Introduction
Atrial fibrillation (AF) is the most common form of
cardiac arrhythmia and is a major contributor to morbidity,
mortality and health care expenditure (1). Due to abnormal electrical activity
and blood flow, AF is significantly associated with severe cardiac
conditions, including stroke, heart failure and renal diseases
(2). The mechanisms underlying AF
are intricate and are typically classified as triggers or
substrates, which result in electrophysiological remodeling and
structural remodeling. Multiple molecular factors have been
reported to be involved in pathophysiological progression, such as
fibrosis, abnormal Ca2+ handling and inflammation
(3-5). However, the exact mechanisms
underlying the development and progression of AF are incompletely
understood (3-5). In addition, unlike protein-coding
genes, little is known about the long non-coding RNA (lncRNA) in
AF; however, it has been demonstrated in a recent study that lncRNA
may also serve an important role in AF by acting as competing
endogenous RNAs (6). Although a
number of therapies have been used to treat AF to prevent
thrombotic events, cardioversion and control heart rhythm, the
limited efficacy for cardioversion or correction of AF and the
detrimental side effects of antiarrhythmic drugs or anticoagulants
contribute to the challenge faced by clinicians when managing
patients with AF (7). Therefore,
there is an urgent requirement to elucidate the precise molecular
mechanisms underlying AF, in order to identify appropriate
therapeutic targets.
Recently, a number of algorithms and research
methods have been developed to examine the potential mechanisms of
gene networks, which provide comprehensive insights into specific
diseases or conditions. Weighted gene co-expression network
analysis (WGCNA) is one such tool, which divides gene co-expression
networks of complex biological processes into several
characteristic modules (8). The
modules are then used to analyze their association with clinical
traits to find functional key modules (8). WGCNA has been used for analyzing a
number of biological processes, including ontogeny (9), cancer (10-12) and mental disorders (13), and has been validated as a
valuable method to identify underlying mechanisms, potential
biomarkers or therapeutic targets in different types of diseases by
placing a focus on key modules. Previous studies on the mechanisms
of AF have primarily concentrated on specific pathophysiological
functions, with relatively fewer studies identifying comprehensive
regulatory networks. Therefore, in the present study, WGCNA was
used to determine networks associated with AF.
The GSE79768 dataset was used in the present study,
which contained 26 samples from paired left atria (LA) and right
atria (RA) in patients with persistent AF or sinus rhythm (SR)
(14). Two key modules with the
highest level of significance correlation with AF were identified,
and the 3 genes with the highest intramodular connectivity were
selected as the hub genes in the respective modules for AF. As LA
is more likely to serve as a driver of AF and AF-associated stroke
than RA (15-17), the blue module was selected as a
module of interest, as it was significantly correlated with both AF
and LA, to further examine potential associations. Differentially
expressed gene (DEG) analysis was additionally used, and the
results were overlapped with WGCNA to further supplement and
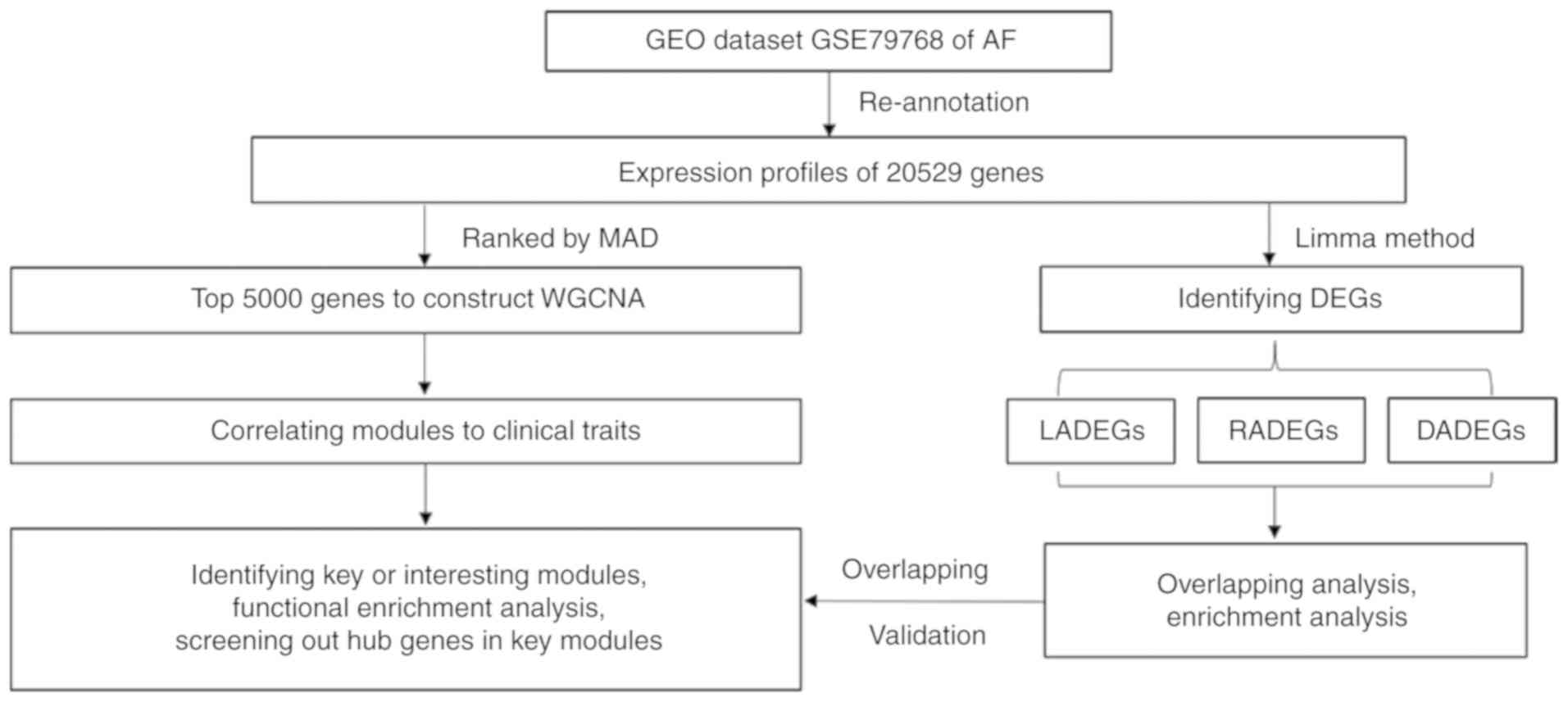
validate the results. The workflow used in the present study is
presented in Fig. 1. The results
may provide novel insight into the underlying mechanisms of AF and
may assist the identification of potential biomarkers for diagnosis
and treatment of AF.
