Introduction
Diabetic nephropathy (DN) is a leading cause of
chronic kidney disease (CKD) and end-stage renal failure,
constituting a major health concern worldwide (1). Damage to the glomer-ular filtration
unit is one of the most important pathological characteristics of
DN. The filtration unit comprises capillary endothelial cells, the
glomerular basement membrane and podocytes, which are specialized
epithelial cells. Once considered to be primarily quiescent and
terminally differentiated cells, podocytes have been identified as
a key factor in DN (2). Podocyte
injury is characterized by the pathological loss of regularity in
branching and widening of the foot processes, termed 'foot process
effacement'. Severe insults lead to podo-cyte loss by apoptosis or
detachment. Numerous studies have reported that disruption of
multiple renal signaling pathways is crucial for the progression of
such pathological damage (3,4).
However, there are currently no effective therapeutic options for
DN in the clinical setting. Indeed, the precise regulatory
signaling pathways through which podocytes are injured in DN are
largely unclear.
Autophagy is a major catabolic pathway through which
mammalian cells degrade and recycle macromolecules and organelles.
The major roles of autophagy are the removal of damaged organelles,
degradation of proteins, and reconstitution of intracellular
metabolism for the maintenance of intracellular homeostasis and
cell health (5). Generally,
autophagy is upregulated under conditions of oxidative stress and
hypoxia in podocytes. Recently, convincing evidence indicates that,
in the case of diabetes, autophagy plays a pivotal role in
maintaining the homeostasis of lysosomes in podocytes. The
impairment of autophagy, as it is implicated in the pathogenesis of
podocyte loss, may lead to massive overflow proteinuria in DN
(6-8). However, the exact role of autophagy
in podocytes remains elusive.
Connexins (Cxs) are a multigenic family of
transmem-brane proteins that form gap junctions and participate in
the exchange of information between cells. Connexin 43 (Cx43) is
considered to be the most abundant and widely expressed gap
junction protein. Extensive studies have revealed that, in addition
to its role in intercellular communication, Cx43 mediates gene
transcription, cytoskeleton dynamics, ATP exocytosis, vesicle
release and cell stress (9).
Sawai et al (10) reported
the presence of Cx43 in normal podocytes in a linear pattern, and
demonstrated a shift in this linear distribution in patients with
DN. Our previous studies also indicated that upregulation of Cx43
is involved in podocyte injury (11), suggesting that Cx43 may be a
critical regulator in podo-cytes under DN conditions. Furthermore,
Cx43 has recently been implicated in inflammation and fibrosis.
Inhibiting Cx43 may alleviate kidney damage and maintain renal
function. Therefore, new therapies targeting Cx43 blockade in ideal
cell populations may be a viable option for effectively inhibiting
the progression of CKD (12).
Interestingly, Cx43 rapidly modulates autophagy response, playing a
critical role in cell apoptosis (13). However, the effect of Cx43 on the
regulation of podocyte autophagy under DN conditions remains
unclear.
The aim of the present study was to determine the
effect of Cx43 on impaired autophagic flux, and to determine
whether the regulation of Cx43 can protect podocytes under DN
conditions.
Materials and methods
Antibodies and reagents
Rapamycin (RP) and chloroquine (CQ) were purchased
from Sigma-Aldrich; Merck KGaA. Antibodies against LC3, mammalian
target of rapamycin (mTOR) and p-mTOR were acquired from Cell
Signaling Technology, Inc. Anti-Cx43, anti-podocin, anti-nephrin
and anti-p62 antibodies were obtained from Abcam. Anti-GAPDH was
purchased from CWBio.
Animals
The study protocols were reviewed and approved by
the Institutional Animal Care and Use Committee of Nanjing Medical
University. A total of 24 male Sprague-Dawley rats (aged 5-6 weeks
and weighing ~190 g) were housed under specific pathogen-free
conditions at optimal temperature with a 12-h light/dark cycle, and
were allowed free access to standard food and water. The rats were
randomly divided into four groups: Group 1, PBS-infused rats
(control, n=6); group 2, streptozocin (STZ; 60 mg/kg)-infused rats
(n=6); group 3, STZ (60 mg/kg)-infused rats with scrambled siRNA
(SCR, n=6); and group 4, STZ (60 mg/kg)-infused rats with Cx43
siRNA [oligodeoxynucleotide antisense (AS), n=6]. At the end of the
28-day infusion period, the rats were weighed and blood and urine
samples were collected. The blood urea nitrogen and urine protein
levels were analyzed according to the manufacturer's protocol
(R&D Systems, Inc.). Tail capillary blood glucose levels were
monitored with a glucometer (Accu-Chek Performa; Roche Diagnostics
GmbH).
Cell culture
The immortalized mouse podocyte cell line MPC5 was
kindly provided by Dr Junwei Yang (Nanjing Medical University) and
the cells were cultured as previously described (14). Podocytes were differentiated
without interferon-γ at 37°C for 14 days prior to the experiments.
Differentiated podocytes were incubated in medium containing 0.1%
fetal bovine serum for 24 h. The podocytes exposed to HG (30 mM)
were then cultured for various time periods.
Transfection of small interference
RNA
Podocytes were transfected with Cx43 siRNA (50 nM)
(sense, 5′-AAAGUUGCUGCUGGACAU GAATT-3′ and antisense,
5′-UUCAUGUCCAGCAGCAACUUUTT-3′) or negative control siRNA (sense,
5′-UUCUCCGAACGUGUCACGUTT-3′ and anti-sense,
5′-ACGUGACACGUUCGGAGAATT-3′) for 24 h using
Lipofectamine® 3000 (Invitrogen; Thermo Fisher
Scientific, Inc.) according to the manufacturer's protocol.
Thereafter, the level of targeting protein with knockdown of Cx43
was detected by western blotting.
Western blotting
The cells were harvested after treatment with the
different compounds for the indicated times. Protein levels were
detected by western blotting according to established protocols
(15). Primary antibodies against
Cx43 (1:1,000), LC3 (1:1,000), p62 (1:2,000), podocin (1:1,000),
synaptopodin (1:1,000), mTOR (1:1,000), p-mTOR (1:1,000) and GAPDH
(1:2,000) were used.
