1. Introduction
Over 450 types of protein post-translational
modifications have been discovered, including phosphorylation,
glycosylation, ubiquitination, acetylation, nitrosylation,
methylation and ADP-ribosylation (1). Such modifications dramatically
expand the repertoire of potential protein functions and contribute
to the high diversity of cellular activities due to their
reversible nature, relatively small metabolic cost, and their
ability to extensively modulate the functions of target proteins
(2,3). Among all post-translational
modifications, phosphorylation and glycosylation are considered the
most common and the best investigated.
Glycosylation was, until recently, considered a
stable and conserved post-translational modification, while
phosphorylation was generally considered as a dynamic and
reversible modification. A specialized form of O-glycosylation
refers to the presence of β-D-N-acetylglucosamine (GlcNAc)
monosaccharide units linked O-glycosidically to intracellular
proteins (O-GlcNAcylation). This type of modification was not
discovered until 1984 and was found to be directly involved in the
modulation of a number of cellular processes (4). O-GlcNAcylation differs from
N-glycosylation and other O-glycosylation like O-GalNAcylation by
occurring mainly on nuclear, cytoplasmic and mitochondrial
proteins, and by being temporally dynamic. Unlike N-glycosylation,
O-GlcNAcylation is more analogous to phosphorylation as it occurs
rapidly and reversibly. Interestingly, O-GlcNAcylation can either
compete or co-operate with phosphorylation since it targets either
the same or the proximal serine/threonine (Ser/Thr) residue sites
required for phosphorylation (5).
However, unlike phosphorylation that is regulated by a diverse
array of kinases and phosphatases, O-GlcNAcylation is controlled by
the action of a single pair of enzymes, OGT and OGA (6). OGT is the specific enzyme
responsible for catalyzing the reaction between a single GlcNAc
moiety and the hydroxyl group of a ser or thr residue on proteins,
while OGA is the specific glycoside hydrolase that removes it
(6,7). The substrate of the transferase
reaction is uridine diphosphate N-acetylglucosamine (UDP-GlcNAc),
which is synthesized by the hexosamine biosynthetic pathway (HBP).
HBP co-ordinates with almost every other metabolic cellular pathway
since O-GlcNAcylation is sensitive to the levels of insulin,
glucose, amino acids (glutamine), fatty acid (acetyl-CoA),
nucleotide (UDP) and cellular stress. Therefore, O-GlcNAcylation
has mainly been considered to serve in the regulation of cellular
signaling, transcription and translation in response to nutrients
and stresses (8) (Fig. 1). Signaling pathways convert
environmental cues into intracellular events, such as immune cell
activation and inflammation. Combined aberrant O-GlcNAcylation may
be the cause of excessive nutrient intake, diabetes and/or
autoimmune diseases progressions, including those involved in
inflammatory and immune responses in individual cell types and
tissues (Table I).
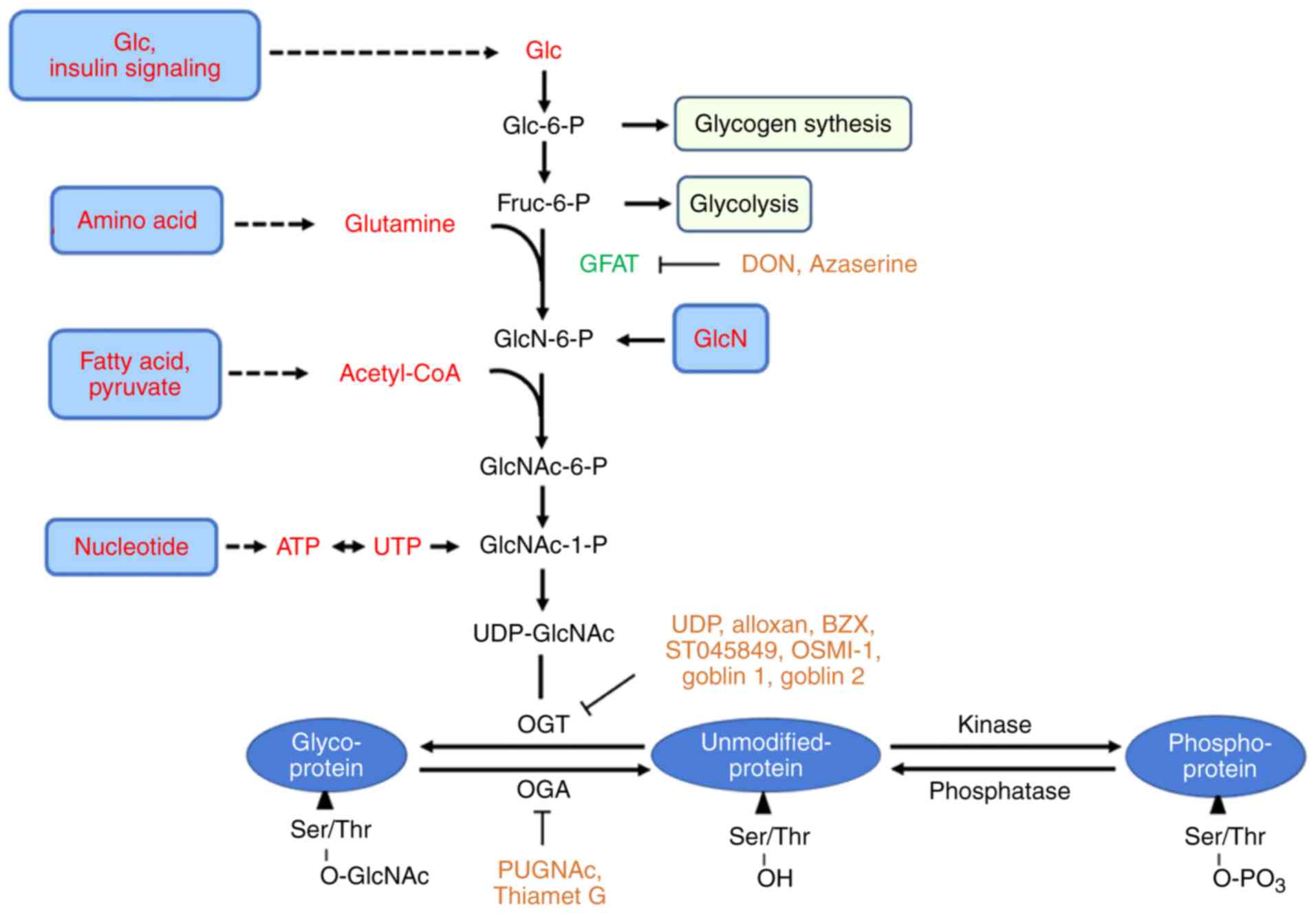 | Figure 1Nutrient flux through the HBP
modulates protein O-GlcNAcylation. The majority of glucose is taken
for glycolysis and glycogen synthesis, but a small percentage
(~2-5%) is funneled directly into the HBP for UDP-GlcNAc synthesis.
OGT catalyzes the addition of GlcNAc from UDP-GlcNAc to serine and
threonine residues, while OGA catalyzes their removal. Together
these two enzymes tune the dynamic addition and removal of O-GlcNAc
for thousands of diverse proteins. O-GlcNAcylation sites are
usually directly at, or close to, the same serine or threonine
residues used by kinases. HBP, hexosamine biosynthetic pathway;
OGT, O-linked N-acetylglucosamine transferase; OGA, O-GlcNAcase;
GlcNAc, N-acetylglucosamine; UDP, uridine diphosphate; GFAT,
glutamine-fructose-6-phosphate aminotransferase; OSMI-1,
(αR)-α-[[(1,2-Dihydro-2-oxo-6-quinolinyl)sulfonyl]amino]-N-(2-furanylmethyl)-2-methoxy-
N-(2-thienylmethyl)-benzeneacetamide. |
 | Table IFunctions of O-GlcNAcylation in
immune and inflammatory responses. |
Table I
Functions of O-GlcNAcylation in
immune and inflammatory responses.