Materials and methods
Dataset information
The AF dataset, GSE79768, was obtained from the NCBI
Gene Expression Omnibus (GEO; https://www.ncbi.nlm.nih.gov/geo/). The dataset
contains data from 13 pairs of left and right atrial appendages
from 7 patients with persistent AF and 6 patients with SR (14). The dataset consisted of patients
with AF whom presented with persistent AF for >6 months, or
patients with SR with no evidence of AF and whom did not use any
anti-arrhythmic drugs (14). The
characteristics of the patients are summarized in Table SI, which may be downloaded from
GEO (ncbi.nlm.nih.gov/geo/).
Microarray probe reannotation
Reannotation of microarray probes improves accuracy
and makes it possible to identify new transcripts (18). Therefore, the probes of HG-U133_
Plus_2.0 array in the dataset was reannotated based on a similar
method used in previous studies (19,20). The probe sequences were downloaded
from Affymetrix (affyme-trix.com), and remapped to the human genome
(GRCh38 release 94 primary assembly; ensembl.org/index.html) using
the R package 'Rsubread' (http://www.bioconductor.org/packages/release/bioc/html/Rsubread.html)
supported by the Bioconductor package (bioconductor.org/) (21). Uniquely mapped probes with no
mismatches were retained. Subsequently, the chromosomal positions
of these probes were matched to the corresponding genome annotation
database in Ensembl using the R package 'GenomicRanges' (http://www.biocon-ductor.org/packages/release/bioc/html/GenomicRanges.html).
Probe sets that were mapped to >1 gene were
removed to ensure the reliability of the reannotation. Following
reannotation and removal of duplicated probes, 20,529 genes were
retained, which included 16,285 protein coding genes and 3,243
lncRNAs according to the biotypes identified by Ensembl. As the
non-coding genes may be associated with AF, WGCNA was performed on
the top 5,000 genes from the reannotation results without
partitions and other exclusions.
WGCNA
RAW data from the GSE79768 dataset were preprocessed
and normalized using the R package 'affy' (http://www.bioconductor.org/packages/release/bioc/html/affy.html)
and the 'rma' method. Subsequently, the genes were ranked by median
absolute deviation from large to small, and the top 5,000 genes
were selected for WGCNA using the R package 'WGCNA' (8,22).
The power parameter ranging from 1-20 was screened out using the
'pickSoftThreshold' (package WGCNA) function. A suitable soft
threshold of 5 was selected, as it met the degree of independence
of 0.85 with the minimum power value. Subsequently, modules were
constructed, and following dynamic branch cutting with a merging
threshold of 0.25, 20 modules were obtained. The resulting gene
network was visualized as a heatmap by randomly selecting 1,000
genes based on Topological Overlap Matrix dissimilarity and their
cluster dendrogram.
Identification of key or modules of
interest
Correlation between module eigengenes and clinical
traits were analyzed to identify modules of interest that were
significantly associated with clinical traits. The correlation
values were displayed within a heatmap. Modules correlated with AF
most significantly were considered as the key modules of AF. Gene
significance (GS) was defined as the correlation between gene
expression and each trait. In addition, module membership (MM) was
defined as the association between gene expression and each module
eigengene. Subsequently, the correlation between GS and MM were
examined to verify certain module-trait associations. The
correlation analyses in this study were performed using the Pearson
correlation as described in the 'WGCNA' package (22).
Functional enrichment analysis of modules
of interest
The genes in each module of interest were extracted
from the network and enrichment analysis was performed to further
explore the functions of the respective modules. The R package
'clusterProfiler' (23)
(http://www.bioconductor.org/packages/release/bioc/html/clusterProfiler.html)
was used to perform Gene Ontology (GO) (24,25) and Kyoto Encyclopedia of Genes and
Genomes (KEGG) (26-28) pathway enrichment analysis.
P<0.05 was set as the significance threshold, and the enrichment
results of GO biological process (BP), GO molecular function (MF),
GO cellular component (CC) and KEGG pathways in each module of
interest module were obtained. Of the results that exceeded the
threshold, the top 6 KEGG pathways and top 6 terms of each GO
domain were identified.
Module visualization and identification
of hub genes
The intramodular connectivity of genes in the
corresponding modules of interest was measured using module
eigengene based connectivity kME. The top 30 genes of each modules
of interest, which represented the central status in the module
gene network, were selected to visualize the subordinate module
using Cytoscape (v.3.7.0; cytoscape.org/) (29). Subsequently, two key modules were
chosen which exhibited the highest levels of positive or negative
correlation with AF to search for hub genes for AF in the modules.
The top three genes with the highest kME were selected as the hub
genes in the corresponding module (30) and their GS for AF and intramodular
connectivity kME were determined to confirm the reliability of
these hub genes.
