Introduction
The symptoms and signs of ketamine-induced
ulcerative cystitis (KIC) include increased voiding frequency,
non-voiding contraction, hematuria and dysuria. Previous
experimental observations revealed that KIC occurred in bladder
mucosa and decreased bladder capacity, enhanced detrusor
hyperactivity and thereafter induced interstitial fibrosis
(1). The symptoms of KIC are
similar to interstitial cystitis (2,3).
Histological features of ketamine-associated damage in a rat
bladder were characterized by an ulcerated urothelium, erythrocyte
accumulation (hemorrhages), mononuclear cell infiltration and an
increased interstitial fibrosis between detrusor smooth muscle
bundles (1,4-6).
Clinical characteristics of ketamine abusers exhibit urinary
frequency, urgency and at times urinary incontinence. Clinical
studies in bladder tissue of KIC patients have shown increases in
the infiltration of eosinophil and mast cells as well as serum
immunoglobulin (Ig)E level, which displayed an association with
hypersensitivity and/or allergic reactions (7,8).
In spite of the above progress, the pathophysiological mechanism of
the bladder voiding dysfunction in KIC patients is still
unclear.
The mechanism of epigenetic regulation involves the
CpG site methylation of promoter regions and the modification of
DNA and histones by altering chromatin structure (9-11).
DNA meth-ylation represses transcription by interfering with
transcription factor binding and indirectly by recruiting
methyl-CpG-binding proteins and reducing chromatin remodeling
activities (12). Moreover, the
CpG site methylation is mediated by DNA methyltransferases (DNMTs),
which catalyze the addition of a methyl group to cytosine (13,14). The DNMT family enzymes include
DNMT1, DNMT3a and DNMT3b. DNMT1 preserves the methyltransferase by
binding to hemi-methylated CpG sites and methylates the cytosine on
the newly synthesized strand after DNA replication, whereas
DNMT3a/DNMT3b are required for the de novo genomic
methylation of DNA (15). In
contrast, the Ten-Eleven-Translocation (TET) dioxygenase family,
including TET1, TET2 and TET3, mediates active DNA demethylation
and hydroxylate-methylated DNA by converting 5-methylcyto-sine to
5-hydroxymethylcytosine to regulate DNA methylation status. The
role of DNMT and TET proteins in regulating the epigenetic
mechanisms includes DNA methylation at CpG sites and histone
methylation, particularly histone H3 lysine-4 (H3K4) and H3K27
(16). Histone-lysine methylation
is associated with either gene activation or repression depending
on the histone residue modification. For example, methylation of
H3K4 is associated with transcriptional activation, whereas H3K9,
H3K27 and H3K36 are related to transcriptional repression (17-19).
DNA methylation plays a critical role in normal
development, while aberrant hypermethylation of 5′ CpG sites are
implicated in the transcriptional silencing of the cyclooxygenase
(COX)-2 gene in the pathogenesis of the inflammatory diseases
(20-22) and neoplastic disorders (23-25). Two types of DNA methylation
changes are observed in cancer: Hypomethylation, linked to
chromosomal instability and activity (26), and hyper-methylation that can lead
to transcriptional silencing (27). In pathological diseases,
overexpression of COX-2 and abnormal production of COX-induced
prostaglandin E2 (PGE2) have been reported (28). Certain periodontal bacteria can
induce epigenetic alterations in the DNA methylation of the COX-2
gene promoter and affect the transcriptional regulation of COX-2 in
chronic periodontitis (20).
Helicobacter pylori infection causes aberrant DNA
methylation of the COX-2 gene promoter in the gastric mucosa of
patients. Treatment with the DNA-demethylating drug
5-aza-deoxycytidine (a DNA methyl-transferase inhibitor) was found
to increase COX-2 expression and prostaglandin synthesis (29,30).
The COX-2 gene promoter has several potential
response elements for transcription factors, such as nuclear factor
(NF)-κB, nuclear factor of activated T cells/NF-interleukin (IL)-6
(NFAT/NF-IL6) and activator protein-1 (AP-1) (6,31).
Inflammatory stimuli cause NF-κB dimers (p65 and p50 subunits) to
dissociate from cytoplasmic inhibitors, followed by NF-κB p65
translocation and binding to specific gene promoter sequences
(32,33). An animal study with
chemically-induced hemorrhagic cystitis revealed that COX-2
upregulation played an important role in bladder inflammation
(34). Moreover, the authors'
previous results demonstrated that ketamine and norketamine
accelerated NF-κB p65 translocation and induced the upregulation of
COX-2 expression in bladder urothelium (6). Promoter-deletion analysis of the rat
COX-2 promoter region ranging from −918 to −250 bp suggested that
NF-κB was a crucial transcription factor for COX-2 gene activation
(6). However, promoter deletion
analysis did not provide any conclusion with respect to which
specific binding sites for NF-κB were involved in the COX-2
modification response to ketamine and metabolites norketamine.
In the present study, specific binding sequences
(sites) of the COX-2 promoter responding to NF-κB were identified
by focusing on the promoter ranging from -1,522 to -71 bp. The
authors hypothesized that ketamine-induced chronic inflammation is
associated with an altered DNA methylation level within the COX-2
promoter and with change in transcriptional modification. To test
this hypothesis, the potential alteration in DNA methylation status
of the COX-2 promoter via NF-κB activation and its effect on the
transcriptional COX-2 expression in KIC were investigated.
Moreover, to improve an understanding of the epigenetic regulation
of the COX-2 gene via NF-κB activation, it was important to
determine specific NF-κB binding sequences within the COX-2
promoter and to investigate methylation associated enzymes
responsible for the promoter COX-2 activity.
Materials and methods
Animals and ketamine administration
A total of 45 adult female Sprague-Dawley (S-D) rats
weighing 250 g were purchased from the Animal Center of BioLASCO.
All S-D rats were housed in a standard room with constant
temperature (23±2°C), a relative humidity of 70%, and a 12 h
light-dark cycle (light on at 07:00 a.m., light off at 07:00 p.m.;
100 lux at cage level) conditions with ad libitum access to
food and water. S-D rats were divided into three experimental
groups: i) The control (saline) group, ii) the ketamine-treated
group and iii) the ketamine combined with COX-2 inhibitor
(ketamine+COX-2 inhibitor) group, which received 0.9% saline,
ketamine (30 mg/kg/day, Pfizer, Inc.) intraperitoneal (IP)
injection, and ketamine combined with COX-2 inhibitor (Parecoxib
sodium, Dynastat®, Pfizer, Inc.; 10 mg/kg/day) IP
injection for 3 months respectively (4-6).
At the end of the experimental period, bladder tissues and whole
blood were collected from all animals. After induction of
anesthesia with 4% isoflurane, rats were subjected to cardiac
puncture or euthanasia and total blood collection. Blood samples
were left to clot for 2 h on the ice, then centrifuged at 1,500 × g
at 4°C for 15 min to separate out the serum samples which were
preserved at −80°C in the refrigerator for further detection. S-D
rats were perfused with 09% saline solution through the left
ventricle and the bladders were removed and washed in ice-cold PBS
and carefully dissected in a horizontal plane into two portions:
One was fixed in 4% paraformaldehyde solution at 4°C for 24 h for
histological analysis; another was placed in liquid nitrogen for
mRNA, protein and chromatin immunoprecipitation (ChIP) analysis.
This study was approved by the Animal Care and Treatment Committee
of Kaohsiung Medical University. All experiments were conducted
according to the guidelines for laboratory animal care.
Western blot analysis
Nuclear and cytoplasmic extracts of rat bladder
tissues were prepared according to the protocols modified from the
method described by NE-PER nuclear and cytoplasmic extraction
reagents (Thermo Fisher Scientific, Inc.) (6). The protein concentration was
determined using Bicinchoninic Acid Protein Assay kits (Pierce
Biotechnology; Thermo Fisher Scientific, Inc.). A total of 50
µg of protein from the bladders were loaded on a 10% SDS
polyacrylamide electrophoresis gel and transferred to PVDF
membranes (Immobilon-P; EMD Millipore). Afterward, the PVDF
membrane was blocked with 5% non-fat-milk in PBS with Tween-20
(PBST) for 2 h at room temperature. Primary antibodies COX-2 (cat.
no. 160126; Cayman Chemical Company; rabbit IgG, 1:1,000; MW, 72
kDa) and NF-κB p65 (cat. no. NBP1-48427; Novus Biologicals LLC;
rabbit IgG; 1:1,000; MW, 65 kDa) were used for determining protein
expression at 4°C overnight, and were incubated with the
horseradish peroxidase-conjugated goat anti-mouse (cat. no.
115-035-003; Jackson ImmunoResearch Laboratories;
1:7,000-1:100,000) and goat anti-rabbit (cat. no. 111-035-003;
Jackson ImmunoResearch Laboratories; 1:7,000) secondary antibody
for 1 h at room temperature. Lamin A/C (cat. no. 2032S; Cell
Signaling Technology, Inc.; mouse IgG; 1:5,000; MW, 70 kDa) served
as the internal control for nuclei extract and β-actin (cat. no.
MAB1501; Upstate Biotechnology, Inc.; mouse IgG; 1:5,000; MW, 41-43
kDa) served as the internal control for total protein and
cytoplasmic extract. The band intensity was quantified by
densitometry using image analysis software (ImageJ, version 1.49;
National Institutes of Health). In each experiment, negative
control without the primary antibody was analyzed.
ELISA
The level of PGE2 production was used to serve as
determining COX activity. Determination of PGE2 levels in serum was
performed by Prostaglandin E2 ELISA kit following the
manufacturer's protocol (Abcam; cat. no. ab133021). A mouse IgG
antibody was precoated onto 96-well plates. Serum samples were
added to the wells, along with an alkaline phosphatase (AP)
conjugated-PGE2 antibody. After reagent incubation, substrate was
added and catalyzed by AP to produce a yellow color. The intensity
of the yellow coloration was inversely proportional to the amount
of PGE2.
Immunofluorescence
For in vivo bladder section, immunostaining
was performed according to published methods (6). The sections were then double-stained
with the primary antibody to NF-κB p65 (cat. no. 6956S; Cell
Signaling Technology, Inc.; mouse IgG2b; 1:50-100) and COX-2 (cat.
no. 4212-1; Epitomics; rabbit IgG; 1:50) at 4°C overnight, then
incubated with goat anti-mouse IgG Alexa Fluor 568 (cat. no.
A-11004; Invitrogen; Thermo Fisher Scientific, Inc.; 1:800) and
goat anti-rabbit IgG Alexa Fluor 488 secondary antibody (cat. no.
A-27034; Invitrogen; Thermo Fisher Scientific, Inc.; 1:800)
conjugated to fluorescein isothiocyanate (FITC) for NF-κB and
rhodamine for COX-2 for 1 h at room temperature. The nuclei of the
cells were counterstained with 4′,6′-diamidino-2-phenylindole
(DAPI; 1:5,000) for 10 min at room temperature. Immunofluorescence
images were observed using a Leica DMI6000 inverted microscope
(Leica Microsystems, Inc.).