Annexin V-fluorescein isothiocyanate
conjugated with propidium iodide (PI) staining
Podocyte injury was quantified by Annexin V/PI
staining (BD Biosciences) following the manufacturer's protocol.
Briefly, cells were harvested and washed twice with PBS.
Subsequently, the cells were resuspended in 100 µl binding buffer,
then incubated with 5 μl Annexin V and 10 μl PI for
15 min at 25°C in the dark. After mixing gently in 400 μl of
binding buffer, the cells were observed under a fluorescence
microscope (DS-Ri1; Nikon Corporation).
Immunofluorescence staining
Paraffin sections (3-μm) were prepared and
incubated with primary antibodies against synaptopodin, Wilms'
tumor-1 (WT-1) and p-mTOR in PBS containing 1% BSA overnight at 4°C
after blocking with 5% BSA for 1 h. Subsequently, the sections were
incubated with secondary antibody in PBS containing 1% BSA in the
dark. After washing with PBS, cell nuclei were stained with DAPI.
Immunostained samples were visualized under a fluorescence
microscope (Nikon Corporation).
Immunohistochemistry
Paraffin-embedded sections were used for
immunohistochemistry. Briefly, the sections were incubated with
primary antibodies against Cx43 and podocin overnight at 4°C. The
sections were then analyzed using a streptavidin peroxidase
detection system (Maixin) according to the manufacturer's protocol.
Reactions were conducted using a DAB substrate kit (Maixin) and
counterstaining was performed using hematoxylin. The sections were
visualized under a microscope (Nikon Corporation).
Statistical analysis
Data are presented as the mean ± the standard error
of the mean. When more than two groups were compared, one-way
analysis of variance followed by Bonferroni's correction was
employed to analyze the differences using SPSS 22.0 statistical
software (IBM Corp.). A two-sided P-value of <0.05 was
considered to indicate statistically significant differences.
Results
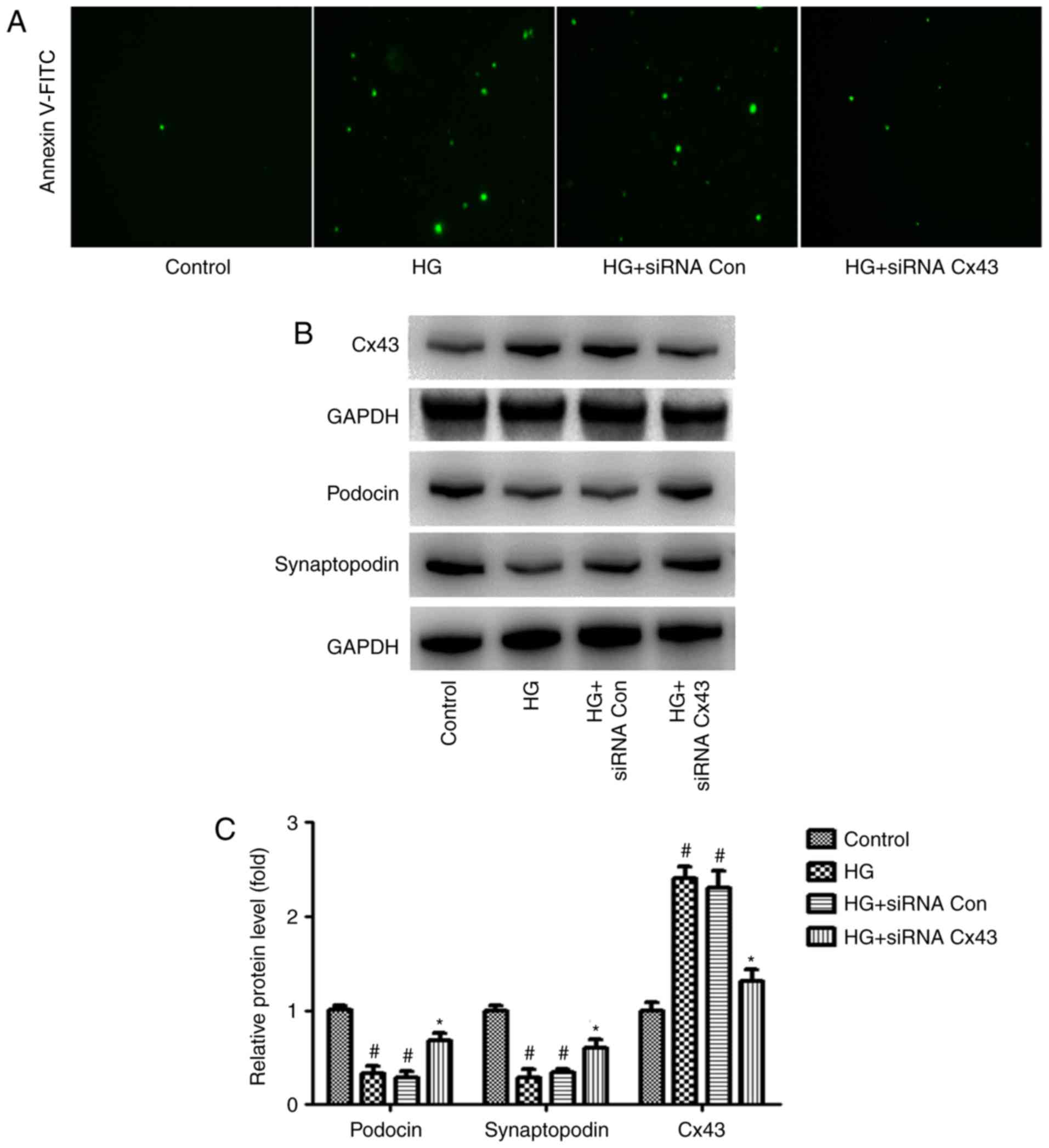
Cx43 silencing attenuates HG-induced
podocyte injury
It was observed that podocyte injury increased
significantly in the HG group, as did the expression of Cx43,
suggesting that abnormal activation of Cx43 may be involved in
HG-induced podocyte injury. To examine this hypothesis, Cx43
function was inhibited with Cx43 siRNA in the podocytes.
siRNA-control served as a negative control. Interestingly,
following transfection with Cx43 siRNA, we observed that, compared
with the siRNA-control group, podocyte injury was markedly
attenuated, as evidenced by the number of Annexin V/PI-positive
cells (green) (Fig. 1A). In
addition, increased levels of podocin and synaptopodin (surrogate
markers for podocytes) were observed in podocytes transfected with
Cx43 siRNA via western blot analysis (Fig. 1B). These findings indicate that
the inhibition of Cx43 plays a protective role against HG-induced
podocyte injury.
