic imprinting in addition to the abnormalities in DNA
methylation events which are associated with disease complications
(16). Among the DNMTs (DNMT1,
DNMT3A, and DNMT3B), DNMT1 is important as it prefers
hemi-methylated DNA strands (17). DNMT1 is closely associated with
newly replicated DNA, suggesting its function to methylate daughter
strands with respect to the methylation patterns of the parental
strand (18). DNMT3A and DNMT3B
are de novo methyltransferases which act during development
and are subsequently replaced with DNMT1 as cell division
progresses (19). DNMT3A and
DNMT3B usually act on unmodified DNA and cause hemi-methylation,
whereas DNMT1 acts on hemi-methylated DNA. The representation of
hemi-methylation and methylation patterns is shown in Fig. 2B.
DNMT3B is functionally active during early stages of
embryogenesis and is known to arrest germ line genes during the
transition from blastocyst to epiblast following implantation. By
contrast, DNMT3A facilitates the retention and establishment of
parental imprints and in differentiating somatic cell DNA
methylation. Collectively, DNMT3A and DNMT3B maintain symmetry in
CpG methylation, particularly at the hemi-methylated sites of
embryonic cells (20,21). DNMT3L, another DNMT, shares
structural similarities with DNMT3A and DNMT3B with regards to the
ATRX-DNMT3-DNMT3-like domain and binds to the lysine (K) residue at
the 4th position of unmethylated H3. Although DNMT3L lacks
catalytic activity, it is reported to function as a cofactor for
DNMT3A and DNMT3B (22).
Unlike humans, mouse embryonic cells are reported to
exhibit methylated genomic DNA at CpA, CpT and CpG dinucleotides,
with CpG being the predominant form in all somatic cells (23,24); this shows that CpG methylation in
the genome is common in the mammalian system. 5-methyl cytosine
(5MeC) accounts for almost 1% of the total DNA bases of the genome,
which constitute >70% of all CpG dinucleotides. Those stretches
of genomic sequences comprising predominantly CpG dinucleotides
concentrated in the genomic DNA are referred to as CpG islands,
where the DNA is heavily methylated. These CpG islands are located
around the promoter regions of several genes, signifying a role in
gene regulation through the methylation of those genes. Due to the
potential mutation resulting from the deamination of methylated
cytosine to thymine, the majority of CpG dinucleotides are confined
to CpG islands (25). The
methylated state of DNA is the result of dynamic, but independent,
methylation and demethylation, which varies between cell types. For
example, mature germ cells and somatic cells are heavily methylated
compared with hypomethylated embryonic cells (26,27). The non-X-linked promoter CpG
islands are reported to be methylated in normal tissues but exempt
from methylation in germ line cells. DNA or histone methylation
recruits repressive machinery and elicits an unfavorable chromatin
conformation to prevent transcription (28). Such alterations in chromatin
structure induce methylation of the adjacent chromatin segments
(29). In addition, the
methylation at H3K4 assists in the assembly of regulatory proteins
of the chromatin-remodeling complex which offers transcriptional
regulation. The mechanism of the regulatory role of H3K4
methylation is described in detail in the following sections.
Transcriptional repression by DNA
methylation
CpG islands are associated with promoter regions of
the majority of housekeeping genes and other genes that are
regulated during developmental stages. Such loci are
transcriptionally inactive irrespective of their hypomethylation
status (30). The hypo-methylated
state is necessary for the binding of transcription factors (TFs),
which prevents DNA methylation and histone methylation machinery
from targeting these loci (31,32). Based on the transcriptional
activity of the CpG islands, the associated promoters can be poorly
methylated, including intermediate CpG density promoters (inactive
on methylation) and low CpG density promoters (hypermethylated), or
transcriptionally active promoters regardless of their methylation
status (31). The intermediate
CpG density promoters can attain hypermethylation upon encountering
differentiation signals. This DNA methylation offers an effective
method of specific gene silencing, which was observed during the
differentiation of germ cells (20). As with promoters, enhancer
methylation can also modulate gene expression, however, the
regulatory role of enhancer methylation remains to be elucidated
(33).
The level of DNMT1 is reported to be increased
during the S phase of the cell cycle, as evident from its high
expression in mitotic cells. Proliferating cell nuclear
factor-interacting binding factor and nuclear protein 95 attract
DNMT1 to the replication fork to ensure its binding to the
unmethylated daughter strand, converting hemi-methylated DNA into
fully methylated DNA (34). The
resulting methyl groups of 5MeC occupy the major groove of the DNA
duplex and inhibit transcription by preventing TF binding or by
interacting with methyl-binding proteins which recruits
transcriptional repressors (35).
In addition, the presence of methylated DNA elements within the
genes facilitates the elongation phase of transcription and
minimizes premature initiation (36). In addition, splicing of the
transcripts is favored by methylation as the exon-intron boundaries
are characterized by transitions in methylation pattern (37).
Demethylation
The reprogramming of 5MeC to reset the methylation
status of cells for each new generation is essential, which
suggests the existence of cellular demethylation machinery. The
zygote presents DNA demethylation as compartmentalized deletion of
5MeC with maternal and paternal chromosomes leading to a
demethylated epigenome. However, imprinted gene loci, certain
maternally-contributed promoters, and several transposable elements
(TEs) are exceptions (20). It
has been reported that the maternal genome retains 5MeC and
depletes during subsequent cell cycles, whereas the paternal genome
demethylates prior to cell division (38). However, it has been reported that
5MeC is globally converted to 5'-hydroxymethyl cytosine (5hMeC),
which is removed in subsequent replications (39). In addition, a second stage of
reprogramming of DNA methylation occurs in primordial germ cells to
decrease global 5MeC levels. The regulation of gene expression in a
locus-specific manner has also been revealed. Reprograming by
demethylation is also vital for cell fusion and the differentiation
of induced pluripotent stem cells (40).
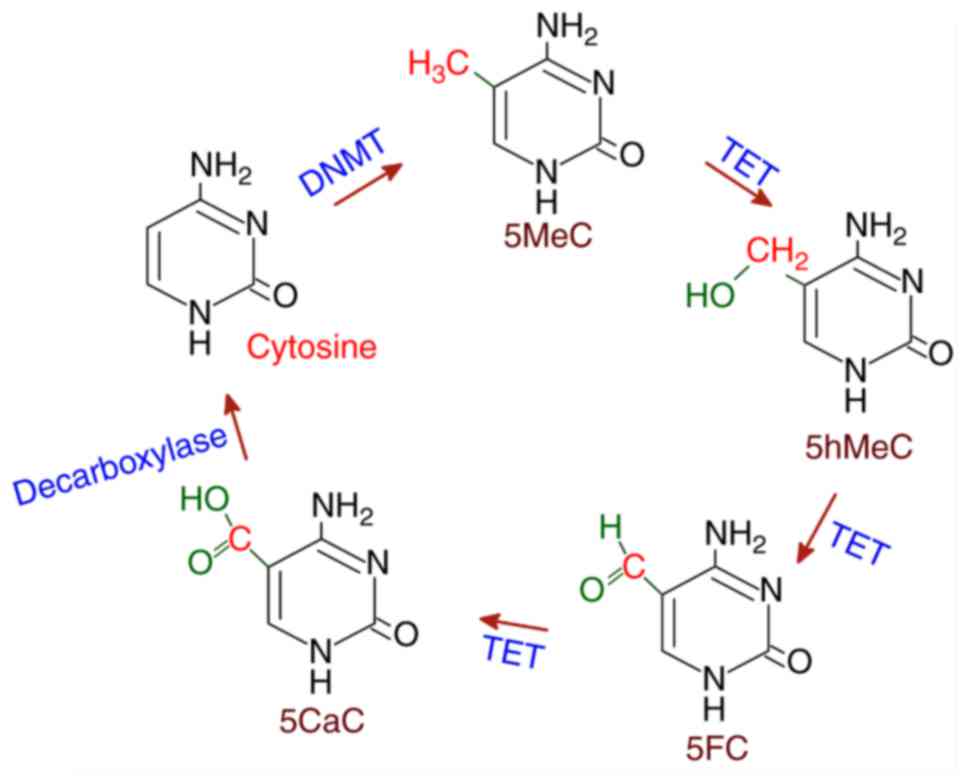
Several proteins that exhibit demethylase activities
have been reported in the mammalian system. The ten-eleven
translocation (TET) family of deoxygenases have been found to
catalyze the conversion of 5MeC to 5hMeC and then 5'-formyl
cytosine (5FC) and finally 5'-carboxyl cytosine (5CaC) in three
sequential oxidation reactions. Subsequently, 5CaC is
decarboxylated to cytosine resulting in demethylation. TET belongs
to the Fe(II)/α-ketoglutarate-dependent dioxygenases, which utilize
Fe(II) as cofactor (41). TET has
been identified in acute myeloid leukemia as part of histone H3 Lys
4 (H3K4). Of the three members in the TET family, TET1-3, TET3
possesses 5MeC hydroxylase activity. TET activity modifies the
existing DNA methylation pattern, which paves the way for
transcriptional regulation (42).