Analysis and design of rat COX-2
promoter
The DNA sequence of COX-2 promoter was analyzed with
MethPrimer software (version 2.0) (35). The promoter regions of the rat
COX-2 gene (accession number NM_017232) ranging from −1,522 to −71
contains 1,452 bp (nt) and 44 CpG sites were identified by
nucleotide sequence analysis for potential methylation (www.genome.ucsc.edu). In this region, a COX-2 promoter
construct was designed for five subregions, including Region I
(−71/−299), II (−286/−551), III (−548/−830), IV (−829/−1,195) and V
(−1,181/−1,522), for bisulfite methylation-specific PCR (MSP),
cloning, and genomic sequencing. Additionally, five potential
motifs similar to the consensus binding sites for NF-κB in the
region were identified by TFBIND software (version 8.3; http://tfbind.hgc.jp/; Table I).
 | Table IPrimer sequences used in
methylation-specific polymerase chain reaction, bisulfite DNA
sequencing, ChIP assay and RT-qPCR. |
Table I
Primer sequences used in
methylation-specific polymerase chain reaction, bisulfite DNA
sequencing, ChIP assay and RT-qPCR.
A, Primers for
methylation-specific transcription PCR
|
|---|
| Name | Primer
sequences | Sequence range | Product | Methylated NF-κB
binding sequence |
|---|
Rat-COX2 region
I
Bisulfite-sequence | F:
5′-TTGTTTTTATGGGTATTATGTAATTGG-3′
R: 5′-ACAAAACACAAAACTAAATTCCTTC-3′ | −299 to −71 | 229 bp | 5′-ggggaaagtcga-3′
(−235 to −224) |
Rat-COX2 region
II
Bisulfite-sequence | F:
5′-AAGGGGATTTTTTTAGTTAGGATTT-3′
R: 5′-ACCCATAAAAACAAACTTTACTCAC-3′ | −551 to −286 | 266 bp | 5′-ggggattttt-3′
(-549 to -540) |
Rat-COX2 region
III
Bisulfite-sequence | F:
5′-TAGGGAGGAAAATATTTTAAAGTAATG-3′
R: 5′-CCTTACCTCTCCCCACTAAAAC-3′ | −830 to −548 | 283 bp | 5′-ggaaaatattt-3′
(−824 to −814) |
Rat-COX2 region
IV
Bisulfite-sequence | F:
5′-AGTTTTTTATTTTTTTGTTTTATT-3′
R: 5′-TACTATTACATAACTTTTATCATTTTAATC-3′ | −1,195 to −829 | 367 bp | - |
Rat-COX2 region
V
Bisulfite-sequence | F:
5′-AGTATGTATATGAAGTAAATAGTTAAAAA-3′
R: 5′-AAAAATAAAAAAACTAAAACATTCAATTAA-3 | −1,522 to
−1,181 | 342 bp | 5′-gtcgattttt-3′
(−1,413 to −1,404)
5′-cggtagttttc-3′ (−1,442 to −1,432) |
|
COX2-Methylation | F:
5′-AAGGGGATTTTTTTAGTTAGGATTTC-3′
R: 5′-TCCAAACGCCCTATAATTCG-3′ | −551 to −393 | 159 bp | |
|
COX2-Unmethylation | F:
5′-AGGGGATTTTTTTAGTTAGGATTTT-3′
R: 5′-TTCCAAACACCCTATAATTCACT-3′ | −550 to −392 | 159 bp | |
B, ChIP primers for NF-κB binding sites of Cox-2 promoter
sequence
|
| Name | Primer
sequences | Sequence range | Product | Predicted NF-κB
binding sequence |
|
| Rat-COX2 -Primer
I | F:
5′-TGCCCCTATGGGTATTATGC-3′
R: 5′-CTGAAGCTCTCCGCTCAGTT-3′ | −298 to −117 | 182 bp | 5′-ggggaaagccga-3′
(−235 to −224) |
| Rat-COX2 -Primer
II | F:
5′-GACAGCAGCCCTCTCATTTC-3′
R: 5′-CGGAGGAGCAAGAGAATGTC-3′ | −660 to −484 | 177 bp | 5′-ggggattccc-3′
(−549 to −540) |
| Rat-COX2 -Primer
III | F:
5′-TGTAAACGTAAACGTGGACAAAA-3′
R: 5′-CCTTTCCCAGAGACAGATGC-3′ | −948 to −751 | 198 bp | 5′-ggaaaatacct-3′
(−824 to −814) |
| Rat-COX2 -Primer
IV | F:
5′-AGCATGCACATGAAGCAAAC-3′
R: 5′-GCCCTGCTCAAAAGAAAACA-3′ | −1,522 to
−1,331 | 192 bp | 5′-gtcgattccc-3′
(−1,413 to −1,404)
5′-cggtagtttcc-3′ (−1,442 to −1,432) |
C, RT-qPCR primers for DNMT and TET methylcytosine dioxygenase
|
| Gene | Primer
sequences | Accession
no.a | Tm (°C) | Product size
(bp) |
|
| Rat-DNMT1 | F:
5′-GGAGGTGTCCTAACTTGGC-3′
R: 5′-GGGTGACGGCAACTCTGGTA-3′ | NM_053354.3 | 60 | 80 |
| Rat-DNMT3a | F:
5′-GTGCTTACCAATACGATGACGA-3′
R: 5′-ATCCACACACTCCACACAAAAG-3′ | NM_001003958.1 | 60 | 122 |
| Rat-DNMT3b | F:
5′-GATGATGGAGATGGCTCTGATA-3′
R: 5′-GGCTGGAGATACTGTTGCTGTT-3′ | NM_001003959.1 | 60 | 121 |
| Rat-TET1 | F:
5′-GAAACCCTGAATTGGCAAAA-3′
R: 5′-GGGTGAGCTTTCTGATCGAC-3′ | XM_017601794.1 | 60 | 157 |
| Rat-TET2 | F:
5′-TCGGAGGAGAAGAGTCAGGA-3′
R: 5′-TAGGGCTTGCATTTTCCATC-3′ | XM_006224264.3 | 60 | 167 |
| Rat-TET3 | F:
5′-ATGGCATGAAACCACCCAAC-3′
R: 5′-ACTTGATCTTCCCCTCCAGC-3′ | XM_008763094.2 | 60 | 81 |
MSP, cloning and genomic sequencing
The DNA methylation pattern in the COX-2 promoter
was analyzed by MSP. Genomic DNAs from rat bladder tissues were
isolated using the PureLink Genomic DNA Mini kit (Invitrogen;
Thermo Fisher Scientific, Inc.). Total extracted DNA was used for
bisulfite-mediated conversion of unmethylated cytosines to uracils
using an EpiTect Bisulfite kit (Qiagen, Inc.). Bisulfite-treated
genomic DNA was amplified with primers (Table I) specific to the CpG sites within
COX-2 promoter sequence. After bisulfite treatment, unmethylated
cytosine residues were changed into uracil residues, whereas
methylated cytosine remained unmodified. The differentiation
between methylated and unmethylated sequences could be amplified
using specific primers that targeted the uracil or the cytosine
nucleotide. COX-2 promoter temperature profiles for amplification
were 5 min at 95°C, 30 cycles of 45 sec at 94°C, 45 sec at 63°C and
45 sec at 72°C. All reactions were repeated three times to ensure
reproducibility of results. Colonies showing positive PCR fragment
insertion were selected and the purified fragments were cloned into
a PCR 2.1 vector (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. PCR products from 4
clones for each individual sample were sequenced. The completeness
of the conversion of unmethylated cytosines to uracils was
confirmed by evaluation of non-CpG cytosine conversion. The DNA
methylation patterns were analyzed by sequencing.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated from bladder tissues with the
use of the RNeasy Mini kit (Qiagen, Inc.). The cDNA was then
synthesized from 1 µg of total RNA using the Omniscript kit
(Qiagen, Inc.) by random decamer primers (Applied
Biosystems/Ambion; Thermo Fisher Scientific, Inc.). The reaction
was incubated at 37°C for 60 min, inactivated by heating at 95°C
for 5 min, and stored at -20°C. An SYBR-Green I kit (Takara
Biotechnology, Co., Ltd.) was used and all the primers of rat DNA
methyltransferase (DNMT1, 3a, and 3b) and TET enzymes (TET1, TET2,
and TET3) were listed in Table I.
RT-qPCR was performed in a 7500 Sequence Detection System apparatus
(Applied Biosystems; Thermo Fisher Scientific, Inc.) using the
following thermocycling conditions: Initial denaturation at 95°C
for 10 min, followed by 40 cycles of 95°C for 15 sec and 60°C for 1
min. The relative expression levels of each targeted gene were
normalized by subtracting the corresponding β-actin threshold cycle
(Cq) values using the ΔΔCq comparative method (36). Six samples for each group were
used and run in triplicate.
ChIP
A total of 70 mg bladder tissues were homogenized
into small pieces and treated with 1% formaldehyde for 10 min at
37°C, followed by sonication of DNAs. ChIP was performed with
primary antibodies for anti-Histone H3K4 (tri methyl K4; cat. no.
AB8580; Abcam; rabbit polyclonal IgG; 1:100), H3K9 (cat. no.
AB8898; Abcam; rabbit polyclonal IgG; 1:100), H3K27 (cat. no.
AB6002; Abcam; mouse monoclonal IgG; 1:100), H3K36 (cat. no.
AB9050; Abcam; rabbit polyclonal IgG; 1:100) and H3K79 (cat. no.
AB2621; Abcam; rabbit poly-clonal IgG; 1:100), NF-κB p65 (cat. no.
6956S; Cell Signaling Technology, Inc.; mouse monoclonal IgG2b;
1:100), and IgG (cat. no. 6180-01; negative control;
SouthernBiotech; rabbit IgG; 1:500) overnight. Immune complexes
were collected using a protein G agarose, Fast flow (50% slurry;
EMD Millipore) and the DNA was reverse cross-linked, extracted, and
quantified on a Taqman SDS 7900HT. All the primers were listed in
Table I. PCR conditions were as
follows: Initial denaturation at 95°C for 5 min followed by 30
cycles of denaturation at 94°C for 45 sec, annealing at 63°C for 45
sec and extension at 72°C for 45 sec; final extension at 72°C; and
holding at 4°C. PCRs run on the ABI 7700 Taqman thermocycler. All
Taqman reagents were purchased from Applied Biosystems; Thermo
Fisher Scientific, Inc. The relative intensities of the amplified
products were normalized according to the input DNAs.
Statistical analysis
Each experiment was carried out at least three times
independently. Data were expressed as the mean ± standard deviation
and subjected to one-way analysis of variance to find the
significant difference in various parameters between the control
and the treated groups. The post hoc Tukey honest significant
difference tests were used to make comparisons between the control
and each of the treated groups and to calculate P-values for
comparison, P<0.05 was considered to indicate a statistically
significant difference. The statistical analyses were implemented
using the IBM SPSS Statistics package (version 21.0; IBM,
Corps.).