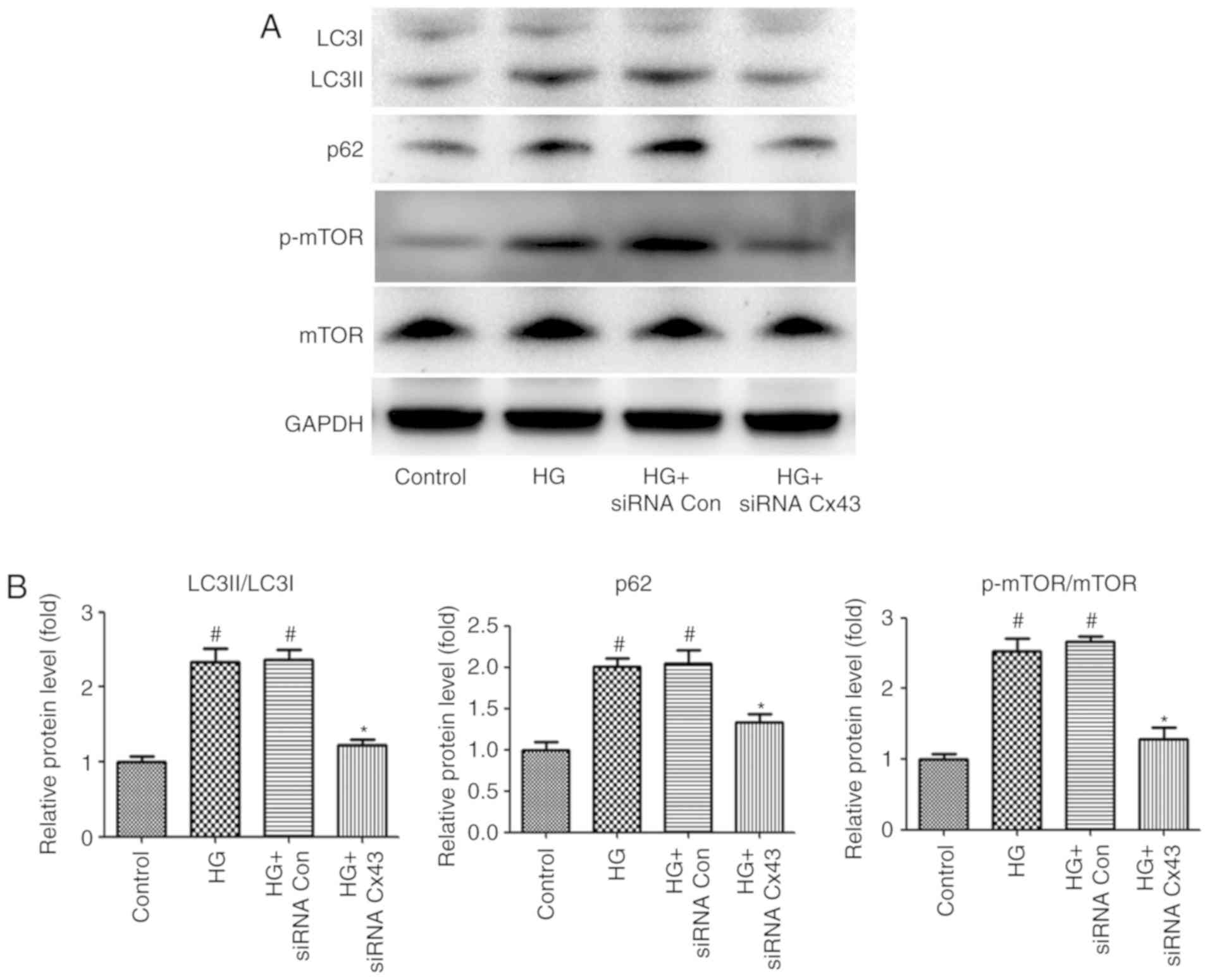
Cx43 negatively regulates autophagic flux
by enhancing mTOR signaling in HG-induced podocytes
In addition, impaired autophagic flux was also
observed in HG-induced podocytes, as demonstrated by the increased
expression levels of p62 and LC3II/LC3I. Convincing evidence has
demonstrated that connexins play a key role in autophago-some
formation (16). To examine
whether Cx43 regulates autophagic flux in HG-induced podocytes,
Cx43 siRNA was also used to inhibit Cx43 expression. Of note,
compared with podocytes transfected with NC-siRNA, the LC3II/LC3I
ratio and the level of p62 expression were markedly decreased in
Cx43 siRNA-treated podocytes, as detected by western blotting,
indicating that Cx43 also plays a key role in regulating autophagic
flux. Subsequently, the exact mechanism of the regulatory role of
Cx43 on autophagic flux was investigated. Accumulating evidence has
demonstrated that mTOR signaling is a pharmacological target for
autophagy regulation (17).
Therefore, attention was focused on mTOR signaling. The results
revealed that the p-mTOR/mTOR protein ratio decreased significantly
in the Cx43 siRNA group (P<0.05; Fig. 2), confirming that Cx43 inhibited
autophagy by activating the mTOR signaling pathway in HG-induced
podocytes.