The TET-mediated demethylation of 5MeC is shown in Fig. 3.
4. Histone modifications
Post-translational modifications in histones,
particularly at the specific amino acid residues of N-terminal
ends, have implications on the extent of gene expression. A wide
array of modification patterns have been established on histones
(Table I) where the
acetylation/deacetylation and methylation of K residues are of
significance in terms of transcriptional regulation (43,44) (Fig.
4). In general, acetylation facilitates the decondensation of
chromatin, which exposes the genes to enable access to the
replication/transcription machinery, and deacetylation causes the
chromatin condensation that represents inactive genes. The
acetylation neutralizes the negative charge density around the
genes and disturbs the secondary structure of DNA, which makes the
genes accessible for interacting with regulatory signals. In short,
the interplay between acetylation and deacetylation determines the
regulation of gene activity (45). The gene regulation by histone
methylation depends on the amino acid residue undergoing
methylation (16). Similarly,
histone modifications caused by phosphorylation can either activate
or inactivate the genes with respect to the inducing signals. The
actual effects of other histone modifications on gene expression
remain to be fully elucidated.
 | Table ITypes of histone modification. |
Table I
Types of histone modification.
| Histone
modification | Residue |
|---|
| Acetylation | Lysine (K) |
| Methylation | Lysine (K) |
| Methylation | Arginine (R) |
| Ubiquitination | Lysine (K) |
|
Phosphorylation | Serine (S) |
|
Phosphorylation | Threonine (T) |
| ADP
ribosylation | Glutamic acid
(E) |
| Sumoylation | Lysine (K) |
| Deimination | Arginine (R) |
| Proline
isomerization | Proline (P) |
The combinations of histone modifications at a
promoter region can determine the epigenetic status of cells, which
facilitates either gene activation or repression and constitutes
the ‘histone code’ hypothesis. The molecular cues for these
modifications are encoded in the tail domains of histones, which
are read by the regulator molecules. The parental histones are
distributed randomly on daughter strands following replication and
retain an epigenetic memory for gene expression (46).
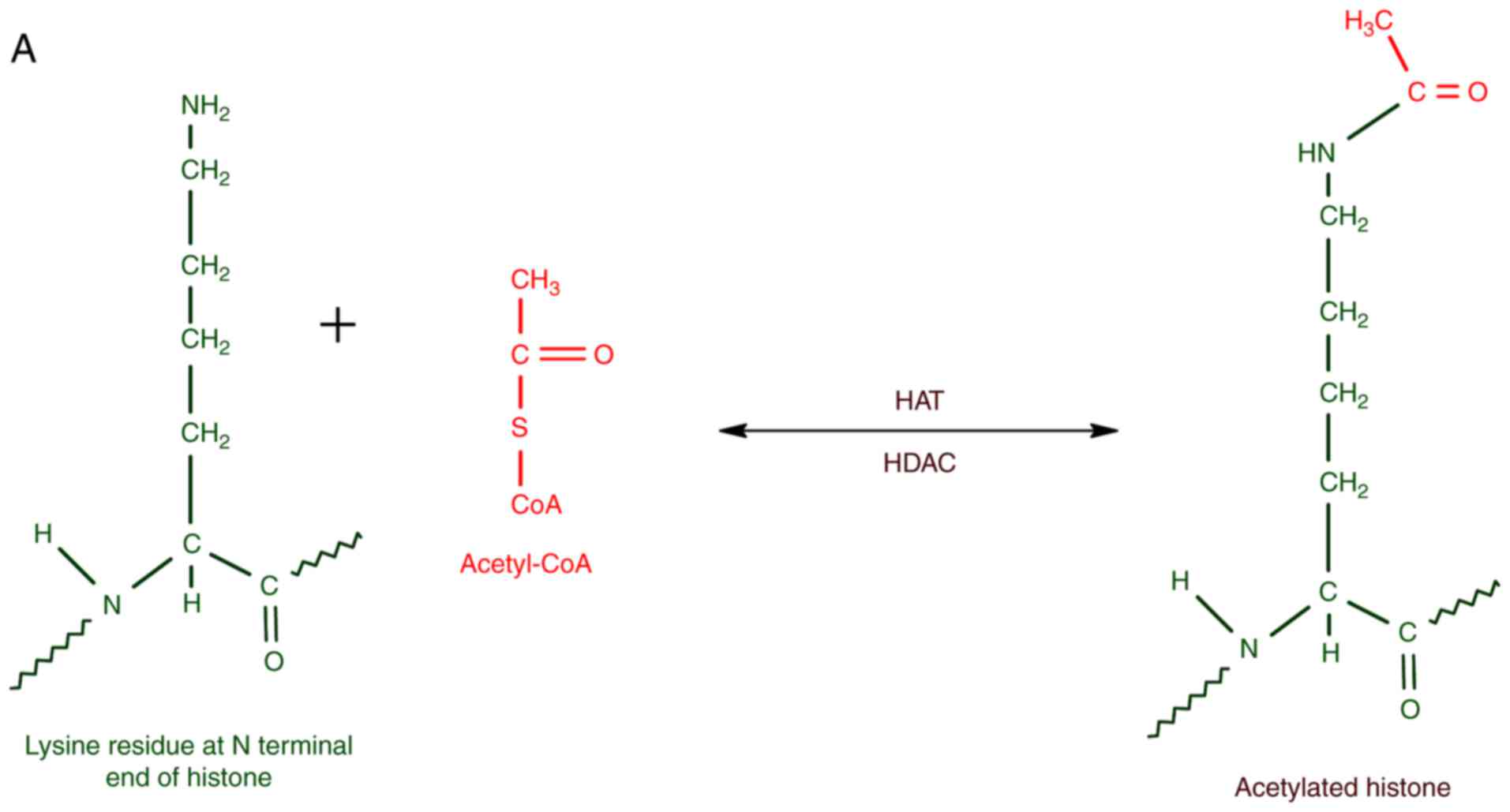
The enzymes that catalyze histone acetylation,
mainly at K residues, can be collectively referred to as histone
acetyl transferases (HATs). The enzymes that remove acetyl groups
from acetylated histones are histone deacetylases (HDACs). HATs are
usually associated with transcriptional activators, whereas HDACs
form part of transcriptional repressors. In short, the balance
between HAT and HDAC activities are significant for the epigenetic
regulation of gene expression. K and arginine (R) residues are
subjected to methylation by histone methyltransferases (HMTs). The
methylation at K residues is generally considered as a stable and
readily reversible histone modification compared with others. The
existence of histone demethylases (HDMs) has also been reported
(45). Transcriptionally active
genes exhibit acetylation/methylation of K4, K36 and K79 of H3 and
vice-versa. The majority of the K residues subjecting to HAT
activity reside in the N-terminal tail of histone, although H3K56
in the core domain is an exception; the acetylated H3K56 residue
exists in the core region of the H3 subunit (43). In addition, certain HMTs have also
been found to be associated with HATs. For example,
coactivator-associated arginine methyltransferase CARM1/PRMT4 is
attached to HAT with physical interactions and acts cooperatively
in mediating nuclear factor-κB (NF-κB) activity (47). G9a, a histone lysine
methyltransferase, also has a dual function as a transcriptional
suppressor and activator (48).