Results
Increases in COX-2 expression and NF-κB
p65 translocation after ketamine treatment
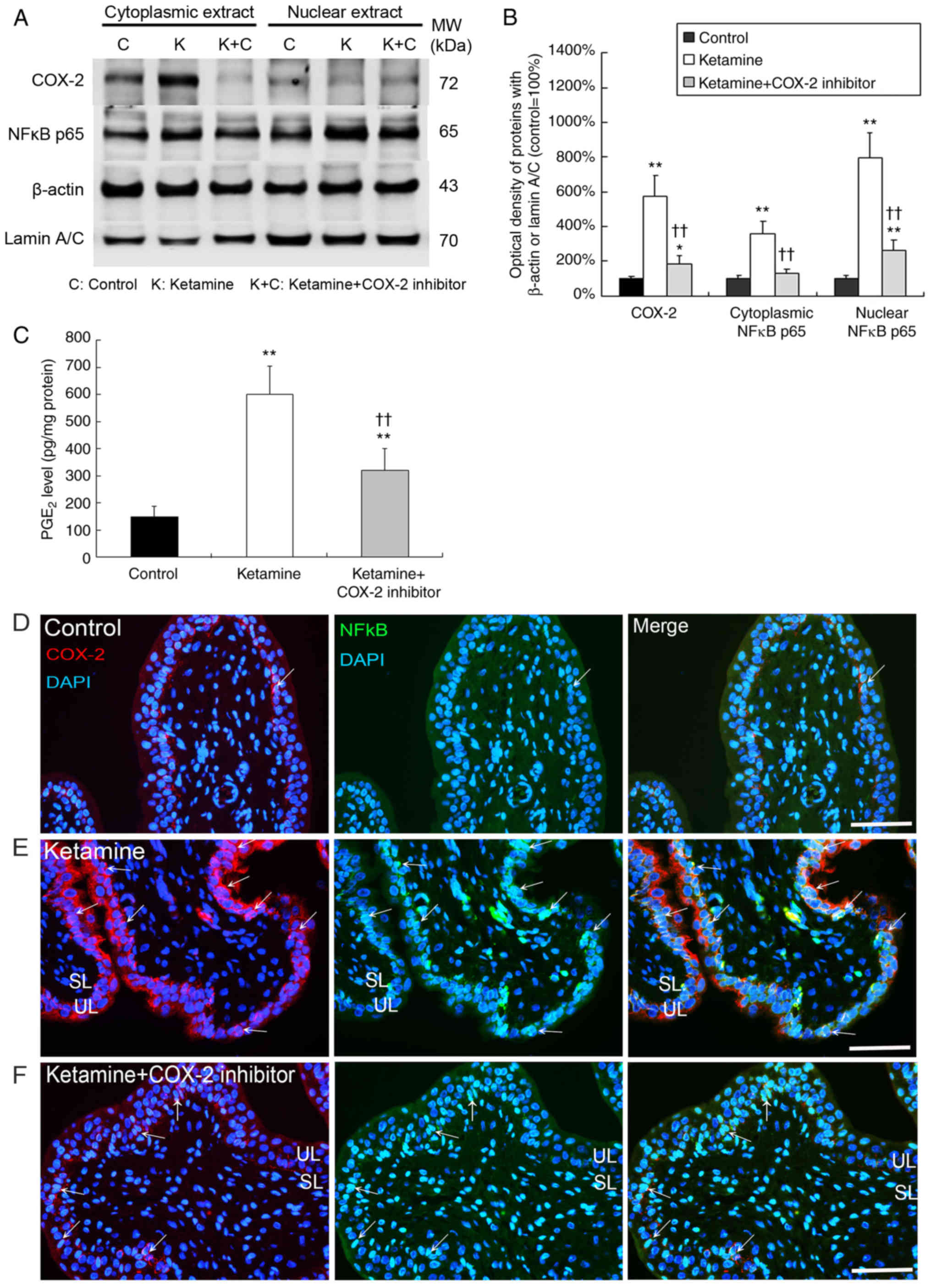
In Fig. 1A and B,
western blot analysis showed that the expression of COX-2,
cytoplasmic NF-κB p65 and nuclear NF-κB p65 was significantly
increased by 5.8-, 3.7- and 8.0-fold in the bladder tissue of the
ketamine groups respectively, as compared with the control group
(P<0.01). In the ketamine+COX-2 inhibitor group, the expression
of COX-2 and nuclear NF-κB p65 was significantly increased by 2.0-
and 2.9-fold respectively, compared with the control group
(P<0.05). These data indicated that the ketamine+COX-2 inhibitor
group resulted in more significant decline in NF-κB p65 expression
in nucleus and cytoplasm compared with the ketamine treatment
group.
In Fig. 1C, PGE2
production in the control group was 150 pg/mg, which was
significantly increased by 3.9- and 2.1-fold in the ketamine and
the ketamine+COX-2 inhibitor groups respectively (P<0.01). These
findings demonstrated that ketamine treatment induced NF-κB p65
translocation and activated COX-2 expression and PGE2 production in
bladder tissue. Whereas the COX-2 inhibitor suppressed the level of
NF-κB p65 translocation and COX-2 expression as well as PGE2
production.
Double-staining for the distribution of COX-2 (red)
and NF-κB p65 (green) proteins was performed. In the control group,
weak staining for the co-labeling of NF-κB p65 and COX-2 was found
(Fig. 1D). However, in the
ketamine group, most NF-κB p65/DAPI cells co-stained with COX-2
expression were enhanced and restricted to the thinner and
disrupted urothelium (Fig. 1E;
arrows). In the ketamine+COX-2 inhibitor group, double-staining of
NF-κB p65 and COX-2 was decreased, as compared to the ketamine
group (Fig. 1F; arrows). These
observations revealed that COX-2 expression coincided with NF-κB
p65 after ketamine treatment, implying that COX-2 and NF-κB p65
were synthesized during the inflammatory process of KIC, whereas
COX-2 inhibitor reduced NF-κB p65 translocation and COX-2
expression.
Methylation sequencing analysis for the
upstream sequences of COX-2 promoter region
The present study also explored whether NF-κB
translocation after ketamine treatment was associated with COX-2
hypomethylation and transcriptional modification in KIC. To
determine which NF-κB binding sites within COX-2 promoter were
involved in COX-2 activation and DNA methylation in KIC, the
methylation level of CpG sites within the COX-2 promoter region
ranging from -1,522 to -71 bp in relation to the transcriptional
starting site was analyzed by bisulfite MSP and genomic sequencing
analysis. A schematic diagram for COX-2 promoter spanning the
region with respect to the ATG start site (+1) is shown in Fig. 2A. Locations of NF-κB binding
sequences within the region for methylation analysis were denoted
in Fig. 2A and Table I. According to rat COX-2 gene
(accession number NM_017232) sequence analysis, bisulfite-COX-2
sequence in the promoter had a size of 1,452 bp and contained 44
CpG sites, as identified by nucleotide sequence analysis for
potential methylation (Fig. 2B
and Table I). The bisulfite
genomic sequence located from -1,522 to -71 bp was denoted as
regions I-V and designed to amplify with the primers that were
specific to the CpG site within COX-2 promoter. The bisulfite
sequencing chromatogram of the methylated cytosine of these CpG
sites for bladder tissue is presented in Fig. 2C.
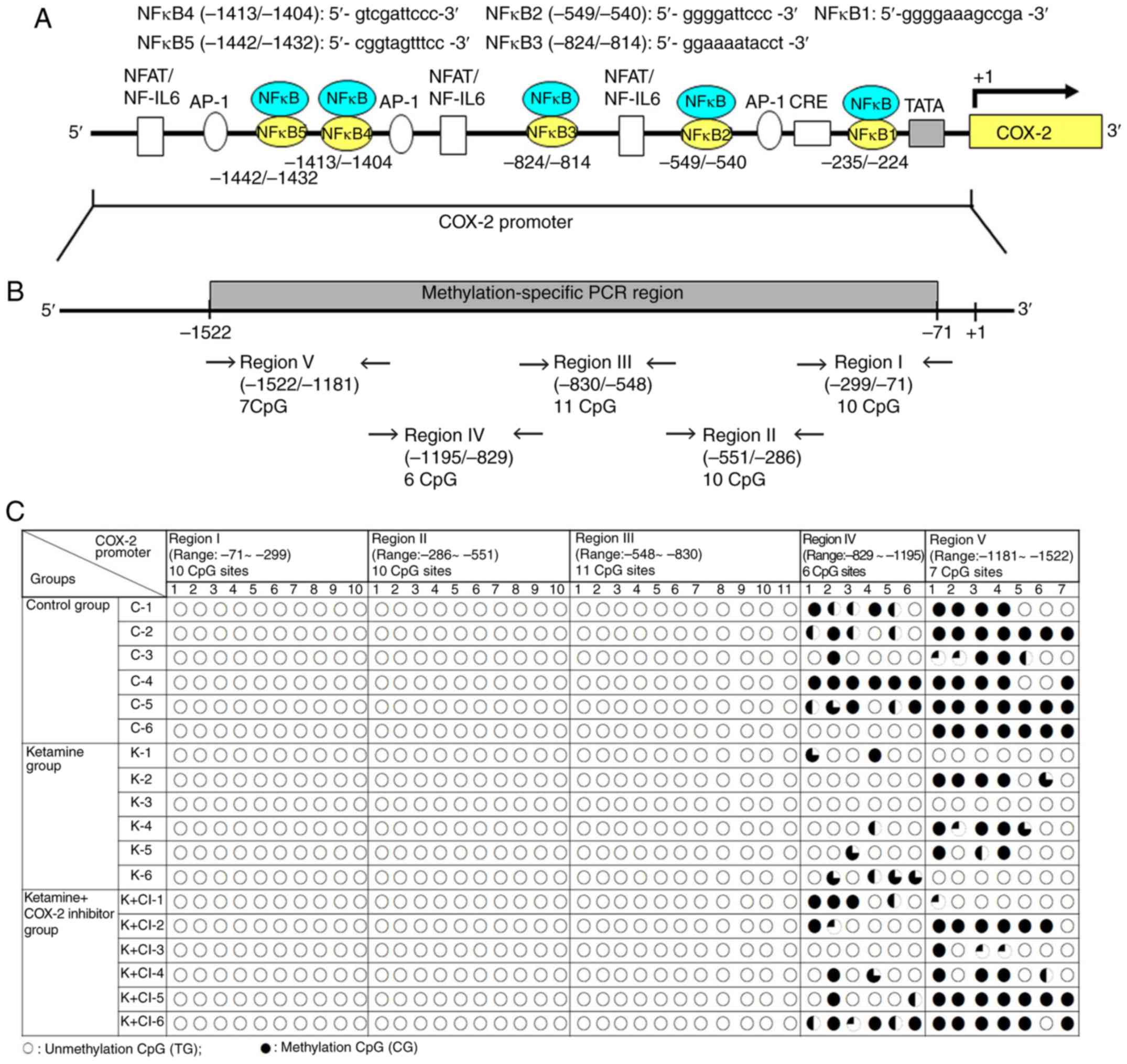 | Figure 2Methylation status of the CpG site in
the COX-2 promoter region in ketamine-induced ulcerative cystitis.
(A) Schematic diagram of COX-2 promoter spanning the region from
-1,522 to -71 bp with respect to the ATG start site (+1)
(translation initiation site) and targeted sites for
methylation-specific PCR region and bisulfite genomic sequencing
analysis are shown. Additionally, five putative NF-κB DNA binding
sites in the region were identified by TFBIND software (http://tfbind.hgc.jp/). The broad arrow indicated the
direction of transcription. A total of five putative NF-κB binding
sequences for methylation analysis are denoted. (B) The
bisulfite-COX-2 sequence in the promoter had a size of 1,452 bp and
contained 44 CpG sites as identified by nucleotide sequence
analysis for potential methylation (http://www.genome.ucsc.edu). There were 10 CpG sites
in the region I fragment of COX-2 promoter ranging from −299 to −71
bp, 10 CpG sites in the region II ranging from −551 to −286 bp, 11
CpG sites in the region III ranging from -830 to -548 bp, 6 CpG
sites in the region IV ranging from -1,195 to -829 bp and 7 CpG
sites in the region V ranging from -1,522 to -1,181 bp for
methylation sequencing analysis. A total of five sets of primers
were designed to amplify the bisulfite genomic sequence. (C)
Bisulfite sequencing chromatogram of CpG sites in bladder tissue.
