Introduction
Chronic obstructive pulmonary disease (COPD)
includes chronic bronchitis and emphysema. COPD is the third
leading cause of mortality worldwide, and is characterized by
partially reversible airflow limitation and persistent alveolar
destruction (1). The increasing
morbidity and mortality of this disease increases the associated
burdens on society (1). Exposure
to first- and second-hand cigarette smoke (CS) is the most
important risk factor for COPD (2,3).
The percentage of elderly smokers who suffer from COPD is almost
50% (4,5). CS damages bronchial and alveolar
epithelial cells by inducing cell death (6). However, the molecular mechanisms
through which CS induces COPD remain incompletely understood.
Nuclear receptor 77 (Nur77), also known as NR4A1,
TR3 or NGFI-B, belongs to the NR4A receptor subfamily which
consists of Nurr1 (NR4A2, NOT) and NOR-1 (NR4A3, MINOR); Nur77 is
also considered an orphan receptor as it lacks identified ligands
(7-9). Similar to other nuclear receptors,
Nur77 contains an activation function (AF)-1 N-terminal
transactivation domain, a central zinc finger DNA-binding domain
and an AF-2 C-terminal segment containing the ligand-binding domain
(LBD) (7-9). In general, Nur77 resides in the cell
nucleus, although some stimulants trigger Nur77 nuclear export
(9). Nur77 plays a critical role
in inflammation and in cellular processes, such as proliferation,
differentiation, survival and death (9). Previous researchers investigating
Nur77 have focused mostly on neurological disorders, cardiovascular
diseases and cancer (10). Nur77
expression has been described in lung tissue and lung epithelial
cells. Notably, Nur77 is overexpressed in the majority of lung
cancer and pulmonary artery smooth muscle cell (PASMC)
proliferation models (11). Nur77
has been shown to exert anti-inflammatory effects on ovalbumin
(OVA)-induced airway allergic inflammation and in a
lipopolysaccharide (LPS)-induced sepsis mouse model (12,13). However, the role of Nur77 in
cigarette smoke extract (CSE)-induced COPD remains unclear.
Autophagy is a cellular lysosomal degradation
process that eliminates damaged organelles or aberrant proteins and
is characterized by the activity of bilayer structures called
autophagosomes (14,15). Emerging evidence suggests that
autophagy plays a deleterious role in CS-induced COPD (14,16). The dysregulation of autophagy is
responsible for CS-induced airway epithelial damage, such as the
shortening of the cilia and the hyperproduction of mucus (17,18). To a certain extent, CS-induced
emphysema is due to an imbalance in autophagy. Recently,
researchers have shown interest in the association between Nur77
and autophagy (19). It has been
shown that when cells are treated with 1-(3,4,5-trihydroxyphenyl)
THPN, a Nur77-targeting compound, Nur77 targets the mitochondria
through contact with the outer membrane protein, Nix, and enters
the mitochondria, in turn contributing to the dissipation of the
mitochondrial membrane potential and to the induction of autophagy
(19). Nur77 has been shown to
promote the autophagy of PC12 cells, resulting in PC12 cell death
(20). In addition, the
interaction between Nur77 and Bcl2 has been shown to induce
apoptosis in various types of cancer (21,22). A recent study revealed that Nur77
binds to Bcl2 family proteins (Bcl-B) and subsequently mediates
autophagy, suggesting that the interaction between Bcl-B and Nur77
affects the stability of the Bcl-B-Beclin-1 complex and triggers
autophagy (23). Bcl2 family
proteins inhibit autophagy by binding to Beclin-1, as well as Bcl2
(24,25). However, whether Nur77-Bcl2
complexes dissociate Beclin-1 from Bcl2 and subsequently induce
autophagy remains unknown, particularly in the context of
CS-induced autophagy.
Increasing evidence indicates that autophagy is
critically involved in the pathogenesis of CS-induced COPD. Given
the roles of Nur77 in autophagy, in this study, we aimed to
determine whether CS triggers Nur77-induced autophagy and to
elucidate the underlying mechanisms. We report that CS-induced
autophagy requires the nuclear export of Nur77, the interaction
between Nur77 and Bcl-2, and the dissociation of Beclin-1 from
Bcl2.
Materials and methods
Animals
Male C57BL/6 mice (28 mice in total, 26-30 days old,
weighing 11-14 g) were purchased from the Changsha SLAC
Experimental Animal Center (Changsha, China). All mice were given
water and food ad libitum under a 12/12-h light-dark cycle
under specific pathogen-free conditions, and were bred to 10-12
weeks for establishing our model. All in vivo experiments
were approved by the local Animal Health Service, the Central South
University Animal Care Committee and were performed according to
the National Institutes of Health guidelines on animal care and
welfare.
Exposure to CS
The mice (28 mice were randomly divided into 4
groups, n=7 per group, 4 groups in total, 10-12 weeks old) were
exposed to air or CS, as previously described (26). Briefly, the whole bodies of the
mice were exposed to CS from 8 cigarettes simultaneously (without a
filter; Baisha, Changsha, China) in a box (40×60×50 cm), 4 times
for 25 min each at 20-min intervals; exposure to CS was carried out
5 days per week for 10 weeks. Baisha Pai cigarettes contain 10 mg
of tar, 1.0 mg of nicotine and 13 mg of carbon monoxide (CO). The
animals in the control group were exposed to room air (RA). All
animals received siRNA-Nur77-lentivirus (siRNA-Nur77 sequence,
5'-CGC CTG GCA TAC CGA TCT AAA -3') or lentiviral vector vehicle
(50 µl containing 1×109/mouse/treatment) on the
first day of each week.
Histopathological analysis and
immunohistochemistry (IHC)
The mice were anesthetized by an intraperitoneal
injection of pentobarbital (40 mg/kg) at 24 h after the final CS
exposure. One main bronchus was ligated, and the lung was perfused
with 4% paraformaldehyde at a constant pressure of 2.45 kPa for 1 h
before being removed from the animal and then was placed into fresh
4% paraformaldehyde for 24 h. The fixed lungs were embedded in
paraffin and then were cut into 4-µm-thick lung tissues
slices. Some lung slices were dewaxed with xylene, hydrated with
ethanol and then stained with 20% hematoxylin for 5 min and 0.5%
eosin for 3 min at room temperature (H&E, G1120, Solarbio
Science & Technology, Beijing, China) for the analysis of
inflammation and emphysema. A total of 10 of random fields were
examined using a Leica digital microscope (Leica DM6Bl Leica,
Wetzlar, Germany). Histopathology was graded by experienced
pathologists using a semi-quantitative histology score system
(27). Nur77 expression in lung
tissues was detected by IHC according to a previously described
protocol (12). Briefly, the lung
tissues slices were incubated with an anti-Nur77 antibody (1:400;
NB100-56745; Novus Biologicals, Centennial, CO, USA) overnight at
4°C, incubated with anti-rabbit IgG (ZB2301,1:1,000) for 30 min at
room temperature, and then stained with 3,3'-diaminobenzidine (DAB,
DA1010, Solarbio Science & Technology) as the chromagen.
Finially, the slides were counterstained with hematoxylin,
dehydrated and mounted. In total, 10 random fields were examined
using a Leica digital microscope.
Reagents and antibodies
Dulbecco's modified Eagle's medium (DMEM) and fetal
bovine serum (FBS) were purchased from Gibco/Thermo Fisher
Scientific (Waltham, MA, USA). Dulbecco's phosphate-buffered saline
(DPBS) was purchased from Invitrogen/Thermo Fisher Scientific. In
addition, 3-methyladenine (3-MA) and rapamycin (RAPA) were
purchased from Selleck Chemicals (Shanghai, China), and leptomycin
B (LMB) was purchased from the Beyotime Institute of Biotechnology
(Shanghai, China). The anti-Nur77 antibody was purchased from Novus
Biologicals, and anti-LC3 (cat. nos. 12741 and 2775) and anti-GAPDH
antibodies (cat. no. 5174) were purchased from Cell Signaling
Technology (Danvers, MA, USA). Anti-Beclin-1 (cat. no. ab210498)
and anti-Bcl2 (cat. no. ab692) antibodies were purchased from Abcam
(Cambridge, MA, USA). Anti-rabbit and mouse IgG (ZB2301, ZB2305,
1:2,000) were purchased from Zhong Shan Golden Bridge Biotechnology
Co., Ltd. (Beijing, China).
Preparation of CSE
CSE was prepared using a smoke device as previously
described (28). A filtered
cigarette (Baisha Pai) was combusted with a vacuum pump (VWR
International, West Chester, PA, USA), and the smoke was directed
via a tube through 5 ml of PBS. This solution, which was considered
to be 100% CSE, was filtered through a 0.22-µm pore filter
(Lida Manufacturing Corp., Kenosha, WI, USA) for sterilization.