DEG analysis and interactions with the
modules of interest
The R package 'limma' was used to screen out DEGs
among three sets of comparisons between AF and SR (31,32); left atria DEGs (LADEGs), right
atria DEGs (RADEGs) and LA/RA ratio DEGs or DEGs of atrial
differences (DADEGs). The cutoff for DEGs was a |fold change
(FC)|>1.5 and P<0.05. The DEGs were visualized using the R
package 'ggplot2' (https://cran.r-project.org/web/packages/ggplot2/)
as a volcano plot (Fig. S1).
Subsequently, the DEGs were overlapped with the modules of interest
to identify potential links, the results of which are presented as
a Venn diagram using the R package 'VennDiagram' (https://cran.r-project.org/web/packages/VennDiagram/).
GO and KEGG enrichment analysis of upregulated or downregulated
genes in each set of DEGs was performed, and their associations
with the genes in the modules of interest were examined.
Validation of hub genes in public
database
The expression profiles of hub genes in the key
modules from the GSE79768 dataset were selected as representative
genes used for validation. To verify these hub genes in an external
dataset, GEO was searched with the key words 'atrial fibrillation'.
The inclusion criteria for the validation dataset were as follows:
i) Atria samples from individuals with persistent AF or SR; and ii)
the sample size of each group was >5. Based on these criteria, 2
appropriate datasets were identified; GSE2240 and GSE115574.
GSE2240 included data from 30 RA tissues obtained from 10 cardiac
patients with persistent AF and 20 cardiac patients with SR.
GSE115574 contained data from 14 LA tissues and 14 RA tissues from
patients with AF and severe mitral regurgitation, and 15 LA tissues
and 16 RA tissues from patients with AF and severe mitral
regurgitation. Hub genes in the key modules were assessed for
further validation. The expression profiles of these two microarray
datasets were examined using the same methods as aforementioned for
GSE79768.
Animal models
A total of 16 Sprague-Dawley male rats (body weight,
200-250g) were obtained from the Experimental Animal Center of
Medical School, Xi'an Jiaotong University, and were kept in a
specific-pathogen-free grade and temperature- and
humidity-controlled facility on a 12:12 h light: Dark cycle with
ad libitum access to food and water. This animal study was
approved by the Institutional Ethics Committee for Animal
Experiments of Xi'an Jiaotong University. All procedures conformed
to the Guide for the Care and Use of Laboratory Animals published
by the National Institutes of Health. Following acclimation under
normal conditions for 1 week, the 16 rats were randomly assigned
into 2 groups (control and AF). As described in a previous study
(33), AF was induced by tail
vein injection with acetylcholine (ACh) + CaCl2 (60
µg/ml Ach; Sigma-Aldrich + 10 mg/ml CaCl2;
Beijing Solarbio Science & Technology Co., Ltd.) daily at 1
ml/kg for 1 week in the AF group (n=8). Rats in the control group
(n=8) were injected with 0.9% normal saline daily via the tail vein
at 1 ml/kg for 1 week. At baseline, all the rats were anesthetized
with sodium pentobarbital [40 mg/kg; intraperitoneal (IP)].
Following anesthesia, the limb lead II electrocardiogram was
recorded with limb leads inserted into the rat's limbs to ensure
that all the rats had sinus rhythm at baseline. After 7 days of
treatment, electrocardiogram test was performed on the rats again
following anesthesia with sodium pentobarbital; 40 mg/kg IP). AF
was induced successfully in all the rats in the AF group.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) assay of atrial tissues
Total RNA was extracted from both left atrial and
right atrial tissues using TRIzol® Reagent (Thermo
Fisher Scientific, Inc.) following the manufacturer's protocol.
Purified RNA was reverse-transcribed into cDNA using PrimeScript™
RT Master Mix (Takara Bio, Inc.). Newly synthesized cDNA was
analyzed by RT-qPCR using an iQ5 Multicolor Real-Time PCR detection
system (Bio-Rad Laboratories, Inc.) with FastStart Essential DNA
Green Master (Roche Diagnostics, Ltd.) according to the
manufacturer's protocol. All the experiments were repeated in
triplicate. The housekeeping gene β-actin was selected as the
endogenous reference. The following primers were used in this
study: β-actin forward, CTAAGGCCAACCGTGAAAAG; β-actin reverse,
ACCAGAGGCATACAGGGACA; acetyl-CoA Acetyltransferase 1 (ACAT1)
forward, GAGCAGAGGAGCAACACCATA; ACAT1 reverse,
CTCAGCTTCTTCGCGGTGTT; death domain-containing protein CRADD (CRADD)
forward, GTGGTACAGGTTCCCTATCAG; CRADD reverse,
AGTGAGCGGAGAACTTGCTT; gypsy retrotransposon integrase 1 (GIN1)
forward, ATCGTGGCTGCGGTTAGAG; GIN1 reverse, GCCATCCTTCCACCAGTTCTT;
FTX transcript, XIST regulator (FTX) forward, CAGCAACACGCCAAGATGAA;
FTX reverse, TGGGCAGGTTTGTGCGTAT; minichromosome maintenance
complex component 3 associated protein (MCM3AP) forward,
CTGGACCTGCCATCCTTTGT; and MCM3AP reverse, GCTGCCATCTCCTGTGAACT. The
relative mRNA expression levels were then calculated using the
2−ΔΔCq method (34).
Statistical analysis
Comparisons among groups were analyzed using
analysis of variance followed by Tukey Honest Significant
Differences post hoc test. The statistical analyses were performed
in R v.3.5.1, and P<0.05 was considered to indicate a
statistically significant difference.