Each circle denoted as one CpG site and represented the methylation
status of the CpGs. The clone was sequenced from PCR products
generated from amplification of bisulfite-treated DNA. The
percentage of methylation at a single CpG site was calculated from
the sequencing results of four independent clones. ○, unmethylated
cytosines; •, methylated cytosines. NF, nuclear factor; IL,
interleukin; NFAT/NF-IL6, nuclear factor of activated T
cells/NF-IL-6; AP-1, activator protein 1; CRE, cAMP-response
element; SP-1, specificity protein 1; TATA, TATA box motif; COX,
cyclooxygenase. |
The present study's data showed DNA unmethylation
(white circles) in regions I-III of COX-2 promoter fragments in
three experimental groups. However, in regions IV and V, the CpG
site methylation in the control group, and hypomethylation in the
ketamine and the ketamine+COX-2 group was found. Although the
cloned sequences exhibited considerable heterogeneity in
methylation, it was also demonstrated that bisulfite MSP and
genomic sequencing data displayed the hypomethylation at the CpG
sites in the ketamine and the ketamine+COX-2 inhibitor groups.
Methylation analysis of rat COX-2
promoter ranging from −830 to −71 bp by methylation-specific PCR
and bisulfite genomic sequencing
Schematic diagrams of the promoter fragment region I
ranging from −299 to −71 bp and region II ranging from −551 to −286
bp in the COX-2 promoter are presented in Figs. S1A and S2A. Both regions I and II contained 10
methylatable CpG sites marked with red numbers. A total of two
potential NF-κB binding sequences spanning from −235 to −224 and
−549 to −540 bp were marked with a square box (Figs. S1B and S2B). Sequence traces were obtained from
the PCR products from bisulfite-treated DNA using primers (blue
color in bisulfate sequence) and detailed normal and bisulfite
genomic sequences of regions I and II were presented (Figs. S1C and D, S2C and D). Consistent with the data, it
was found that the CpG sites displayed unmethylation at the CpG
sites in the control, the ketamine and the ketamine+COX-2
groups.
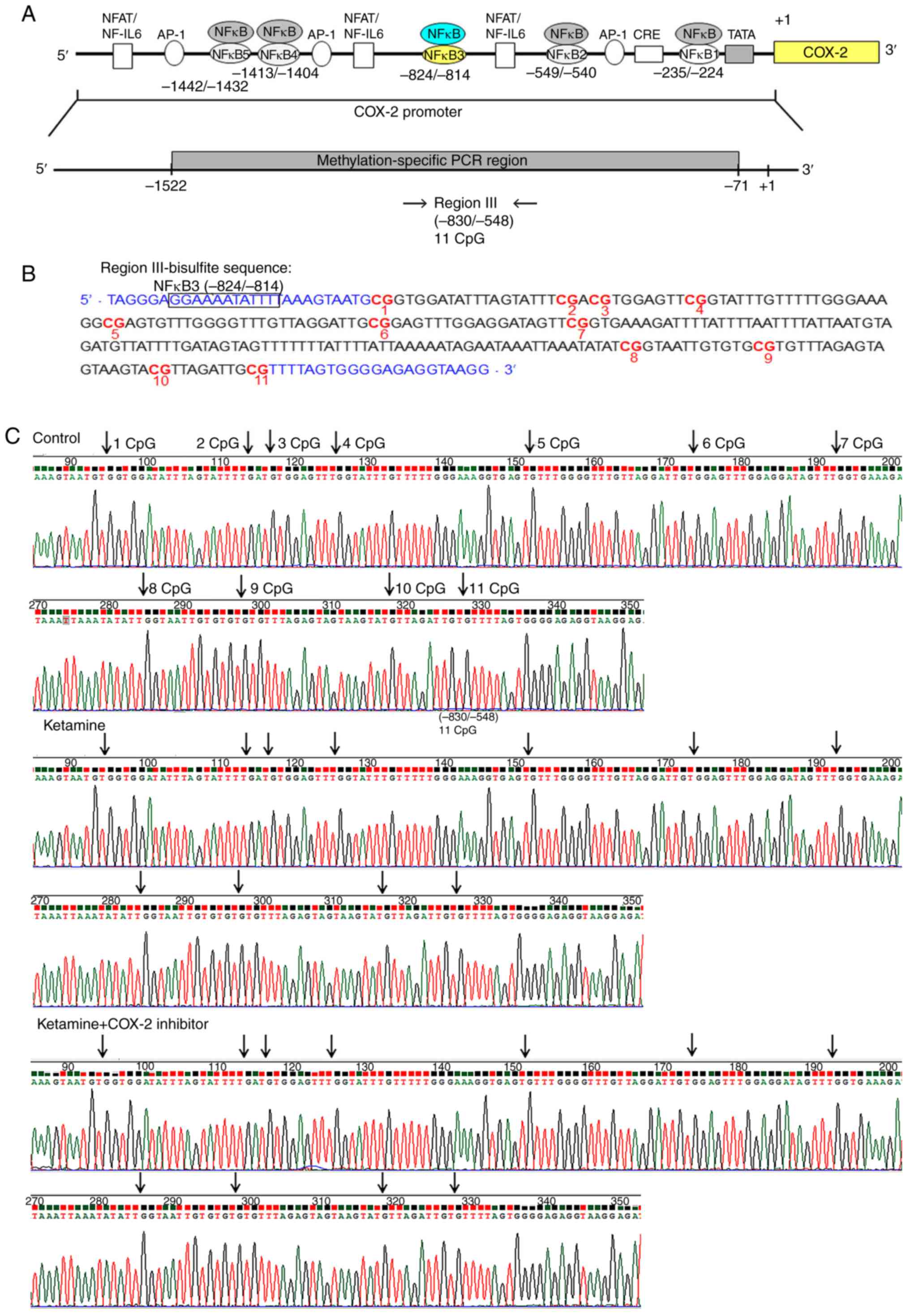
A schematic diagram of the fragment region III
spanning from −830 to −548 bp containing 11 methylated CpG sites
and the potential NF-κB binding sequence spanning from −824 to −814
bp was marked with square box. Bisulfite methylated NFκB3 (mNFκB3)
sequence was located at −824 to −814 bp, 5′-GGA AAA TAT TT-3′.
Sequence traces were obtained from the PCR products from
bisulfite-treated DNA (Fig. 3A and
B), and the CpG sites were highlighted as black arrows
(Fig. 3C). Detailed normal and
bisulfite genomic sequences of region III in COX-2 promoter were
presented (Fig. S3). Based on
the above findings, the control, the ketamine and the
ketamine+COX-2 inhibitor groups all displayed unmethylation at the
CpG sites in regions I-III ranging from −830 to −71 bp.
Ketamine treatment caused hypomethylation
of the COX-2 promoter ranging from −1,195 to −829 bp
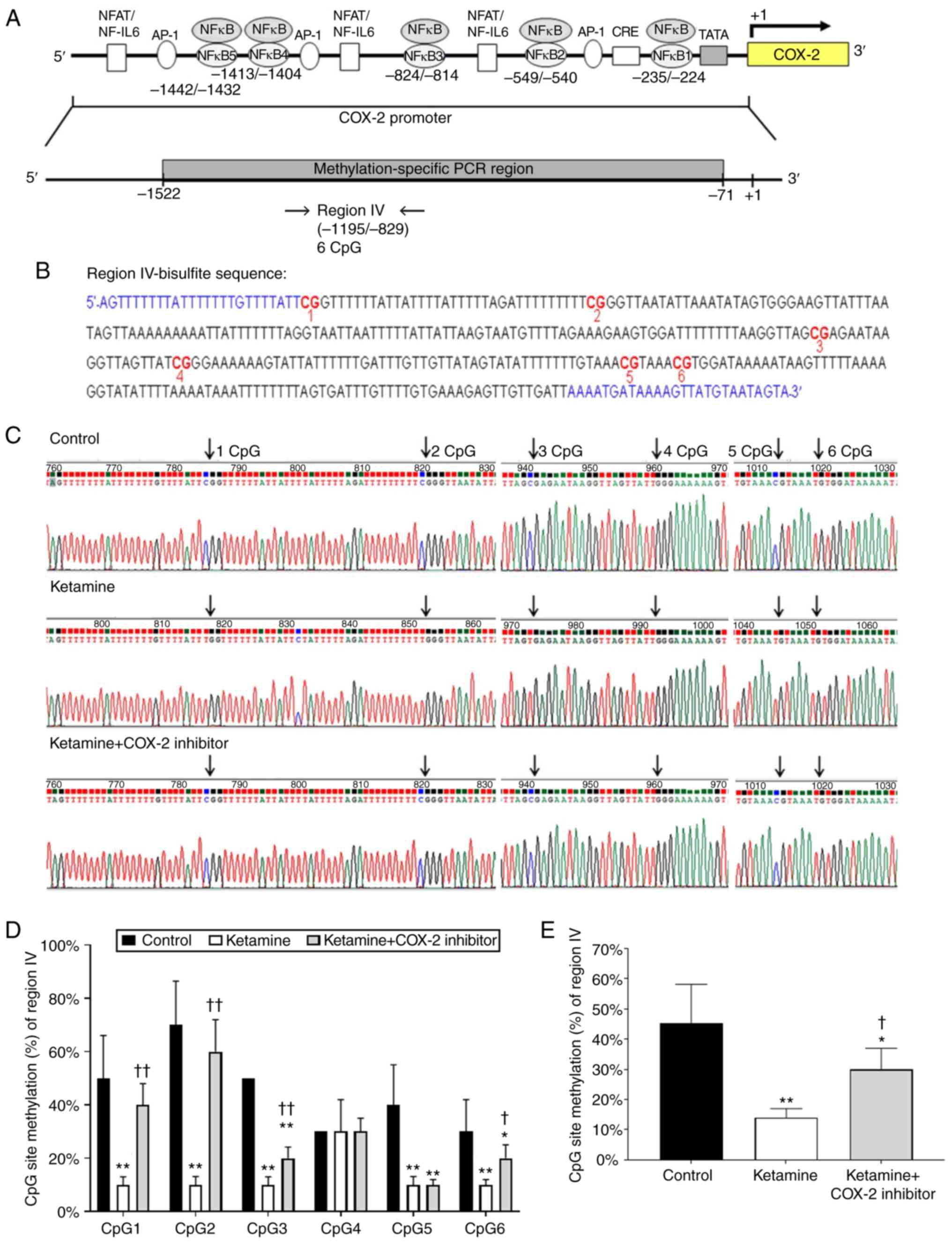
Diagrammatic presentation of the region IV ranging
from −1,195 to −829 bp is shown in Fig. 4A. The promoter fragment region IV
contained 6 methylatable CpG sites marked with red numbers
(Fig. 4B). There was no potential
sequence for NF-κB binding site in region IV fragment. Sequence
traces were obtained from the PCR products of bisulfite-treated DNA
using primers marked with blue color in bisulfite sequence
(Fig. 4B) and the CpG sites were
highlighted as black arrows in Fig.