Different preparations of CSE were standardized by measuring the
absorbance at a wavelength of 320 nm using a NanoDrop 2000
spectrophotometer (Thermo Fisher Scientific). Freshly prepared CSE
was diluted with culture medium supplemented with 10% FBS
immediately prior to use and was used within 30 min.
Cells and cell culture
Human bronchial epithelial (HBE) cells and A549
cells were kindly provided by the Central South University Advanced
Research Center (Changsha, China) and maintained in DMEM
supplemented with 10% FBS, 100 U/ml penicillin and 100 mg/ml
streptomycin in a humidified atmosphere containing 5%
CO2 at 37°C. CSE and other reagents (rapamycin, 3-MA,
LMB, siRNA) were added to the HBE or A549 cells at the indicated
concentrations and for the indicated periods of time (see
corresponding figure legends for details).
Cell transfection
siRNA-Nur77 (sequence, 5′-TCGAGGACTTCCAGGTGTA-3′)
and a negative control sequence (cat. no. siN05815) were obtained
from RiboBio (riboFect™CP; Guangzhou, China). The cells in the
exponential growth phase were seeded in 6-well plates at
4×105 cells per well, grown to 40-50% density for 24 h,
and transfected with siRNA-Nur77 or negative control for a further
36 h according to the manufacturer's recommended protocol.
siRNA-Nur77, pre-mixed with riboFECT™CP buffer and riboFECT™CP
reagent, was added to the medium at a final concentration of 100
nM. Western blot analysis was performed to evaluate the
transfection efficiency.
Cell proliferation assay
The inhibition ratio of CSE was measured with a Cell
Counting kit-8 (CCK-8; Dojindo, Kumamoto, Japan) assay, as
specified. A total of 1×104 HBE cells or
5×103 A549 cells were seeded into 96-well plates, grown
to nearly 40% confluence and transfected with siRNA or negative
control for 36 h. Subsequently, the cells were treated with or
without rapamycin (1 mM) or 3-MA (2.5 mM) or left untreated for 2 h
and then exposed to 5% CSE for a further 24 h. After all the
treatments were performed, 10 µl of CCK-8 were added to each
well for 1 h at 37°C, and the absorbance was detected at 450 nm
with a microplate reader (BioTek Instruments, Winooski, VT,
USA).
Immunofluorescence
A total of 1×105 HBE cells or
5×104 A549 cells were seeded in 6-well glass-bottom
dishes and subjected to various treatments for the indicated
periods of time. The treated cells were washed and fixed in 4%
paraformaldehyde for 20 min, treated with TritonX-100 for 15 min,
and blocked with 5% bovine serum albumin (BSA) for 30 min. The
cells were then incubated with primary antibodies (anti-Nur77,
1:200; anti-Bcl2, 1:200; anti-LC3, 1:200) overnight at 4°C, washed
with PBS, and then incubated with fluorescent secondary antibodies
(anti-rabbit IgG: 711-025-152, anti-mouse IgG: 715-545-150; 1:200
dilution; Jackson ImmunoResearch Laboratories, Inc., West Grove,
PA, USA) for 1 h at 37°C; after washing with PBS, and the cells
were stained with DAPI (C1005, Beyotime Institute of Biotechnology,
Haimen, China) for 5 min at room temperature. Following washing
with PBS, the coverslips were sealed by anti-fade mounting Medium
(P0126-5 ml, Beyotime Institute of Biotechnology). Images were
captured under a fluorescence microscope (Leica, Buffalo Grove, IL,
USA).
Western blot analysis
Total proteins were extracted with
radioimmunoprecipitation assay (RIPA) lysis buffer (Beyotime
Institute of Biotechnology) and quantified with the bicinchoninic
acid (BCA) method to ensure equal concentration. Loading buffer and
total proteins were mixed and heated at 95°C for 10 min to denature
albumin. Equal quantities of the samples (30-50 µg/well)
were separated by sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE, 10-15%) and transferred to
polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA,
USA). After blocking with 5% non-fat milk, the membranes were
incubated with corresponding primary antibodies (anti-Nur77,
1:1,000; anti-LC3, 1:1,000; anti-Beclin-1, 1:1,000; anti-GAPDH,
1:1,000) at 4°C overnight. Subsequently, the membranes were
incubated with secondary antibodies (anti-rabbit IgG: 1:2,000) for
a further 1 h at 37°C, and the proteins were detected with an
enhanced chemiluminescence (ECL) reagent (Millipore) and an ECL
system (Syngene, Cambridge, England, UK). ImageJ 1.48v software
(National Institutes of Health) was used to measure the gray value
of each band.
Transmission electron microscopy
(TEM)
The HBE and A549 cells were collected and fixed with
2.5% glutaradehyde overnight at at 4°C; they were then washed, and
fixed with 1% osmium tetroxide for 1 h. Subsequently, the cells
were dehydrated with a graded ethanol series, and embedded in epoxy
resin (CAS 25068-38-6; Santa Cruz Biotechnology, Santa Cruz, CA,
USA). Ultrathin sections were detected using a transmission
electron microscope (Hitachi Ltd., Tokyo, Japan).
Co-immunoprecipitation
The HBE cells were treated with LMB (10 ng/ml) for 2
h or left untreated, and were then exposed to 5% CSE for a further
24 h. The cells were collected, lysed in immunoprecipitation
(IP)-RIPA lysis buffer, and then centrifuged at 10,000 × g for 10
min at 4°C. The supernatants were collected and quantified with the
BCA method to ensure equal concentration; 10% and another 3% of
total proteins were used as input experiments. The remaining were
incubated with anti-Nur77 (1:50), anti-Bcl2 (1:20), or
anti-Beclin-1 (1:30) antibodies or with the same species IgG
(anti-GAPDH rabbit antibodies, cat. no. 5174; and anti-GAPDH mouse
antibodies, cat. no. 51332; 1:20 dilution; Cell Signaling
Technology) as a negative control overnight at 4°C. Protein G
agarose beads were pre-washed 3 times in IP-RIPA lysis buffer and
then incubated with the protein/antibody mixtures at 4°C for 4 h on
shaking tables. The supernatant was discarded and the precipitate
was retained and washed 4 times with IP-RIPA lysis buffer. Loading
buffer was added to the precipitate; and the mixture was then
heated at 95°C for 5 min. After the beads were discarded, the
supernatant was analyzed by western blot analysis.
Statistical analysis
The data are expressed as the means ± standard error
of the mean (SEM). The data were analyzed using one-way analysis of
variance (ANOVA) followed by Bonferroni's multiple comparison tests
with GraphPad Prism 5.0 software (GraphPad Software, San Diego, CA,
USA). Differences with P-values <0.05 were regarded as
statistically significant.