The role of histone modifications is also central in
responses to DNA damage for marking the damage site. For example,
the methylation of H4K20 and action of cell cycle checkpoint
protein Crb2 result in cell cycle arrest at the G2/M transition
(49). Histone acetyltransferase
binding to origin recognition complex HBO1 assists in chromatin
remodeling during replication by coordinating with the ING family
of tumor suppressors. H4 acetylation by HBO1 and the association of
ING4, ING5 and p53 in the protein complex containing HBO1 are
evident in S phase cells; the p53-HBO1 interaction occurs through
physical binding and this interaction decreases the activity of HAT
(50). Histone modification by
phosphorylation/dephosphorylation is also significant in mammalian
cell replication, mitosis, apoptosis and gametogenesis as these
modifications can induce compaction/decompaction of DNA by altering
the charge density (51).
Regulation of histone modifications
Histone modifications rely on unraveling chromosomal
DNA and the recruitment and assembly of regulatory signals to the
target site. Such regulatory signals possess catalytic domains, the
activities of which can trigger a cascade of events that result in
transcription, replication or repair depending on cellular status.
The regulatory proteins associated with histone modifications are
characterized by specific domains. For example, chromo-like domains
of the Royal family recognize methylation and plant homeodomain
(PHD), acetylation by bromodomain and phosphorylation by 14-3-3
domain (43). The recruitment of
basal transcription machinery by proteins with HAT activities,
including p300 and CREB-binding protein (CBP), and chromatin
remodeling complex components, including Brahma-related gene-1 are
facilitated by acetylated histones. These proteins bind to
acetylated nucleosomes or acetylated histone chains with higher
affinity. In addition, TATA box binding protein-associated factor 1
binds to acetylated histone and promotes the ubiquitination and
phosphorylation of H1, by which transcription is activated
(52).
Methylation at H3K4 assists in assembling regulatory
proteins by binding to bromodomain PHD finger transcription factor
via the PHD domain, a key component of the nucleosome remodeling
factor chromatin-remodeling complex. This assembly is followed by
the recruitment of sucrose non-fermenting homologue 2-like ATPase,
resulting in activation of the homeobox C8 gene (53). Similarly, Jumonji domain (JMJD)2A,
a lysine demethylase, and chromodomain helicase DNA binding protein
1, an ATPase, bind to methylated H3K4 with the tudor domain and
chromodomain, respectively (54).
The structural basis of these interactions remains to be fully
elucidated and the binding is reported to occur preceding H3R2
(55). These protein domains act
as adaptors for binding and a remodeling complex through methylated
H3.
Methylated H3L27 recruits polycomb protein PC2,
which exhibits ubiquitin ligase activity. Similarly, HP1 protein
interacts with H3K9 and exhibits methyltransferase and deacetylase
activities. The methylation of H3K4 and phosphorylation of H3T3
interfere with the binding of the transcriptionally repressive NuRD
complex and inhibitor of acetyltransferase complex, respectively
(52,56). The binding of a protein to histone
may also disrupt the modification in adjacent residues, which is
evident from altered heterochromatin protein 1 (HP1) binding to
methylated H3K9 upon the phosphorylation of H3S10 (51). In addition, a modification in one
histone tail can alter the modification on a different histone
tail. For example, the ubiquitination of H2B is required for the
methylation of H3K4 (43).
Suppressor of variegation 3-9 homolog 1 (SUV39H1) is a lysine
methyltransferase recruited by trimethylated H3K9-bound HP1 that
methylates adjacent histones. SUV39H1 activity facilitates further
binding of HP1 resulting in opening of the heterochromatin region.
Similarly, Enhancer of zeste homolog 2 possesses methyltransferase
activity specific to H3K27 and recruits the dimeric protein PC,
which is a component of the polycomb repressive complex 1. The
binding of PC to histones prevents access of the SWI/SNF remodeling
complex to chromatin, preventing transcription (57).
According to ‘histone code hypothesis’, histone
modification at specific residues determines subsequent
modifications in the same or a different histone (52). These modifications are recognized
by regulatory proteins resulting in chromatin remodeling and
transcription. For example, the methyl/phospho binary switch
hypothesis states that the combination of several modifications
results in the transition of a stable methyl/lysine state to a
dynamic transcriptional state, and the phosphorylation of S/T
residues adjacent to methylation site channels the downstream
proteins to facilitate transcription. This type of regulation by
phosphorylation depends on the position of phosphorylated residues,
if a phosphorylated residue occurs prior to methylation and
transcription is activated and vice versa (52).
Investigations on yeast chromosome mutations have
revealed the ubiquitination of H2B-K123 occurred previous to H3-K4
and that H3-K79 methylation facilitates chromatin opening and
transcription (58). In addition,
the dynamics of H2B ubiquitination/de-ubiquitination determines the
transcription of a certain set of genes. The de-ubiquitination
activity of ubiquitin-specific processing protease 8 of
Spt-Ada-Gcn5-acetyltransferase is essential for the methylation of
H3-K36, which in turn results in the gene expression of
galactokinase 1 in budding yeast (59). The proteosomal ATPases, regulatory
particle triple-a protein, or regulatory particle triphosphatase
(Rpt)4 and Rpt6 activate the methylation of H3-K4 and H3-K79 by
altering the chromatin structure in the vicinity of ubiquitination
(by Rad6). The 19S proteasome component ATPases independent of 20S
facilitates the initiation and elongation phases of transcription
and the 20S sub-complex is associated with RNA polymerase III
(60,61).
In higher eukaryotes, the impact of histone
modifications on transcription is complex and contrasting effects
exist in the modification patterns of histone residues. In
addition, the actual role and effect of modifications including
sumoylation, ubiquitination and de-ubiquitination remain to be
fully elucidated. Genome-wide analysis of H2B ubiquitination and
H3K4 methylation revealed the occurrence of ~5% of ubiquitination,
which is less adequate in mediating the methylation of H3K4
(62). The de-ubiquitination of
previously ubiquitinated H2B, which is usually ~35%, is attained
following the methylation of H3K4, which in turn facilitates the
methylation of H3K36 and thereby promotes transcription (63). The mechanism of the activation of
methyltransferase activities, including SET and MYND domain
containing 3 (SMYD3), which also has a DNA binding domain, requires
detailed investigation. SMYD3 can behave as a transcription factor
and as a DNA binding enzyme, however, its epigenetic implications
remain to be fully elucidated.
The epigenetics of histone ubiquitination in
eukaryotes remains unclear, however, the ubiquitin conjugating
enzyme Ubc2 facilitates the methylation of H3K4 by the
ubiquitination of H2B-K123 in Saccharomyces cerevisiae. In
addition, investigations of mutations have shown that the
ubiquitination of H2B-K123 is a prerequisite for H3K4 methylation
(62). The involvement of
ubiquitination and SUMOylation in the epigenetic regulation of
inflammatory responses remains to be fully elucidated, and there is
scope for extensive investigation. The ubiquitination and
SUMOylation of NF-κB activates inflammatory responses, but not in
an epigenetic manner (64,65).
Further investigation of these aspects in relation to inflammation
and inflammatory disorders is warranted.
5. RNA-mediated epigenetics
The emergence of long non-coding RNAs and small
non-coding RNAs through either intergenic or antisense
transcription has been shown to be involved in regulating chromatin
organization, translational repression and gene expression in
eukaryotic cells. The small RNAs exert their effect via RNA
interference (RNAi) pathways (66,67). The implications of DNA methylation
and subsequent gene silencing mediated through RNAi has been
established in Caenorhabditis, Schizosaccharomyces, and
Tetrahymena, and also in somatic and germ cells (68,69). Elucidation of the RNAi-mediated
downregulation of genes in human diseases offers potential for
RNA-based therapeutic strategies. However, the selection of small
interfering RNAs (siRNAs) or microRNAs (miRNAs), and knowledge of
multiple targets and binding efficiency/strength are crucial to
minimize unwanted reactions.
In general, small RNAs that regulate gene expression
in the cytoplasm can be either miRNAs, which are derived from a
hairpin loop, cause translational repression and usually carry
non-complimentary sequences to the target (70); siRNAs, which cause the degradation
of the transcript owing to its sequence complementarity; or
piwiRNAs (piRNAs), which mediate transposon transcripts. These RNAs
are capable of guiding chromatin modifications mediated through
histone/DNA methyltransferases (71).