4C. Detailed normal and bisulfite genomic sequences of region
IV were shown in Fig. S4. The
association between the percentage of methylation at CpG sites
within the fragment region and the COX-2 transcriptional level was
evaluated. The percentage of methylation level for each individual
CpG site from bladder tissue was presented in Fig. 4D. The percentage of methylation at
a single CpG site was calculated from the sequencing results of
four independent clones. The percentage of CpG sites in the control
group was 50.0±16.1% in CpG1, 70.0±16.5% in CpG2, 50.0±0% in CpG3,
30.0±0% in CpG4, 40.0±15.1% in CpG5 and 30.0±12.0% in CpG6. In
comparison, the percentage of CpG sites in the ketamine group was
significantly decreased: 10.0±3.0% in CpG1, 10.0±3.1% in CpG2,
10.0±2.0% in CpG3, 30.0±12.0% in CpG4, 10.0±3.1% in CpG5, and
10.0±2.0% in CpG6. However, such ketamine-induced percentage
decrease was largely restored by COX-2 inhibitor as revealed by the
percentage of CpG sites in the ketamine+COX-2 inhibitor group:
40.0±8.0% in CpG1, 60.0±12.0% in CpG2, 20.0±4.2% in CpG3, 30.0±5.0%
in CpG4, 10.0±2.0% in CpG5 and 20.0±5.0% in CpG6. Moreover, the
methylated percentage of total six CpG sites in COX-2 promoter
located from −1,195 to −829 bp was 45.2±13.0% in the control group,
14.0±3.0% in the ketamine group and 30.0±7.0% in the ketamine+COX-2
inhibitor group. Similarly, ketamine caused significant reduction
(P<0.01) in the methylated percentage, while such reduction was
largely restored by COX-2 inhibitor (Fig. 4E). These observations demonstrated
that ketamine reduced the methylated level of region IV and such
the reduction was largely restored by the COX-2 inhibitor.
Genomic sequencing ranging from −1,522 to
−1,181 bp influences the COX-2 transcriptional level
Diagrammatic representation of the region V ranging
from −1,522 to −1,181 bp is shown in Fig. 5A. The fragment region V containing
7 CpG sites was marked with a red number (Fig. 5B). There are two potential binding
sequences for NF-κB transcriptional factor spanning from -1,442 to
−1,432 and −1,413 to −1,404 bp marked with a square box (Fig. 5B). The CpG sites by bisulfite
genomic sequencing data are highlighted as arrows (Fig. 5C). Detailed normal and bisulfite
genomic sequences for region V are shown in Fig. S5. The percentage of methylation
level for each individual CpG site is presented in Fig. 5D. The percentage of CpG sites in
the control group was 90.0±16.3% in CpG1, 90.0±16.1% in CpG2,
100.0±0% in CpG3, 100.0±0% in CpG4, 60.0±25.9% in CpG5, 52.0±25.8%
in CpG6, and 70.0±23.0% in CpG7. In comparison, the percentage of
CpG sites in the ketamine group was significantly decreased
(P<0.01): 50.0±20.0% in CpG1, 20.0±10.5% in CpG2, 40.0±19.5% in
CpG3, 50.0±19.6% in CpG4, 10.0±5.0% in CpG5, 10.0±4.8% in CpG6 and
unmethylation in CpG7. However, such ketamine-induced percentage
decrease was largely restored by COX-2 inhibitor as revealed by the
percentage of CpG sites: 90.0±16.0% in CpG1, 50.0±19.5% in CpG2,
70.0±24.0% in CpG3, 70.0±24.0% in CpG4, 50.0±9.6% in CpG5,
40.0±15.0% in CpG6 and 30.0±14.0% in CpG7 in the ketamine+COX-2
inhibitor group. Additionally, the methylated percentage of total
seven CpG sites in the COX-2 promoter region located from −1,522 to
−1,181 bp was 80.0±15.4% in the control group, 25.7±10.0% in the
ketamine group and 57.1±3.7% in the ketamine+COX-2 inhibitor group.
Similarly, ketamine caused a significant reduction in the
methylated percentage, while such reduction was largely restored by
COX-2 inhibitor (P<0.01; Fig.
5E). The above findings implied that the hypo-methylation of
CpG sites in the region ranging from −1,522 to −829 bp might
increase the sequence-specific binding affinity of NF-κB and
enhance the COX-2 gene activation.
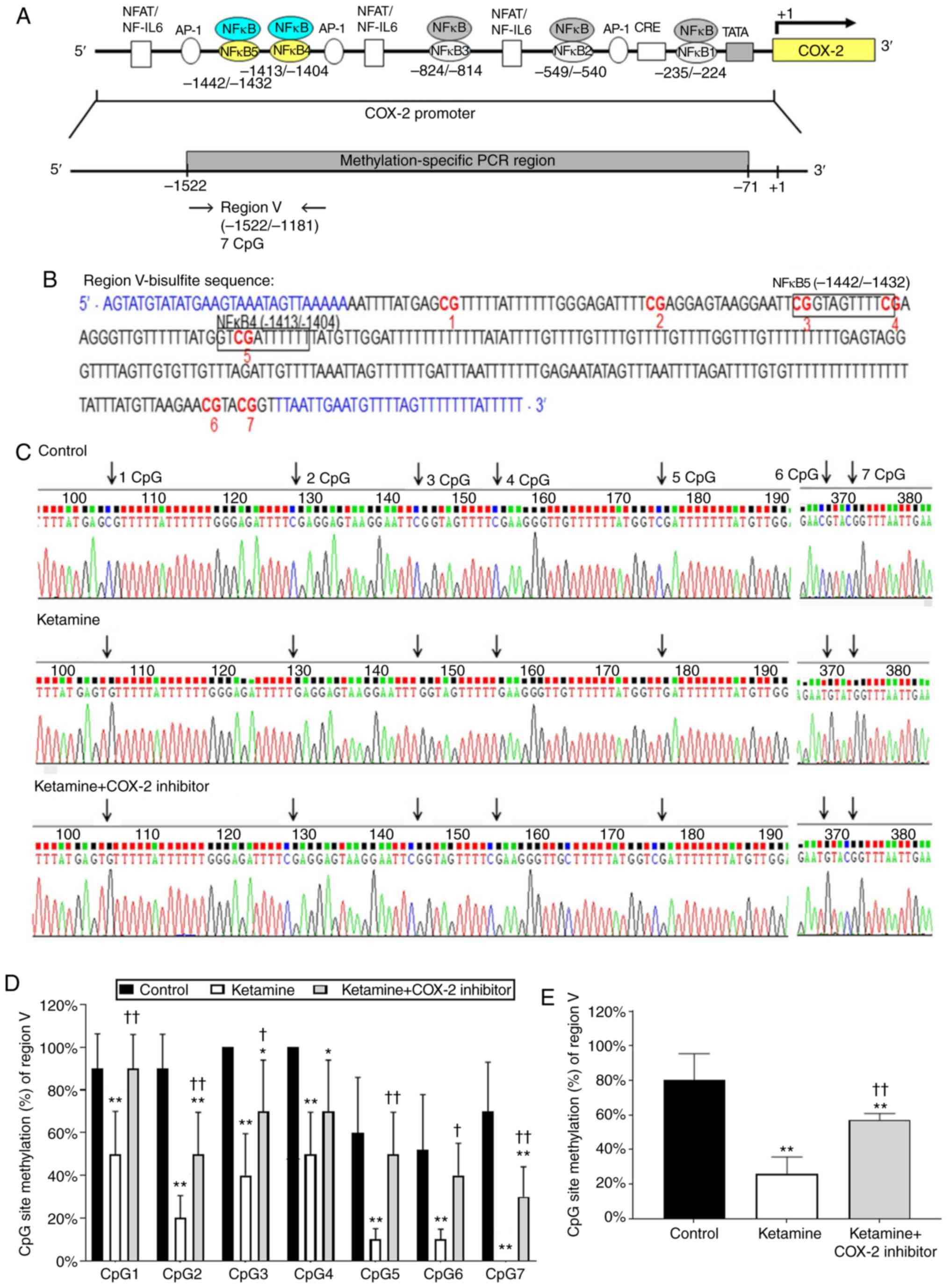 | Figure 5Methylation status of the CpG site in
COX-2 promoter region ranging from −1,181 to −1,522 in
ketamine-induced ulcerative cystitis. (A) A schematic depiction of
region V and sequence ranging from -1,181 to −1,522 relative to the
transcription start site (+1) is presented. (B) Bisulfite genomic
sequencing analysis for region V in the COX-2 promoter. Genomic
sequencing of bisulfite-COX-2 region V-associated 7 CpG sites was
obtained from the PCR products from bisulfite-treated DNA using
primers (blue color). The site-specific methylation of the CpG
sites (dinucleotides) at −1,442, −1,432 and −1,411 bp located the
sequence of NF-κB binding sites in the COX- promoter regions
ranging from -1,442 to -1,404 bp. The potential site for NF-κB
binding was marked with black square box and the genomic NFκB4 was
located at -1,413 to -1,404 bp, 5′-GTC GAT TCC C-3′ (Bisulfite
sequence of mNFκB4: 5′-GTC GAT TTT T-3′) and NFκB5 located at
-1,442 to -1,432 bp, 5′-CGG TAG TTT CC-3′ (Bisulfite sequence of
mNFκB5: 5′-CGG TAG TTT TC-3′). (C) Methylation patterns of the
bisulfite-COX-2 region V in three groups. CpG sites were
highlighted as arrows. (D) The methylation level of region V in the
COX-2 promoter. The methylated percentage of each individual CpG
site from bladder tissue was presented. (E) The percentage of total
seven CpG sites methylation in region IV. The methylation level of
region V from the control group showed a 3.11-fold increase
compared with in the ketamine group and a 1.40-fold increase
compared with in the ketamine+COX-2 group. Values were the mean ±
standard deviation for n=6. *P<0.05 and
**P<0.01 vs. the control group. †P<0.05
and ††P<0.01 vs. the ketamine group. COX,
cyclooxygenase; NF-κB4, nuclear factor-κB binding site. |
Analysis of NF-κB binding sequences and
DNA methylation in association with enzymes responsible for COX-2
promoter activity
To determine which NF-κB binding sequence within the
COX-2 promoter was involved in the COX-2 expression in KIC, the
potential NF-κB binding sequence was analyzed by ChIP assay.
Schematic diagram for COX-2 promoter region ranging from −1,522 to
−71 bp is shown (Fig. 6A). These
data suggest that ketamine treatment was able to increase the
binding of NF-κB on the COX-2 promoter region ranging from -1,522
to -1,331 bp as compared with the control group. However, the
ketamine+COX-2 inhibitor group reduced the level of NF-κB binding
in COX-2 promoter in comparison with the ketamine group (Fig. 6B and D).