Results
Nur77 is overexpressed in lung tissues
and pulmonary cells following exposure to CS and CSE
administration
Nur77, which plays a critical role in inflammation
and in cell proliferation, survival and death, is overexpressed in
the majority of lung cancer and PASMC proliferation models
(11). However, limited
information of the mechanisms through which CS affects the
expression of Nur77 is available. Thus, in this study, in order to
shed light into this matter, C57BL/6 mice were bred to 10-12 weeks
and were randomly divided into 4 groups, and the weight growth of
each group did not differ significantly (compared to weight at
purchase, data not shown). The C57BL/6 mice were exposed to RA or
CS for 10 weeks, and lung tissues were collected the day after the
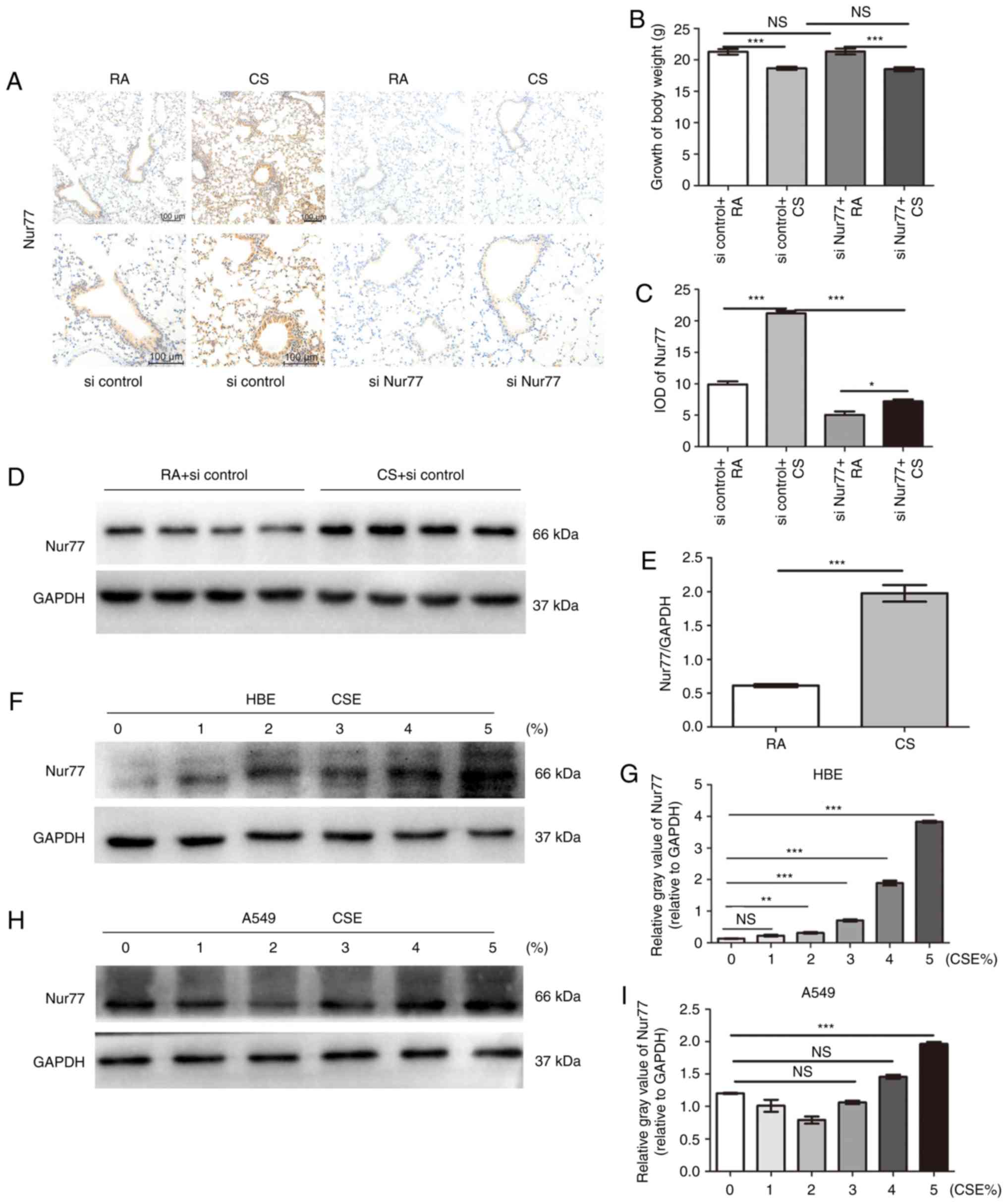
final CS exposure. We found that exposure to CS significantly
decreased the weight gain of the mice; however, lentivirus
infection did not (Fig. 1B,
compared to initial weight: Weight of 26-30-day-old mice, 11-14 g).
Nur77 expression in the lung tissues was examined by
immunohistochemical staining and western blot analysis. The number
of Nur77-positive pulmonary epithelial cells in the CS-exposed
group was higher than that in the RA group (Fig. 1A and C). Western blot analysis
confirmed that CS increased the Nur77 levels in the lung tissues
(Fig. 1D and E). We also examined
the effects of various concentrations of CSE on Nur77 expression at
the cellular level. The Nur77 levels significantly increased with
the increasing CSE concentrations, and the group of HBE cells
treated with 5% CSE, expressed higher Nur77-levels than the other
groups (Fig. 1F and G). To
further confirm the upregulation of Nur77 by CSE, we also examined
the Nur77 levels in A549 cells exposed to various concentrations of
CSE. Consistent with our findings with the HBE cells, the Nur77
levels were elevated with the increasing CSE concentrations
(Fig. 1H and I). These results
demonstrated that CS and CSE increased the Nur77 levels in
pulmonary epithelial cells.
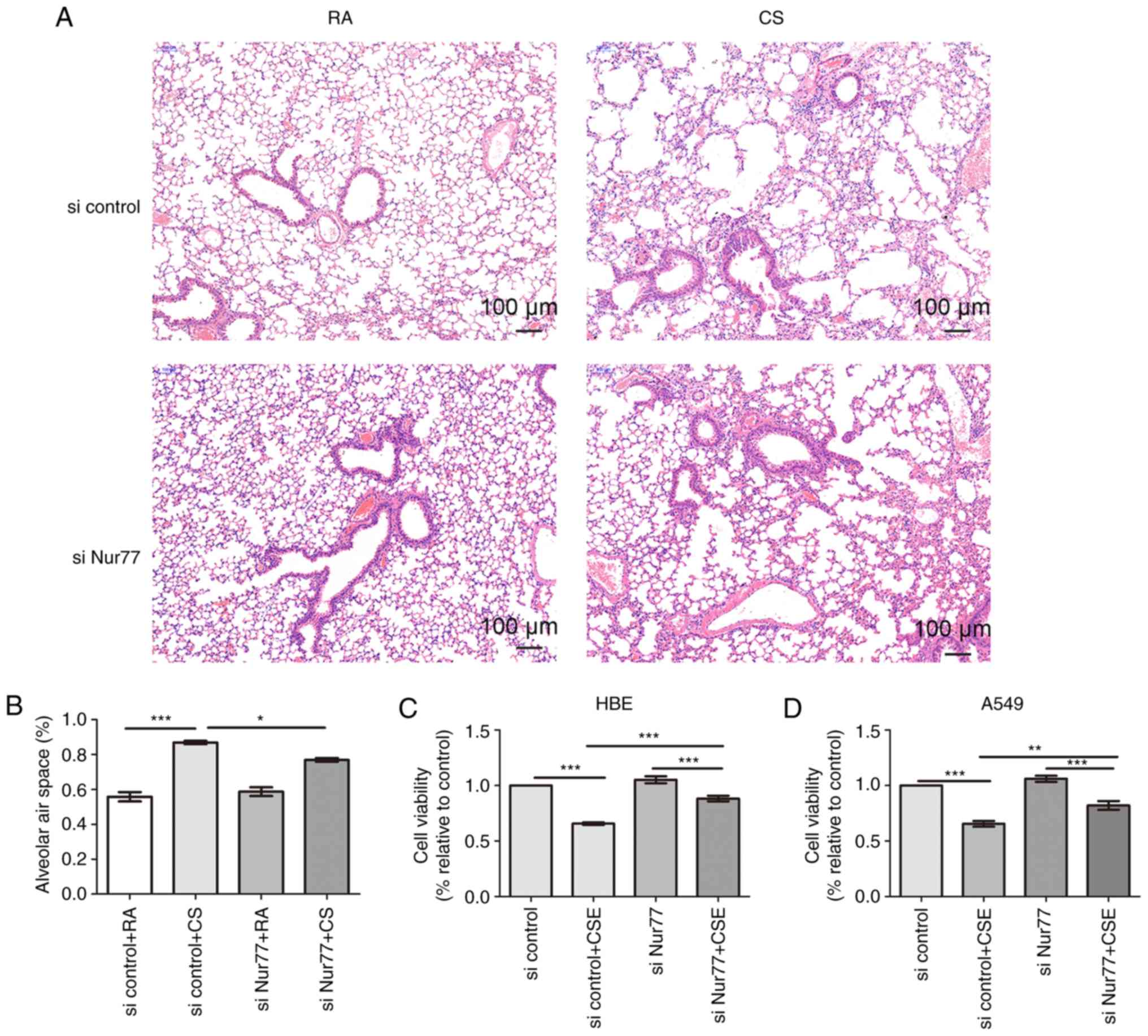
Knockdown of Nur77 attenuates CS-induced
lung injury and CSE-induced cell death
Exposure to CS results in pulmonary epithelial cell
dysfunction and eventual cell death. Lung tissues and pulmonary
epithelial cells treated with CS and CSE overexpressed Nur77;
however, whether the high Nur77 levels participate in the
pathophysiological processes of pulmonary epithelial cell
dysfunction and cell death remained unclear. The results of this
study revealed that CS-exposed mice exhibited more severe
destruction of the pulmonary alveoli and bronchial tubes than
RA-exposed mice; however, intratracheal inoculation with
siRNA-Nur77 lentivirus mitigated the observed CS-induced lung
injury (Fig. 2A and B). We
further found that CSE reduced the viability of the HBE and A549
cells, an effect that was attenuated by the knockdown of Nur77
(Fig. 2C and D).