The biogenesis of siRNAs is predominantly elicited
by double-stranded RNAs (dsRNAs) and is mediated by the Argonaute
(Ago) family of proteins. Ago proteins are ubiquitous effectors for
miRNAs and siRNAs whereas piwi proteins are responsible for piRNAs
(72). The precursor dsRNA is
cleaved by the Dicer family of enzymes, including RNAse III-forming
20-25 nucleotide (nt) duplexes where 3'OH overhangs
5'PO4 (73). Evidence
for the existence of several Dicer-independent mechanisms of siRNA
synthesis is also available in the literature (74). The cellular location of siRNA
biogenesis is debated; however, a large body of evidence suggests
that the cytoplasm is the possible site. By contrast, in S.
pompe, Ago-mediated cleavage and RNA-dependent RNA polymerase
amplification occurs in the nucleus (71). The generated duplexes are then
loaded to the effector Ago protein, which requires heat shock
protein 90. However, the mechanism, location and regulation of
these proteins in regard to RNAi remain to be elucidated. Gene
silencing by the miRNA interference pathway is shown in Fig. 5.
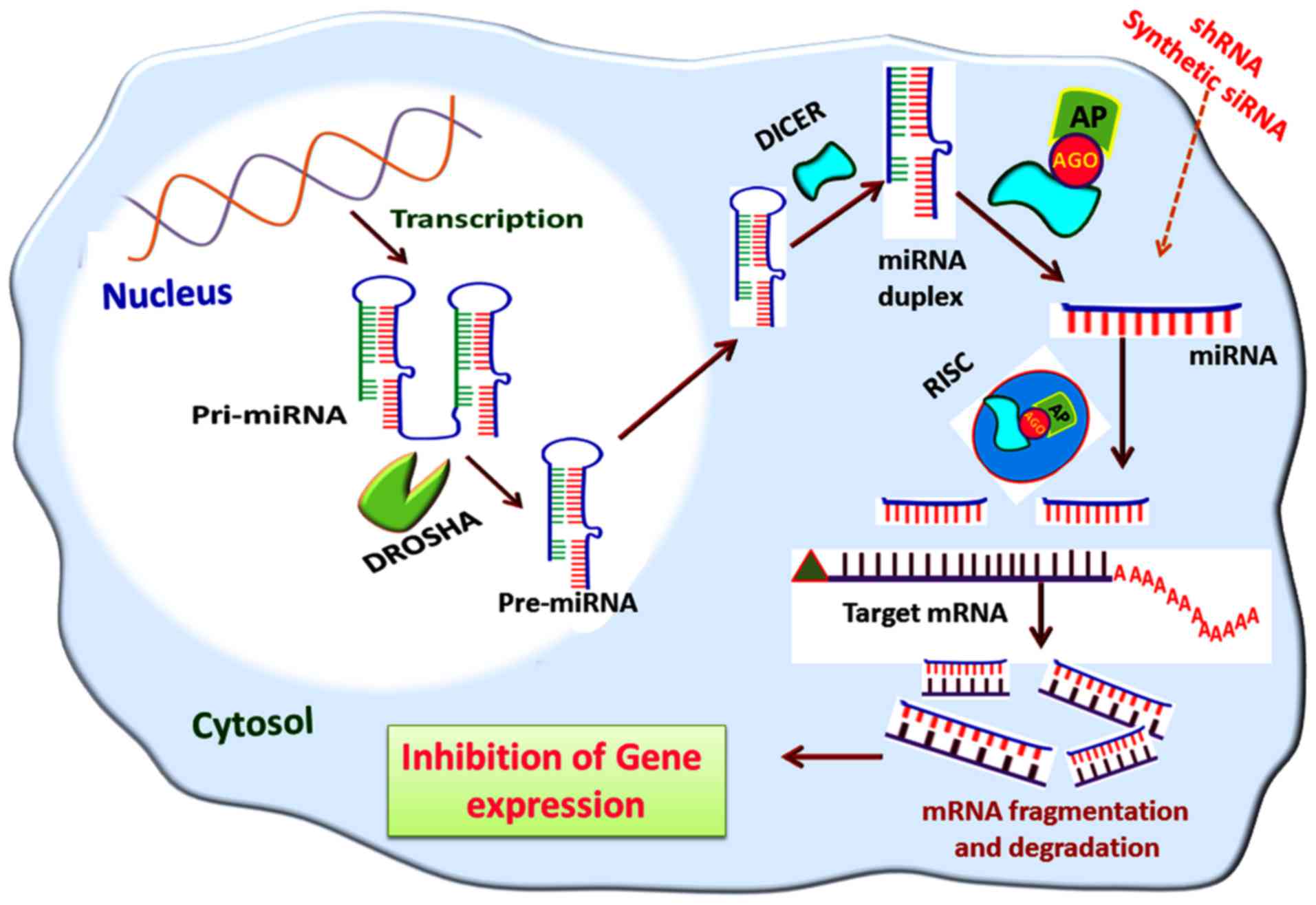 | Figure 5Gene silencing via the miRNA
interference pathway. The pre-miRNA formed in the nucleus by DROSHA
activity is transported to the cytosol mediated by exportin-5; in
the cytosol, the pre-miRNAs are cleaved by nucleases, including
DICER, to miRNA duplexes from which miRNAs are formed. The miRNAs
are then assembled to the RISC and leads to the degradation of
target mRNA transcripts. A similar mode of action is elicited by
shRNA and synthetic siRNA, if delivered to the cytosol. miRNA,
microRNA; pri-miRNA, primary miRNA; pre-miRNA, precursor miRNA;
RISC, RNA-induced silencing complex; AGO, argonaute; AP, effector
AGO protein; shRNA, short hairpin RNA; siRNA, small interfering
RNA. |
The link between the RNAi pathway and nucleosome
structure has been revealed by the identification of
heterochromatic chromodomain protein calcineurin-like EF-hand
protein 1 as a component of RNA-induced transcriptional silencing
(RITS) associated with Ago1 and small RNAs. Trans-acting siRNA Tas3
associates with RITS through Ago1 and the functional RITS complex
comprises an RNA-dependent RNA polymerase complex (RDRC; e.g.
Rpd1), helicases (e.g. helicase required for RNAi-mediated
heterochromatin assembly), and poly(A) polymerase CTC-interacting
domain 12) (75). The RITS
complex interacts with the heterochromatin and small RNAs, although
the exact function and mechanism of this interaction remains to be
elucidated; however, by binding to euchromatic mRNAs, RITS leads to
the methylation of H3K9 (76).
The RNAs associated with chromatin can act as scaffolds for the
organization of RITS and assists in the recognition of chromatin
modifications which ultimately lead to H3K9-mediated gene silencing
(77).
The Clr4 subunit of the Clr4-Rik1-Cul4 (CLRC)
complex, a protein complex that assembles at the target nucleosome,
possesses methyltransferase activity which methylates H3K9. Rik1
links with the RDRC and RITS complex, and the Stc1protein of the
CLRC interacts with Ago and Tas3, thereby linking the RNAi
machinery (77). These
interactions are reported to occur through physical means, which
suggests feedback among the RITS complex, the RDRC and the CLRC
complex (78,79). Rik1-associated factor 2 (also
known as Cmc2) and Rik1, components of the CLRC complex, associated
with Cdc20 (the catalytic subunit of the leading-strand DNA
polymerase-ε) and Mms19 (a regulator of the TFIIH) coordinate DNA
replication and the RNAi-mediated release of RNA polymerase II for
heterochromatin-mediated inheritance (80,81). These results were the outcome of
studies performed in Saccharomyces species and the evidence
for the coordination between RNAi and DNA replication requires
detailed investigation, particularly in higher eukaryotes.
6. Tendon disorders and epigenetic
regulation of inflammation
Tendon disorders, particularly rotator cuff
tendinopathies, are one of the most common musculoskeletal
disorders of which pain and inflammation are the major symptoms.
Shoulder tendinopathies constitute >30% of referrals for
musculoskeletal disorders. Despite the increased incidence of
tendinopathies, the exact mechanisms underlying the etiology and
pathogenesis of shoulder tendinopathies remain to be fully
elucidated (82). The expression
of inflammatory cytokines and mediators associated with
tendinopathies has been reported in several studies. Tenocytes upon
inflammatory trigger/stimuli tend to express inflammatory
cytokines, including tumor necrosis factor (TNF)-α, interleukin
(IL)-1β, IL-6, IL-21, and transforming growth factor (TGF)-β
(83,84).