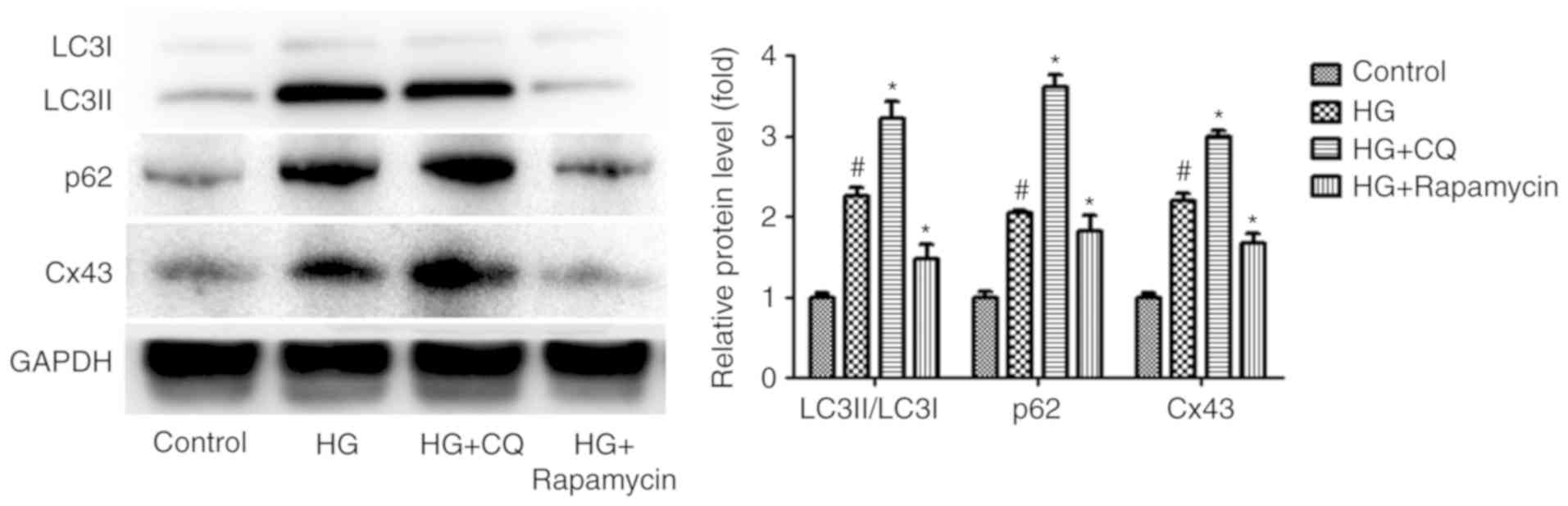
Impaired autophagic flux blocks the
degradation of Cx43 in HG-induced podocytes
Accumulating evidence demonstrates that Cx43 is also
regulated by autophagy under certain pathological conditions
(18,19), suggesting that autophagy may
regulate Cx43 expression in HG-induced podocytes. To examine this
hypothesis, CQ and RP, acting as an autophagy inhibitor and an
autophagy inducer, respectively (20,21), were used to regulate autophagic
flux. Interestingly, compared with the HG group, the level of Cx43
increased significantly in the HG + CQ group (P<0.05). By
contrast, the level of Cx43 decreased significantly in the HG + RP
group compared with the HG group (P<0.05; Fig. 3). These results indicated that
Cx43 activation is also regulated by impaired autophagic flux in
HG-induced podocytes.
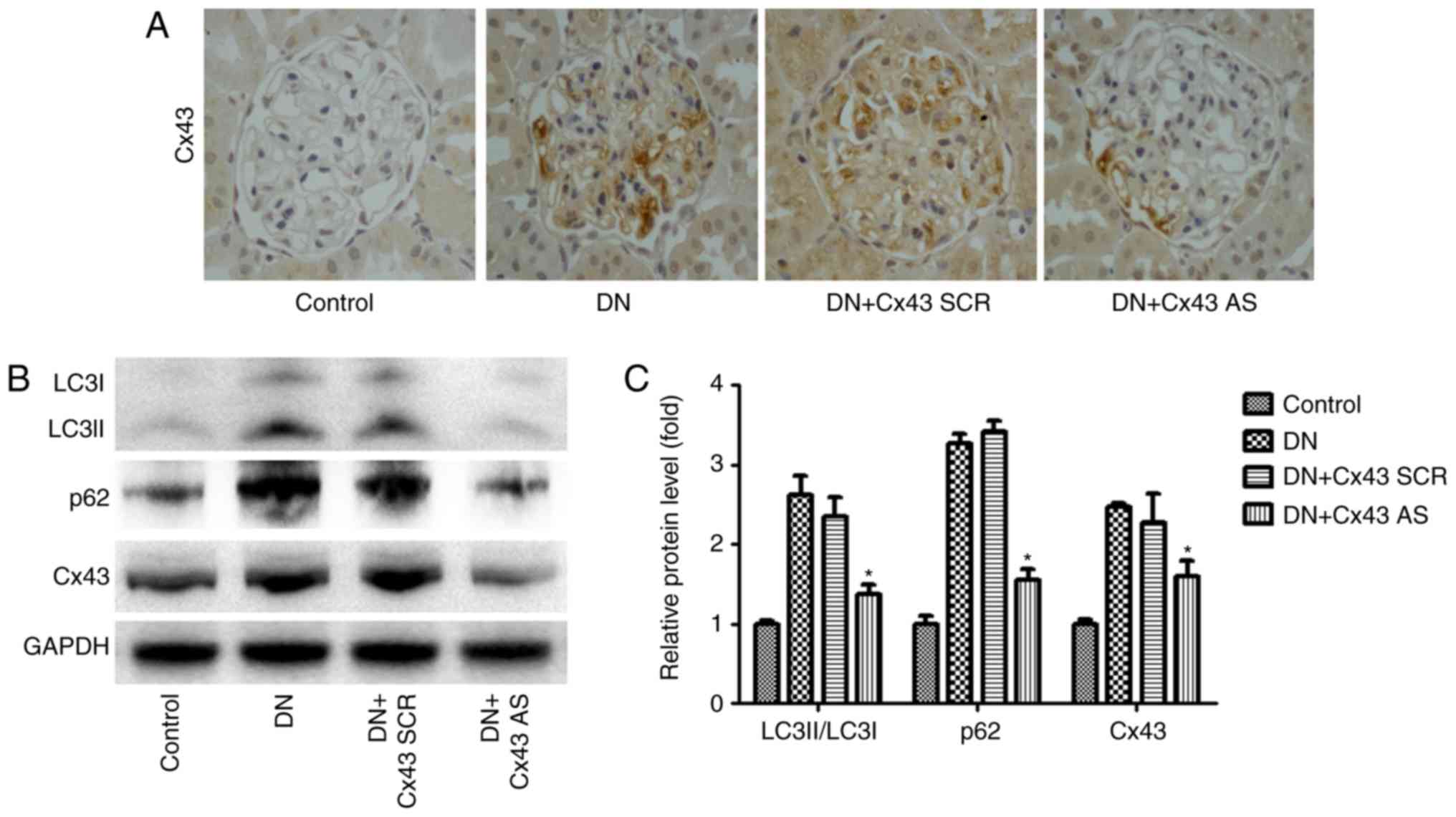
Cx43 silencing improves autophagic flux
in the glomeruli of DN rats
To determine the effect of Cx43 on autophagic flux
in the glomeruli in a DN model in vivo, Cx43 siRNA or
scrambled negative control siRNA was administered to the rats via
the tail vein. After induction of diabetes, the DN rats and Cx43
siRNA-treated DN rats were compared in terms of body weight, blood
urea nitrogen (BUN), urine protein and blood glucose levels. Of
note, body weight was higher while blood glucose, urine protein and
BUN levels were lower in Cx43 siRNA-treated diabetic rats compared
with their control diabetic littermates (Table I). Immunohistochemical analysis
revealed that Cx43 siRNA treatment significantly reduced Cx43
expression in the glomeruli (Fig.