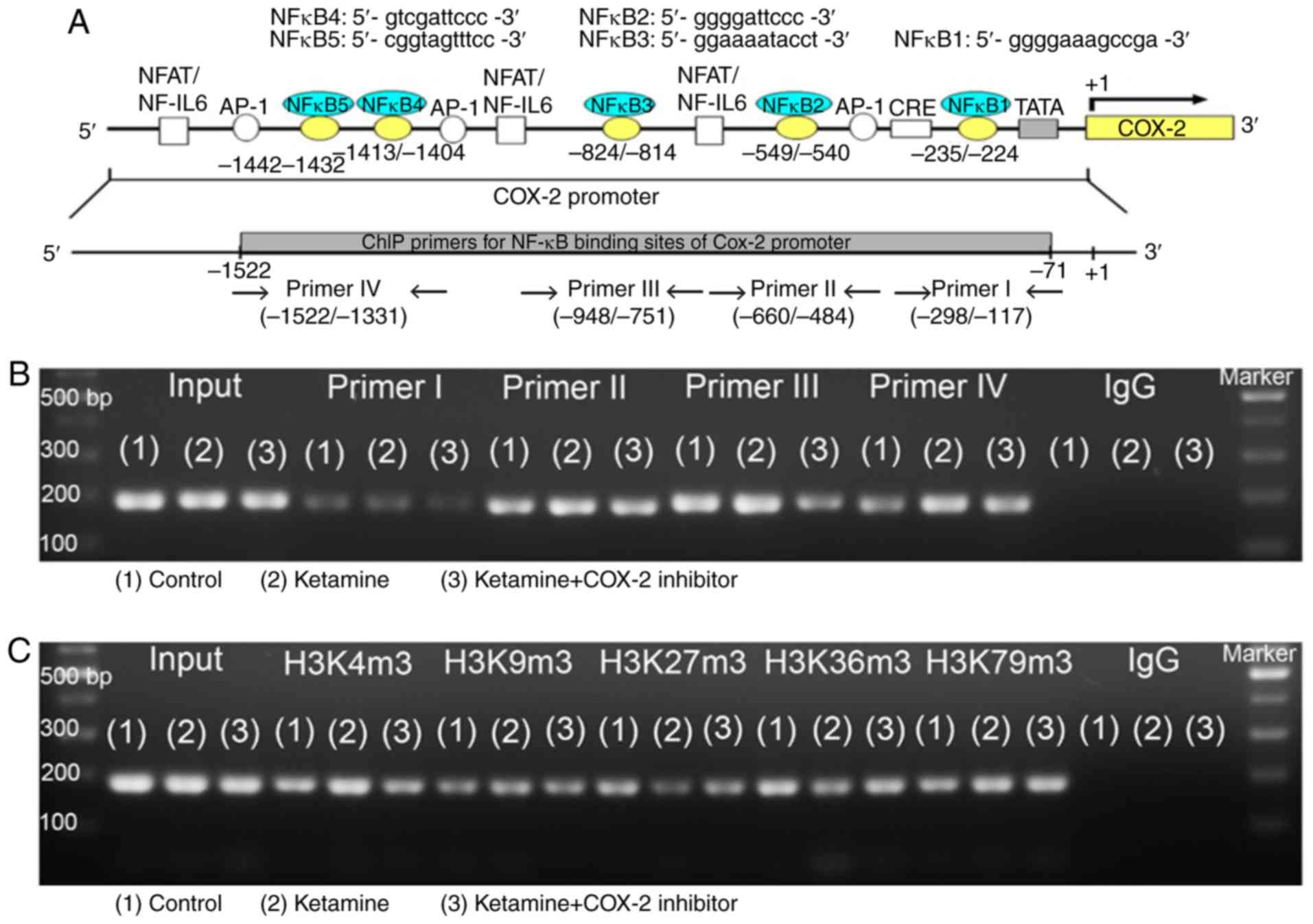 | Figure 6Analyses of NF-κB binding sequences
and DNA methylation-associated enzymes responsible for the promoter
activity of COX-2 gene. (A) Schematic diagram of the COX-2 promoter
and targeted sites ranging from -1,522 to -71 bp for ChIP analysis
was presented. A total of five potential NF-κB binding sequences in
the COX-2 promoter are denoted in the diagram. DNA fragments from
ChIP assay were amplified using primer sets to amplify the sequence
of the ChIP product. A total of four sets of primers were denoted
as primer I-IV, which included five NF-κB binding sequences. ChIP
analysis of the potential NF-κB binding sequence in the COX-2
promoter is shown. Formaldehyde cross-linked protein chromatin
complexes were immunoprecipitated and genomic DNA was analyzed by
(B) PCR and (D) qPCR using primers that recognized the COX-2
promoter region. Input for each reaction was used for internal
control of samples loading. IgG was employed as negative control.
ChIP analysis of histone H3 methylation on the COX-2 promoter was
analyzed by (C) PCR and (E) qPCR. ChIP assay was also applied to
anti-tri methyl histone H3 (H3K4m3, H3K9m3, H3K27m3, H3K36m3 and
H3K79m3) specific for histone H3 tri-methylated at Lysine (K4, K9,
K27, K36, and K79). These antibodies were used to provide direct
evidence that histone H3 modulated COX-2 transcriptional regulation
by modulating methylation of the COX-2 promoter region ranging from
-1,522 to -1,331 bp. Results were normalized as the control
group=100%. The mRNA levels of DNMTs (DNMT1, 3a and 3b) for
evaluation of the DNA methylation (F) and TETs (TET1-3) for
evaluation of the DNA demethylation (G) by reverse
transcription-qPCR. The level of TETs expression in the ketamine
group was represented as the number of fold increases compared with
the control group. Data were expressed as the mean ± standard
deviation for n=8. *P<0.05 and **P<0.01
vs. the control group. ††P<0.01 vs. the ketamine
group. ChIP, chromatin immunoprecipitation; TET,
Ten-Eleven-Translocation; DNMT, DNA methyltransferase; COX,
cyclooxygenase; q, quantitative. |
A ChIP assay using anti-tri methyl histone H3
(H3K4m3, H3K9m3, H3K27m3, H3K36m3 and H3K79m3) antibodies was used
to provide evidence to demonstrate that histone H3 methylation on
the COX-2 promoter region ranging from −1,522 to −1,331 bp affected
the COX-2 mRNA expression. A significant amount of methylated
histone H3 associated with the COX-2 promoter was detected
(Fig. 6C and E). In the ketamine
group, the level of histone H3K4m3 was increased compared with the
control group, but the level of histone H3K27m3 and H3K36m3 was
decreased. However, in the ketamine+COX-2 inhibitor group, the
level of H3K4m3 was reduced in comparison with in the control
group. However, there was no difference in the expression level of
H3K79m3 among these three groups.
To determine which enzyme was involved in COX-2 the
methylation and transcription, RT-qPCR was performed to analyze the
mRNA expression levels of rat DNMT1, 3a and 3b and TET dioxygenase
(TET1, TET2, and TET3) for the evaluation of the
methylation/demethylation (Fig.
6F-G) isolated from bladders. In the ketamine group, the level
of TETs was increased compared with in the control group, but not
for DNMTs. However, in the ketamine+COX-2 inhibitor group, the
level of DNMT3b and TET2 was increased compared with that in the
control group. These observations demonstrated that ketamine
induced NF-κB transcriptional regulation of COX-2 expression by
enhancing TET dioxygenase-mediated demethylation and increasing the
histone H3K4m3 binding affinity of COX-2 promoter.
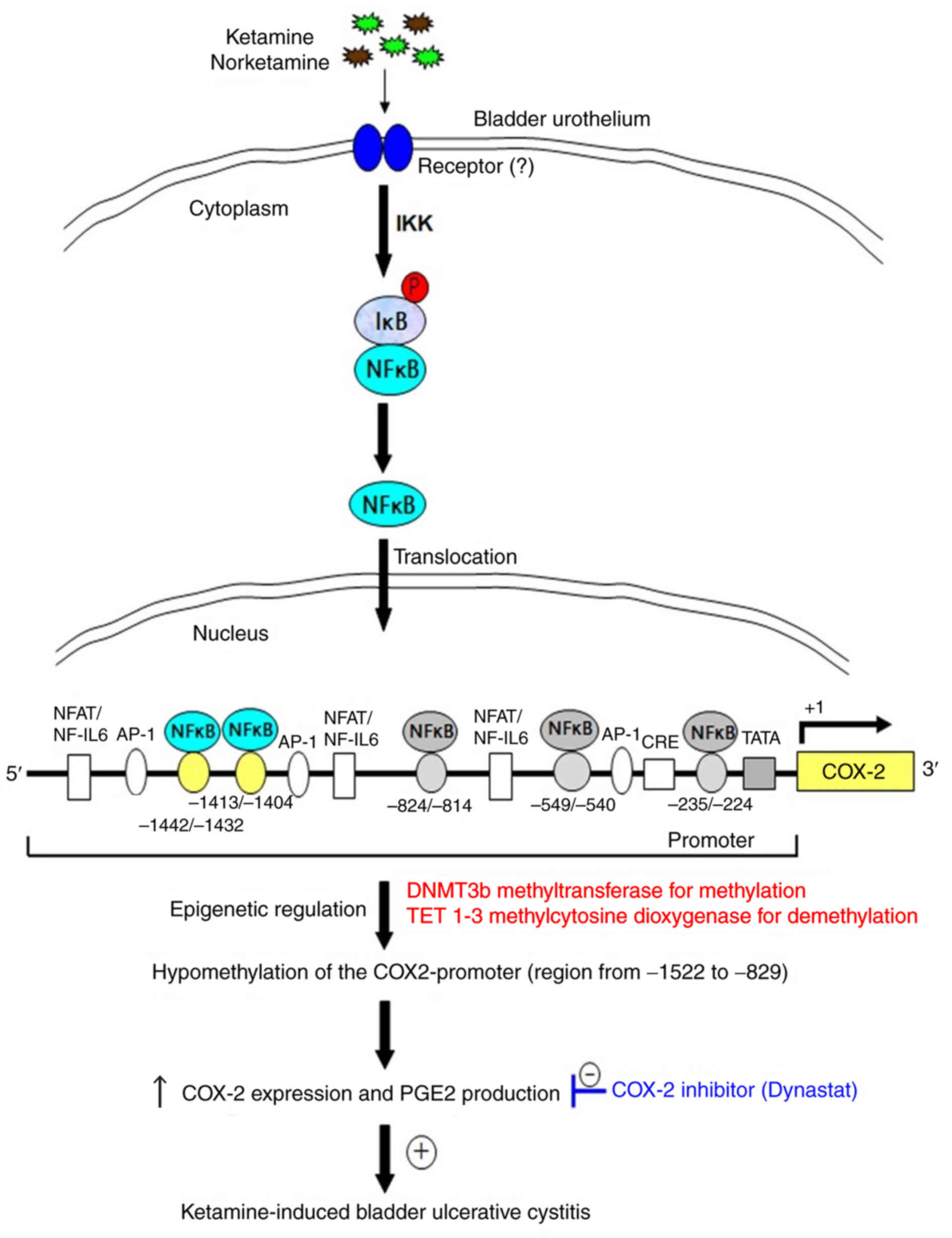
Proposed model for epigenetic regulation
of COX-2 expression by DNA methylation via NF-κB activation in
KIC
The present results revealed that ketamine treatment
caused significant hypomethylation of the COX-2 promoter, whereas
ketamine+COX-2 inhibitor improved the inflammatory effect (Figs. 1, 4 and 5). The hypomethylation of CpG sites in
the COX-2 promoter region ranging from −1,522 to −829 bp increased
the sequence-specific binding affinity of NF-κB transcriptional
factor and enhanced the COX-2 gene activation. The COX-2 promoter
hypomethylation was modulated by increasing TET enzymes for
demethylation. Based on the above findings, a potential model for
illustrating epigen-etic regulation of COX-2 expression by DNA
methylation mediated via NF-κB activation in KIC was proposed in
Fig. 7. Moreover, using TFBIND
software and MethPrimer software analysis showed that two potential
NF-κB binding sites of human COX-2 promoter had potential
methylation sites (Fig. S6A),
which implied that the transcriptional silencing of COX-2 by the
epigenetic mechanisms might have potential applications for the
identification of molecular biomarkers and clinical therapy for
KIC.