Results
Construction of co-expression
modules
The top 5,000 genes in 26 samples of paired left and
right atrial appendages from 6 patients with SR and 7 patients with
AF were used to construct the co-expression network. The results of
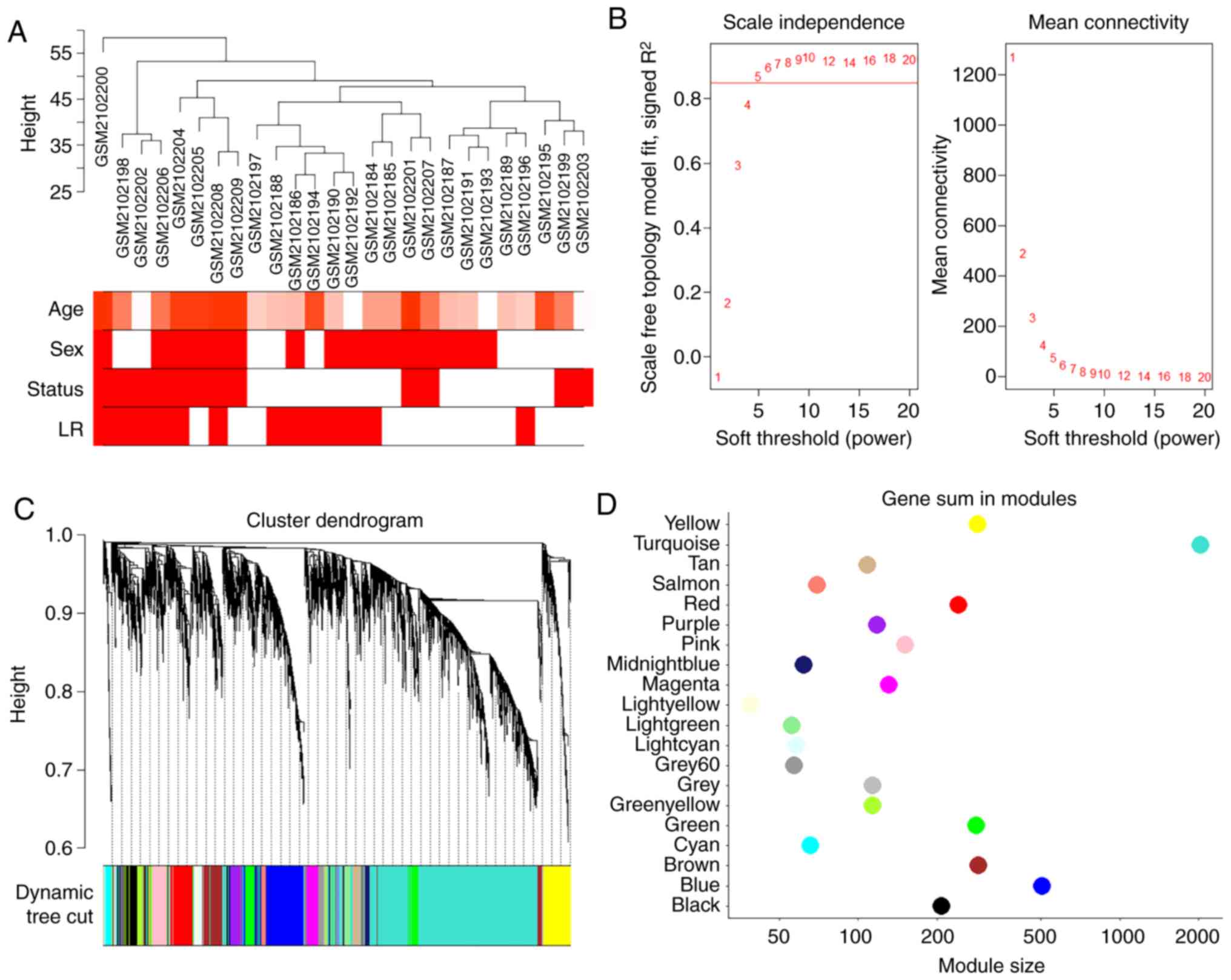
cluster analysis of the samples are demonstrated in Fig. 2A. A soft-threshold power was
introduced into the network topology, which affected the scale
independence and mean connectivity of the network. Following
screening, a soft-threshold of 5 was used to obtain the approximate
scale-free topology with a scale-free topology fit index >0.85
and the lowest power (Fig. 2B).
As indicated in Fig. 2C, 20
modules were identified, in which genes had similar co-expression
traits. The size of these 20 modules are presented in Fig. 2D.
The interaction associations among these modules
were analyzed. The network heatmap of a 1,000 randomly selected
genes indicates a relatively high level of independence among these
clusters (Fig. 3A). Furthermore,
both the eigengene adjacency heatmap and the eigengene dendrogram
indicated that these 20 modules could be divided into several
smaller groups (Fig. 3B and C),
suggesting that co-expression clusters with varying functions were
present in the genetic networks of AF.