4A). Interestingly, compared with scrambled negative
control-injected mice, Cx43 siRNA administration efficiently
reversed the enhanced autophagic flux, as detected by western
blotting (Fig. 4B). Thus, the
inhibition of Cx43 attenuated the enhanced autophagic flux in the
glomeruli of rats with DN.
 | Table IComparison of body weight, BUN, urine
protein and blood glucose levels among different rat groups after
induction of diabetes. |
Table I
Comparison of body weight, BUN, urine
protein and blood glucose levels among different rat groups after
induction of diabetes.
| Groups | Body weight
(g) | Blood glucose
(mmol/l) | BUN (mmol/l) | Urine protein
(mg/24 h) |
| Control | 501.6±14.1 | 6.6±0.3 | 4.6±0.6 | 8.2±0.5 |
| DN | 310.5±7.9a | 30.5±2.3a | 9.7±1.3a | 24.6±4.3a |
| DN + Cx43 SCR | 292.3±7.3a | 29.8±2.1a | 8.7±1.1a | 25.8±5.1a |
| DN + Cx43 AS | 433.5±8.5b | 28.7±2.3 | 5.6±0.7b | 12.8±3.1b |
Cx43 negatively regulates autophagic flux
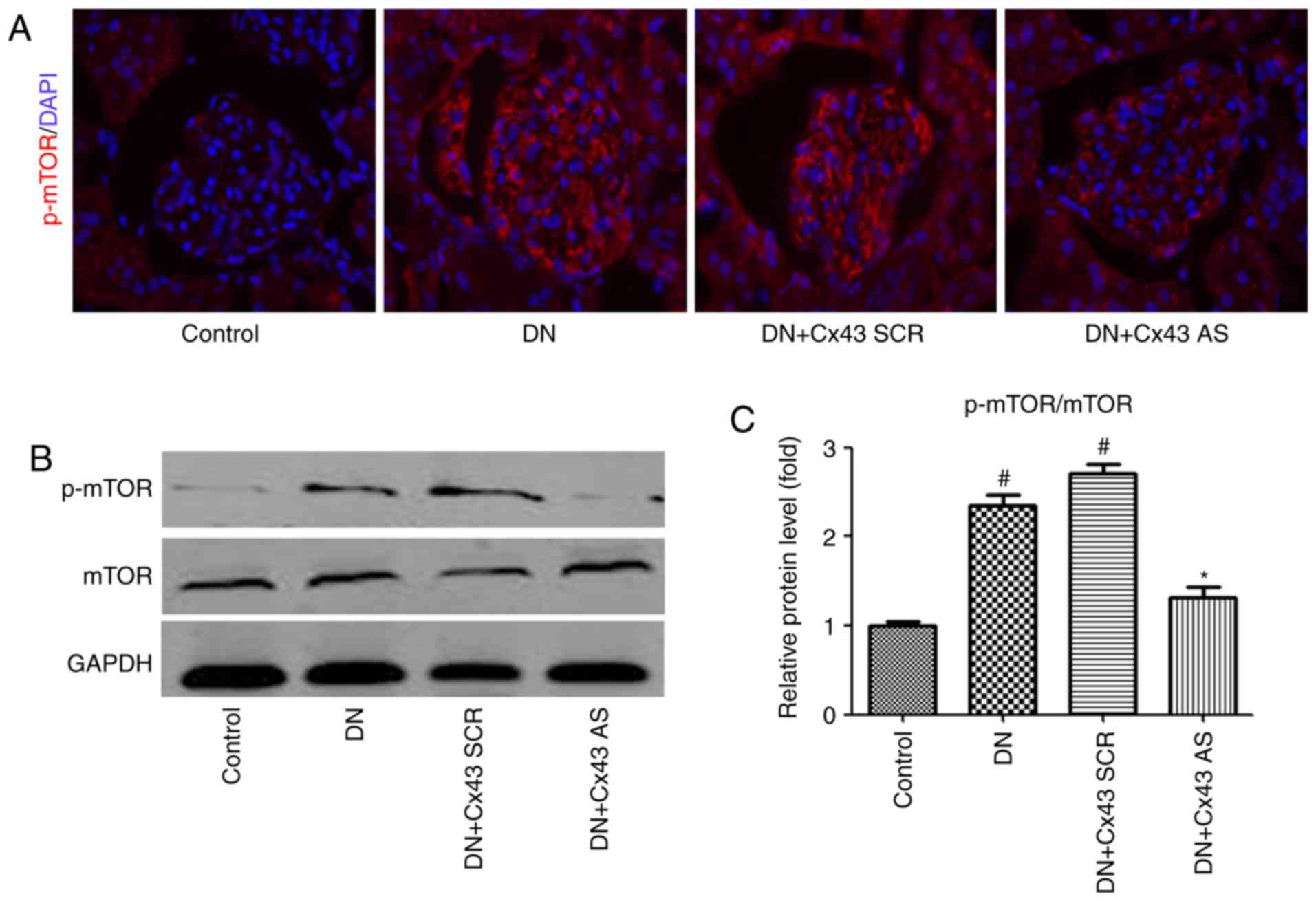
by enhancing mTOR signaling in the glomeruli of DN rats
To examine whether mTOR signaling is involved in the
regulatory effect of Cx43 on autophagic flux in vivo, mTOR
levels were measured. Consistently with the results in
vitro, immunofluorescence analysis revealed that the number of
p-mTOR-positive cells decreased in the glomeruli of DN rats
following treatment with Cx43 siRNA (Fig. 5A). Furthermore, the p-mTOR/mTOR
protein ratio was markedly decreased, as confirmed by western
blotting (Fig. 5B). Therefore,
activation of the mTOR pathway may negatively contribute to
Cx43-regulated autophagic flux.