Of note, the shoulder tendon tissues in the majority
of cases do not show the classical histology of inflammation,
including the infiltration of macrophages or neutrophils and
cytokine production (84). By
contrast, reports showing the presence of the infiltration and
activation of immune cells are also available; however, this
depends on the severity of the injury (85). Our previous study described a
novel dual-mechanism of the inflammatory status of injured rotator
cuff tendon tissue in relation to triggering receptor expressed on
myeloid cells-1 (TREM1). During asymptomatic tendinopathies, the
tenocytes express the TREM1 molecule and function like immune
cells, which in turn are regulated by high mobility group protein 1
and receptor for advanced glycation end products (1,86).
So far, the epigenetics of inflammation associated
with tendon disorders has not been investigated, however, the basic
cellular mechanisms appear to be applicable to tenocytes. The
trigger for the epigenetic switch and the pathological aspects of
epigenetic alterations in tendon cells remain to be fully
elucidated, however, several miRNAs have been reported to be
involved in tendon disorders and inflammation (87). For example, microRNA (miR)-29a is
involved in the regulation of IL-33-mediated inflammation in
rotator cuff tendons (88). In
our previous studies, the screening of alterations of miRNAs
associated with shoulder tendon matrisome disorganization (89), and the Janus kinase 2/signal
transducer and activator of transcription (STAT)3 pathway of
inflammation (90) was performed.
These studies revealed the alteration of hundreds of miRNAs which
are considered to be associated with the pathological changes in
the tendon. Among them, miR-145-5p, miR-100-5p, miR-195-5p, and
let-7 were found to be the key miRNAs, and warrant further detailed
investigations.
Due to the limited availability of literature
regarding the epigenetic regulations of tendon inflammation, the
following section mainly describes the epigenetics of general
inflammation irrespective of specific tissue; the extrapolation of
such knowledge to tendon tissues may have an impact on tendon
disorders.
The inflammatory response is complex and is elicited
by signal-specific or gene-specific cascade mechanisms which
usually result in antimicrobial defense, immune response and
repair, and regeneration of the affected tissue (91). A wide array of transcription
factors associated with inflammatory responses, including NF-κB,
forkhead box P3 (FOXP3), and STAT2 are regulated by epigenetic
changes including DNA methylation and/or histone modifications
(92). For example, JMJD3
regulates the methylation status of H3K27 and controls the
differentiation and phenotype identity of macrophage cells. The
IL-4 mediated activation of JMJD3 removes the repressive
methylation tag from H3K27 of the STAT6 promoter, which in turn
activates the downstream inflammatory genes. In addition, activated
STAT6 positively regulates JMJD3 by binding to its promoter
(93).
The impairment of DNA methylation following chronic
inflammation has led to an understanding of the role of the
polycomb group proteins in the epigenetic regulation of
inflammatory responses (94).
NF-κB/reticuloendotheliosis B (RelB)-mediated gene repression is
another classical example. RelB induction, resulting from the
actions of endotoxins or during sepsis, represses a number of
proinflammatory genes by enhancing heterochromatin formation
through interacting with H3K9 methyltransferase G9a. The subsequent
trimethylation of H3K9 recruits HP1 to form a repressive complex at
RelB promoters, and this recruits DNMT3a and DNMT3b to CpG
methylation. Therefore, RelB links histone modifications and DNA
methylations which have implications in inflammation (95,96).
The activation of HAT results in the transcription
of inflammatory genes whereas HDAC represses inflammation.
Proinflammatory cytokines, including IL-1, IL-8, IL-2 and IL-12,
are acetylated by CBP/p300 at their promoter regions leading to
their transcription. In addition, the activity of HDAC represses
the transcription of these genes. Proteins with HDAC activity also
regulate the transcription of proinflammatory and anti-inflammatory
cytokines by recruiting co-repressors and transcription factors,
including FOXP3, STATs, GATA, zinc finger E-box-binding homeobox 1
and NF-κB (97). Following
challenge with cytokines, NF-κB is regulated by inhibitor of NF-κB
kinase subunit α (IKK-α) of inhibitor of NF-κB (IκB) kinase, and
forms a complex by binding with the promoter region. This binding
is facilitated by the RNA polymerase II complex and CBP, where
IKK-α promotes the acetylation of H3K9 and phosphorylation of
H3S10. The phosphorylation of H3S10 leads to CBP-dependent H3K14
acetylation, which channels NF-κB flux. HDAC and glucocorticoid
receptor activation can reverse these processes leading to the
transcriptional repression of NF-κB-dependent inflammatory genes
(98).
The epigenetic regulation of inflammatory genes by
DNA methylation has also been reported. The hypomethylation of
Toll-like receptor (TLR)2 results in the aggravation of
inflammation, as reported in bronchial epithelial cells following
challenge with bacterial peptidoglycan (99). The combined effects of DNA
methylation and histone acetylation result in regulation of the
TLR4 gene in the epithelial lining of the intestine, suggesting the
role of epigenetic DNA methylation on microbial defense (100). The increased methylation of
interferon-γ promoter cells and the demethylation of IL-2 and IL-6
promoters on CD4+ T cells upon allergen challenge on
experimental asthma models provide further evidence (101,102). Helicobacter pylori
induces inflammation of the gastric mucosa and activates oncogenic
pathways by altering the methylation patterns of gastric epithelial
cells (103). In addition, the
inflammatory cytokine TGF-β suppresses CD133 stem cell/cancer
biomarker by methylation, with demethylation at the promoter
regions inducing resistance to apoptosis and anticancer drugs
(104,105). In addition, the hypermethylation
of suppressor of cytokine signaling 1 in macrophages regulates
cytokine signaling, particularly TNF-α and IL-6, and thereby
inflammation (106,107).
Chemokines of the (C-X-C motif) ligand (CXCL) family
are reported to be involved in various carcinogenic and
anti-carcinogenic signaling pathways and in inflammatory responses.
CXCL1/growth regulated oncogene-α (GROα) enhances angiogenesis and
inhibits extracellular matrix (ECM) synthesis in prostate cancer
cells. CXCL1/GROα acts by the activation of NF-κB and its
subsequent interaction with HDAC1 to repress the ECM protein,
fibulin-1D (108). Similarly,
the expression of CXCL14 is suppressed by the hypermethylation of
CpG, and this hypomethylation enhances its expression. CXCL14
exerts its anticancer function by preventing cell migration and
invasion by inhibiting NF-κB signaling (109,110). CXCL12 functions by binding to
the receptor CXCR4, where it is downregulated by aberrant
methylation in breast cancer cells. As the existence of
demethylated CXCR4 and hypermethylated CXCL12 has been established
in tumor cells, the methylation status of the cells can be used as
a diagnostic tool for various types of cancer (111,112).
7. Summary and future directions
The epigenetic regulation of gene expression has
been established in mammalian systems, including humans. The
structures, localization, mechanism of recruitment and regulation
of several proteins with DNA methyltransferase activities, HAT and
HDAC activities, and their roles in DNA replication and mRNA
transcription have been reported. RNAi-mediated by miRNAs is also
considered to be involved in epigenetics. The implications of
epigenetics in diseases including cancer, cardiovascular disease,
diabetes and neurological disorders have led to the understanding
of their molecular pathology and disease management approaches.
Inflammation is a well-established epigenetically-regulated
biological process and the major genes involved are well described.
There is a lack of sufficient evidence in the literature regarding
the epigenetic regulation of gene expression in tendon tissues.
Inflammation is a generalized occurrence associated with all
tendinopathies, including rotator cuff insults. Conventional
treatment strategies mainly deal with the management of pain and
inflammation. However, the sustained disorganization of ECM
increases the risk of recurrence of tendon damage. The
possibilities of epigenetic regulation for the expression of
inflammatory genes and the genes associated with tendon repair
cannot be neglected. Additionally, extrapolation of the epigenetic
changes in inflammation may offer opportunities to establish the
same understanding in tendons.