| Author, year | Cell type and
tissue | Stimuli and
treatment | Target protein and
O-GlcNAcylation site | Function | (Refs.) |
|---|
| Golks et al,
2007; Lund et al, 2016; Bunting et al, 2007; Swamy
et al, 2016 | T cells | Anti-CD3/CD28
Ab | NFAT, p65, c-Rel
(S350)
c-Myc (T58) | T cell activation,
IL-2, IFNG, and CSF2 ↑
T cell clonal expansion | (52)
(40)
(53)
(41) |
| Golks et al,
2007; Wu et al, 2017 | B cells | Anti-IgM Ab - | NFAT, p65 Lyn
(S19) | B cell
activation
B-cell activation and expansion | (52)
(56) |
| Kneass and
Marchase, 2005; Kneass and Marchase, 2004 | Neutrophils | fMLF/PMA | - | Cellular
migration | (66)
(67) |
| James et al,
2002 | Mesangial
cells | High glucose,
GlcN | p65 | VCAM-1 ↑ | (73) |
| Dela et al,
2017 | Placentas
cells | High glucose | p65 | TNF-α and IL-6
↑ | (74) |
| Krick et al,
2018 | Epithelial
cells | FGF23 | - | NFAT activation ↑
IL-6 ↑ | (71) |
| Donovan et
al, 2014 | | high glucose | Sp1 | VEGF-A ↑ | (81) |
| Zhang et al,
2015 | Pancreatic acinar
cells | caerulein | p65, IKKα | TNF-α and NO ↑ | (75) |
| Li et al,
2014; Donovan et al, 2014; Zhang et al, 2017 | Endothelial
cells | LPS
High glucose | p65
Sp1 | Inflammatory
mediators ↑
VEGF-A, ICAM-1 ↑ | (76)
(81,82) |
| Allison et
al, 2012; Ma et al, 2017; Pathak et al, 2012 | 293T | TNF-α IL-1/osmotic
stress | p65 (T305, S319)
TAB1 (S395) | IL-6 and TNF-α
↑ | (78,79)
(85,88) |
| Hou et al,
2016 | | MDP | Nod2 | NF-κB activity
↑ | |
| Hwang et al,
2017; Ryu and Do, 2011; Li et al, 2017; Pathak et al,
2012; Zhang et al, 2016 | Macrophages | LPS IL-1/osmotic
stress |
STAT3(T717)
TAK1(S427) | NO/iNOS ↑; IL-12,
CXCL1 and CXCL2 ↑; IL-10 ↓; polarization | (62,63,84
85,87) |
| Hwang et al,
2010; Hwang et al, 2014 | Macrophages smooth
Muscle cells | GlcN with LPS | p65 and c-Rel,
RNAPII | COX-2, iNOS, IL-1β,
IL-6, TNF-α ↓ | (64,65) |
| He et al,
2017 | | Thiamet G with MCAO
or LPS | - | Iba+ cells ↓ iNOS
and COX2 ↓ p65 translocation ↓ M1 polarization ↓ | (103) |
| Yang et al,
2008 | | High glucose | p65 (T352) | VCAM-1 ↑ | (77) |
| Yang et al,
2010; Xing et al, 2011; Yao et al, 2018 | Smooth muscle cells
Colon epithelial cell | High glucose
GlcN/OGA inhibitor with TNF-α | PGX1 p65 (S536),
A20 | anti-oxidant
activity ↑ MCP-1, P-selectin, VCAM-1 ↓ | (112) (98,111) |
| Hirata et
al, 2018 | | dextran sodium
sulfate | - | phosphor-p65
↓
IL-1β ↓ | (99) |
| Zou et al,
2007 | Heart | GlcN with trauma-
hemorrhage | - | cardiac output and
organ perfusion recovered | (91) |
| Guo et al,
2015 | Heart | Intermittent
hypoxia | - | MAPK activity ↑ p65
levels ↑ inflammatory mediators ↑ | (72) |
2. Enzymes controlling O-GlcNAc cycling
O-linked N-acetylglucosamine transferase
(OGT)
OGT is encoded by a single gene in mammals, however,
its transcript produces multiple variants after selective splicing
which are as follows: Nucleocytoplasmic, (ncOGT; 116 kDa);
mitochondrial, (mOGT; 103 kDa) and short, (sOGT; 78 kDa) (9). They share a common catalytic and
phosphorylase-derived C-terminal domain but differ in length owing
to variable numbers of the N-terminal tetratricopeptide repeat
(TPR) domain (10), separated by
a spacer region. It had long been believed that OGT isoforms are
present in the cytoplasm and nucleus, such as ncOGT and sOGT; and
in mitochondria, such as mOGT (7). However, an atypical OGT termed
epidermal growth factor domain-specific O-GlcNAc transferase (EOGT)
was identified, which transfers GlcNAc to Ser or Thr in secreted
and membrane proteins containing the epidermal growth factor (EGF)
repeat with a specific consensus sequence. Interestingly, OGT and
EOGT exist in separate cellular compartments and have mostly
distinct substrates (11,12).
OGT modulates a variety of cellular processes like
transcription, protein synthesis (9), protein degradation (13,14), protein-protein interaction or
localization (7,15,16) and stress response (17) using several substrates. However,
it has been difficult to elucidate the mechanisms by which OGT
recognizes its substrates since it targets only a fraction of
Ser/Thr in different biological situations for glycosylation. There
have been several suggested potential mechanisms. OGT primarily
recognizes most of its substrates through asparagine ladder binding
in the TPR domain (18,19). The interaction between OGT and its
substrates also contributes to the OGT catalytic mechanism. The
N-acyl group in UDP-GlcNAc is crucial for its affinity to sOGT. In
this process, the backbone carbonyl oxygen of L653 and the hydroxyl
group of T560 in sOGT are important for the recognition of the
UDP-GlcNAc (20). Additionally,
OGT can also non-specifically modify proteins which contain regions
of intrinsic disorder (e.g. tau and nuclear pore proteins) without
recognizing any specific sequences or structures (7). Different UDP-GlcNAc concentrations
or varying UDP-GlcNAc gradients may influence OGT binding
specificity (21). Finally, OGT
is also regulated by phosphorylation. The phosphorylation of OGT at
T444 by AMP-activated protein kinase (AMPK) is closely associated
with OGT nuclear localization in myotubes and phosphomimetic
T444E-OGT exhibits altered substrate selectivity. Acute treatment
(2 h) with a highly specific activator of AMPK (A-769662) induced
significant global changes in O-GlcNAcylated proteins bound to
wild-type OGT compared with T444E-ncOGT. It demonstrated the
placement of a large highly charged moiety on residue T444 is
sufficient to dramatically alter the substrate selectivity of OGT
(22).
Genetic and chemical approaches are used to inhibit
both OGT expression and activity to comprehensively study the
biological role of O-GlcNAc and assess the catalytic vs.
non-catalytic roles of OGT. Complete OGT knockout is embryo-lethal
in a range of animal models. As such, tissue-specific OGT knockout
mice have been employed (23).
Consequently, chemical inhibitors are a worthwhile means to explore
the biological role of O-GlcNAcylation without affecting OGT
protein levels. UDP, alloxan, compound 4 (ST045849), compound 5 and
(αR)-α-[[(1,2-Dihydro-2-oxo- 6- quinolinyl)sulfonyl]
amino]-N-(2-furanylmethyl)-2-methoxy-N-(2-hienylmethyl)-benzeneacetamide
are small molecules that compete with the donor substrate (24-26) (Fig.
1). In addition, a new series of competitive inhibitors,
goblin1 (OGT bisubstrate-linked inhibitor 1) and goblin 2, derived
from both OGT and the substrate, were designed based on the
understanding of the OGT catalytic mechanism (27). However, off-target effects, lack
of specificity and low-grade cell permeability are limitations that
need to be resolved (28).
O-GlcNAcase (OGA)
The human OGA (hOGA) gene MGEA5 is alternatively
spliced to generate nucleocytoplasmic (nc; 130 kDa) and short (s;
75 kDa) isoforms. The ncOGA contains both an N-terminal O-GlcNAc
hydrolase and a C-terminal histone acetyltransferase-like
(HAT-like) domain; while sOGA lacks the HAT-like domain and is
localized to the endoplasmic reticulum and lipid droplets (29,30).
hOGA belongs to the carbohydrate-active enzymes
database glycoside hydrolase 84 family of enzymes (31). The substrate recognition mechanism
of hOGA was not considered to be sequence specific until the
crystal structure for hOGA was reported (32). The crystallographic data for hOGA
suggested that the full-length and nuclear spliced isoforms could
be structurally distinguished by their ability to form an
interlinked dimer. Based on the crystal structure, novel inhibitors
targeting hOGA other than
O-(2-acetamido-2-deoxy-D-glucopyranosylidene)
amino-N-phenylcarbamate (PUGNAc) and thiamet G could be designed
(32,33) (Fig.
1). Notably, OGA can be O-GlcNAcylated by OGT at S405; however,
the implications of O-GlcNAcylation of OGA have remained elusive
(1,34).
3. Immunoregulatory role of
O-GlcNAcylation
O-GlcNAcylation activates T cells
The T cell receptor (TCR) is a molecule present on
the surface of T cells that is responsible for recognizing antigens
as peptides which are bound to major histocompatibility complex II
molecules on the surface of antigen presenting cells. This triggers
the initial activation of cluster of differentiation
(CD)4+ T cells (35)
and directs CD4+ T cell differentiation into four
effectors: T helper 1 (Th1), which target intracellular bacteria
and viruses; Th2, which target extracellular parasites; Th17, which
target fungi and extracellular pathogens and inducible regulatory T
cells (iTregs), which have the opposing function of decreasing an
inflammatory immune response. T cell activation induces expression
of GLUT1 and amino acid transporters on the cell surface which
transport adequate glucose and glutamine for the differentiation of
Th1, Th2 and Th17 cells. Moreover, the conversion of glutamine to
acetyl CoA boosts fatty acid synthesis during T cell activation
(36,37). Taken together, glucose, glutamine
and acetyl CoA are all substrates that participate in the HBP,
driving an increase in UDP-GlcNAc synthesis and thus protein
O-GlcNAcylation by OGT (37,38) (Fig.
2).
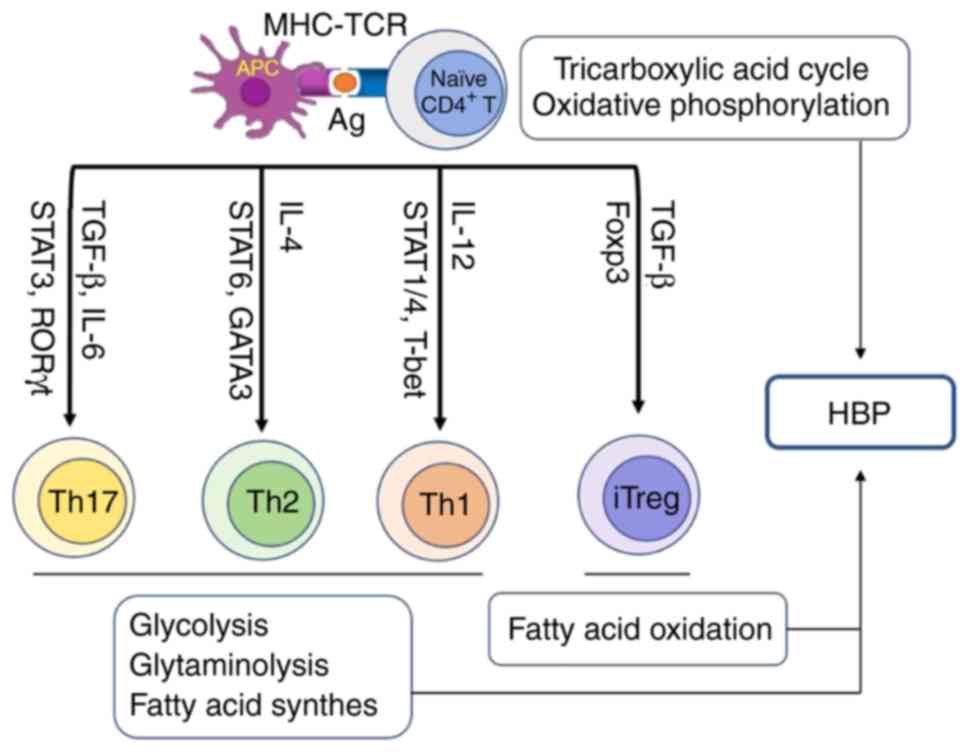 | Figure 2Connection between helper T cell
differentiation and HBP. After T cell antigen receptor engagement,
naïve CD4+ T cells differentiate into effector T cells
including Th1, Th2 and Th17 cells, as well as iTreg. Effector T
cells utilize glucose through glycolysis and amino acids through
glutaminolysis to meet their energy need for differentiation,
whereas regulatory T cells use energy from fatty acid oxidation.