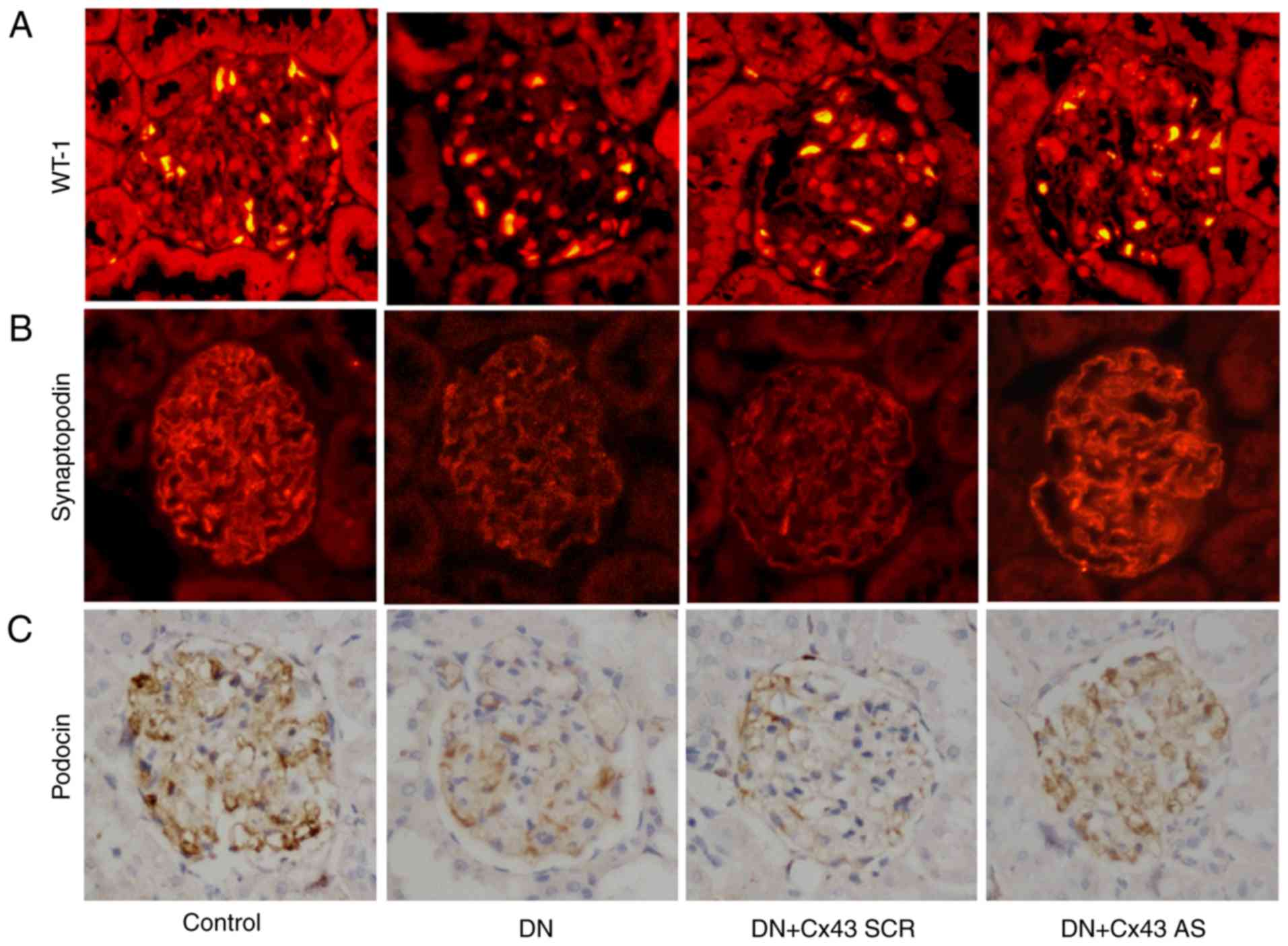
Cx43 silencing attenuates podocyte injury
in DN rats
To determine the role of Cx43 in podocyte injury
in vivo, immunostaining for WT-1 and synaptopodin was
performed. The results revealed that the number of podocytes was
significantly increased following treatment with Cx43 siRNA
(P<0.05; Fig. 6A and B).
Similarly, compared with the NC-siRNA-treated DN rats, podocin (a
marker for podocytes) also increased after knockdown of Cx43, as
assessed by immunohistochemical staining analysis (Fig. 6C). Thus, Cx43 activation appears
to promote podocyte injury.
Discussion
The critical role of podocyte injury, the
characteristic pathological change of DN, in the initiation and
progression of DN has been recognized for several years, and
impaired autophagy appears to play a key role in podocyte injury
(22,23). It was recently reported that Cx43
is involved in the autophagic process (24). However, whether Cx43 plays a
critical role in podocyte autophagy in DN remains largely unknown.
The present study demonstrated the involvement of increases in Cx43
expression and impaired autophagic flux in HG-induced podocyte
injury. The silencing of Cx43 expression ameliorated impaired
autophagic flux and reduced podocyte injury by suppressing the mTOR
pathway. In vivo studies indicated that the inhibition of
Cx43 improved impaired autophagic flux in STZ-induced DN animal
models. Furthermore, the pathogenic effect of Cx43 on podocyte
injury was also confirmed. These findings may facilitate the
identification of novel therapeutic targets for the treatment of
podocyte injury in DN.
Autophagy ('self-eating') is a tightly regulated
process that delivers senescent intracellular constituents to
lysosomes for degradation in order to maintain intracellular
homeostasis. An emerging body of research demonstrated that
autophagy regulation is closely linked to human health (25,26). Consistently with previous studies
on aldosterone-induced podocyte injury (27), we herein observed that impaired
autophagic flux was involved in HG-induced podocyte injury.