Understanding the mechanism of DNA methylation
patterns and histone modifications of tendon specific inflammatory
genes can assist in improving current understanding of tendon
pathology and may lead to the development of novel
therapeutic/management strategies. The screening and validation of
miRNA-mediated RNAi in tendon pathology can form another promising
approach. Therapeutics based on miRNAs or miRNA-inhibitors possess
appreciable therapeutic potential in several diseases, including
cancer, however, none have been reported for rotator cuff tendons.
Those miRNAs targeting multiple mRNAs in the same or different
pathways can provide a promising approach, however, the selection
of pathways and consideration of non-specific targets require
careful evaluation. The mode of delivery of such therapeutic miRNAs
is also a challenge. Further investigations are warranted in the
field of tendon physiology/pathology for the elucidation of novel
and promising epigenetic therapeutic targets and their clinical
application.
Funding
This review was supported by a research grant from
the State of Nebraska to DKA (grant no. LB506), grants from the
National Heart, Lung and Blood Institute, National Institutes of
Health (Bethesda, MD, USA; grant nos. R01 HL104516, R01 HL116042
and R01 HL120659) and from the George F. Haddix Faculty Grant from
Creighton University (grant no. CU-240083).
Availability of data and materials
Not applicable.
Authors' contributions
FGT, CSB and DKA conceived the review and analyzed
the relevant literature. FGT and CSB sourced the literature and
wrote the first draft of the manuscript. FGT, MFD and DKA
critically revised the manuscript. FGT and CSB produced the
figures. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors confirm that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Thankam FG, Dilisio MF, Dougherty KA,
Dietz NE and Agrawal DK: Triggering receptor expressed on myeloid
cells and 5'adenosine monophosphate-activated protein kinase in the
inflammatory response: A potential therapeutic target. Expert Rev
Clin Immunol. 12:1239–1249. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Thankam FG, Dilisio MF and Agrawal DK:
Immunobiological factors aggravating the fatty infiltration on
tendons and muscles in rotator cuff lesions. Mol Cell Biochem.
417:17–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Page P: Shoulder muscle imbalance and
subacromial impingement syndrome in overhead athletes. Int J Sports
Phys Ther. 6:51–58. 2011.PubMed/NCBI
|
|
4
|
Waddington CH: Towards a theoretical
biology. Nature. 218:525–527. 1968. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kornberg RD: Chromatin structure: A
repeating unit of histones and DNA. Science. 184:868–871. 1974.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Weintraub H and Groudine M: Chromosomal
subunits in active genes have an altered conformation. Science.
193:848–856. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Narlikar GJ, Fan HY and Kingston RE:
Cooperation between complexes that regulate chromatin structure and
transcription. Cell. 108:475–487. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Felsenfeld G and Groudine M: Controlling
the double helix. Nature. 421:448–453. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Surani MA, Hayashi K and Hajkova P:
Genetic and epigenetic regulators of pluripotency. Cell.
128:747–762. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Human Genome Structural Variation Working
Group; Eichler EE, Nickerson DA, Altshuler D, Bowcock AM, Brooks
LD, Carter NP, Church DM, Felsenfeld A, Guyer M, et al: Completing
the map of human genetic variation. Nature. 447:161–165. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sebat J: Major changes in our DNA lead to
major changes in our thinking. Nat Genet. 39(Suppl 7): S3–S5. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jin B, Li Y and Robertson KD: DNA
Methylation: Superior or subordinate in the epigenetic hierarchy.
Genes Cancer. 2:607–617. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Herman JG and Baylin SB: Gene silencing in
cancer in association with promoter hypermethylation. N Engl J Med.
349:2042–2054. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Voelter-Mahlknecht S: Epigenetic
associations in relation to cardiovascular prevention and
therapeutics. Clin Epigenetics. 8:42016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Shea JM, Serra RW, Carone BR, Shulha HP,
Kucukural A, Ziller MJ, Vallaster MP, Gu H, Tapper AR, Gardner PD,
et al: Genetic and epigenetic variation, but not diet, shape the
sperm methylome. Dev Cell. 35:750–758. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Lachner M and Jenuwein T: The many faces
of histone lysine methylation. Curr Opin Cell Biol. 14:286–298.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Pradhan S, Bacolla A, Wells RD and Roberts
RJ: Recombinant human DNA (cytosine-5) methyltransferase. I.
Expression, purification, and comparison of de novo and maintenance
methylation. J Biol Chem. 274:33002–33010. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Leonhardt H, Page AW, Weier HU and Bestor
TH: A targeting sequence directs DNA methyltransferase to sites of
DNA replication in mammalian nuclei. Cell. 71:865–873. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Okano M, Bell DW, Haber DA and Li E: DNA
methyltransferases Dnmt3a and Dnmt3b are essential for de novo
methylation and mammalian development. Cell. 99:247–257. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Borgel J, Guibert S, Li Y, Chiba H,
Schübeler D, Sasaki H, Forné T and Weber M: Targets and dynamics of
promoter DNA methylation during early mouse development. Nat Genet.
42:1093–1100. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wu H, Coskun V, Tao J, Xie W, Ge W,
Yoshikawa K, Li E, Zhang Y and Sun YE: Dnmt3a-dependent nonpromoter
DNA methylation facilitates transcription of neurogenic genes.
Science. 329:444–448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ooi SK, Qiu C, Bernstein E, Li K, Jia D,
Yang Z, Erdjument-Bromage H, Tempst P, Lin SP, Allis CD, et al:
DNMT3L connects unmethylated lysine 4 of histone H3 to de novo
methylation of DNA. Nature. 448:714–717. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ramsahoye BH, Biniszkiewicz D, Lyko F,
Clark V, Bird AP and Jaenisch R: Non-CpG methylation is prevalent
in embryonic stem cells and may be mediated by DNA
methyltransferase 3a. Proc Natl Acad Sci USA. 97:5237–5242. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pinney S: Mammalian Non-CpG methylation:
Stem cells and beyond. Biology (Basel). 3. pp. 739–751. 2014
|
|
25
|
Suzuki MM and Bird A: DNA methylation
landscapes: Provocative insights from epigenomics. Nat Rev Genet.
9:465–476. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ehrlich M, Gama-Sosa MA, Huang LH, Midgett
RM, Kuo KC, McCune RA and Gehrke C: Amount and distribution of
5-methylcytosine in human DNA from different types of tissues of
cells. Nucleic Acids Res. 10:2709–2721. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Reik W, Dean W and Walter J: Epigenetic
reprogramming in mammalian development. Science. 293:1089–1093.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Shen L, Kondo Y, Guo Y, Zhang J, Zhang L,
Ahmed S, Shu J, Chen X, Waterland RA and Issa JP: Genome-wide
profiling of DNA methylation reveals a class of normally methylated
CpG island promoters. PLoS Genet. 3:2023–2036. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Henderson IR and Jacobsen SE: Tandem
repeats upstream of the Arabidopsis endogene SDC recruit non-CG DNA
methylation and initiate siRNA spreading. Genes Dev. 22:1597–1606.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lienert F, Wirbelauer C, Som I, Dean A,
Mohn F and Schübeler D: Identification of genetic elements that
autonomously determine DNA methylation states. Nat Genet.
43:1091–1097. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
31
|
Meissner A, Mikkelsen TS, Gu H, Wernig M,
Hanna J, Sivachenko A, Zhang X, Bernstein BE, Nusbaum C, Jaffe DB,
et al: Genome-scale DNA methylation maps of pluripotent and
differentiated cells. Nature. 454:766–770. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Smith ZD, Chan MM, Mikkelsen TS, Gu H,
Gnirke A, Regev A and Meissner A: A unique regulatory phase of DNA
methylation in the early mammalian embryo. Nature. 484:339–344.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aran D, Sabato S and Hellman A: DNA
methylation of distal regulatory sites characterizes dysregulation
of cancer genes. Genome Biol. 14:R212013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Sharif O and Knapp S: From expression to
signaling: Roles of TREM-1 and TREM-2 in innate immunity and
bacterial infection. Immunobiology. 213:701–713. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jones PA: Functions of DNA methylation:
Islands, start sites, gene bodies and beyond. Nat Rev Genet.
13:484–492. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Maunakea AK, Nagarajan RP, Bilenky M,
Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C,
Zhao Y, et al: Conserved role of intragenic DNA methylation in
regulating alternative promoters. Nature. 466:253–257. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Laurent L, Wong E, Li G, Huynh T, Tsirigos
A, Ong CT, Low HM, Kin Sung KW, Rigoutsos I, Loring J and Wei CL:
Dynamic changes in the human methylome during differentiation.