Finally, HBP integrates glucose, amino acid and fatty acid
metabolism to generate UDP-GlcNAc for O-GlcNAc modification. CD,
cluster of differentiation; HBP, hexosamine biosynthetic pathway;
iTregs, inducible regulatory T cells; MHC, major
histo-compatibility complex; TCR, T cell receptor; IL, interleukin;
UDP-GlcNAc, uridine diphosphate N-acetylglucosamine; APC, antigen
presenting cell. |
T cell activation enhances both OGT expression and
overall protein O-GlcNAylation levels. More than 2,000 intact
O-linked glycopeptides have been identified on activated primary
human T cells via a technique termed as isotope targeted
glycoproteomics. A large portion (>45%) of identified O-GlcNAc
sites lie in proximity to or coincide with a known phosphorylation
site, indicating a possibility of post-translational modification
crosstalk (39). It has been
reported that deficiency in protein O-GlcNAcylation results in the
blocking of T cell proliferation, development, transformation and
differentiation (40,41). Importantly, Notch, TCR and c-Myc
(T58) have been identified as key controllers of T cell protein
O-GlcNAcylation by regulation of glucose and glutamine transport
(41,42).
The nuclear factor (NF)-κB transcription factor
family consists of five proteins, including p65 (RelA), RelB,
c-Rel, p105/p50 (NF-κB1) and p100/p52 (NF-κB2). NF-κB p65/p50 is
the most commonly occurring heterodimer complex among NF-κB
homodimers and heterodimers and is the functional component
involved in nuclear translocation and activation of NF-κB (43). Under quiescent conditions, the
inactive NF-κB p65/p50 heterodimer is mainly sequestered in the
cytoplasm and is associated with inhibitor of NF-κB α (IκBα).
However, upon stimulation by pro-inflammatory cytokines,
lipopoly-saccharide (LPS) or glucose, IκBα is phosphorylated by the
IκB kinase (IKK) on S32 and S36. This phosphorylation results in
IκB ubiquitination and subsequent degradation by the 26S
proteasome. NF-κB p65/p50 then detaches from IκBα and translocates
to the nucleus where it binds to NF-κB promoter/enhancer sites.
Through its acidic transactivation domain, p65 has the capacity to
interact with several different transcriptional regulatory
proteins, such as CREB-binding protein (CBP)/p300, to initiate
transcription of NF-κB target genes (44,45). NF-κB is primarily modulated by
post-translational modifications, such as phosphorylation,
acetylation and glycosylation. It regulates several cellular
processes like innate immunity, adaptive immunity, inflammation,
cell apoptosis, cell survival and differentiation. The activity of
NF-κB is closely associated with the pathogenesis of metabolic
syndrome (46). Metabolic
syndrome comprises hyperglycemia, hyperlipidemia, insulin
resistance, obesity and hepatic steatosis which result from
nutrient excess (47). Mounting
an immune response to infection resulting in the release of
cytokines is energy intensive. NF-κB is activated by cytokines to
promote re-localization, activation and differentiation of
macrophages at the site of infection. These activated macrophages
defend against invading microorganisms by producing antimicrobial
molecules and recruiting leukocytes, subsequently removing
pathogens and clearing dead cells (48,49). NF-κB-regulated expression of the
inflammatory mediators drives the differentiation of monocytes into
either M1 or M2 macrophages which are vital to the development of
the inflammation-associated metabolic disease. M1 macrophages
produce and release IL-1, IL-6, tumor necrosis factor (TNF)-α, and
other pro-inflammatory cytokines, while M2 macrophages secrete the
anti-inflammatory cytokine IL-10 (49,50). Therefore, NF-κB controls the
expression of the inflammatory mediators that recruit monocytes,
drive differentiation to macrophages and direct macrophage cell
fate. NF-κB therefore modulates inflammation in the liver, adipose
tissue and central nervous system in the development of metabolic
diseases (49,51). Furthermore, an increase in NF-κB
p65 O-GlcNAcylation contributes to T cell activation. It has been
reported that OGT is required for the O-GlcNAcylation and
transcriptional activity of both p65 as well as nuclear factor of
activated T cells (NFAT). This leads to IL-2 production which is
consistent with T cell activation (52). Additionally, NF-κB subunit c-Rel
is modified and activated by O-GlcNAcylation at S350. Conversely,
mitigating the O-GlcNAcylation of this residue disturbs the
c-Rel-mediated expression of IL-2, IFN-γ and colony stimulating
factor (CSF)2 in response to TCR activation. However, mutating the
O-GlcNAcylation site of c-Rel or treating cells with PUGNAc has no
effect on TCR or TNF-α induced expression of TNFAIP3
(encodes TNF-α induced protein 3) and NFKBIA (encodes NF-κB
inhibitor α), genes that are considered to contain c-Rel binding
sites in their promoters (53).
Although TCR and TNF both elevate the nuclear translocation of
c-Rel, only TCR activity resulted in the O-GlcNAcylation of c-Rel
in the nucleus. These results suggest a stimulus-specific role or
O-GlcNAcylation of c-Rel in promoting T cell-mediated autoimmunity.
It is the generation of these autoimmune T helper cell cytokines
which result in pancreatic β-cell injury leading to type 1 diabetes
(54,55).
O-GlcNAcylation activates B-cells
B cell activation, like T cell activation, is
triggered by the specific recognition of antigens by the B cell
receptor (BCR). In activated B cells, NF-κB and NFAT are
O-GlcNAcylated by OGT through direct interaction and this
correlates strongly with their translocation to the nucleus
(52). Intriguingly,
O-GlcNAcylation of Lyn at S19 is critical for Lyn activation and
Syk interaction in BCR-mediated B-cell activation and expansion
(56). Taken together,
O-GlcNAcylation mediated by OGT contributes to maintain
homeostasis, transduce BCR-mediated activation signals and activate
humoral immunity.
O-GlcNAcylation modulates the activation
of monocytes and macrophages
Monocytes and macrophages play vital roles in acute
and chronic inflammation. Obesity, diabetes and insulin resistance
are concomitant with the aggregation of pro-inflammatory monocytes
and macrophages in different organs, such as the pancreas, adipose
tissue, liver and blood vessel walls (57-61). O-GlcNAcylation is implicated in
the inflammatory reaction in monocytes and macrophages, such as
c-Rel. At normal glucose concentrations, glucosamine (GlcN)
dose-dependently increases LPS-stimulated c-Rel O-GlcNAcylation and
production of NO/inducible nitric oxide synthase (6,62).
Of interest, OGT de-nitrosylation is triggered by LPS in macrophage
cells and results in p62 and p65 hyper-O-GlcNAcylation and
NO/cytokine production. As mentioned previously, this study
supports a role for de-nitrosylation of S-nitrosylated OGT in
controlling the LPS-stimulated innate immune response due to
upregulation of OGT activity (63).
Numerous previous studies (64,65), on the other hand, indicate that
O-GlcNAcylation participates in anti-inflammatory responses in
monocytes and macrophages and have tried to uncover the mechanisms
by which O-GlcNAcylation protects monocytes and macrophages from
inflammatory stresses. GlcN may serve as a novel neuroprotective or
anti-inflammatory agent, not only because it reduces infarct volume
and affords a reduction in motor impairment and neurological
deficits in a rat middle cerebral artery occlusion model, but also
because it suppresses LPS-induced upregulation of pro-inflammatory
mediators both in microglia (BV2) and macrophages (RAW264.7)
(64). GlcN is a substrate for
glutamine: Fructose-6-phosphate amidotransferase (GFAT), the
rate-limiting enzyme in the HBP. Thus, when the glucose and
glutamine influx rapidly increases, UDP-GlcNAc levels and
subsequent O-GlcNAcylation can also rise. However, in this case,
GlcN inhibits the O-GlcNAcylation of NF-κB, probably by disturbing
the association between OGT and NF-κB (64).
O-GlcNAcylation activates
neutrophils
Neutrophils (polymorphonuclear leukocytes) are a key
member of the innate immune system and are activated when presented
with a large and diverse group of stimuli. The process of
activation initiates a cascade of events that lead to spreading and
finally migration. It has been reported that O-GlcNAcylation
mediates the activation of the small GTPase Rac and the downstream
p38 and p44/42 mitogen-activated protein kinase (MAPK) signaling
pathways in neutrophils. These MAPKs are known to modulate cellular
chemotaxis leading to cell movement and inflammatory response
(66-68).
4. Pro-inflammatory role of
O-GlcNAcylation
Positive regulation of the transcription
factors by O-GlcNAcylation
A total of ~4,000 proteins have been identified as
OGT targets to-date and this will continue to increase as the
technology for mapping and quantifying O-GlcNAc sites improves
(69,70). O-GlcNAcylation affects NF-κB, NFAT
and specificity protein 1 (Sp1) which are related to inflammation
and immune reactions. For example, fibroblast growth factor 23
augments the global changes in the O-GlcNAc modification of
proteins in human bronchial epithelial cells to regulate airway
inflammation through NFAT activation and IL-6 upregulation
(71). Intermittent hypoxia
raises the O-GlcNAc in proteins, accompanied by an increase in the
levels of myocardial NF-κB, inflammatory cytokines, caspase-3 and
cardiomyocyte apoptosis (72).
The most widely and intensively studied
O-GlcNAcylated transcription factor is NF-κB (7). High glucose and pro-inflammatory
factors, risks for metabolic syndrome and diabetes mellitus are
deemed to activate the NF-κB pathway and initiate the transcription
of relevant downstream target genes. Therefore, the role of
O-GlcNAcylation in NF-κB activation has gained more attention.