Discussion
The results of the present study demonstrated that
ketamine treatment induced the translocation of NF-κB p65 to
activate COX-2 expression and promote PGE2 production in bladder
tissue in KIC, whereas the COX-2 inhibitor suppressed the
inflammatory effect mediated by KIC. Furthermore, whether
epigenetic regulation of COX-2 promoter by DNA hypomethylation
could alter NF-κB translocation and transcriptional signaling of
COX-2 expression was also determined in KIC. Bisulfite MSP and
genomic sequencing data showed that all the three experimental
groups exhibited un-methylation at CpG sites within the COX-2
promoter region ranging from -830 to -71 bp. However, ketamine
treatment caused COX-2 promoter hypomethylation ranging from -1,522
to -829 bp, which contributed to the COX-2 transcriptional
regulation, whereas ketamine combined with COX-2 inhibitor improved
such alterations. Ketamine treatment increased the binding of NF-κB
and permissive histone H3K4m3, but decreased the repressive histone
H3K27m3 and H3K36m3 binding on the COX-2 promoter region ranging
from -1,522 to -1,331 bp as determined by ChIP assay. These
observations suggested that there was a hypomethylation pattern of
the COX-2 promoter via NF-κB p65 translocation in association with
the level of COX-2 transcription after ketamine treatment.
Following ketamine injection, translocated NF-κB p65
bound to the specific sequence of COX-2 promoter region ranging
from −1,522 to −829 bp, which transcriptionally induced COX-2
expression to activate PGE2 production. Synthesized PGE2 might act
on its specific receptor subtype (EP2) and further feedback
activate NF-κB (37-39). The treatment of COX-2 inhibitor
reduced COX-2 transcriptional expression and decreased PGE2
production, leading to an inhibited inflammatory reaction (40). Additionally, previous studies
indicated that NF-κB could control COX-2 transcription (6,41),
however cyclopentenone prostaglandins could inhibit NF-κB
activation via IκB kinase (IKK) inhibition (42), suggesting cyclopentenone feedback
could inhibit continued nuclear accumulation of NF-κB (38). Following ketamine injection
combined with the COX-2 inhibitor, the COX-2 inhibitor was
converted to an anti-inflammatory compound (valdecoxib). The
pharmacological effect of valdecoxib was to inhibit COX-2-mediated
prostaglandin synthesis. In the present study, it was proposed that
the pharmacological effect of COX-2 inhibitor might suppress PGE2
synthesis and block the nuclear accumulation of NF-κB, leading to
inhibition of the positive feedback loop of the COX-2-PGE2-EP2-NFκB
pathway. The prostaglandins might inhibit NF-κB activity through
IKK inhibition, restrict NF-κB to the cytoplasm and this negative
feedback loop might result in reducing NF-κB-dependent COX-2 gene
expression in KIC. COX-2 overexpression affected the activity of
TET (43), which implied that
COX-2 inhibitor might affect the methylation status within COX-2
promoter.
Previous studies reported that COX-2 expression
might be related to the methylation status of 5′-CpG site of COX-2
gene in cancer cell lines (25,44), which exhibited hypermethylation of
CpG sites in the promoter or exon 1 region and
methylation-inhibiting agents restored the expression of COX-2
(28). Toyota et al
(23) suggested that aberrant
methylation of the 5′ CpG island to reduce COX-2 expression was
presented in colorectal cancer and a detailed methylated mapping of
24 CpG sites revealed the methylated region closest to the
transcription start site. However, the results of the present study
indicated that ketamine treatment induced the translocation of
NF-κB p65 to activate COX-2 expression and PGE2 production in
bladder tissue. Meanwhile, bisulfite MSP and genomic sequencing
data showed that DNA hypomethylation at CpG sites within COX-2
promoter region located from −1,522 to −829 bp might contribute to
the COX-2 transcriptional regulation in the ketamine group.
Ketamine treatment was able to increase the binding of NF-κB and
transcriptionally permissive histone H3K4m3, but decrease the
repressive histone H3K27m3 and H3K36m3 on the COX-2 promoter
ranging from −1,522 to −1,331 bp as determined by ChIP assay. Thus,
such epigenetic regulation of COX-2 activity for PGE2 production is
interesting and deserves further investigations.
The hypermethylation of CpG sites in the COX-2
promoter for the suppression of gene expression was shown to reduce
the binding affinity of sequence-specific transcription factors
(30,45). Kelavkar et al (11) reported that the increase in the
methylation status of specific CpG sites of COX-2 promoter might
affect NF-κB binding affinity. In the present study, there were 7
CpG sites and two potential sequences for NF-κB transcriptional
factor binding within the COX-2 promoter located from -1,522 to
-1,181 bp. Two potential genomic sequences for NFκB4 binding site
in the COX-2 promoter were located at -1,413 to -1,404 bp, 5′-GTC
GAT TCC C-3′ and NFκB5 located at −1,442 to −1,432 bp, 5′-CGG TAG
TTT CC-3′. The site-specific methylation of 5′-CpG dinucleotides at
sites -1,442, 1,432, -1,411 bp (3, 4 and 5 CpG site of region V)
was located at the COX-2 promoter sequence ranging from −1,442 to
1,404 bp. The observed increase in the methylation at these
specific CpG sites might impair NF-κB binding affinity to affect
COX-2 expression in the control group. The potential NF-κB binding
sequence involved in the COX-2 expression in KIC by ChIP analysis
was also analyzed. The results of the present study suggested that
the NF-κB binding sequence ranging from -1,522 to -1,331 bp
modulated the COX-2 mRNA expression as observed. ChIP assay used
anti-trimethyl histone H3 antibodies to provide direct evidence
that histone H3 methylation on the COX-2 promoter affected the
COX-2 mRNA expression in KIC.
The double helical strands forming the DNA backbone
contained a major groove and a minor groove and the major groove
interacted with more functional groups than the minor groove, which
could interact with sequence-specific DNA-binding proteins.
Moreover, transcription factors were composed of DNA-binding
domains and had a specific affinity for single- or double-stranded
DNA (46). Cytosine methylation
could either influence the interactions between promoter DNA and
transcription factors or exhibit no effect, depending on the
context (14). Despite being only
a minor chemical change, addition of a methyl group to cytosine
could affect nucleotide readout via hydrophobic contacts in the
major groove and shape readout via electrostatic contacts in the
minor groove (47,48). 5-methylcytosines changed the
nucleosome stability and affected the chromatin structure and the
affinity of transcription factor bound to genomic DNA (14,49,50). The results of the present study
demonstrated that ketamine treatment caused significant COX-2
hypomethylation ranging from −1,522 to −829 bp, which contributed
to the COX-2 transcriptional regulation and increased NF-κB binding
to the promoter region located from −1,442 to −1,404 bp. It was
also found that the NF-κB binding sequence ranging from -1,522 to
-1,331 bp modulated the COX-2 mRNA expression as observed by ChIP
assay. These data indicated that the COX-2 promoter region located
from −1,522 to −829 bp resided in the major groove of DNA and
played a pivotal role for regulation COX-2 gene expression in KIC.
Furthermore, the authors' previous study demonstrated the
involvement of the regulation of COX-2 via the NF-κB pathway in the
inflammatory signaling of KIC in rat urinary bladder (6). Misoprostol, the FDA approved
clinical medication, is beneficial to various immunological
diseases, e.g., interstitial cystitis (51). It attenuates the transcriptional
activity of NF-κB and recruitment of p65 (52). The combination of those studies
with the present study's findings suggested that the inhibitor of
NF-κB transcription factor might provide a potential application
for therapeutic strategy for KIC.
The COX-2 promoter region, NF-κB binding sites and
the potential methylation of CpG sites were compared between rat
and human species. In rats, the promoter of COX-2 gene (accession
number NM_017232) ranging from -1,522 to -71 including 1,452 bp and
44 CpG sites were identified by nucleotide sequence analysis for
potential methylation. There were 7 CpG sites and 2 potential
sequences for NF-κB transcriptional factor binding within the COX-2
promoter located from -1,522 to −1,181 bp. A total of two potential
genomic sequences for NF-κB binding sites in the COX-2 promoter
were located at -1,413 to −1,404 bp, 5′-GTC GAT TCC C-3′ and at
-1,442 to -1,432 bp, 5′-CGG TAG TTT CC-3′. The site-specific
methylation of 5′-CpG dinucleotides at sites -1,442, -1,432 and
-1,411 bp was located the COX-2 promoter sequence ranging from
-1,442 to -1,404 bp (Table I).
Moreover, in humans the promoter regions of the COX-2 gene
(accession number NM_000963) ranging from -2,000 to -1 including
2,000 bp and 59 CpG sites were identified by nucleotide sequence
analysis for potential methylation. Seven potential motifs similar
to the consensus binding sites for NF-κB in the region were
identified by TFBIND software. Two potential genomic sequences for
NF-κB binding sites in the COX-2 promoter were located at -320 to
-310 bp, 5′-CTG GGT TTC CG-3′ and at -640 to -631 bp, 5′-GTG ACT T
CC T-3′. The site-specific methylation of 5′-CpG dinucleotides at
sites -640, -310 bp was located in the COX-2 promoter. The present
study showed that NF-κB binding sequence ranging from -1,442 to
1,404 modulated the COX-2 mRNA expression as observed in rats.
These findings suggested that the transcriptional silencing of
COX-2 by epigenetic mechanisms might be involved in the
pathogenesis of KIC. However, further investigations of the
epigenetic mechanisms in the pathogenesis of KIC in human are
needed.
DNA hypomethylation of COX-2 promoter region might
contribute to COX-2 transcriptional regulation and induce a
pro-inflammatory response in KIC. However, COX-2 inhibitor
treatment increased the methylation of COX-2 promoter and reduced
COX-2 transcriptional expression. These results implied that the
transcriptional silencing of COX-2 by the epigenetic mechanisms
might be involved in the pathogenesis of KIC, which could have
potential application for the molecular biomarker and clinical
therapy of KIC.
Supplementary Materials
Funding
The present study was supported by the Ministry of
Science and Technology NSC (grant no. 105-2314-B-037-043-MY3);
Ministry of Health and Welfare (grant no.
MOHW107-TDU-B-212-123006); Department of Medical Research,
Kaohsiung Medical University Hospital (grant nos. KMUH105-5R44 and
KMUH106-6R60); and from the Chi-Mei Medical Center and the
Kaohsiung Medical University Research (grant no. 107CM-KMU-12).
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
SMC, JHL, CYL, KLL, YCL, HPH, CCT, WJW, HJY and YSJ
designed the study; SMC, JHL, CYL, KLL, YCL, HPH, CCT, WJW, and YSJ
conducted review and editing; SMC, JHL and YSJ had full access to
all the data in the study and took responsibility for the integrity
of the data and the accuracy of the data analysis. SMC, JHL and YSJ
wrote the paper. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
This study was approved by the Animal Care and
Treatment Committee of Kaohsiung Medical University. All
experiments were conducted according to the guidelines for
laboratory animal care.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Professor Chang-Hwei
Chen of the University at Albany, State University of New York for
his valuable comments on this manuscript.