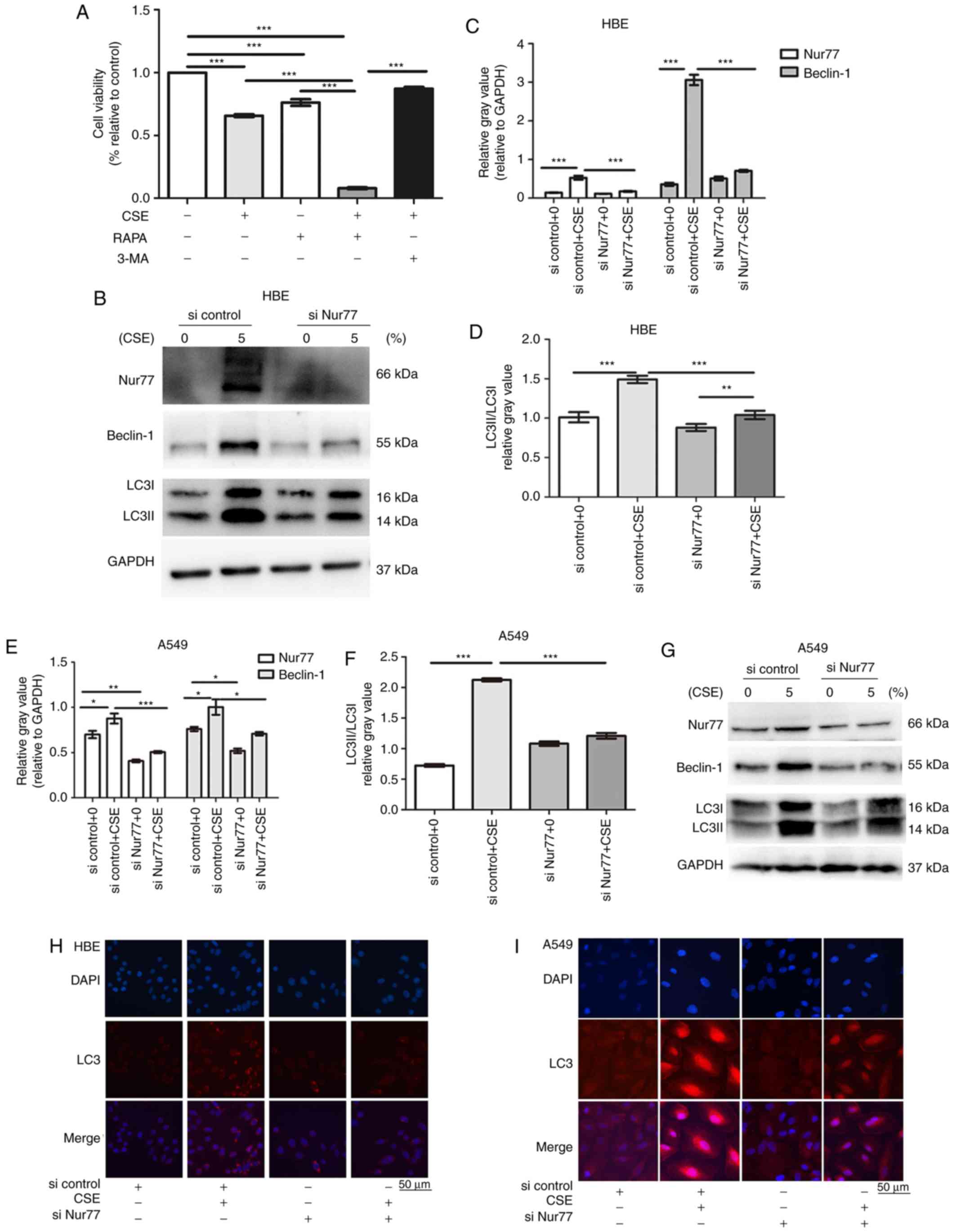
Nur77 mediates CS-induced autophagy in
vitro and in vivo
The autophagy of lung epithelial cells is involved
in the pathological process of CS-induced COPD. In this study,
combined treatment with CSE and RAPA (an autophagy activator)
accelerated cell death, while 3-MA (an autophagy inhibitor)
effectively attenuated CSE-induced cell death (Fig. 3A). It has been demonstrated that
Nur77 mediates autophagic cell death in mammalian cells (29). Hence, we hypothesized that Nur77
may be involved in CSE-induced autophagy. To investigate the role
of Nur77 in CSE-induced autophagy, the HBE cells were treated with
5% CSE for 24 h, and the expression levels of LC3 (an autophagy
marker; an increasing LC3II/LC3I ratio indicates autophagy
induction) and Beclin-1 (responsible for autophagy initiation) were
then examined. We found that the LC3I to LC3II conversion and
Beclin-1 expression increased synchronously with Nur77 upregulation
(Fig. 3B-D). siRNA-Nur77
inhibited the expression of Nur77 (Fig. 3B), and the knockdown of Nur77 with
siRNA-Nur77 attenuated the ability of CSE to upregulate LC3II/LC3I
and Beclin-1 expression (Fig.
3B-D). These data indicate that the CSE-induced upregulation of
Nur77 is linked to CSE-induced autophagy. A similar result was
observed in the A549 cells co-treated with siRNA-Nur77 and CSE
(Fig. 3E-G). Moreover, we found
that punctate LC3 staining was greater in the CSE-treated HBE and
A549 cells than in the control cells, as indicated by
immunofluorescence staining, while transfection with siRNA-Nur77
diminished punctate LC3 staining, and the conversion of LC3-I to
LC3-II (Fig. 3H and I).
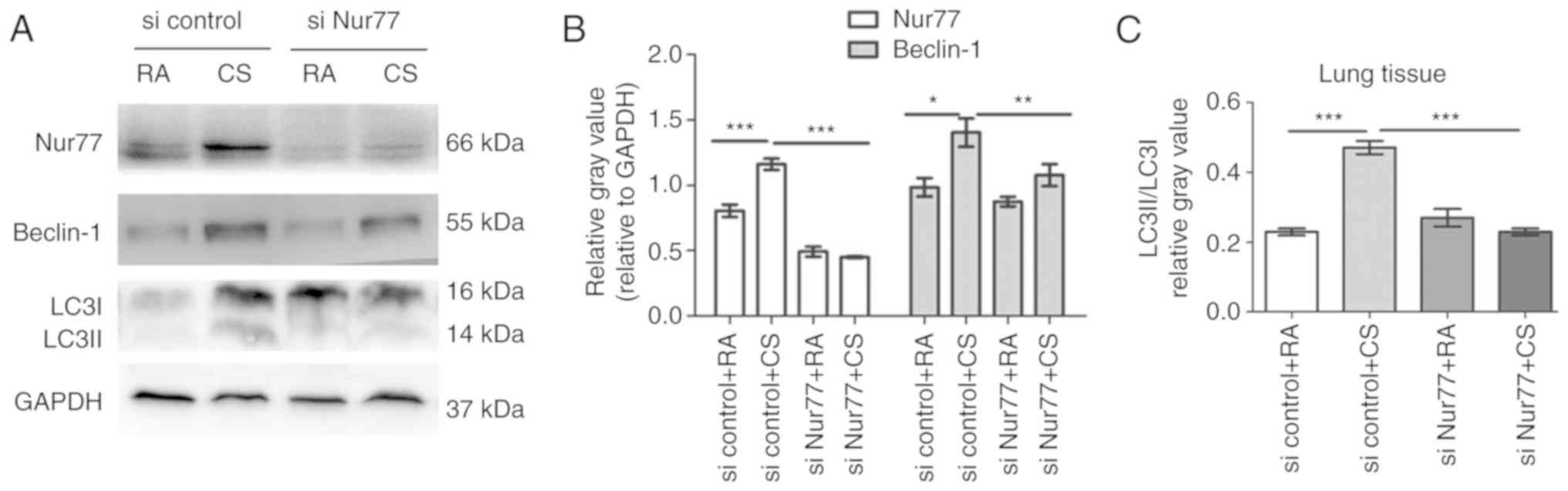
To determine whether Nur77 plays a key role in
CS-induced autophagy in vivo, mice were exposed to CS and
treated with siRNA-Nur77 lentivirus for 10 weeks, and LC3, and
Beclin-1 protein levels were detected in the lung tissues by
western blot analysis. The lung tissues from the group of mice
co-treated with siRNA-Nur77 lentivirus and CS demonstrated lower
levels of LC3I to LC3II conversion and a lower Beclin-1 protein
expression than those from the group exposed to CS only, indicating
that autophagy was inhibited (Fig.
4).