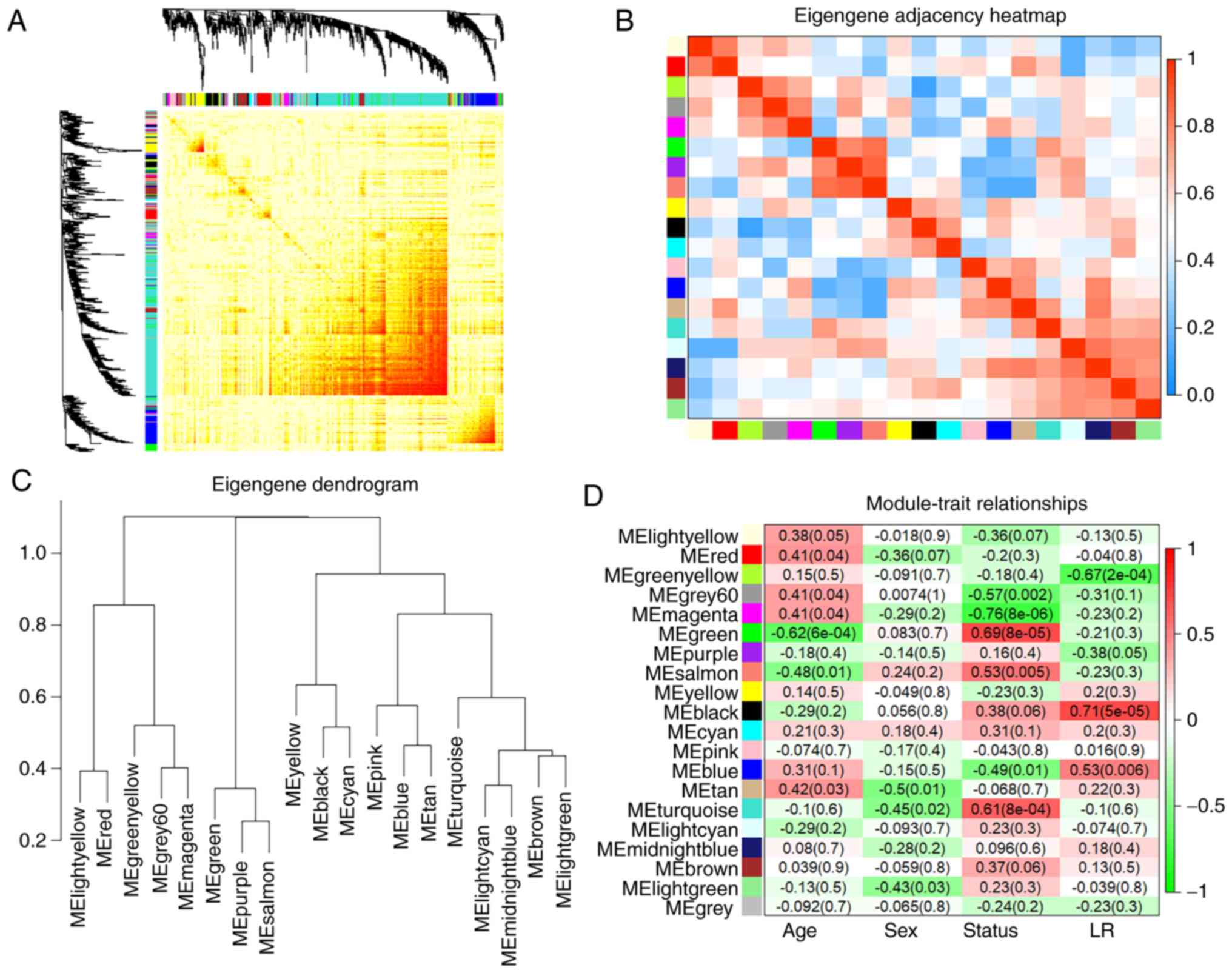 | Figure 3WGCNA module analysis. (A)
Interaction of co-expression genes based on TOM dissimilarity and
the cluster dendrogram of 1,000 randomly selected genes. The colors
of the axes represent respective modules. The intensity of the
yellow inside the heatmap represents the overlap degree of overlap,
with a darker yellow representing an increased overlap. (B)
Eigengene adjacency heatmap of different gene co-expression
modules. (C) An eigengene dendrogram identified groups of
correlated modules. (D) Heatmap of the correlation between clinical
traits including age, sex, status (AF and SR), the left or right
atria, and module eigengenes. Each column corresponds to a clinical
trait, and each row corresponds to a module. Each cell contains the
correlation coefficients which correspond to the cell color; green
represents negative correlation and red represents positive
correlation. The P-values are stated in the brackets. WGCNA,
weighted gene co-expression network analysis; TOM, Topological
Overlap Matrix; AF, atrial fibrillation; SR, sinus rhythm. |
Correlation between modules and clinical
traits
The module-trait associations were analyzed by
correlating module-sample eigengenes with clinical traits to
identify significant associations. The colors of all the modules
were selected at random to distinguish between modules. Focusing on
the status trait (AF and SR), the green module exhibited the
highest positive correlation (r=0.69; P<0.001) and the magenta
module (r=−0.76; P<0.001) exhibited the highest negative
correlation (Fig. 3D). Therefore,
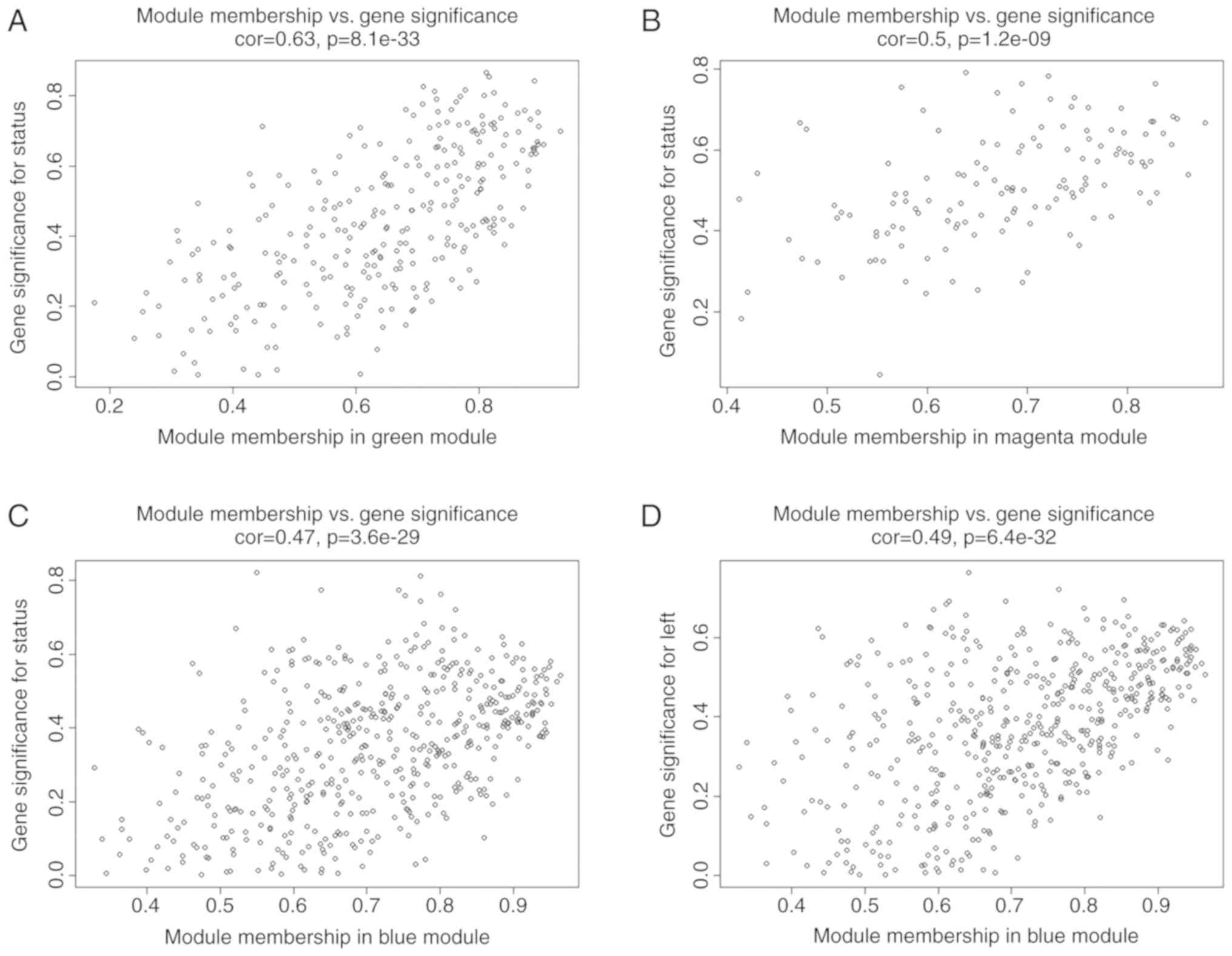
these 2 modules were identified as key modules for AF. Additional
associations were identified between MM and GS for specific traits.
The significant correlations between green and magenta MM and GS
for AF are presented in Fig. 4A and
B. As previous studies demonstrated that differential
left-to-right atria gene expression ratio may affect
arrhythmogenesis and thrombogenesis of AF (14), which may suggest a specific
co-expression pattern, the blue module was analyzed further as it
exhibited a significant and relatively substantial correlation with
both the LA (r=0.53; P=0.006) and AF (r=−0.49; P=0.01). Fig. 4C and D confirmed a significant
association between the blue MM with GS for both AF and LA.
Functional enrichment analysis in modules
of interest
GO and KEGG pathway enrichment analyses were
performed on the green, magenta and blue gene clusters, and the top
terms of each category are presented in Tables I and II. The KEGG pathway enrichment results