Genome Res. 20:320–331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Mayer W, Niveleau A, Walter J, Fundele R
and Haaf T: Demethylation of the zygotic paternal genome. Nature.
403:501–502. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Inoue A and Zhang Y: Replication-dependent
loss of 5- hydroxymethylcytosine in mouse preimplantation embryos.
Science. 334:1942011. View Article : Google Scholar
|
|
40
|
Guo JU, Su Y, Zhong C, Ming GL and Song H:
Hydroxylation of 5-methylcytosine by TET1 promotes active DNA
demethylation in the adult brain. Cell. 145:423–434. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yang H, Lin H, Xu H, Zhang L, Cheng L, Wen
B, Shou J, Guan K, Xiong Y and Ye D: TET-catalyzed 5-methylcytosine
hydroxylation is dynamically regulated by metabolites. Cell Res.
24:1017–1020. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Wu H and Zhang Y: Mechanisms and functions
of Tet protein-mediated 5-methylcytosine oxidation. Genes Dev.
25:2436–2452. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gill G: SUMO and ubiquitin in the nucleus:
Different functions, similar mechanisms. Genes Dev. 18:2046–2059.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Tsankova N, Renthal W, Kumar A and Nestler
EJ: Epigenetic regulation in psychiatric disorders. Nat Rev
Neurosci. 8:355–367. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Ragunathan K, Jih G and Moazed D:
Epigenetic inheritance uncoupled from sequence-specific
recruitment. Science. 348:12586992015. View Article : Google Scholar :
|
|
47
|
Covic M, Hassa PO, Saccani S, Buerki C,
Meier NI, Lombardi C, Imhof R, Bedford MT, Natoli G and Hottiger
MO: Arginine methyltransferase CARM1 is a promoter-specific
regulator of NF-kappaB-dependent gene expression. EMBO J. 24:85–96.
2005. View Article : Google Scholar
|
|
48
|
Tachibana M, Sugimoto K, Nozaki M, Ueda J,
Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H and Shinkai Y:
G9a histone methyltransferase plays a dominant role in euchromatic
histone H3 lysine 9 methylation and is essential for early
embryogenesis. Genes Dev. 16:1779–1791. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Botuyan MV, Lee J, Ward IM, Kim JE,
Thompson JR, Chen J and Mer G: Structural basis for the methylation
state-specific recognition of histone H4-K20 by 53BP1 and Crb2 in
DNA repair. Cell. 127:1361–1373. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Techima M: Inquiring for new approach to
nursing education. III. A case in clinical practicum (2). Kango
Kenkyu. 24:366–372. 1991.In Japanese.
|
|
51
|
Fischle W, Tseng BS, Dormann HL,
Ueberheide BM, Garcia BA, Shabanowitz J, Hunt DF, Funabiki H and
Allis CD: Regulation of HP1-chromatin binding by histone H3
methylation and phosphorylation. Nature. 438:1116–1122. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Margueron R, Trojer P and Reinberg D: The
key to development: Interpreting the histone code. Curr Opin Genet
Dev. 15:163–176. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Wysocka J, Swigut T, Xiao H, Milne TA,
Kwon SY, Landry J, Kauer M, Tackett AJ, Chait BT, Badenhorst P, et
al: A PHD finger of NURF couples histone H3 lysine 4 trimethylation
with chromatin remodelling. Nature. 442:86–90. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Andrews FH, Gatchalian J, Krajewski K,
Strahl BD and Kutateladze TG: Regulation of methyllysine readers
through phosphorylation. ACS Chem Biol. 11:547–553. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Couture JF, Collazo E and Trievel RC:
Molecular recognition of histone H3 by the WD40 protein WDR5. Nat
Struct Mol Biol. 13:698–703. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kadota S and Nagata K: pp32, an INHAT
component, is a transcription machinery recruiter for maximal
induction of IFN-stimulated genes. J Cell Sci. 124:892–899. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Shao Z, Raible F, Mollaaghababa R, Guyon
JR, Wu CT, Bender W and Kingston RE: Stabilization of chromatin
structure by PRC1, a Polycomb complex. Cell. 98:37–46. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Chandrasekharan MB, Huang F and Sun ZW:
Ubiquitination of histone H2B regulates chromatin dynamics by
enhancing nucleosome stability. Proc Natl Acad Sci USA.
106:16686–16691. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Canzonetta C, Vernarecci S, Iuliani M,
Marracino C, Belloni C, Ballario P and Filetici P: SAGA DUB-Ubp8
deubiquitylates centromeric histone variant Cse4. G3 (Bethesda).
6:287–298. 2015. View Article : Google Scholar
|
|
60
|
Sun L, Johnston SA and Kodadek T: Physical
association of the APIS complex and general transcription factors.
Biochem Biophys Res Commun. 296:991–999. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Muratani M and Tansey WP: How the
ubiquitin-proteasome system controls transcription. Nat Rev Mol
Cell Biol. 4:192–201. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Sun ZW and Allis CD: Ubiquitination of
histone H2B regulates H3 methylation and gene silencing in yeast.
Nature. 418:104–108. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Henry KW, Wyce A, Lo WS, Duggan LJ, Emre
NC, Kao CF, Pillus L, Shilatifard A, Osley MA and Berger SL:
Transcriptional activation via sequential histone H2B
ubiquitylation and deubiquitylation, mediated by SAGA-associated
Ubp8. Genes Dev. 17:2648–2663. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Gao C, Huang W, Kanasaki K and Xu Y: The
role of ubiquitination and sumoylation in diabetic nephropathy.
Biomed Res Int. 2014.160692:2014.
|
|
65
|
Collins P, Mitxitorena I and Carmody R:
The ubiquitination of NF-κB subunits in the control of
transcription. Cells. 5:pii: E232016. View Article : Google Scholar
|
|
66
|
Moazed D: Small RNAs in transcriptional
gene silencing and genome defence. Nature. 457:413–420. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Cech TR and Steitz JA: The noncoding RNA
revolution-trashing old rules to forge new ones. Cell. 157:77–94.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fire A, Xu S, Montgomery MK, Kostas SA,
Driver SE and Mello CC: Potent and specific genetic interference by
double-stranded RNA in caenorhabditis elegans. Nature. 391:806–811.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Matzke M, Matzke AJ and Kooter JM: RNA:
Guiding gene silencing. Science. 293:1080–1083. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Boosani C and Agrawal D: Epigenetic
regulation of innate immunity by microRNAs. Antibodies. 5:82016.
View Article : Google Scholar
|
|
71
|
Castel SE and Martienssen RA: RNA
interference in the nucleus: Roles for small RNAs in transcription,
epigenetics and beyond. Nat Rev Genet. 14:100–112. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Kim VN, Han J and Siomi MC: Biogenesis of
small RNAs in animals. Nat Rev Mol Cell Biol. 10:126–139. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Pak J and Fire A: Distinct populations of
primary and secondary effectors during RNAi iC elegans. Science.
315:241–244. 2007. View Article : Google Scholar
|
|
74
|
Halic M and Moazed D: Dicer-independent
primal RNAs trigger RNAi and heterochromatin formation. Cell.
140:504–516. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Motamedi MR, Verdel A, Colmenares SU,
Gerber SA, Gygi SP and Moazed D: Two RNAi complexes, RITS and RDRC,
physically interact and localize to noncoding centromeric RNAs.
Cell. 119:789–802. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Bühler M, Verdel A and Moazed D: Tethering
RITS to a nascent transcript initiates RNAi- and
heterochromatin-dependent gene silencing. Cell. 125:873–886. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Holoch D and Moazed D: RNA-mediated
epigenetic regulation of gene expression. Nat Rev Genet. 16:71–84.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Bayne EH, White SA, Kagansky A, Bijos DA,
Sanchez-Pulido L, Hoe KL, Kim DU, Park HO, Ponting CP, Rappsilber J
and Allshire RC: Stc1: A critical link between RNAi and chromatin
modification required for heterochromatin integrity. Cell.