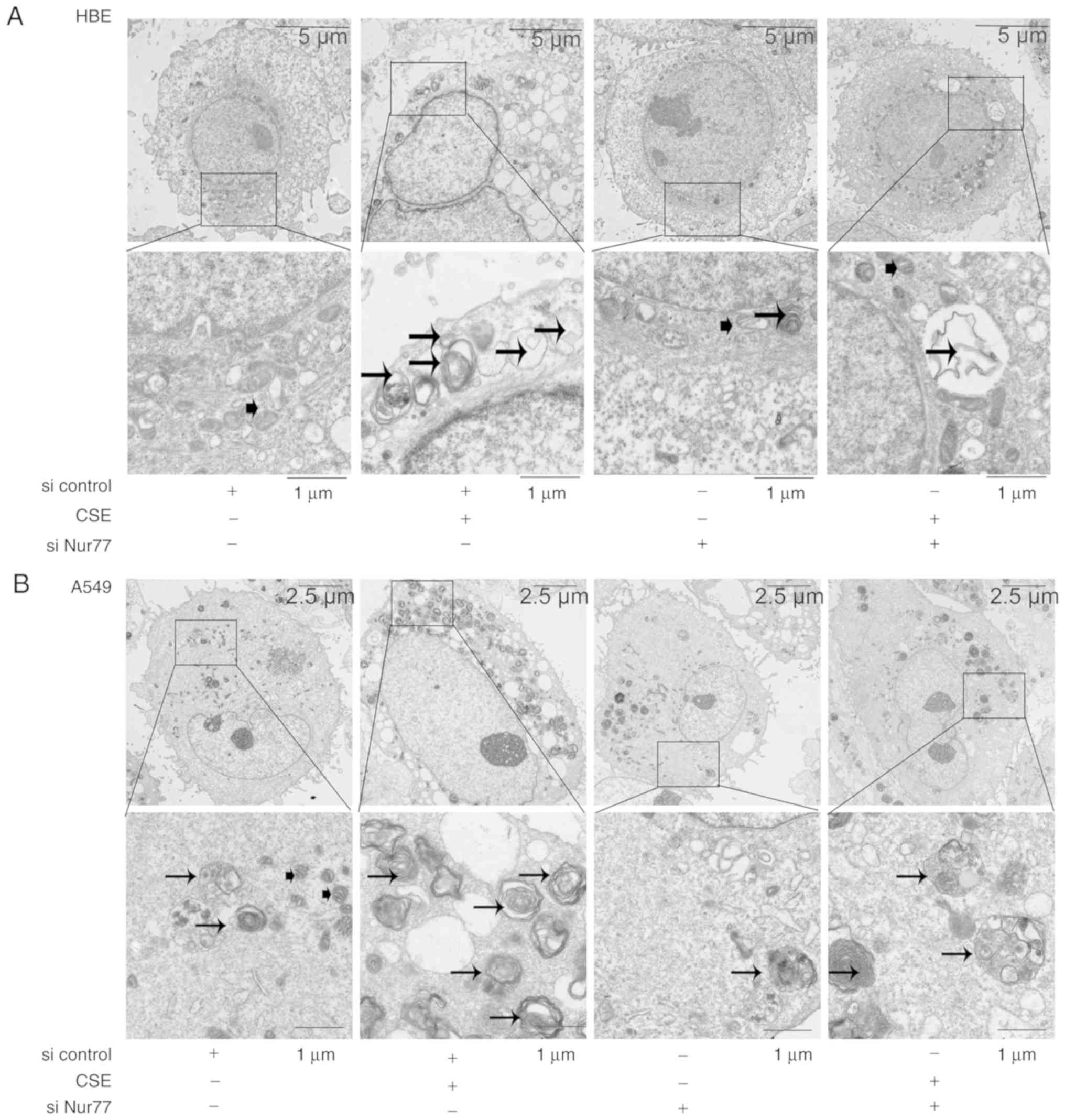
Knockdown of Nur77 decreases CSE-induced
autophagosome formation at the ultrastructural level
To confirm that autophagy is indeed induced by CSE,
the HBE and A549 cells treated with 5% CSE were analyzed by TEM for
the evidence of autophagy (Fig.
5). A greater abundance of double-membrane structures
containing organelles and more mature autophagosomes containing
electron-dense regions were evident in the CSE-exposed HBE cells
than in the control cells (Fig.
5A). Similar to the HBE cells, the CSE-exposed A549 cells
exhibited much more autophagic features compared to those of the
untreated group (Fig. 5B). We
then examined whether Nur77 is involved in morphological changes
consistent with autophagy. The presence of autophagic structures
was markedly reduced in the CSE-exposed HBE and A549 cells
following transfection with siRNA-Nur77 (Fig. 5). This finding supports our
hypothesis that CSE-induced Nur77 upregulation is responsible for
CSE-induced autophagy.
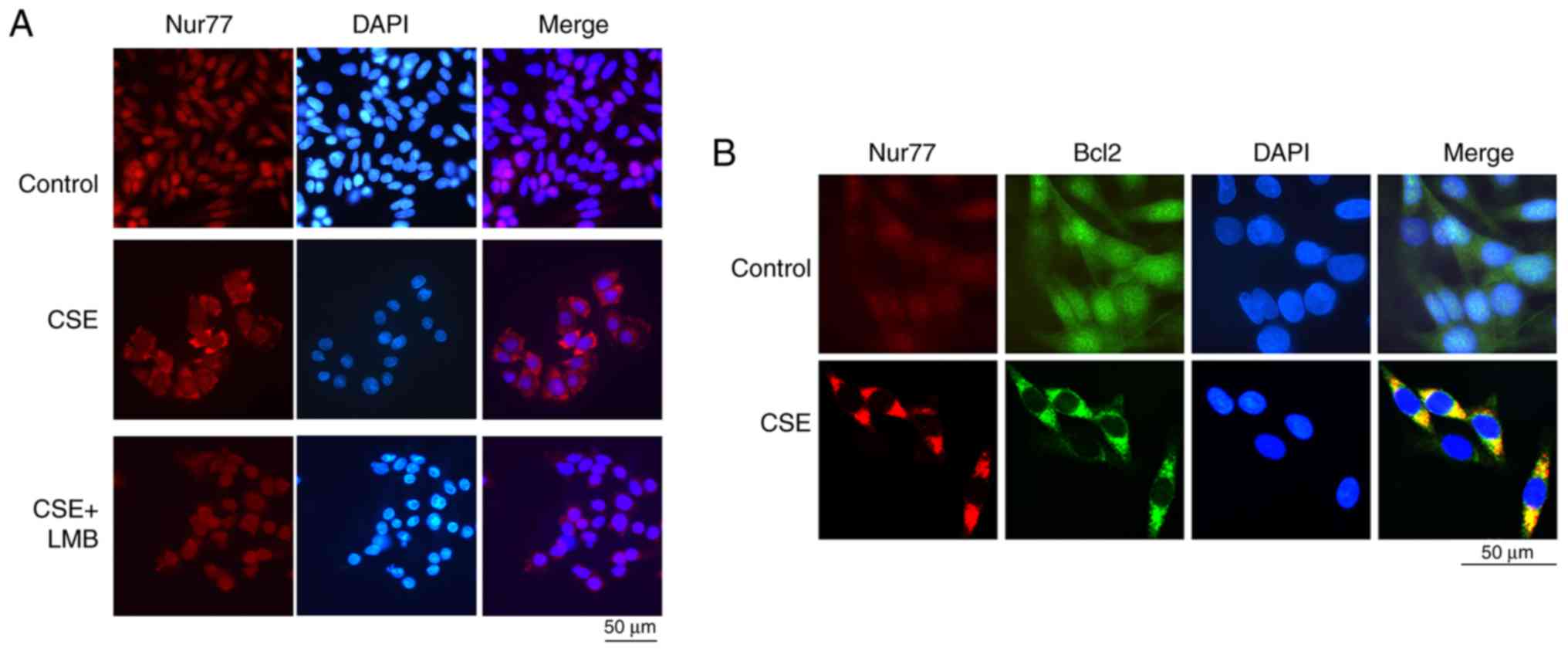
The interaction between Nur77 and Bcl2 is
enhanced by CSE
Bcl-2 family proteins are key regulators of
programmed cell death. It has been previously clarified that Nur77
interacts with Bcl2 and can convert the function of Bcl2 from a
protective function to a pro-death function; in addition, some
stimuli can regulate the interaction of Nur77 and Bcl2 to induce
cell apoptosis (30). The
Nur77-Bcl2 complex also induces autophagy (30). In this study, we observed the
Nur77 transfer to the cytoplasm in CSE-exposed HBE cells, while LMB
prevented the CSE-induced Nur77 nuclear export (Fig. 6A). To clarify whether CSE promotes
the interaction between Nur77 and Bcl2, double immunofluorescence
was first used to examine whether Nur77 co-localizes with Bcl2 in
the cytoplasm of HBE cells that were exposed or not to CSE. When
the HBE cells were exposed to CSE, Nur77 and Bcl2 co-localized in
the cytoplasm, while in the control cells, Nur77 existed mainly in
the nucleus, and Bcl2 was distributed in the nucleus and cytoplasm
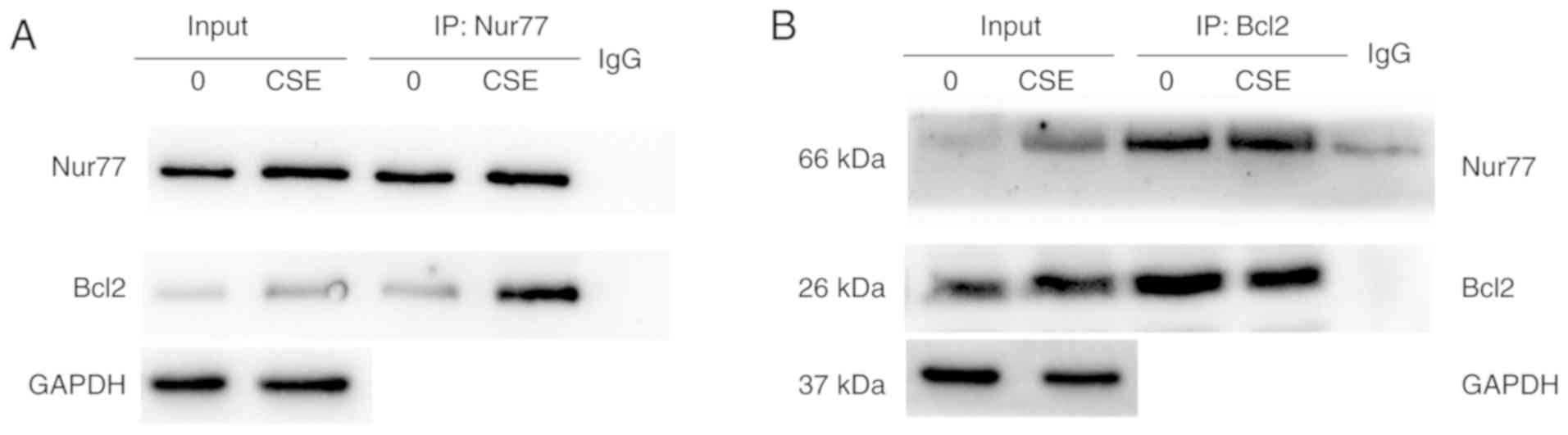
(Fig. 6B). Subsequently, we
investigated whether the interaction between Nur77 and Bcl2 was
enhanced in CSE-exposed cells with a co-immunoprecipitation assay.