Furthermore, increased Cx43 expression was also observed in
association with this pathogenic process. However, whether Cx43
plays a critical role in impaired autophagic flux remains
elusive.
Accumulating evidence has demonstrated that Cx43 may
be involved in the process of podocyte injury in various kidney
diseases (10,11,28). Our in vitro and in
vivo data demonstrated that the expression of Cx43 was also
elevated by treatment with HG, which may facilitate podocyte
injury. Interestingly, after silencing Cx43 expression, HG-induced
injury in cultured podocytes was partially attenuated by improving
impaired autophagic flux, suggesting that Cx43 was partially
involved in HG-induced podocyte injury and that the amelioration of
impaired autophagic flux represents a novel mechanism mediating the
effect of Cx43 on podocyte injury. However, the exact regulatory
mechanisms of Cx43 on impaired autophagic flux have yet to be fully
elucidated.
mTOR is a well-known master regulator of cellular
metabolism, growth and proliferation in response to a wide range of
triggers. The deregulation of mTOR signaling, which plays a key
role in the regulation of autophagy, has been implicated in a
number of human diseases, including diabetes, neurodegenerative
diseases and cancer (29,30). Interestingly, in the present
study, the mTOR signaling pathway acted as a critical regulator
involved in Cx43/autophagy-mediated podocyte injury in DN. It was
observed that mTOR signaling was activated after stimulation with
HG in vivo and in vitro, indicating that an
mTOR-dependent mechanism may be involved in Cx43-mediated podocyte
injury in DN, which is consistent with previous results (31,32). Furthermore, after inhibition of
Cx43 function by Cx43 siRNA, the p-mTOR/mTOR protein ratio
decreased markedly, followed by a decrease in autoph-agic flux,
indicating that Cx43 may regulate autophagy flux by negatively
affecting mTOR signaling, which may be partially responsible for
podocyte injury in DN.
In addition, Cx43 was found to be regulated by
autophagic flux. It has been reported that the initiation of
autophagy includes the formation of the phagophore, the initial
sequestering compartment, which expands into an autophagosome.
Completion of the autophagosome is followed by fusion with
lysosomes and degradation of its contents, allowing complete flux,
or flow, through the entire pathway. As RP promotes initiation of
the autophagic flux, LC3I to LC3II turnover is increased in the
glomeruli. Of note, LC3II is also a selective substrate of
autophagy. Hence, LC3II degradation is blocked in the presence of
CQ, which inhibits the acidification of organelles and,
subsequently, autophagosome-lysosome fusion (33). This may explain why CQ treatment
caused a greater accumulation of LC3II in the HG + CQ group when
compared with the HG + RP group in the present study (even though
CQ is an inhibitor of autophagy). Accordingly, compared with the HG
group, the level of Cx43 increased significantly in the HG + CQ
group. On the contrary, the level of Cx43 decreased significantly
in the HG + RP group, indicating that a negative feedback mechanism
is involved in Cx43-modulated podocyte injury to maintain the
homeostasis of podocytes.
In summary, the findings of the present study
uncovered a previously unknown mechanism implicated in podocyte
injury under DN conditions. It was confirmed that upregulation of
Cx43 expression negatively regulates autophagy by activating the
mTOR signaling pathway in DN, while inhibiting Cx43 expression
improved impaired autophagic flow and reduced podocyte damage.
Hence, the Cx43-autophagy loop may be used as a novel therapeutic
target for the development of new drugs for DN.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant. no. 81670650); the
Natural Science Foundation of Jiangsu Province Grant (grant. nos.
BK20161071 and BK20161599); the Jiangsu Province Women and Children
Health Key Talents (grant. no. FRC201737); the Nanjing Medical
Science and Technique Development Foundation (grant. no. QRX17106);
and the Health Commission Foundation of Jiangsu Province (grant.
no. H2017002).
Availability of data and materials
The datasets analyzed during the present study are
available from the corresponding author on reasonable request.
Authors' contributions
JJ, WG and AZ designed the study, performed the
experiments, analyzed the results, and wrote and edited the
manuscript. YZ, CN and MY performed the experiments and discussed
the results. XZ and HS performed the experiments and analyzed the
data. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The experimental procedures in the present study
were approved by Nanjing Medical University (Nanjing, China).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Gnudi L, Coward RJ and Long DA: Diabetic
nephropathy: Perspective on novel molecular mechanisms. Trends
Endocrinol Metab. 27:820–830. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Susztak K, Raff AC, Schiffer M and
Böttinger EP: Glucose- induced reactive oxygen species cause
apoptosis of podocytes and podocyte depletion at the onset of
diabetic nephropathy. Diabetes. 55:225–233. 2006. View Article : Google Scholar
|
|
3
|
Madhusudhan T, Wang H, Dong W, Ghosh S,
Bock F, Thangapandi VR, Ranjan S, Wolter J, Kohli S, Shahzad K, et
al: Defective podocyte insulin signalling through p85-XBP-1
promotes ATF6-dependent maladaptive ER-stress response in diabetic
nephropathy. Nat Commun. 6:64962015. View Article : Google Scholar
|
|
4
|
Long J, Badal SS, Ye Z, Wang Y, Ayanga BA,
Galvan DL, Green NH, Chang BH, Overbeek PA and Danesh FR: Long
noncoding RNA Tug1 regulates mitochondrial bioenergetics in
diabetic nephropathy. J Clin Invest. 126:4205–4218. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mizushima N and Komatsu M: Autophagy:
Renovation of cells and tissues. Cell. 147:728–741. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tagawa A, Yasuda M, Kume S, Yamahara K,
Nakazawa J, Chin-Kanasaki M, Araki H, Araki S, Koya D, Asanuma K,
et al: Impaired podocyte autophagy exacerbates proteinuria in
diabetic nephropathy. Diabetes. 65:755–767. 2016. View Article : Google Scholar
|
|
7
|
Liu Y, Zhang J, Wang Y and Zeng X: Apelin
involved in progression of diabetic nephropathy by inhibiting
autophagy in podocytes. Cell Death Dis. 8:e30062017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kume S, Yamahara K, Yasuda M, Maegawa H
and Koya D: Autophagy: Emerging therapeutic target for diabetic
nephrop-athy. Semin Nephrol. 34:9–16. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ribeiro-Rodrigues TM, Martins-Marques T,
Morel S, Kwak BR and Girão H: Role of connexin 43 in different
forms of intercel-lular communication-gap junctions, extracellular
vesicles and tunneling nanotubes. J Cell Sci. 130:3619–3630. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sawai K, Mukoyama M, Mori K, Yokoi H,
Koshikawa M, Yoshioka T, Takeda R, Sugawara A, Kuwahara T, Saleem
MA, et al: Redistribution of connexin43 expression in glomerular
podocytes predicts poor renal prognosis in patients with type 2
diabetes and overt nephropathy. Nephrol Dial Transplant.