It has been reported that glucosamine, high glucose
and overexpression of GFAT are employed to prompt the binding
between nuclear proteins and NF-κB consensus sequences. This
enhances vascular cell adhesion molecule-1 (Vcam-1) promoter
activity in glomerulus cells isolated from male Sprague-Dawley rats
(73). This set of data suggest
that NF-κB O-GlcNAcylation can participate in inflammation
(73). Furthermore, high glucose
has been found to augment O-GlcNAcylation of NF-κB and production
of cytokines TNF-α and IL-6 in rat placenta (74). However, little is known about the
mechanisms through which O-GlcNAcylation upregulates NF-κB
activity. In the caerulein-stimulated acute pancreatitis model,
OGT-mediated O-GlcNAcylation of NF-κB p65 and IKKα promotes NF-κB
signaling activation, TNF-α secretion and nitric oxide (NO)
production in AR42 J rat pancreatic acinar cells which might
exacerbate the progression of acute pancreatitis (75). In addition, the authors' previous
study has confirmed that LPS triggers the OGT-dependent
O-GlcNAcylation of NF-κB and thereby induces a vascular endothelial
inflammatory response (76).
However, the O-GlcNAc modification sites on NF-kB have not been
confirmed and the mechanism by which O-GlcNAc modification promotes
NF-kB activation has not been completely elucidated.
An increasing amount of research is focused today on
demonstrating the pro-inflammatory role of O-GlcNAcylation in the
NF-κB signaling pathway. In rat vascular smooth muscles,
O-GlcNAcylation of NF-κB p65 on T352 inhibits the interaction
between NF-κB p65 and IκB, thus elevating the translocation of
NF-κB to the nucleus and increasing the transcription of VCAM-1
under hyperglycemic conditions (77). These results suggest that a
specific O-GlcNAcylation site at T352 on NF-κB p65 may contribute
to a prolonged increase in NF-κB activity during the progression of
diabetes (77). Moreover, the
transcriptional activity of NF-κB seems to increase its concomitant
phosphorylation and acetylation, which may interact with
O-GlcNAcylation in 293T cells (78,79). Under the stimulation of TNF-α, OGT
enhances the CBP/p300-dependent acetylation of NF-κB p65 on K310 by
combining with NF-κB regulated promoters and subsequently promotes
full NF-κB transcription. Previous mapping studies reveal T305 to
be an important residue required for attachment of the O-GlcNAc
moiety on p65 (78,79). Similarly, Ma et al
(79) have verified that
O-GlcNAcylation of p65 on T305 and S319 increases
CBP/p300-dependent acetylation of p65 on K310, facilitating NF-κB
transcriptional activation. In addition to enhancing
O-GlcNAcylation, OGT increases the expression of p300, IKKα and
IKKβ, and elevates IKK-induced p65 phosphorylation on S536, leading
to NF-κB activation. However, O-GlcNAcylation of p65 at T305 is
likely impaired by the phosphorylation of p65 at T308 (79). In addition, O-GlcNAcylation of
IKKβ occurring at S733 has been discovered to prompt the NF-κB
activity by heightening IKK activity, IκB phosphorylation and IκB
degradation in cultured mouse and human fibroblasts (80).
Sp1, another transcription factor, besides NF-κB, is
also O-GlcNAcylated under high glucose concentration. Donovan et
al (81) indicated that
hyperglycemia significantly increases the binding of Sp1 to the
vascular endothelial growth factor (VEGF)-A promoter, while the
downregulation of OGT or Sp1 by shRNA significantly abro gates
glucose-induced changes in proangiogenic protein VEGF-A in ARPE-19
(human retinal pigment epithelial cells) and TR-iBRB (rat retinal
microendothelial cells). This suggests that hyperglycemia-driven
elevation of O-GlcNAcyaltion of Sp1 mediates VEGF-A production in
the retinal endothelium and pigment epithelium. Furthermore, Zhang
et al (82) confirmed that
hyperglycemia also stimulated intercellular adhesion molecule
(ICAM)-1 expression by O-GlcNAc modification of Sp1 in human
umbilical vein endothelial cells and rat retinal capillary
endothelial cells. Taken together, this group of studies helps to
enhance understanding of how O-GlcNAcylation modulates inflammatory
factors during diabetic retinopathy.
Signal transducer and activator of transcription 3
(STAT3) plays a key role in the cytokine-cytokine receptor
signaling pathway in colitis (83). Nevertheless, the critical
mechanism that regulates STAT3 phosphorylation and its target
pro-inflammatory cytokine production remains unclear. Li et
al (84) demonstrated that
modification of STAT3 with O-GlcNAc at T717 suppresses its
phosphorylation and the expression of the downstream
anti-inflammatory gene, IL-10, in bone marrow-derived macrophages
(BMMs) isolated from cullin (Cul) 3-deficient mice led to
increased disease severity in azoxymethane (AOM) induced colitis
and a colitis-associated cancer model. Meanwhile, the expression of
pro-inflammatory cytokines, such as IL-12, CXCL1 and CXCL2 are
upregulated by O-GlcNAcylated STAT3 in BMMs generated from
Cul 3-deficient mice make them more susceptible to
AOM-induced colitis and colitis-associated cancer. Furthermore, the
researchers found that myeloid-derived CUL3, a cullin family E3
ubiquitin ligase, attenuated OGT expression and downregulated STAT3
O-GlcNAc modification by facilitating the interaction between the
Ogt promoter region and nuclear factor-erythroid-2 related
transcription factor-2. The depletion of STAT3 O-GlcNAcylation
mediated by CUL3 is concomitant with the upregulation of STAT3
phosphorylation and IL-10 gene expression.
Pro-inflammatory effects of other
O-GlcNAcylated proteins
In addition to transcription factors, other
functional proteins are found to be O-GlcNAcylated in
pro-inflammatory reaction. Transforming growth factor-β activated
kinase (TAK) 1 is an important serine/threonine protein kinase that
mediates signals transduced by pro-inflammatory cytokines such as
TGF-β, TNF-α and IL-1. A number of experimental data suggest that
O-GlcNAcylation of TAK1-binding protein 1 (TAB1) at S395 is
required for full TAK1 activation upon stimulation with
IL-1/osmotic stress, for the downstream activation of NF-κB and
finally for the production of IL-6 and TNF-α in IL-1R cells 293
(cells stably expressing the IL-1 receptor) and immortalized
Tab1-deficient MEFs (85).
Another study indicated that TAK1 is O-GlcNAcylated as well. Upon
stimulation with IL-1 and NaCl, O-GlcNAcylation of TAK1 at S427 is
required for T187/S192 phosphorylation and full activation of TAK1
in RAW264.7 cells (86). Thus,
TAK1 O-GlcNAcylation has been found to activate downstream JNK and
NF-κB signaling pathways and increase pro-inflammatory cytokine
production, and finally facilitate M1 polarization of macrophages
which is closely associated with acute inflammatory responses in
RAW264.7 cells (87).
Nucleotide-binding oligomerization domain 2 (Nod2)
is a cytoplasmic human NOD-like receptor that recognizes bacterial
components. Both wild-type Nod2 and a Nod2 Crohn's-associated
variant are O-GlcNAcylated and this modification affects Nod2's
ability to produce various inflammatory molecules such as cytokines
and chemokines via NF-κB activation in 293T cells (88).
5. Anti-inflammatory role of
O-GlcNAcylation
In terms of cardiac and vasculature diseases,
augmenting O-GlcNAc modification of proteins in the vasculature may
represent a novel anti-inflammatory and vasoprotective mechanism
(89,90). On one hand, acute GlcN and PUGNAc
treatment increases O-GlcNAc-modified protein levels and
ameliorates acute inflammatory responses in balloon-injured rat
carotid arteries; a 14-day GlcN treatment attenuates neointima
formation (89). On the other
hand, acute increases in protein O-GlcNAcylation relieve TNF-α
triggered vascular dysfunction, at least partly, via mitigating the
expression of iNOS (90).
Furthermore, GlcN treatment and OGT overexpression attenuate the
trauma-hemorrhage induced increase in cardiac levels of TNF-α and
IL-6 mRNA, IκB α phosphorylation, NF-κB, NF-κB DNA binding
activity, ICAM-1, and myeloperoxidase activity. The enhanced
O-GlcNAcylation triggered by both GlcN and OGT overexpression
mediates improvement in cardiac function after hemorrhagic shock
occurred (91,92).
Negative regulation of the transcription
factors by O-GlcNAcylation
Aberrant regulation of NF-κB activity has been
associated with a variety of disorders such as autoimmune disease,
cancer and diabetes (93).
Chronic inflammation is the main culprit of the diseases mentioned
above (94-96). Therefore, fully elucidating the
molecular mechanisms that precisely tune NF-κB activity may have
great biological and clinical significance. Although increasing
O-GlcNAc levels are usually accompanied by NF-κB activation in
diabetes and insulin resistance, O-GlcNAcylation inducing
treatments appear to have anti-inflammatory and pro-survival
effects during acute injuries like myocardial infarction, burns,
trauma and sepsis (91,92,97). For instance, GlcN or PUGNAc
administration after trauma-hemorrhage improve organ perfusion and
function, and this is associated with increased protein O-GlcNAc
levels (91). Moreover, GlcN and
PUGNAc treatment protect against TNF-α induced inflammatory stress
by enhancing O-GlcNAcylation at S536 of p65 and by inhibiting TNF-α
induced phosphorylation of NF-κB p65, thus inhibiting NF-κB
signaling in rat aortic smooth muscle cells (98). In vivo, dextran sodium
sulfate-induced NF-κB p65 phosphorylation and IL-1β mRNA expression
are significantly lower in Ogt-transgenic than in wild type mice.
This suggests that O-GlcNAcylation could prevent acute colitis by
reducing acute maximum inflammation (99).
Another study indicated that GlcN alleviates the
O-GlcNAcylation of both nuclear and cytosolic forms of c-Rel and
inhibits the binding of c-Rel to the NF-κB site in the iNOS
promoter upon stimulation by LPS. The mechanism by which GlcN
exerts these effects involves the suppression of the interaction
between c-Rel and OGT (100).