In the absence CSE, only endogenous Nur77 and a small amount of
Bcl2 were pulled down by an anti-Nur77 antibody. Upon CSE exposure,
a greater amount of Nur77 co-immunoprecipitated with Bcl2 (Fig. 7A). The same outcome was detected
when the cell lysates were immunoprecipitated with an anti-Bcl2
antibody (Fig. 7B). These results
indicate that CSE promotes the interaction between Nur77 and
Bcl2.
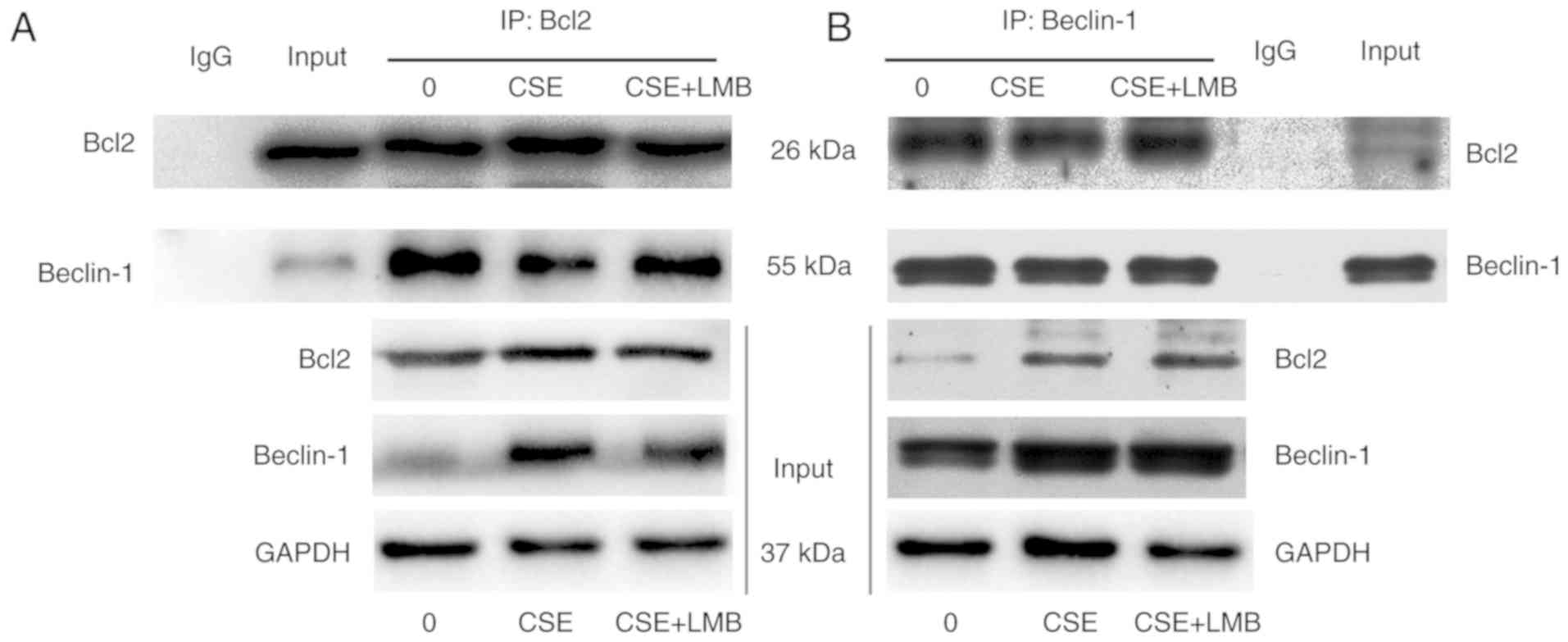
Nur77 plays a role in the CSE-induced
dissociation of Bcl2 from Beclin-1
The autophagic function of Beclin-1 is inhibited by
Bcl-2 and is induced when Bcl-2 dissociates from Beclin-1 (24,30). It has been verified that the
interaction of Nur77 and Bcl2 family proteins augments autophagy
(30). The results of this study
indicated that CSE induced autophagy by the elevating Nur77 levels
and increasing the interaction between Nur77 and Bcl2. To determine
whether the CSE-induced increase in Nur77-Bcl-2 complexes increases
dissociation of Beclin-1 from Bcl2, we then triggered
Beclin-1-dependent autophagy. We first performed
co-immunoprecipitation experiments with the HBE cells with or
without CSE exposure to detect the endogenous levels of both Bcl2
and Beclin-1. We observed that an anti-Bcl2 antibody and an
anti-Beclin-1 antibody pulled down both Bcl2 and Beclin-1 in the
absence of CSE; in addition, smaller amounts of Bcl2 and Beclin-1
were pulled down following exposure to CSE (Fig. 8). We then sought to determine
whether the binding of Nur77 to Bcl2 prompts the dissociation of
the Bcl2-Beclin-1 complex in HBE cells exposed to CSE. LMB inhibits
Nur77 nuclear export and then prevents Nur77 from interacting with
Bcl2. HBE cells were thus treated with CSE in the presence or
absence of LMB. Through co-immunoprecipitation assays, we
discovered that the amounts of Bcl-2 and Beclin-1-pulled down by
the anti-Bcl2 antibody were lower in the CSE-exposed group than in
the control and LMB groups (Fig.
8). These observations confirm that CSE induces the interaction
between Nur77 and Bcl-2, which leads to the dissociation of Bcl-2
from Beclin-1.
Discussion
COPD is characterized by progressive, partially
reversible airflow obstruction, and its pathologic features include
chronic bronchitis, airway remodeling and emphysema (3). Cigarette smoking is the strongest
risk factor for COPD (2,31). Autophagy plays a detrimental role
in CS-induced COPD by damaging lung epithelial cells (6). However, the mechanisms through which
CS triggers autophagy remain unclear. In this study, we explored
the role of Nur77 in CS-induced autophagy and demonstrated that CS
induced Nur77 expression and nuclear export, and then promoted the
interaction between Nur77 and Bcl2, which dissociated Bcl2 from
Beclin-1 and consequently activated autophagy.
The orphan nuclear receptor Nur77 serves mainly as a
transcription factor to regulate the expression of multiple genes
in the nucleus. Nur77 also translocates from the nucleus to the
cytoplasm to exert biological effects. Nur77 exerts critical
effects on inflammation, and on the proliferation, differentiation
and apoptosis of cancer cells (32-35). There is evidence to indicate that
Nur77 can act as a therapeutic target to induce cell death in
various types of cancer, such as lymphoma and melanoma, in which
Nur77 is downregulated (19,36). Several studies have also explored
the significance of Nur77-mediated cell survival in breast cancer,
pancreatic tumors and lung cancer, in which Nur77 is upregulated
(37-39). Nur77 also has dual effects in
non-tumoral diseases. For example, Nur77 can inhibit PASMC
proliferation through the inhibition of the Axin2-β-catenin
signaling pathway, exerting a protective function against hypoxic
pulmonary hypertension (11).
Nur77 may be a molecular target for the prevention of sepsis, as it
interacts with TRAF6 and regulates its autoubiquitination (13). However, the expression of Nur77
family genes is induced by cadmium exposure, which leads to lung
cell death, and may thus cause pulmonary toxicity (40). Nicotine, an ingredient found in
tobacco, induces Nur77 expression in human lung cancer cells
(41). Accumulating evidence
suggests that Nur77 is widely expressed in the lungs, although the
nature of the expression and function of Nur77 in CS-induced COPD
remains unclear. In the present study, we observed that Nur77
expression was upregulated following exposure to CSE in a
concentration-dependent manner in pulmonary epithelial cells and
that CSE accelerated HBE and A549 cell death, while siRNA-Nur77
partially alleviated this effect.