21:2472–2477. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yang M, Wang B, Li M and Jiang B: Connexin
43 is involved in aldosterone-induced podocyte injury. Cell Physiol
Biochem. 34:1652–1662. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Prakoura N, Kavvadas P and Chadjichristos
CE: Connexin 43: A new therapeutic target against chronic kidney
disease. Cell Physiol Biochem. 49:9852018. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Murphy SF, Varghese RT, Lamouille S, Guo
S, Pridham KJ, Kanabur P, Osimani AM, Sharma S, Jourdan J, Rodgers
CM, et al: Connexin 43 inhibition sensitizes chemoresistant
glioblastoma cells to temozolomide. Cancer Res. 76:139–149. 2016.
View Article : Google Scholar :
|
|
14
|
Zhang A, Han Y, Wang B, Li S and Gan W:
Beyond gap junction channel function: The expression of Cx43
contributes to aldosterone-induced mesangial cell proliferation via
the ERK1/2 and PKC pathways. Cell Physiol Biochem. 36:1210–1222.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang B, Xu X, He X, Wang Z and Yang M:
Berberine improved aldo-induced podocyte injury via inhibiting
oxidative stress and endoplasmic reticulum stress pathways both in
vivo and in vitro. Cell Physiol Biochem. 39:217–228. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bejarano E, Yuste A, Patel B, Stout RF Jr,
Spray DC and Cuervo AM: Connexins modulate autophagosome
biogenesis. Nat Cell Biol. 16:401–414. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kim YC and Guan KL: mTOR: A pharmacologic
target for autophagy regulation. J Clin Invest. 125:25–32. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lichtenstein A, Minogue PJ, Beyer EC and
Berthoud VM: Autophagy: A pathway that contributes to connexin
degradation. J Cell Sci. 124:910–920. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Martins-Marques T, Catarino S, Zuzarte M,
Marques C, Matafome P, Pereira P and Girão H: Ischaemia-induced
autophagy leads to degradation of gap junction protein connexin43
in cardiomyocytes. Biochem J. 467:231–245. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Li C, Ye L, Yang L, Yu X, He Y, Chen Z, Li
L and Zhang D: Rapamycin promotes the survival and adipogenesis of
ischemia-challenged adipose derived stem cells by improving
autophagy. Cell Physiol Biochem. 44:1762–1774. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kimura T, Takabatake Y, Takahashi A and
Isaka Y: Chloroquine in cancer therapy: A double-edged sword of
autophagy. Cancer Res. 73:3–7. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Yasuda-Yamahara M, Kume S, Tagawa A,
Maegawa H and Uzu T: Emerging role of podocyte autophagy in the
progression of diabetic nephropathy. Autophagy. 11:2385–2386. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu M, Liang K, Zhen J, Zhou M, Wang X,
Wang Z, Wei X, Zhang Y, Sun Y, Zhou Z, et al: Sirt6 deficiency
exacerbates podo-cyte injury and proteinuria through targeting
Notch signaling. Nat Commun. 8:4132017. View Article : Google Scholar
|
|
24
|
Kim SN, Kwon HJ, Im SW, Son YH, Akindehin
S, Jung YS, Lee SJ, Rhyu IJ, Kim IY, Seong JK, et al: Connexin 43
is required for the maintenance of mitochondrial integrity in brown
adipose tissue. Sci Rep. 7:71592017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kroemer G: Autophagy: A druggable process
that is deregulated in aging and human disease. J Clin Invest.
125:1–4. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Choi AM, Ryter SW and Levine B: Autophagy
in human health and disease. N Engl J Med. 368:651–662. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang B, Ding W, Zhang M, Li H, Guo H, Lin
L, Chen J and Gu Y: Role of FOXO1 in aldosterone-induced autophagy:
A compensatory protective mechanism related to podocyte injury.
Oncotarget. 7:45331–45351. 2016.PubMed/NCBI
|
|
28
|
Haefliger JA, Demotz S, Braissant O, Suter
E, Waeber B, Nicod P and Meda P: Connexins 40 and 43 are
differentially regulated within the kidneys of rats with
renovascular hypertension. Kidney Int. 60:190–201. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Laplante M and Sabatini DM: mTOR signaling
in growth control and disease. Cell. 149:274–293. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Shimobayashi M and Hall MN: Making new
contacts: The mTOR network in metabolism and signalling crosstalk.
Nat Rev Mol Cell Biol. 15:155–162. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gödel M, Hartleben B, Herbach N, Liu S,
Zschiedrich S, Lu S, Debreczeni-Mór A, Lindenmeyer MT, Rastaldi MP,
Hartleben G, et al: Role of mTOR in podocyte function and diabetic
nephrop-athy in humans and mice. J Clin Invest. 121:2197–2209.
2011. View
Article : Google Scholar
|
|
32
|
Cinà DP, Onay T, Paltoo A, Li C, Maezawa
Y, De Arteaga J, Jurisicova A and Quaggin SE: Inhibition of MTOR
disrupts autophagic flux in podocytes. J Am Soc Nephrol.
23:412–420. 2012. View Article : Google Scholar :
|
|
33
|
Klionsky DJ, Abdelmohsen K, Abe A, Abedin
MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD,
Adeli K, et al: Guidelines for the use and interpretation of assays
for monitoring autophagy (3rd edition). Autophagy. 12:1–222. 2016.
View Article : Google Scholar : PubMed/NCBI
|