140:666–677. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Gerace EL, Halic M and Moazed D: The
methyltransferase activity of Clr4Suv39h triggers RNAi
independently of histone H3K9 methylation. Mol Cell. 39:360–372.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Zaratiegui M, Castel SE, Irvine DV, Kloc
A, Ren J, Li F, de Castro E, Marí L, Chang AY, Goto D, et al: RNAi
promotes heterochromatic silencing through replication-coupled
release of RNA Pol II. Nature. 479:135–138. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Li F, Martienssen R and Cande WZ:
Coordination of DNA replication and histone modification by the
Rik1-Dos2 complex. Nature. 475:244–248. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Millar NL, Murrell GA and McInnes IB:
Inflammatory mechanisms in tendinopathy-towards translation. Nat
Rev Rheumatol. 13:110–122. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Campbell AL, Smith NC, Reilly JH, Kerr SC,
Leach WJ, Fazzi UG, Rooney BP, Murrell GA and Millar NL: IL-21
receptor expression in human tendinopathy. Mediators Inflamm.
2014.481206:2014.
|
|
84
|
Legerlotz K, Jones ER, Screen HR and Riley
GP: Increased expression of IL-6 family members in tendon
pathology. Rheumatology (Oxford). 51:1161–1165. 2012. View Article : Google Scholar
|
|
85
|
Dean BJ, Gettings P, Dakin SG and Carr AJ:
Are inflammatory cells increased in painful human tendinopathy? A
systematic review. Br J Sports Med. 50:216–220. 2016. View Article : Google Scholar
|
|
86
|
Thankam FG, Dilisio MF, Dietz NE and
Agrawal DK: TREM-1, HMGB1 and RAGE in the shoulder tendon: Dual
mechanisms for inflammation based on the coincidence of
glenohumeral arthritis. PLoS One. 11:e01654922016. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Guo J, Zhang JF, Li G and Chan KM: A
Mini-review: MicroRNA in tendon injuries. J Stem Cell Res Ther.
5:3032015.
|
|
88
|
Millar NL, Gilchrist DS, Akbar M, Reilly
JH, Kerr SC, Campbell AL, Murrell GA, Liew FY, Kurowska-Stolarska M
and McInnes IB: MicroRNA29a regulates IL-33-mediated tissue
remodelling in tendon disease. Nat Commun. 6:67742015. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Thankam FG, Boosani CS, Dilisio MF, Dietz
NE and Agrawal DK: MicroRNAs associated with shoulder tendon
matrisome disorganization in glenohumeral arthritis. PLoS One.
11:e01680772016. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Thankam FG, Boosani CS, Dilisio MF and
Agrawal DK: MicroRNAs associated with inflammation in shoulder
tendinopathy and glenohumeral arthritis. Mol Cell Biochem.
437:81–97. 2018. View Article : Google Scholar
|
|
91
|
Medzhitov R and Horng T: Transcriptional
control of the inflammatory response. Nat Rev Immunol. 9:692–703.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Wierda RJ, Geutskens SB, Jukema JW, Quax
PH and van den Elsen PJ: Epigenetics in atherosclerosis and
inflammation. J Cell Mol Med. 14:1225–1240. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
De Santa F, Totaro MG, Prosperini E,
Notarbartolo S, Testa G and Natoli G: The histone H3 lysine-27
demethylase Jmjd3 links inflammation to inhibition of
polycomb-mediated gene silencing. Cell. 130:1083–1094. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Hahn MA, Hahn T, Lee DH, Esworthy RS, Kim
BW, Riggs AD, Chu FF and Pfeifer GP: Methylation of polycomb target
genes in intestinal cancer is mediated by inflammation. Cancer Res.
68:10280–10289. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Chen X, El Gazzar M, Yoza BK and McCall
CE: The NF-kappaB Factor RelB and Histone H3 lysine
methyltransferase G9a directly interact to generate epigenetic
silencing in endotoxin tolerance. J Biol Chem. 284:27857–27865.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
96
|
Gazzar ME, Yoza BK, Chen X, Hu J, Hawkins
GA and McCall CE: G9a and HP1 couple histone and DNA methylation to
TNF transcription silencing during endotoxin tolerance. J Biol
Chem. 283:32198–32208. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Villagra A, Sotomayor EM and Seto E:
Histone deacetylases and the immunological network: Implications in
cancer and inflammation. Oncogene. 29:157–173. 2010. View Article : Google Scholar
|
|
98
|
Bayarsaihan D: Epigenetic Mechanisms in
Inflammation. J Dent Res. 90:9–17. 2011. View Article : Google Scholar :
|
|
99
|
Shuto T, Furuta T, Oba M, Xu H, Li JD,
Cheung J, Gruenert DC, Uehara A, Suico MA, Okiyoneda T and Kai H:
Promoter hypomethylation of Toll-like receptor-2 gene is associated
with increased proinflammatory response toward bacterial
peptidoglycan in cystic fibrosis bronchial epithelial cells. FASEB
J. 20:782–784. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Takahashi N: Microbial ecosystem in the
oral cavity: Metabolic diversity in an ecological niche and its
relationship with oral diseases. Int Congr Ser. 1284:103–112. 2005.
View Article : Google Scholar
|
|
101
|
Brand RA: Surgical anatomy of the rotator
cuff and the natural history of degenerative periarthritis. Clin
Orthop Related Res. 466:543–551. 2008. View Article : Google Scholar
|
|
102
|
Claycombe KJ, Brissette CA and Ghribi O:
Epigenetics of inflammation, maternal infection, and nutrition. J
Nutr 145 (Suppl):. 1109S–1115S. 2015.
|
|
103
|
Niwa T, Tsukamoto T, Toyoda T, Mori A,
Tanaka H, Maekita T, Ichinose M, Tatematsu M and Ushijima T:
Inflammatory processes triggered by Helicobacter pylori infection
cause aberrant DNA methylation in gastric epithelial cells. Cancer
Res. 70:1430–1440. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
104
|
Ding W, Mouzaki M, You H, Laird JC, Mato
J, Lu SC and Rountree CB: CD133+ liver cancer stem cells
from methionine adenosyl transferase 1A-deficient mice demonstrate
resistance to transforming growth factor (TGF)-beta-induced
apoptosis. Hepatology. 49:1277–1286. 2009. View Article : Google Scholar
|
|
105
|
Wang YQ, Li YM, Li X, Liu T, Liu XK, Zhang
JQ, Guo JW, Guo LY and Qiao L: Hypermethylation of TGF-β1 gene
promoter in gastric cancer. World J Gastroenterol. 19:5557–5564.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Boosani CS, Dhar K and Agrawal DK:
Down-regulation of hsa-miR-1264 contributes to DNMT1-mediated
silencing of SOCS3. Mol Biol Rep. 42:1365–1376. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Cheng C, Huang C, Ma TT, Bian EB, He Y,
Zhang L and Li J: SOCS1 hypermethylation mediated by DNMT1 is
associated with lipopolysaccharide-induced inflammatory cytokines
in macrophages. Toxicol Lett. 225:488–497. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Kuo PL, Shen KH, Hung SH and Hsu YL:
CXCL1/GROα increases cell migration and invasion of prostate cancer
by decreasing fibulin-1 expression through NF-κB/HDAC1 epigenetic
regulation. Carcinogenesis. 33:2477–2487. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Chen S, Boosani C and Agrawal DK: Abstract
12818: KPNA4 mediates vitamin D-dependent inhibition of NF-κB
activity in swine epicardial preadipocytes. Circulation.
130:A128182014.
|
|
110
|
Cao B, Yang Y, Pan Y, Jia Y, Brock MV,
Herman JG and Guo M: Epigenetic silencing of CXCL14 induced
colorectal cancer migration and invasion. Discov Med. 16:137–147.
2013.PubMed/NCBI
|
|
111
|
Ramos EA, Grochoski M, Braun-Prado K,
Seniski GG, Cavalli IJ, Ribeiro EM, Camargo AA, Costa FF and
Klassen G: Epigenetic changes of CXCR4 and its ligand CXCL12 as
prognostic factors for sporadic breast cancer. PLoS One.
6:e294612011. View Article : Google Scholar
|
|
112
|
Yasmin R, Siraj S, Hassan A, Khan AR,
Abbasi R and Ahmad N: Epigenetic regulation of inflammatory
cytokines and associated genes in human malignancies. Mediators
Inflamm. 2015.201703:2015.
|