demonstrated that the genes in the green module were primarily
enriched in pathways associated with energy substance metabolism
including amino acid metabolism, glycometabolism and lipid
metabolism, in agreement with GO, BP and MF enrichment analysis.
Results of GO CC analysis indicated that genes in the green module
were primarily enriched in components associated with the
mitochondria such as the mitochondrial membrane and matrix. Genes
in the magenta module were enriched in several pathways primarily
associated with apoptosis and inflammatory infiltration, including
the Hippo signaling pathway, binding to receptor for advanced
glycation end products receptor and positive regulation of NF-κB
transcription factor activity. KEGG analysis indicated that the
genes in the blue module were primarily enriched in complement and
coagulation cascades. Furthermore, GO enrichment results
demonstrated that blue module genes were significantly associated
with complement activity and extracellular matrix (ECM) formation,
such as complement activation, cell-cell adhesion mediator activity
and ECM organization, which are all closely associated with
coagulation, nonspecific immunity and cardiac remodeling.
 | Table IKEGG enrichment analysis in
interesting modules. |
Table I
KEGG enrichment analysis in
interesting modules.
| Module | ID | Description | P-value | Count |
|---|
| Green | hsa00280 | Valine, leucine and
isoleucine degradation | 0.00013 | 5 |
| Green | hsa00250 | Alanine, aspartate
and glutamate metabolism | 0.003972 | 3 |
| Green | hsa01200 | Carbon
metabolism | 0.00591 | 5 |
| Green | hsa03420 | Nucleotide excision
repair | 0.008537 | 3 |
| Green | hsa00071 | Fatty acid
degradation | 0.009175 | 3 |
| Green | hsa01212 | Fatty acid
metabolism | 0.013585 | 3 |
| Magenta | hsa04390 | Hippo signaling
pathway | 0.000993 | 5 |
| Magenta | hsa04060 | Cytokine-cytokine
receptor interaction | 0.01245 | 5 |
| Magenta | hsa04550 | Signaling pathways
regulating pluripotency of stem cells | 0.033364 | 3 |
| Magenta | hsa00590 | Arachidonic acid
metabolism | 0.034535 | 2 |
| Magenta | hsa05224 | Breast cancer | 0.039373 | 3 |
| Magenta | hsa05226 | Gastric cancer | 0.040072 | 3 |
| Blue | hsa04610 | Complement and
coagulation cascades |
6.51×10−7 | 12 |
| Blue | hsa05133 | Pertussis | 0.000178 | 9 |
| Blue | hsa04976 | Bile secretion | 0.003199 | 7 |
| Blue | hsa05222 | Small cell lung
cancer | 0.005002 | 8 |
| Blue | hsa05150 | Staphylococcus
aureus infection | 0.009243 | 5 |
| Blue | hsa04530 | Tight junction | 0.017169 | 10 |
| Table IIGO enrichment analysis in interesting
modules. |
Table II
GO enrichment analysis in interesting
modules.