Autophagy is an evolutionarily conserved catabolic
pathway that delivers long-lived proteins and damaged organelles to
lysosomes for degradation (42).
Moderate autophagy facilitates the maintenance of cellular
homeostasis, although excessive autophagy leads to cell death
(43,44). For example, in CS-induced COPD,
augmented autophagy has been reported to promote CS-induced
pulmonary emphysema and to regulate the secretion and senescence of
airway epithelial cells (42-44). Nur77 can interact with p53 to
promote autophagic cell death, which is involved in regulating gene
expression in the nucleus (29).
It has also been demonstrated that Nur77 is translocated to the
cytoplasm and targets the mitochondria, dissipating the
mitochondrial membrane potential and inducing the autophagy pathway
to ultimately lead to malignant melanoma cell death (19). Dendrogenin A (DDA), a natural
metabolite of cholesterol and histamine, induces Nur77-dependent
lethal autophagy in melanoma and acute myeloid leukemia (36). In this study, we discovered that
Nur77 plays an important role in CSE-induced autophagy and promotes
cell death by autophagy. The increased conversion of LC3II to LC3I
was accompanied by Nur77 overexpression in the HBE and A549 cells
exposed to CSE. We further investigated the association between
Nur77 and CSE-induced autophagy by knocking down Nur77 with siRNA.
The knockdown of Nur77 impaired CSE-induced autophagy, confirming
that the functional induction of autophagy by CSE is partly
dependent on Nur77.
Pro-apoptotic and anti-apoptotic Bcl-2 family
proteins share a motif known as the Bcl-2 homology 3 (BH3) motif,
which is involved in regulating apoptosis (22,30). Nur77, through its LBD, binds
adjacent to the BH3 peptide-binding crevice of Bcl-2 family
proteins and converts anti-apoptotic proteins to pro-apoptotic
proteins (22,23,30). The autophagy-related protein,
Beclin-1, binds to the BH3-binding pocket on anti-apoptotic Bcl-2
family proteins though its BH3-like domain, which inhibits
autophagy (24,45). The dissociation of Bcl2 from
Beclin-1 has been verified to trigger autophagy (45,46). The interaction between Nur77 and
Bcl2 family proteins is known to be associated with autophagy and
cell death regulation. We thus hypothesized that the Nur77-Bcl2
complex dissociates Beclin-1 from Bcl2 to mediate CSE-induced
autophagy. In this study, we found that CSE increased Nur77-Bcl2
interactions from the control levels. We also observed that CSE
induced the dissociation of Beclin-1 from Bcl2, enhancing
autophagy. The limitation of Nur77 nuclear export re-stabilized the
Bcl2-Beclin-1 heterodimer.
In conclusion, the data from this study support the
hypothesis that Nur77 is overexpressed in pulmonary tissue and in
HBE and A549 cells exposed to CS, and that Nur77 promotes autophagy
by binding to Bcl2 and weakening the affinity of Beclin-1 for Bcl2.
These results may provide novel insight into the pathogenesis of
COPD and may into the mechanisms underlying CSE-induced COPD. Our
data may also aid in the development of novel treatment strategies
for COPD.
Funding
The present study was supported in part by the
National Natural Science Foundation of China (grant no.
81660008).
Availability of data and materials
The datasets used during the present study are
available from the corresponding author on reasonable request.
Authors' contributions
HQ participated in the experimental design,
completed the animal experiments and the cell experiments,
performed the data analysis, and drafted and edited the manuscript.
FG and YW assisted with the animal experiments and drafted the
manuscript. BH contributed to the immunofluorescence assay and the
immunoassays. LP carried out the electron microscopy analysis. BM
performed the data analysis and statistical analysis. CW designed
the experiments, drafted and edited the manuscript, and supervised
the completion of the experiment. All authors have read and
approved the final manuscript.
Ethics approval and consent to
participate
All the animal experiments were approved by the
Central South University Animal Care Committee.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
Nur77
|
nuclear receptor 77
|
|
CS
|
cigarette smoke
|
|
CSE
|
cigarette smoke extract
|
|
COPD
|
chronic obstructive pulmonary
disease
|
|
RA
|
room air
|
|
LMB
|
leptomycin B
|
|
RAPA
|
rapamycin
|
|
3-MA
|
3-methyladenine
|
|
siRNA
|
small interfering RNA
|
|
HBE cells
|
human bronchial epithelial cells
|
Acknowledgments
Not applicable.
References
|
1
|
Vogelmeier CF, Criner GJ, Martinez FJ,
Anzueto A, Barnes PJ, Bourbeau J, Celli BR, Chen R, Decramer M,
Fabbri LM, et al: Global strategy for the diagnosis, management and
prevention of chronic obstructive lung disease 2017 Report: GOLD
executive summary. Respirology. 22:575–601. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rosenberg SR, Kalhan R and Mannino DM:
Epidemiology of chronic obstructive pulmonary disease: Prevalence,
morbidity, mortality, and risk factors. Semin Respir Crit Care Med.
36:457–469. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bagdonas E, Raudoniute J, Bruzauskaite I
and Aldonyte R: Novel aspects of pathogenesis and regeneration
mechanisms in COPD. Int J Chron Obstruct Pulmon Dis. 10:995–1013.
2015.PubMed/NCBI
|
|
4
|
He Y, Jiang B, Li LS, Li LS, Ko L, Wu L,
Sun DL, He SF, Liang BQ, Hu FB and Lam TH: Secondhand smoke
exposure predicted COPD and other tobacco-related mortality in a
17-year cohort study in China. Chest. 142:909–918. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen Z, Peto R, Zhou M, Iona A, Smith M,
Yang L, Guo Y, Chen Y, Bian Z, Lancaster G, et al: Contrasting male
and female trends in tobacco-attributed mortality in China:
Evidence from successive nationwide prospective cohort studies.
Lancet. 386:1447–1456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vij N, Chandramani-Shivalingappa P, Van
Westphal C, Hole R and Bodas M: Cigarette smoke-induced autophagy
impairment accelerates lung aging, COPD-emphysema exacerbations and
pathogenesis. Am J Physiol Cell Physiol. 314:C73–C87. 2018.
View Article : Google Scholar :
|
|
7
|
Pawlak A, Strzadala L and Kalas W:
Non-genomic effects of the NR4A1/Nur77/TR3/NGFIB orphan nuclear
receptor. Steroids. 95:1–6. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wang Z, Benoit G, Liu J, Prasad S,
Aarnisalo P, Liu X, Xu H, Walker NP and Perlmann T: Structure and
function of Nurr1 identifies a class of ligand-independent nuclear
receptors. Nature. 423:555–560. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kurakula K, Koenis DS, van Tiel CM and de
Vries CJ: NR4A nuclear receptors are orphans but not lonesome.
Biochim Biophys Acta. 1843:2543–2555. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Niu G, Lu L, Gan J, Zhang D, Liu J and
Huang G: Dual roles of orphan nuclear receptor TR3/Nur77/NGFI-B in
mediating cell survival and apoptosis. Int Rev Cell Mol Biol.
313:219–258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nie X, Tan J, Dai Y, Mao W, Chen Y, Qin G,
Li G, Shen C, Zhao J and Chen J: Nur77 downregulation triggers
pulmonary artery smooth muscle cell proliferation and migration in
mice with hypoxic pulmonary hypertension via the Axin2-β-catenin
signaling pathway. Vascul Pharmacol. 87:230–241. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kurakula K, Vos M, Logiantara A, Roelofs
JJ, Nieuwenhuis MA, Koppelman GH, Postma DS, van Rijt LS and de
Vries CJ: Nuclear receptor Nur77 attenuates airway inflammation in
mice by suppressing NF-κB activity in lung epithelial cells. J
Immunol. 195:1388–1398. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li XM, Zhang S, He XS, Guo PD, Lu XX, Wang
JR, Li JM and Wu H: Nur77-mediated TRAF6 signalling protects
against LPS-induced sepsis in mice. J Inflamm (Lond). 13:42016.
View Article : Google Scholar
|
|
14
|
Mizumura K, Cloonan S, Choi ME, Hashimoto
S, Nakahira K, Ryter SW and Choi AM: Autophagy: Friend or foe in
lung disease? Ann Am Thorac Soc. 13(Suppl 1): S40–S47.