Moreover, OGT plays a key role in the inflammatory responses of
macrophages. It has been observed that in N9 microglial cells, in
response to LPS, the enhanced expression of iNOS, NO and ROS is
mediated via the downregulation of OGT and protein O-GlcNAcylation,
or via the upregulation of MAPKs phosphorylation and NF-κB
translocation (101). In
addition, overexpression of OGT exhibits inhibitory effects on the
LPS-driven activation of NF-κB and iNOS through modulation of
histone acetylation either directly or indirectly (102). In addition to GlcN and OGT
overexpression, thiamet G, an OGA inhibitor, has been found to
dramatically reduce the infarct volume and ameliorate the
neurological deficits either before or after middle cerebral artery
occlusion (MCAO). Moreover, thiamet G administration reduces the
number of Iba1+ cells in MCAO mice and decreases the
expression of iNOS and cyclooxygenase 2 mainly by suppressing NF-κB
p65 pathway (103).
Sp1 is a zinc finger transcription factor. As
described above, elevated Sp1 activity upon O-GlcNAcylation could
play a role in hyperglycemia-induced pro-inflammatory and
pro-fibrotic factors involved in diabetic retinopathy (81,82). Alternatively, O-GlcNAc of Sp1 may
also reduce its transcriptional activity, possibly by disturbing
its interaction with its cooperative factors, such as Elf-1
(104), NF-Y (105), Ying-Yang 1 (106) and sterol regulatory element
binding protein 2 (107). Thus,
O-GlcNAc modification of Sp1 could be a participant in negative
regulation of placental and embryonic expression of oncofetal
protein gene (Pem) (104),
hyaluronan synthesis (106) and
lipid synthesis (107).
Interestingly, Suh et al (108) and Lee et al (109) showed that O-GlcNAcylation of Sp1
protects against hypoxia-induced dysfunction of
Na+/glucose cotransporter (SGLT) in renal proximal
tubule cells and hypoxia-induced apoptosis of mouse embryonic stem
cells. A weakening association between Sp1 and its co-operative
factors caused by O-GlcNAcylated Sp1 could be a mechanism
postulated to explain these phenomena.
Anti-inflammatory effects of other
O-GlcNAcylated proteins
The zinc finger protein A20 (also known as TNFAIP3)
has been identified as an inhibitory effector on NF-κB
over-activation by using its deubiquitinase activity (110). However, the regulation of A20
activity remains poorly understood. GlcN and thiamet G
significantly increase A20 O-GlcNAcylation and enhance binding to
Tax1 binding protein 1, a key regulatory protein for A20 activity.
These data suggest that O-GlcNAcylation is a critical regulatory
mechanism for A20 activity, which in turn negatively regulates the
NF-κB signaling cascade in TNF-α injured acute vascular smooth
muscle cells (111).
Glutathione peroxidase 1 (GPX1), an anti-oxidant
enzyme, is critical for cell survival during hyperglycemia and
oxidative stress. Yang et al (112) discovered that hyperglycemia
induces GPX1 activity by enhancing the O-GlcNAcylation of GPX1 and
subsequently increases the association between non-receptor
tyrosine kinase c-Abl and Arg in rat vascular smooth muscle cells.
Also, pharmacological administration of the OGA inhibitor NTZ has
been found to induce GPX1 activation in the mouse liver.
RNA polymerase II (RNAPII) catalyzes the
transcription of all protein-coding genes and a number of
non-coding RNAs. Hwang et al (65) discovered that GlcN relieves basal
activities of transcription induced by LPS through the upregulation
of RNAPII O-GlcNAcylation and DNA binding which are inhibited by
LPS.
6. Conclusions and future perspectives
Since O-GlcNAcylation's involvement in various
immune pathways appears complicated, the present review proposed
several reasons why O-GlcNAcylation may exert different effects on
inflammation and autoimmune diseases.
First, the O-GlcNAc modification as a
dual-directional regulator of inflammation was discussed. On one
hand, different stimuli lead to distinct outcomes, as exhibited by
O-GlcNAcylation modifications in inflammatory responses. The key
factor which determines the pro- or anti-inflammatory role of
particular O-GlcNAcylation modifications is the type of insult
(chronic hyperglycemia in diabetes vs. acute vascular injury). On
the other hand, GlcN regulates inflammation by sensing energy
states of normal and excess nutrients. At normal glucose
concentrations, GlcN dose-dependently enhances LPS-triggered
inflammation in macrophages. However, GlcN suppresses macrophage
inflammation upon high glucose cell culture conditions. In
addition, LPS-stimulates an increase in O-GlcNAcylation as well as
an increase in DNA binding of c-Rel to the iNOS promoter by GlcN in
normal glucose conditions, but a decrease in high glucose
conditions (62). Moreover,
hyperglycemia makes a pregnancy highly risky and may even have
negative effects on the fetus. Since O-GlcNAcylation is believed to
be a nutritionally responsive modification, it provides one means
of explaining how excess nutrients in the intrauterine environment
may affect metabolic deregulation of the offspring (113). Furthermore, GlcN-mediated
O-GlcNAcylation participates in the inhibition of TNF-α and IL-8
gene expression in osteoarthritis (114). It therefore appears that,
depending on the cellular nutrition state, the type of insult
(chronic hyperglycemia in diabetes vs. acute vascular injury) and
the cell state (inflammatory state vs. non-inflammatory state), HBP
may quickly switch the management pattern to regulate inflammation,
resulting in either pro- or anti-inflammatory outcomes (62). These results also suggest that
O-GlcNAc may create a negative feedback loop between pro- and
anti-inflammation. Thus, proper adjustment of O-GlcNAc presents a
potential therapeutic strategy for combating metabolic
dysregulation and inflammatory diseases such as diabetes, sepsis,
and osteoarthritis.
Second, the present review considered O-GlcNAc as a
target in relieving over-nutrition-related chronic inflammation and
autoimmune diseases. The pathogenesis of obesity and type 2
diabetes are accompanied by long-term, low-grade, chronic
inflammation. This inflammation has been implicated in much of the
downstream pathology, including atherosclerosis, insulin resistance
and a high risk for autoimmunity, which are associated with
over-nutrition and adiposity (115,116). In addition, disruption in the
metabolic pathways of effector T cells is integral to the progress
of atherosclerosis and insulin resistance, which may result in
enhanced shunting of metabolites into the HBP, fueling O-GlcNAc
modification. Moreover, effector T cells such as Th1 and Th17 cells
are critical for a number of autoimmune diseases, including
multiple sclerosis (MS), rheumatoid arthritis, inflammatory bowel
disease, and systemic lupus erythematosus (SLE) (117). SLE presents a reactivation of
the silenced X-chromosome due to CD4+ T cell DNA
demethylation and diet is thought to promote disease progression.
The degree of overexpression of OGT in CD4+ T cells
could potentially contribute to SLE in women (118). Additionally, Th17 cells are
likely the most critical pathogenic factor of human MS. It was
shown that miR-15b suppressed Th17 differentiation and the
pathogenesis of MS by decreasing OGT expression in an NF-κB p65-
and c-Rel-dependent manner (119). Thus, these observations
logically led to the investigation of the link between
O-GlcNAcylation of proteins and T cell activation that are involved
in metabolic and autoimmune diseases. Further characterization of
the role of OGT and O-GlcNAc in autoimmunity may suggest new
therapeutic targets for autoimmune diseases. Collectively, the
recent explosion of research in this field indicates that the
mechanisms by which O-GlcNAcylation regulates immune and
inflammation responses will soon become clear.
Abbreviations:
|
O-GlcNAc
|
O-linked N-acetylglucosamine
|
|
OGT
|
O-linked N-acetylglucosamine
transferase
|
|
OGA
|
O-GlcNAcase
|
|
GlcNAc
|
β-D-N-acetylglucosamine
|
|
UDP-GlcNAc
|
uridine diphosphate
N-acetylglucosamine
|
|
HBP
|
hexosamine biosynthetic pathway
|
|
TPR
|
tetratricopeptide repeat
|
|
EGF
|
epidermal growth factor
|
|
AMPK
|
AMP-activated protein kinase
|
|
HAT
|
histone acetyltransferase
|
|
TCR
|
T cell receptor
|
|
Th
|
T helper
|
|
iTregs
|
inducible regulatory T cells
|
|
IL
|
interleukin
|
|
NF-κB
|
nuclear factor-κB
|
|
IκBα
|
inhibitor of NF-κB α
|
|
LPS
|
lipopolysaccharide
|
|
IKK
|
IκB kinase
|
|
CBP
|
CREB-binding protein
|
|
NFAT
|
nuclear factor of activated T
cells
|
|
TNF-α
|
tumor necrosis factor-α
|
|
TNFAIP3
|
TNF-α induced protein 3
|
|
BCR
|
B cell receptor
|
|
TAK1
|
transforming growth factor-β activated
kinase 1
|
|
VCAM-1
|
vascular cell adhesion molecule-1
|
|
NO
|
nitic oxide
|
|
iNOS
|
inducible nitric oxide synthase
|
|
STAT3
|
signal transducer and activator of
transcription 3
|
|
CUL3
|
cullin 3
|
|
TAB1
|
TAK1-binding protein 1
|
|
GlcN
|
glucosamine
|
|
PUGNAc
|
O-(2-acetamido-2-deoxy-D-glucopyranosylidene) amino-
N-phenylcarbamate
|
|
MCAO
|
middle cerebral artery occlusion
|
|
ICAM-1
|
intercellular adhesion molecule
|
|
MPO
|
myeloperoxidase
|
|
GPX1
|
glutathione peroxidase 1
|
|
Glc
|
Glucose
|
|
Fruc
|
fructose
|
|
Nod2
|
Nucleotide-binding oligomerization
domain 2
|
|
Sp1
|
specificity protein 1
|
Acknowledgments
Not applicable.
Funding
The present review was supported by the National
Natural Science Foundation of China (grant no. 81700726), the
Doctoral Starting Grant of Liaoning Province (grant no.
20170520401) and the Dalian Medical Science Research Program (grant
no. 1712006) to Y.L. The present review was also supported by the
National Natural Science Foundation of China [XM (grant no.
81603428), JD (grant no. 81570727) and LM (grant no.
81700747)].
Availability of data and materials
Not applicable.