| Module | Category | ID | Description | P-value | Count |
|---|
| Green | BP | GO:0006520 | Cellular amino acid
metabolic process |
4.71×10−5 | 14 |
| Green | BP | GO:0009083 | Branched-chain
amino acid catabolic process | 0.0001 | 4 |
| Green | BP | GO:1901605 | Alpha-amino acid
metabolic process | 0.000106 | 11 |
| Green | BP | GO:0009063 | Cellular amino acid
catabolic process | 0.000125 | 8 |
| Green | BP | GO:0009081 | Branched-chain
amino acid metabolic process | 0.00019 | 4 |
| Green | BP | GO:1901606 | Alpha-amino acid
catabolic process | 0.000258 | 7 |
| Green | MF | GO:0016790 | Thiolester
hydrolase activity | 0.000692 | 4 |
| Green | MF | GO:0005496 | Steroid
binding | 0.000985 | 6 |
| Green | MF | GO:0016289 | CoA hydrolase
activity | 0.001351 | 3 |
| Green | MF | GO:0015485 | Cholesterol
binding | 0.002827 | 4 |
| Green | MF | GO:0032934 | Sterol binding | 0.004602 | 4 |
| Green | MF | GO:0048037 | Cofactor
binding | 0.009342 | 13 |
| Green | CC | GO:0005743 | Mitochondrial inner
membrane |
4.77×10−7 | 20 |
| Green | CC | GO:0044455 | Mitochondrial
membrane part |
2.56×10−6 | 13 |
| Green | CC | GO:0019866 | Organelle inner
membrane |
3.83×10−6 | 20 |
| Green | CC | GO:0005759 | Mitochondrial
matrix |
1.42×10−5 | 18 |
| Green | CC | GO:0098800 | Inner mitochondrial
membrane protein complex |
2.25×10−5 | 9 |
| Green | CC | GO:0098798 | Mitochondrial
protein complex |
5.73×10−5 | 12 |
| Magenta | BP | GO:0051091 | Positive regulation
of DNA binding transcription factor activity | 0.000163 | 8 |
| Magenta | BP | GO:0051092 | Positive regulation
of NF-kappaB transcription factor activity | 0.000236 | 6 |
| Magenta | BP | GO:0030901 | Midbrain
development | 0.000261 | 5 |
| Magenta | BP | GO:0061564 | Axon
development | 0.000312 | 11 |
| Magenta | BP | GO:0030593 | Neutrophil
chemotaxis | 0.000397 | 5 |
| Magenta | BP | GO:1990266 | Neutrophil
migration | 0.000662 | 5 |
| Magenta | MF | GO:0050786 | RAGE receptor
binding |
4.83×10−7 | 4 |
| Magenta | MF | GO:0005504 | Fatty acid
binding | 0.00122 | 3 |
| Magenta | MF | GO:0017147 | Wnt-protein
binding | 0.002081 | 3 |
| Magenta | MF | GO:0035325 | Toll-like receptor
binding | 0.002156 | 2 |
| Magenta | MF | GO:0036041 | Long-chain fatty
acid binding | 0.003685 | 2 |
| Magenta | MF | GO:0004115 | 3′,5′-cyclic-AMP
phosphodiesterase activity | 0.004916 | 2 |
| Magenta | CC | GO:0030017 | Sarcomere | 0.007341 | 5 |
| Magenta | CC | GO:0044449 | Contractile fiber
part | 0.010578 | 5 |
| Magenta | CC | GO:0030016 | Myofibril | 0.010794 | 5 |
| Magenta | CC | GO:0043025 | Neuronal cell
body | 0.011374 | 8 |
| Magenta | CC | GO:0034774 | Secretory granule
lumen | 0.01299 | 6 |
| Magenta | CC | GO:0043292 | Contractile
fiber | 0.013378 | 5 |
| Blue | BP | GO:0006958 | Complement
activation, classical pathway |
1.45×10−8 | 10 |
| Blue | BP | GO:0030198 | Extracellular
matrix organization |
9.86×10−8 | 30 |
| Blue | BP | GO:0043062 | Extracellular
structure organization |
2.34×10−7 | 32 |
| Blue | BP | GO:0002455 | Humoral immune
response mediated by circulating immunoglobulin |
3.57×10−7 | 10 |
| Blue | BP | GO:0006956 | Complement
activation |
1.54×10−6 | 11 |
| Blue | BP | GO:0030449 | Regulation of
complement activation |
2.89×10−6 | 9 |