2016.PubMed/NCBI
|
|
15
|
Haspel JA and Choi AM: Autophagy: A core
cellular process with emerging links to pulmonary disease. Am J
Respir Crit Care Med. 184:1237–1246. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Araya J, Hara H and Kuwano K: Autophagy in
the pathogenesis of pulmonary disease. Intern Med. 52:2295–2303.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lam HC, Cloonan SM, Bhashyam AR, Haspel
JA, Singh A, Sathirapongsasuti JF, Cervo M, Yao H, Chung AL,
Mizumura K, et al: Histone deacetylase 6-mediated selective
autophagy regulates COPD-associated cilia dysfunction. J Clin
Invest. 123:5212–5230. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Dickinson JD, Alevy Y, Malvin NP, Patel
KK, Gunsten SP, Holtzman MJ, Stappenbeck TS and Brody SL: IL13
activates autophagy to regulate secretion in airway epithelial
cells. Autophagy. 12:397–409. 2016. View Article : Google Scholar :
|
|
19
|
Wang WJ, Wang Y, Chen HZ, Xing YZ, Li FW,
Zhang Q, Zhou B, Zhang HK, Zhang J, Bian XL, et al: Orphan nuclear
receptor TR3 acts in autophagic cell death via mitochondrial
signaling pathway. Nat Chem Biol. 10:133–140. 2014. View Article : Google Scholar
|
|
20
|
Gao H, Chen Z, Fu Y, Yang X, Weng R, Wang
R, Lu J, Pan M, Jin K, McElroy C, et al: Nur77 exacerbates PC12
cellular injury in vitro by aggravating mitochondrial impairment
and endoplasmic reticulum stress. Sci Rep. 6:344032016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liang B, Song X, Liu G, Li R, Xie J, Xiao
L, Du M, Zhang Q, Xu X, Gan X and Huang D: Involvement of TR3/Nur77
trans-location to the endoplasmic reticulum in ER stress-induced
apoptosis. Exp Cell Res. 313:2833–2844. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Thompson J and Winoto A: During negative
selection, Nur77 family proteins translocate to mitochondria where
they associate with Bcl-2 and expose its proapoptotic BH3 domain. J
Exp Med. 205:1029–1036. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Godoi PH, Wilkie-Grantham RP, Hishiki A,
Sano R, Matsuzawa Y, Yanagi H, Munte CE, Chen Y, Yao Y, Marassi FM,
et al: Orphan nuclear receptor NR4A1 Binds a novel protein
interaction site on anti-apoptotic B cell lymphoma gene 2 family
proteins. J Biol Chem. 291:14072–14084. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Robert G, Gastaldi C, Puissant A, Hamouda
A, Jacquel A, Dufies M, Belhacene N, Colosetti P, Reed JC, Auberger
P and Luciano F: The anti-apoptotic Bcl-B protein inhibits
BECN1-dependent autophagic cell death. Autophagy. 8:637–649. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Levine B, Sinha SC and Kroemer G: Bcl-2
family members: Dual regulators of apoptosis and autophagy.
Autophagy. 4:600–606. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
D'Hulst AI, Vermaelen KY, Brusselle GG,
Joos GF and Pauwels RA: Time course of cigarette smoke-induced
pulmonary inflammation in mice. Eur Respir J. 26:204–213. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Crowe CR, Chen K, Pociask DA, Alcorn JF,
Krivich C, Enelow RI, Ross TM, Witztum JL and Kolls JK: Critical
role of IL-17RA in immunopathology of influenza infection. J
Immunol. 183:5301–5310. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hodge S, Hodge G, Ahern J, Jersmann H,
Holmes M and Reynolds PN: Smoking alters alveolar macrophage
recognition and phagocytic ability: Implications in chronic
obstructive pulmonary disease. Am J Respir Cell Mol Biol.
37:748–755. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bouzas-Rodríguez J, Zárraga-Granados G,
Del Rayo Sánchez-Carbente M, Rodríguez-Valentín R, Gracida X,
Anell-Rendón D, Poksay KS, Madden DT, Covarrubias L and
Castro-Obregón S: Correction: The nuclear receptor NR4A1 induces a
form of cell death dependent on autophagy in mammalian cells. PLoS
One. 10:e01187182015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin B, Kolluri SK, Lin F, Liu W, Han YH,
Cao X, Dawson MI, Reed JC and Zhang XK: Conversion of Bcl-2 from
protector to killer by interaction with nuclear orphan receptor
Nur77/TR3. Cell. 116:527–540. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Allinson JP, Hardy R, Donaldson GC,
Shaheen SO, Kuh D and Wedzicha JA: Combined impact of smoking and
early-life exposures on adult lung function trajectories. Am J
Respir Crit Care Med. 196:1021–1030. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hu M, Luo Q, Alitongbieke G, Chong S, Xu
C, Xie L, Chen X, Zhang D, Zhou Y, Wang Z, et al: Celastrol-induced
Nur77 interaction with TRAF2 alleviates inflammation by promoting
mitochondrial ubiquitination and autophagy. Mol Cell.
66:141–153.e6. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hamers AA, Uleman S, van Tiel CM,
Kruijswijk D, van Stalborch AM, Huveneers S, de Vries CJ and van 't
Veer C: Limited role of nuclear receptor Nur77 in Escherichia
coli-induced peritonitis. Infect Immun. 82:253–264. 2014.
View Article : Google Scholar :
|
|
34
|
Myers DR, Lau T, Markegard E, Lim HW,
Kasler H, Zhu M, Barczak A, Huizar JP, Zikherman J, Erle DJ, et al:
Tonic LAT-HDAC7 signals sustain Nur77 and Irf4 expression to tune
naive CD4 T cells. Cell Rep. 19:1558–1571. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Yu C, Cui S, Zong C, Gao W, Xu T, Gao P,
Chen J, Qin D, Guan Q, Liu Y, et al: The orphan nuclear receptor
NR4A 1 p rotects pancreatic β-cells from endoplasmic reticulum (ER)
stress-mediated apoptosis. J Biol Chem. 290:20687–20699. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Segala G, David M, de Medina P, Poirot MC,
Serhan N, Vergez F, Mougel A, Saland E, Carayon K, Leignadier J, et
al: Dendrogenin A drives LXR to trigger lethal autophagy in
cancers. Nat Commun. 8:19032017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Hedrick E, Lee SO, Doddapaneni R, Singh M
and Safe S: Nuclear receptor 4A1 as a drug target for breast cancer
chemotherapy. Endocr Relat Cancer. 22:831–840. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee SO, Jin UH, Kang JH, Kim SB, Guthrie
AS, Sreevalsan S, Lee JS and Safe S: The orphan nuclear receptor
NR4A1 (Nur77) regulates oxidative and endoplasmic reticulum stress
in pancreatic cancer cells. Mol Cancer Res. 12:527–538. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Lee SO, Andey T, Jin UH, Kim K, Singh M
and Safe S: The nuclear receptor TR3 regulates mTORC1 signaling in
lung cancer cells expressing wild-type p53. Oncogene. 31:3265–3276.
2012. View Article : Google Scholar :
|
|
40
|
Shin HJ, Lee BH, Yeo MG, Oh SH, Park JD,
Park KK, Chung JH, Moon CK and Lee MO: Induction of orphan nuclear
receptor Nur77 gene expression and its role in cadmium-induced
apoptosis in lung. Carcinogenesis. 25:1467–1475. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Chen GQ, Lin B, Dawson MI and Zhang XK:
Nicotine modulates the effects of retinoids on growth inhibition
and RAR beta expression in lung cancer cells. Int J Cancer.
99:171–178. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Chen ZH, Kim HP, Sciurba FC, Lee SJ,
Feghali-Bostwick C, Stolz DB, Dhir R, Landreneau RJ, Schuchert MJ,
Yousem SA, et al: Egr-1 regulates autophagy in cigarette
smoke-induced chronic obstructive pulmonary disease. PLoS One.
3:e33162008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Wang G, Zhou H, Strulovici-Barel Y,
Al-Hijji M, Ou X, Salit J, Walters MS, Staudt MR, Kaner RJ and
Crystal RG: Role of OSGIN1 in mediating smoking-induced autophagy
in the human airway epithelium. Autophagy. 13:1205–1220. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li Y, Yu G, Yuan S, Tan C, Xie J, Ding Y,
Lian P, Fu L, Hou Q, Xu B and Wang H: 14,15-Epoxyeicosatrienoic
acid suppresses cigarette smoke condensate-induced inflammation in
lung epithelial cells by inhibiting autophagy. Am J Physiol Lung
Cell Mol Physiol. 311:L970–L980. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Wei Y, Pattingre S, Sinha S, Bassik M and
Levine B: JNK1-mediated phosphorylation of Bcl-2 regulates
starvation-induced autophagy. Mol Cell. 30:678–688. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Liu J, Liu W, Lu Y, Tian H, Duan C, Lu L,
Gao G, Wu X, Wang X and Yang H: Piperlongumine restores the balance
of autophagy and apoptosis by increasing BCL 2 p hosphorylation in
rotenone-induced Parkinson disease models. Autophagy. 14:845–861.
2018. View Article : Google Scholar :
|