Authors' contributions
YL and JD contributed to the conception of the
study, wrote the manuscript, performed the literature search and
prepared the original draft preparation. YL wrote, reviewed and
edited the manuscript. MX and LM performed the literature search
and constructed the figures. All the authors have read and approved
the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ong Q, Han W and Yang) X: O-GlcNAc as an
integrator of signaling pathways. Front Endocrinol (Lausanne).
9:5992018. View Article : Google Scholar
|
|
2
|
D'Hondt C, Iyyathurai J, Vinken M, Rogiers
V, Leybaert L, Himpens B and Bultynck G: Regulation of connexin-
and pannexin-based channels by post-translational modifications.
Biol Cell. 105:373–398. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xie Y, Kang R, Sun X, Zhong M, Huang J,
Klionsky DJ and Tang D: Posttranslational modification of
autophagy-related proteins in macroautophagy. Autophagy. 11:28–45.
2015. View Article : Google Scholar :
|
|
4
|
Schedin-Weiss S, Winblad B and Tjernberg
LO: The role of protein glycosylation in Alzheimer disease. FEBS J.
281:46–62. 2014. View Article : Google Scholar
|
|
5
|
Gurel Z and Sheibani N: O-Linked
β-N-acetylglucosamine (O-GlcNAc) modification: A new pathway to
decode pathogenesis of diabetic retinopathy. Clin Sci (Lond).
132:185–198. 2018. View Article : Google Scholar
|
|
6
|
Hurtado-Guerrero R, Dorfmueller HC and van
Aalten DM: Molecular mechanisms of O-GlcNAcylation. Curr Opin
Struct Biol. 18:551–557. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang X and Qian K: Protein
O-GlcNAcylation: Emerging mechanisms and functions. Nat Rev Mol
Cell Biol. 18:452–465. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Myslicki JP, Belke DD and Shearer J: Role
of O-GlcNAcylation in nutritional sensing, insulin resistance and
in mediating the benefits of exercise. Appl Physiol Nutr Metab.
39:1205–1213. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Levine ZG and Walker S: The biochemistry
of O-GlcNAc transferase: Which functions make it essential in
mammalian cells? Annu Rev Biochem. 85:631–657. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Aquino-Gil M, Pierce A, Perez-Cervera Y,
Zenteno E and Lefebvre T: OGT: A short overview of an enzyme
standing out from usual glycosyltransferases. Biochem Soc Trans.
45:365–370. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ogawa M, Furukawa K and Okajima T:
Extracellular O-linked β-N-acetylglucosamine: Its biology and
relationship to human disease. World J Biol Chem. 5:224–230.
2014.PubMed/NCBI
|
|
12
|
Varshney S and Stanley P: EOGT and
O-GlcNAc on secreted and membrane proteins. Biochem Soc Trans.
45:401–408. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ruan HB, Nie Y and Yang X: Regulation of
protein degradation by O-GlcNAcylation: Crosstalk with
ubiquitination. Mol Cell Proteomics. 12:3489–3497. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhu Y, Liu TW, Cecioni S, Eskandari R,
Zandberg WF and Vocadlo DJ: O-GlcNAc occurs cotranslationally to
stabilize nascent polypeptide chains. Nat Chem Biol. 11:319–325.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Sayat R, Leber B, Grubac V, Wiltshire L
and Persad S: O-GlcNAc-glycosylation of beta-catenin regulates its
nuclear localization and transcriptional activity. Exp Cell Res.
314:2774–2787. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Skorobogatko Y, Landicho A, Chalkley RJ,
Kossenkov AV, Gallo G and Vosseller K: O-linked
β-N-acetylglucosamine (O-GlcNAc) site thr-87 regulates synapsin I
localization to synapses and size of the reserve pool of synaptic
vesicles. J Biol Chem. 289:3602–3612. 2014. View Article : Google Scholar
|
|
17
|
Butkinaree C, Park K and Hart GW: O-linked
beta-N-acetylglu-cosamine (O-GlcNAc): Extensive crosstalk with
phosphorylation to regulate signaling and transcription in response
to nutrients and stress. Biochim Biophys Acta. 1800:96–106. 2010.
View Article : Google Scholar
|
|
18
|
Levine ZG, Fan C, Melicher MS, Orman M,
Benjamin T and Walker S: O-GlcNAc transferase recognizes protein
substrates using an asparagine ladder in the tetratricopeptide
repeat (TPR) superhelix. J Am Chem Soc. 140:3510–3513. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rafie K, Raimi O, Ferenbach AT, Borodkin
VS, Kapuria V and van Aalten DMF: Recognition of a glycosylation
substrate by the O-GlcNAc transferase TPR repeats. Open Biol.
7:1700782017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ma X, Liu P, Yan H, Sun H, Liu X, Zhou F,
Li L, Chen Y, Muthana MM, Chen X, et al: Substrate specificity
provides insights into the sugar donor recognition mechanism of
O-GlcNAc transferase (OGT). PLoS One. 8:e634522013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nagel AK and Ball LE: O-GlcNAc transferase
and O-GlcNAcase: Achieving target substrate specificity. Amino
Acids. 46:2305–2316. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bullen JW, Balsbaugh JL, Chanda D,
Shabanowitz J, Hunt DF, Neumann D and Hart GW: Cross-talk between
two essential nutrient-sensitive enzymes: O-GlcNAc transferase
(OGT) and AMP-activated protein kinase (AMPK). J Biol Chem.
289:10592–10606. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Watson LJ, Long BW, DeMartino AM, Brittian
KR, Readnower RD, Brainard RE, Cummins TD, Annamalai L, Hill BG and
Jones SP: Cardiomyocyte Ogt is essential for postnatal viability.
Am J Physiol Heart Circ Physiol. 306:H142–H153. 2014. View Article : Google Scholar :
|
|
24
|
Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman
M, Janetzko J, Fan C, Duveau DY, Tan ZW, Thomas CJ and Walker S: A
small molecule that inhibits OGT activity in cells. ACS Chem Biol.
10:1392–1397. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gross BJ, Kraybill BC and Walker S:
Discovery of O-GlcNAc transferase inhibitors. J Am Chem Soc.
127:14588–14589. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jiang J, Lazarus MB, Pasquina L, Sliz P
and Walker S: A neutral diphosphate mimic crosslinks the active
site of human O-GlcNAc transferase. Nat Chem Biol. 8:72–77. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Borodkin VS, Schimpl M, Gundogdu M, Rafie
K, Dorfmueller HC, Robinson DA and van Aalten DM: Bisubstrate
UDP-peptide conjugates as human O-GlcNAc transferase inhibitors.
Biochem J. 457:497–502. 2014. View Article : Google Scholar :
|
|
28
|
Trapannone R, Rafie K and van Aalten DM:
O-GlcNAc transferase inhibitors: Current tools and future
challenges. Biochem Soc Trans. 44:88–93. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Ruan HB, Singh JP, Li MD, Wu J and Yang X:
Cracking the O-GlcNAc code in metabolism. Trends Endocrinol Metab.
24:301–309. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hanover JA, Krause MW and Love DC:
Bittersweet memories: Linking metabolism to epigenetics through
O-GlcNAcylation. Nat Rev Mol Cell Biol. 13:312–321. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
He Y, Roth C, Turkenburg JP and Davies GJ:
Three-dimensional structure of a Streptomyces sviceus GNAT
acetyltransferase with similarity to the C-terminal domain of the
human GH84 O-GlcNAcase. Acta Crystallogr D Biol Crystallogr.
70:186–195. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Elsen NL, Patel SB, Ford RE, Hall DL, Hess
F, Kandula H, Kornienko M, Reid J, Selnick H, Shipman JM, et al:
Insights into activity and inhibition from the crystal structure of
human O-GlcNAcase. Nat Chem Biol. 13:613–615. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Keembiyehetty CN, Krzeslak A, Love DC and
Hanover JA: A lipid-droplet-targeted O-GlcNAcase isoform is a key
regulator of the proteasome. J Cell Sci. 124:2851–2860. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Khidekel N, Ficarro SB, Clark PM, Bryan
MC, Swaney DL, Rexach JE, Sun YE, Coon JJ, Peters EC and
Hsieh-Wilson LC: Probing the dynamics of O-GlcNAc glycosylation in
the brain using quantitative proteomics. Nat Chem Biol. 3:339–348.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Crotty S: Follicular helper CD4 T cells
(TFH). Annu Rev Immunol. 29:621–663. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang R, Dillon CP, Shi LZ, Milasta S,
Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger
J and Green DR: The transcription factor Myc controls metabolic
reprogramming upon T lymphocyte activation. Immunity. 35:871–882.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
MacIver NJ, Michalek RD and Rathmell JC:
Metabolic regulation of T lymphocytes. Annu Rev Immunol.
31:259–283. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Machacek M, Slawson C and Fields PE:
O-GlcNAc: A novel regulator of immunometabolism. J Bioenerg
Biomembr. 50:223–229. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Woo CM, Lund PJ, Huang AC, Davis MM,
Bertozzi CR and Pitteri SJ: Mapping and quantification of over
2,000 O-linked glycopeptides in activated human T cells with
isotope-targeted glycoproteomics (IsoTaG). Mol Cell Proteomics.
17:764–775. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lund PJ, Elias JE and Davis MM: Global
analysis of O-GlcNAc glycoproteins in activated human T cells. J
Immunol. 197:3086–3098. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Swamy M, Pathak S, Grzes KM, Damerow S,
Sinclair LV, van Aalten DM and Cantrell DA: Glucose and glutamine
fuel protein O-GlcNAcylation to control T cell self-renewal and
malignancy. Nat Immunol. 17:712–720. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Hao X, Li Y, Wang J, Ma J, Zhao S, Ye X,
He L, Yang J, Gao M, Xiao F and Wei H: Deficient O-GlcNAc
glycosylation impairs regulatory T cell differentiation and notch
signaling in autoimmune hepatitis. Front Immunol. 9:20892018.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ghosh S, May MJ and Kopp EB: NF-kappa B
and Rel proteins: Evolutionarily conserved mediators of immune
responses. Annu Rev Immunol. 16:225–260. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Karin M and Ben-Neriah Y: Phosphorylation
meets ubiquitination: The control of NF-[kappa]B activity. Annu Rev
Immunol. 18:621–663. 2000. View Article : Google Scholar
|
|
45
|
Lecoq L, Raiola L, Chabot PR, Cyr N,
Arseneault G, Legault P and Omichinski JG: Structural
characterization of interactions between transactivation domain 1
of the p65 subunit of NF-κB and transcription regulatory factors.