References
|
1
|
Lee YL, Lin KL, Chuang SM, Lee YC, Lu MC,
Wu BN, Wu WJ, Yuan SF, Ho WT and Juan YS: Elucidating mechanisms of
bladder repair after hyaluronan instillation in ketamine-induced
ulcerative cystitis in animal model. Am J Pathol. 187:1945–1959.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shahani R, Streutker C, Dickson B and
Stewart RJ: Ketamine-associated ulcerative cystitis: A new clinical
entity. Urology. 69:810–812. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Middela S and Pearce I: Ketamine-induced
vesicopathy: A literature review. Int J Clin Pract. 65:27–30. 2011.
View Article : Google Scholar
|
|
4
|
Chuang SM, Liu KM, Li YL, Jang MY, Lee HH,
Wu WJ, Chang WC, Levin RM and Juan YS: Dual involvements of
cyclooxygenase and nitric oxide synthase expressions in
ketamine-induced ulcerative cystitis in rat bladder. Neurourol
Urodyn. 32:1137–1143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu KM, Chuang SM, Long CY, Lee YL, Wang
CC, Lu MC, Lin RJ, Lu JH, Jang MY, Wu WJ, et al: Ketamine-induced
ulcerative cystitis and bladder apoptosis involve oxidative stress
mediated by mitochondria and the endoplasmic reticulum. Am J
Physiol Renal Physiol. 309:F318–331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Juan YS, Lee YL, Long CY, Wong JH, Jang
MY, Lu JH, Wu WJ, Huang YS, Chang WC and Chuang SM: Translocation
of NF-κB and expression of cyclooxygenase-2 are enhanced by
ketamine-induced ulcerative cystitis in rat bladder. Am J Pathol.
185:2269–2285. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jhang JF, Hsu YH, Jiang YH and Kuo HC:
Elevated serum IgE may be associated with development of ketamine
cystitis. J Urol. 192:1249–1256. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee CL, Jiang YH and Kuo HC: Increased
apoptosis and suburothelial inflammation in patients with
ketamine-related cystitis: A comparison with non-ulcerative
interstitial cystitis and controls. BJU Int. 112:1156–1162. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Jones PL and Wolffe AP: Relationships
between chromatin organization and DNA methylation in determining
gene expression. Semin Cancer Biol. 9:339–347. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jenuwein T and Allis CD: Translating the
histone code. Science. 293:1074–1080. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Kelavkar UP, Harya NS, Hutzley J, Bacich
DJ, Monzon FA, Chandran U, Dhir R and O'Keefe DS: DNA methylation
paradigm shift: 15-lipoxygenase-1 upregulation in prostatic
intraepithelial neoplasia and prostate cancer by atypical promoter
hypermethylation. Prostaglandins Other Lipid Mediat. 82:185–197.
2007. View Article : Google Scholar
|
|
12
|
Curradi M, Izzo A, Badaracco G and
Landsberger N: Molecular mechanisms of gene silencing mediated by
DNA methylation. Mol Cell Biol. 22:3157–3173. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Urnov FD and Wolffe AP: Chromatin
remodeling and transcriptional activation: The cast (in order of
appearance). Oncogene. 20:2991–3006. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dantas Machado AC, Zhou T, Rao S, Goel P,
Rastogi C, Lazarovici A, Bussemaker HJ and Rohs R: Evolving
insights on how cytosine methylation affects protein-DNA binding.
Brief Funct Genomics. 14:61–73. 2015. View Article : Google Scholar
|
|
15
|
Jin B and Robertson KD: DNA
methyltransferases, DNA damage repair, and cancer. Adv Exp Med
Biol. 754:3–29. 2013. View Article : Google Scholar
|
|
16
|
Deneberg S, Guardiola P, Lennartsson A, Qu
Y, Gaidzik V, Blanchet O, Karimi M, Bengtzén S, Nahi H, Uggla B, et
al: Prognostic DNA methylation patterns in cytogenetically normal
acute myeloid leukemia are predefined by stem cell chromatin marks.
Blood. 118:5573–5582. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kouzarides T: Chromatin modifications and
their function. Cell. 128:693–705. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Völkel P and Angrand PO: The control of
histone lysine methylation in epigenetic regulation. Biochimie.
89:1–20. 2007. View Article : Google Scholar
|
|
19
|
Sims RJ III and Reinberg D: Histone H3 Lys
4 methylation: Caught in a bind? Genes Dev. 20:2779–2786. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang S, Barros SP, Niculescu MD, Moretti
AJ, Preisser JS and Offenbacher S: Alteration of PTGS2 promoter
methylation in chronic periodontitis. J Dent Res. 89:133–137. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Loo WT, Jin L, Cheung MN, Wang M and Chow
LW: Epigenetic change in E-cadherin and COX-2 to predict chronic
periodontitis. J Transl Med. 8:1102010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pedre X, Mastronardi F, Bruck W,
Lopez-Rodas G, Kuhlmann T and Casaccia P: Changed histone
acetylation patterns in normal-appearing white matter and early
multiple sclerosis lesions. J Neurosci. 31:3435–3445. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Toyota M, Shen L, Ohe-Toyota M, Hamilton
SR, Sinicrope FA and Issa JP: Aberrant methylation of the
Cyclooxygenase 2 CpG island in colorectal tumors. Cancer Res.
60:4044–4048. 2000.PubMed/NCBI
|
|
24
|
Kikuchi T, Itoh F, Toyota M, Suzuki H,
Yamamoto H, Fujita M, Hosokawa M and Imai K: Aberrant methylation
and histone deacetylation of cyclooxygenase 2 in gastric cancer.
Int J Cancer. 97:272–277. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ma X, Yang Q, Wilson KT, Kundu N, Meltzer
SJ and Fulton AM: Promoter methylation regulates cyclooxygenase
expression in breast cancer. Breast Cancer Res. 6:R316–R321. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Feinberg AP and Vogelstein B:
Hypomethylation distinguishes genes of some human cancers from
their normal counterparts. Nature. 301:89–92. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jones PA and Laird PW: Cancer epigenetics
comes of age. Nat Genet. 21:163–167. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chell S, Kaidi A, Williams AC and
Paraskeva C: Mediators of PGE2 synthesis and signalling downstream
of COX-2 represent potential targets for the prevention/treatment
of colorectal cancer. Biochim Biophys Acta. 1766:104–119.
2006.PubMed/NCBI
|
|
29
|
Zhou Y and Hu Z: Genome-wide demethylation
by 5-aza-2′-deoxycytidine alters the cell fate of stem/progenitor
cells. Stem Cell Rev. 11:87–95. 2015. View Article : Google Scholar :
|
|
30
|
Murata H, Tsuji S, Tsujii M, Sakaguchi Y,
Fu HY, Kawano S and Hori M: Promoter hypermethylation silences
cyclooxygenase-2 (Cox-2) and regulates growth of human
hepatocellular carcinoma cells. Lab Invest. 84:1050–1059. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liou JT, Chen ZY, Ho LJ, Yang SP, Chang
DM, Liang CC and Lai JH: Differential effects of triptolide and
tetrandrine on activation of COX-2, NF-kappaB, and AP-1 and virus
production in dengue virus-infected human lung cells. Eur J
Pharmacol. 589:288–298. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Poligone B and Baldwin AS: Positive and
negative regulation of NF-kappaB by COX-2: Roles of different
prostaglandins. J Biol Chem. 276:38658–38664. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Glinghammar B and Rafter J: Colonic
luminal contents induce cyclooxygenase 2 transcription in human
colon carcinoma cells. Gastroenterology. 120:401–410. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Hu VY, Malley S, Dattilio A, Folsom JB,
Zvara P and Vizzard MA: COX-2 and prostanoid expression in
micturition pathways after cyclophosphamide-induced cystitis in the
rat. Am J Physiol Regul Integr Comp Physiol. 284:R574–R585. 2003.
View Article : Google Scholar
|
|
35
|
Li LC and Dahiya R: MethPrimer: Designing
primers for methylation PCRs. Bioinformatics. 18:1427–1431. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
37
|
Aoki T, Nishimura M, Matsuoka T, Yamamoto
K, Furuyashiki T, Kataoka H, Kitaoka S, Ishibashi R, Ishibazawa A,
Miyamoto S, et al: PGE(2) -EP(2) signalling in endothelium is
activated by haemodynamic stress and induces cerebral aneurysm
through an amplifying loop via NF-κB. Br J Pharmacol.
163:1237–1249. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Paul AG, Chandran B and Sharma-Walia N:
Cyclooxygenase-2-prostaglandin E2-eicosanoid receptor inflammatory
axis: A key player in Kaposi's sarcoma-associated herpes virus
associated malignancies. Transl Res. 162:77–92. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Rundhaug JE, Simper MS, Surh I and Fischer
SM: The role of the EP receptors for prostaglandin E2 in skin and
skin cancer. Cancer Metastasis Rev. 30:465–480. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Müller N: COX-2 inhibitors as
antidepressants and antipsychotics: Clinical evidence. Curr Opin
Investig Drugs. 11:31–42. 2010.PubMed/NCBI
|
|
41
|
Ackerman WE IV, Summerfield TL, Vandre DD,
Robinson JM and Kniss DA: Nuclear factor-kappa B regulates
inducible pros-taglandin E synthase expression in human amnion
mesenchymal cells. Biol Reprod. 78:68–76. 2008. View Article : Google Scholar
|
|
42
|
Rossi A, Kapahi P, Natoli G, Takahashi T,
Chen Y, Karin M and Santoro MG: Anti-inflammatory cyclopentenone
prostaglandins are direct inhibitors of IkappaB kinase. Nature.
403:103–108. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
43
|
Chen H, Cai W, Chu ESH, Tang J, Wong CC,
Wong SH, Sun W, Liang Q, Fang J, Sun Z and Yu J: Hepatic
cyclooxygenase-2 overexpression induced spontaneous hepatocellular
carcinoma formation in mice. Oncogene. 36:4415–4426. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Akhtar M, Cheng Y, Magno RM, Ashktorab H,
Smoot DT, Meltzer SJ and Wilson KT: Promoter methylation regulates
Helicobacter pylori-stimulated cyclooxygenase-2 expression in
gastric epithelial cells. Cancer Res. 61:2399–2403. 2001.PubMed/NCBI
|
|
45
|
Clark SJ, Harrison J and Molloy PL: Sp1
binding is inhibited by (m)Cp(m)CpG methylation. Gene. 195:67–71.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Pabo CO and Sauer RT: Protein-DNA
recognition. Annu Rev Biochem. 53:293–321. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Li E and Zhang Y: DNA methylation in
mammals. Cold Spring Harb Perspect Biol. 6:pp. a0191332014,
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Chuang TJ and Chen FC: DNA methylation is
associated with an increased level of conservation at nondegenerate
nucleotides in mammals. Mol Biol Evol. 31:387–396. 2014. View Article : Google Scholar :
|
|
49
|
Kass SU, Pruss D and Wolffe AP: How does
DNA methylation repress transcription? Trends Genet. 13:444–449.
1997. View Article : Google Scholar
|
|
50
|
Bestor TH: Gene silencing. Methylation
meets acetylation. Nature. 393:311–312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Davies NM, Longstreth J and Jamali F:
Misoprostol therapeutics revisited. Pharmacotherapy. 21:60–73.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Gobejishvili L, Ghare S, Khan R, Cambon A,
Barker DF, Barve S, McClain C and Hill D: Misoprostol modulates
cytokine expression through a cAMP pathway: Potential therapeutic
implication for liver disease. Clin Immunol. 161:291–299. 2015.
View Article : Google Scholar : PubMed/NCBI
|