Nucleic Acids Res. 45:5564–5576. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yi H, Peng R, Zhang LY, Sun Y, Peng HM,
Liu HD, Yu LJ, Li AL, Zhang YJ, Jiang WH and Zhang Z:
LincRNA-Gm4419 knockdown ameliorates NF-κB/NLRP3
inflammasome-mediated inflammation in diabetic nephropathy. Cell
Death Dis. 8:e25832017. View Article : Google Scholar
|
|
47
|
Grundy SM: Overnutrition, ectopic lipid
and the metabolic syndrome. J Investig Med. 64:1082–1086. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Zhang Q, Lenardo MJ and Baltimore D: 30
years of NF-κB: A blossoming of relevance to human pathobiology.
Cell. 168:37–57. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Baker RG, Hayden MS and Ghosh S: NF-κB,
inflammation, and metabolic disease. Cell Metab. 13:11–22. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hayes JB, Sircy LM, Heusinkveld LE, Ding
W, Leander RN, McClelland EE and Nelson DE: Modulation of
macrophage inflammatory nuclear factor κB (NF-κB) signaling by
intracellular cryptococcus neoformans. J Biol Chem.
291:15614–15627. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Esser N, Paquot N and Scheen AJ:
Anti-inflammatory agents to treat or prevent type 2 diabetes,
metabolic syndrome and cardiovascular disease. Expert Opin Investig
Drugs. 24:283–307. 2015. View Article : Google Scholar
|
|
52
|
Golks A, Tran TT, Goetschy JF and Guerini
D: Requirement for O-linked N-acetylglucosaminyltransferase in
lymphocytes activation. EMBO J. 26:4368–4379. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bunting K, Rao S, Hardy K, Woltring D,
Denyer GS, Wang J, Gerondakis S and Shannon MF: Genome-wide
analysis of gene expression in T cells to identify targets of the
NF-kappa B transcription factor c-Rel. J Immunol. 178:7097–7109.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Ramakrishnan P, Clark PM, Mason DE, Peters
EC, Hsieh- Wilson LC and Baltimore D: Activation of the
transcriptional function of the NF-κB protein c-Rel by O-GlcNAc
glycosylation. Sci Signal. 6:ra752013. View Article : Google Scholar
|
|
55
|
Baudoin L and Issad) T: O-GlcNacylation
and inflammation: A vast territory to explore. Front Endocrinol
(Lausanne). 5:2352014.
|
|
56
|
Wu JL, Chiang MF, Hsu PH, Tsai DY, Hung
KH, Wang YH, Angata T and Lin KI: O-GlcNAcylation is required for B
cell homeostasis and antibody responses. Nat Commun. 8:18542017.
View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Zanni MV, Burdo TH, Makimura H, Williams
KC and Grinspoon SK: Relationship between monocyte/macrophage
activation marker soluble CD163 and insulin resistance in obese and
normal-weight subjects. Clin Endocrinol (Oxf). 77:385–390. 2012.
View Article : Google Scholar
|
|
58
|
Nagareddy PR, Kraakman M, Masters SL,
Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES,
Abdel-Latif A, Smyth SS, et al: Adipose tissue macrophages promote
myelopoi-esis and monocytosis in obesity. Cell Metab. 19:821–835.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mauldin JP, Nagelin MH, Wojcik AJ,
Srinivasan S, Skaflen MD, Ayers CR, McNamara CA and Hedrick CC:
Reduced expression of ATP-binding cassette transporter G1 increases
cholesterol accumulation in macrophages of patients with type 2
diabetes mellitus. Circulation. 117:2785–2792. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Westerbacka J, Kolak M, Kiviluoto T,
Arkkila P, Sirén J, Hamsten A, Fisher RM and Yki-Järvinen H: Genes
involved in fatty acid partitioning and binding, lipolysis,
monocyte/macrophage recruitment, and inflammation are overexpressed
in the human fatty liver of insulin-resistant subjects. Diabetes.
56:2759–2765. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Vasamsetti SB, Karnewar S, Kanugula AK,
Thatipalli AR, Kumar JM and Kotamraju S: Metformin inhibits
monocyte-to-macrophage differentiation via AMPK-mediated inhibition
of STAT3 activation: Potential role in atherosclerosis. Diabetes.
64:2028–2041. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Hwang JS, Kwon MY, Kim KH, Lee Y, Lyoo IK,
Kim JE, Oh ES and Han IO: Lipopolysaccharide (LPS)-stimulated iNOS
induction is increased by glucosamine under normal glucose
conditions but is inhibited by glucosamine under high glucose
conditions in macrophage cells. J Biol Chem. 292:1724–1736. 2017.
View Article : Google Scholar :
|
|
63
|
Ryu IH and Do SI: Denitrosylation of
S-nitrosylated OGT is triggered in LPS-stimulated innate immune
response. Biochem Biophys Res Commun. 408:52–57. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Hwang SY, Shin JH, Hwang JS, Kim SY, Shin
JA, Oh ES, Oh S, Kim JB, Lee JK and Han IO: Glucosamine exerts a
neuro-protective effect via suppression of inflammation in rat
brain ischemia/reperfusion injury. Glia. 58:1881–1892. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Hwang JS, Hwang SY and Han IO: Basal
transcription is regulated by lipopolysaccharide and glucosamine
via the regulation of DNA binding of RNA polymerase II in RAW264.7
cells. Life Sci. 110:93–98. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kneass ZT and Marchase RB: Protein
O-GlcNAc modulates motility-associated signaling intermediates in
neutrophils. J Biol Chem. 280:14579–14585. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Kneass ZT and Marchase RB: Neutrophils
exhibit rapid agonist-induced increases in protein-associated
O-GlcNAc. J Biol Chem. 279:45759–45765. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Madsen-Bouterse SA, Xu Y, Petty HR and
Romero R: Quantification of O-GlcNAc protein modification in
neutrophils by flow cytometry. Cytometry A. 73:667–672. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Hart GW, Slawson C, Ramirez-Correa G and
Lagerlof O: Cross talk between O-GlcNAcylation and phosphorylation:
Roles in signaling, transcription, and chronic disease. Annu Rev
Biochem. 80:825–858. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Ma J and Hart GW: O-GlcNAc profiling: From
proteins to proteomes. Clin Proteomics. 11:82014. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Krick S, Helton ES, Hutcheson SB, Blumhof
S, Garth JM, Denson RS, Zaharias RS, Wickham H and Barnes JW: FGF23
Induction of O-linked N-acetylglucosamine regulates IL-6 secretion
in human bronchial epithelial cells. Front Endocrinol (Lausanne).
9:7082018. View Article : Google Scholar
|
|
72
|
Guo X, Shang J, Deng Y, Yuan X, Zhu D and
Liu H: Alterations in left ventricular function during intermittent
hypoxia: Possible involvement of O-GlcNAc protein and MAPK
signaling. Int J Mol Med. 36:150–158. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
James LR, Tang D, Ingram A, Ly H, Thai K,
Cai L and Scholey JW: Flux through the hexosamine pathway is a
determinant of nuclear factor kappaB- dependent promoter
activation. Diabetes. 51:1146–1156. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Dela Justina V, Goncalves JS, de Freitas
RA, Fonseca AD, Volpato GT, Tostes RC, Carneiro FS, Lima VV and
Giachini FR: Increased O-linked N-acetylglucosamine modification of
NF-κB and augmented cytokine production in the placentas from
hyperglycemic rats. Inflammation. 40:1773–1781. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Zhang D, Cai Y, Chen M, Gao L, Shen Y and
Huang Z: OGT-mediated O-GlcNAcylation promotes NF-κB activation and
inflammation in acute pancreatitis. Inflamm Res. 64:943–952. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Li Y, Liu H, Xu QS, Du YG and Xu J:
Chitosan oligosaccharides block LPS-induced O-GlcNAcylation of
NF-κB and endothelial inflammatory response. Carbohydr Polym.
99:568–578. 2014. View Article : Google Scholar
|
|
77
|
Yang WH, Park SY, Nam HW, Kim DH, Kang JG,
Kang ES, Kim YS, Lee HC, Kim KS and Cho JW: NFkappaB activation is
associated with its O-GlcNAcylation state under hyperglycemic
conditions. Proc Natl Acad Sci USA. 105:17345–17350. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
78
|
Allison DF, Wamsley JJ, Kumar M, Li D,
Gray LG, Hart GW, Jones DR and Mayo MW: Modification of RelA by
O-linked N-acetylglucosamine links glucose metabolism to NF-κB
acetylation and transcription. Proc Natl Acad Sci USA.
109:16888–16893. 2012. View Article : Google Scholar
|
|
79
|
Ma Z, Chalkley RJ and Vosseller K:
Hyper-O-GlcNAcylation activates nuclear factor
κ-light-chain-enhancer of activated B cells (NF-κB) signaling
through interplay with phosphorylation and acetylation. J Biol
Chem. 292:9150–9163. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Kawauchi K, Araki K, Tobiume K and Tanaka
N: Loss of p53 enhances catalytic activity of IKKbeta through
O-linked beta-N-acetyl glucosamine modification. Proc Natl Acad Sci
USA. 106:3431–3436. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Donovan K, Alekseev O, Qi X, Cho W and
Azizkhan-Clifford J: O-GlcNAc modification of transcription factor
Sp1 mediates hyperglycemia-induced VEGF-A upregulation in retinal
cells. Invest Ophthalmol Vis Sci. 55:7862–7873. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zhang Y, Qu Y, Niu T, Wang H and Liu K:
O-GlcNAc modification of Sp1 mediates hyperglycaemia-induced ICAM-1
up-regulation in endothelial cells. Biochem Biophys Res Commun.
484:79–84. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
O'Shea JJ and Plenge R: JAK and STAT
signaling molecules in immunoregulation and immune-mediated
disease. Immunity. 36:542–550. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Li X, Zhang Z, Li L, Gong W, Lazenby AJ,
Swanson BJ, Herring LE, Asara JM, Singer JD and Wen H:
Myeloid-derived cullin 3 promotes STAT3 phosphorylation by
inhibiting OGT expression and protects against intestinal
inflammation. J Exp Med. 214:1093–1109. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Pathak S, Borodkin VS, Albarbarawi O,
Campbell DG, Ibrahim A and van Aalten DM: O-GlcNAcylation of TAB1
modulates TAK1-mediated cytokine release. EMBO J. 31:1394–1404.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Hirata Y, Takahashi M, Morishita T,
Noguchi T and Matsuzawa A: Post-translational modifications of the
TAK1-TAB complex. Int J Mol Sci. 18:E2052017. View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Zhang D, Xu Z, Tao T, Liu X, Sun X, Ji Y,
Han L, Qiu H, Zhu G, Shen Y, et al: Modification of TAK1 by
O-linked N-acetylglucosamine facilitates TAK1 activation and
promotes M1 macrophage polarization. Cell Signal. 28:1742–1752.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
88
|
Hou CW, Mohanan V, Zachara NE and Grimes
CL: Identification and biological consequences of the O-GlcNAc
modification of the human innate immune receptor, Nod2.
Glycobiology. 26:13–18. 2016.
|
|
89
|
Xing D, Feng W, Not LG, Miller AP, Zhang
Y, Chen YF, Majid-Hassan E, Chatham JC and Oparil S: Increased
protein O-GlcNAc modification inhibits inflammatory and neointimal
responses to acute endoluminal arterial injury. Am J Physiol Heart
Circ Physiol. 295:H335–H342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Hilgers RH, Xing D, Gong K, Chen YF,
Chatham JC and Oparil S: Acute O-GlcNAcylation prevents
inflammation-induced vascular dysfunction. Am J Physiol Heart Circ
Physiol. 303:H513–H522. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
91
|
Zou L, Yang S, Hu S, Chaudry IH, Marchase
RB and Chatham JC: The protective effects of PUGNAc on cardiac
function after trauma-hemorrhage are mediated via increased protein
O-GlcNAc levels. Shock. 27:402–408. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Zou L, Yang S, Champattanachai V, Hu S,
Chaudry IH, Marchase RB and Chatham JC: Glucosamine improves
cardiac function following trauma-hemorrhage by increased protein
O-GlcNAcylation and attenuation of NF-{kappa}B signaling. Am J
Physiol Heart Circ Physiol. 296:H515–H523. 2009. View Article : Google Scholar
|
|
93
|
Yamamoto Y and Gaynor RB: IkappaB kinases:
Key regulators of the NF-kappaB pathway. Trends Biochem Sci.
29:72–79. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
94
|
Meirow Y and Baniyash M: Immune biomarkers
for chronic inflammation related complications in non-cancerous and
cancerous diseases. Cancer Immunol Immunother. 66:1089–1101. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
95
|
Holmdahl R, Sareila O, Olsson LM, Backdahl
L and Wing K: Ncf1 polymorphism reveals oxidative regulation of
autoimmune chronic inflammation. Immunol Rev. 269:228–247. 2016.
View Article : Google Scholar
|
|
96
|
Pietropaolo M, Barinas-Mitchell E and
Kuller LH: The heterogeneity of diabetes: Unraveling a dispute: Is
systemic inflammation related to islet autoimmunity? Diabetes.
56:1189–1197. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Vaidyanathan K and Wells L: Multiple
tissue-specific roles for the O-GlcNAc post-translational
modification in the induction of and complications arising from
type II diabetes. J Biol Chem. 289:34466–34471. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Xing D, Gong K, Feng W, Nozell SE, Chen
YF, Chatham JC and Oparil S: O-GlcNAc modification of NFκB p65
inhibits TNF-α-induced inflammatory mediator expression in rat
aortic smooth muscle cells. PLoS One. 6:e240212011. View Article : Google Scholar
|
|
99
|
Hirata Y, Nakagawa T, Moriwaki K,
Koubayashi E, Kakimoto K, Takeuchi T, Inoue T, Higuchi K and Asahi
M: Augmented O-GlcNAcylation alleviates inflammation-mediated colon
carcinogenesis via suppression of acute inflammation. J Clin
Biochem Nutr. 62:221–229. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
100
|
Hwang SY, Hwang JS, Kim SY and Han IO:
O-GlcNAcylation and p50/p105 binding of c-Rel are dynamically
regulated by LPS and glucosamine in BV2 microglia cells. Br J
Pharmacol. 169:1551–1560. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
101
|
Zheng GM, Yu C and Yang Z: Puerarin
suppresses production of nitric oxide and inducible nitric oxide
synthase in lipopolysac-charide-induced N9 microglial cells through
regulating MAPK phosphorylation, O-GlcNAcylation and NF-κB
translocation. Int J Oncol. 40:1610–1618. 2012.PubMed/NCBI
|
|
102
|
Hwang SY, Hwang JS, Kim SY and Han IO:
O-GlcNAc transferase inhibits LPS-mediated expression of inducible
nitric oxide synthase through an increased interaction with mSin3A
in RAW264.7 cells. Am J Physiol Cell Physiol. 305:C601–C608. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
103
|
He Y, Ma X, Li D and Hao J: Thiamet G
mediates neuroprotection in experimental stroke by modulating
microglia/macrophage polarization and inhibiting NF-κB p65
signaling. J Cereb Blood Flow Metab. 37:2938–2951. 2017. View Article : Google Scholar
|
|
104
|
Lim K and Chang HI: O-GlcNAc inhibits
interaction between Sp1 and Elf-1 transcription factors. Biochem
Biophys Res Commun. 380:569–574. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
105
|
Lim K and Chang HI: O-GlcNAcylation of Sp1
interrupts Sp1 interaction with NF-Y. Biochem Biophys Res Commun.
382:593–597. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
106
|
Jokela TA, Makkonen KM, Oikari S, Kärnä R,
Koli E, Hart GW, Tammi RH, Carlberg C and Tammi MI: Cellular
content of UDP-N-acetylhexosamines controls hyaluronan synthase 2
expression and correlates with O-linked N-acetylglucosamine
modification of transcription factors YY1 and SP1. J Biol Chem.
286:33632–33640. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
107
|
Lim K and Chang HI: O-GlcNAc inhibits
interaction between Sp1 and sterol regulatory element binding
protein 2. Biochem Biophys Res Commun. 393:314–318. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
108
|
Suh HN, Lee YJ, Kim MO, Ryu JM and Han HJ:
Glucosamine- induced Sp1 O-GlcNAcylation ameliorates
hypoxia-induced SGLT dysfunction in primary cultured renal proximal
tubule cells. J Cell Physiol. 229:1557–1568. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
109
|
Lee HJ, Ryu JM, Jung YH, Lee KH, Kim DI
and Han HJ: Glycerol-3-phosphate acyltransferase-1 upregulation by
O-GlcNAcylation of Sp1 protects against hypoxia-induced mouse
embryonic stem cell apoptosis via mTOR activation. Cell Death Dis.
7:e21582016. View Article : Google Scholar : PubMed/NCBI
|
|
110
|
Coornaert B, Carpentier I and Beyaert R:
A20: Central gatekeeper in inflammation and immunity. J Biol Chem.
284:8217–8221. 2009. View Article : Google Scholar :
|
|
111
|
Yao D, Xu L, Xu O, Li R, Chen M, Shen H,
Zhu H, Zhang F, Yao D, Chen YF, et al: O-Linked
β-N-acetylglucosamine modification of A20 enhances the inhibition
of NF-κB (nuclear factor-κB) activation and elicits vascular
protection after acute endoluminal arterial injury. Arterioscler
Thromb Vasc Biol. 38:1309–1320. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
112
|
Yang WH, Park SY, Ji S, Kang JG, Kim JE,
Song H, Mook-Jung I, Choe KM and Cho JW: O-GlcNAcylation regulates
hyperglycemia-induced GPX1 activation. Biochem Biophys Res Commun.
391:756–761. 2010. View Article : Google Scholar
|
|
113
|
Olivier-Van Stichelen S and Hanover JA:
You are what you eat: O-linked N-acetylglucosamine in disease,
development and epigenetics. Curr Opin Clin Nutr Metab Care.
18:339–345. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
114
|
Someya A, Ikegami T, Sakamoto K and
Nagaoka I: Glucosamine downregulates the IL-1β-induced expression
of proinflammatory cytokine genes in human synovial MH7A cells by
O-GlcNAc modification-dependent and -independent mechanisms. PLoS
One. 11:e01651582016. View Article : Google Scholar
|
|
115
|
Chehimi M, Vidal H and Eljaafari A:
Pathogenic role of IL-17-producing immune cells in obesity, and
related inflammatory diseases. J Clin Med. 6:E682017. View Article : Google Scholar : PubMed/NCBI
|
|
116
|
Ridker PM, Everett BM, Thuren T, MacFadyen
JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker
SD, et al: Antiinflammatory therapy with canakinumab for
atherosclerotic disease. N Engl J Med. 377:1119–1131. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
117
|
Carbo A, Hontecillas R, Kronsteiner B,
Viladomiu M, Pedragosa M, Lu P, Philipson CW, Hoops S, Marathe M,
Eubank S, et al: Systems modeling of molecular mechanisms
controlling cytokine-driven CD4+ T cell differentiation
and phenotype plasticity. PLoS Comput Biol. 9:e10030272013.
View Article : Google Scholar
|
|
118
|
Hewagama A, Gorelik G, Patel D,
Liyanarachchi P, McCune WJ, Somers E, Gonzalez-Rivera T, Michigan
Lupus Cohort, Strickland F and Richardson B: Overexpression of
X-linked genes in T cells from women with lupus. J Autoimmun.
41:60–71. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
119
|
Liu R, Ma X, Chen L, Yang Y, Zeng Y, Gao
J, Jiang W, Zhang F, Li D, Han B, et al: MicroRNA-15b suppresses
Th17 differentiation and is associated with pathogenesis of
multiple sclerosis by targeting O-GlcNAc transferase. J Immunol.
198:2626–2639. 2017. View Article : Google Scholar : PubMed/NCBI
|