| Blue | MF | GO:0098632 | Cell-cell adhesion
mediator activity |
7.45×10−5 | 8 |
| Blue | MF | GO:0017171 | Serine hydrolase
activity |
8.63×10−5 | 16 |
| Blue | MF | GO:0004714 | Transmembrane
receptor protein tyrosine kinase activity |
9.32×10−5 | 9 |
| Blue | MF | GO:0005201 | Extracellular
matrix structural constituent | 0.00013 | 14 |
| Blue | MF | GO:0008236 | Serine-type
peptidase activity | 0.000227 | 15 |
| Blue | MF | GO:0098631 | Cell adhesion
mediator activity | 0.000228 | 8 |
| Blue | CC | GO:0031012 | Extracellular
matrix |
4.57×10−5 | 35 |
| Blue | CC | GO:0016324 | Apical plasma
membrane |
1.66×10−5 | 23 |
| Blue | CC | GO:0062023 | Collagen-containing
extracellular matrix |
2.44×10−5 | 24 |
| Blue | CC | GO:0005604 | Basement
membrane |
5.91×10−5 | 11 |
| Blue | CC | GO:0005911 | Cell-cell
junction | 0.000122 | 27 |
| Blue | CC | GO:0045177 | Apical part of
cell | 0.000132 | 24 |
Module visualization and identification
of hub genes
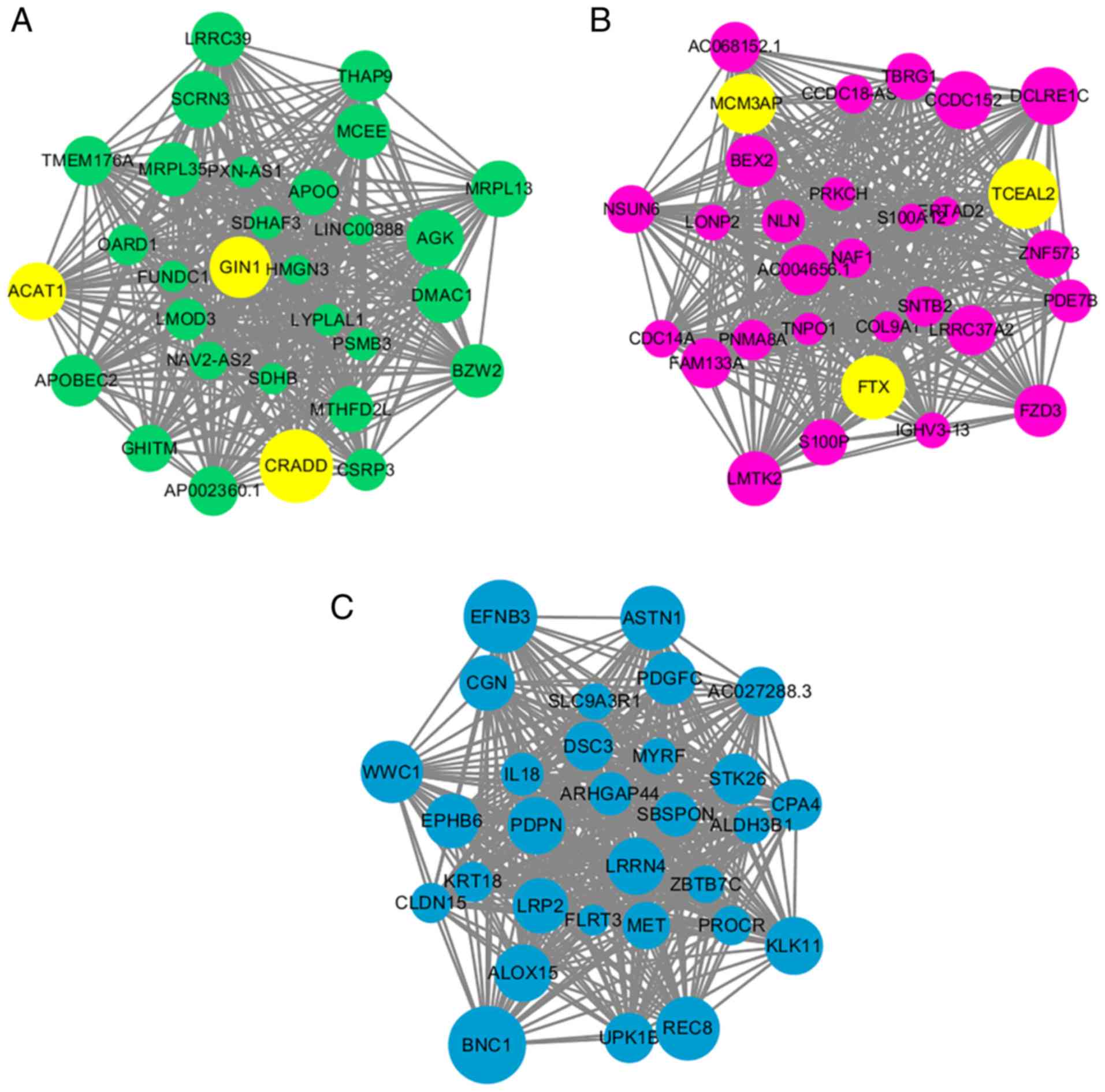
The intramodular connectivity was ranked according
to the kME value, and the top 30 genes of each module of interest
was used to visualize the specific modules (Fig. 5). Subsequently, the top 3 genes of
both the green and magenta modules were labeled as the hub genes
for AF, as they were most positively or negatively statistically
relevant modules with AF. The protein coding genes ACAT1, CRADD and
GIN1 were selected as the hub genes in the green module for AF, and
1 lncRNA, FTX, and 2 protein coding genes, transcription elongation
factor A like 2 (TCEAL2) and MCM3AP, were selected as the hub genes
in the magenta module. All of these hub genes had a kME value
>0.8 and relatively high GS for AF (>0.5; P<0.01), which
established their network centrality and potential vital roles in
AF.
DEG analysis
DEG analysis was performed to validate the WGCNA
results. Setting the cutoff as |FC|>1.5, with a P<0.05, 3
categories of DEG were screened between AF and SR in the LA, RA and
the LA/RA ratio. A total of 497 upregulated (UP) LADEGs and 226
downregulated (DOWN) LADEGs, 104 UP RADEGs and 123 DOWN RADEGs and
203 DADEGs (133 UP and 70 DOWN) were identified. LADEGs had the
highest number of DEGs among the 3 sets of comparisons; whereas
RADEGs and DADEGs had similar sizes but considerably fewer DEGs.
Volcano plots of these DEGs are presented in Fig. S1. The 3 different categories of
DEGs were overlapped to additionally clarify their associations. As
demonstrated in Fig. 6A,
approximately two-thirds of the UP RADEGs and one-fourth of the
DOWN RADEGs were also altered in LADEGs with a similar trend. The
majority of DADEGs exhibited the same differential patterns as the
LADEGs. These results (Fig. 6A and
B) suggested that the sum of the UP and DOWN overlapping genes
between LADEGs and RADEGs were identical with the total number of
overlapping DEGs between LA (69 genes) and RA (32 genes),
suggesting that few genes exhibited reversed expression when
comparing LA to RA. Similarly, DADEGs exhibited a notable overlap
with LADEGs but not with RADEGs, demonstrating that changes of the
LA/RA ratio were primarily due to the overexpression or
under-expression of LA genes. Enrichment analysis of these
different kinds of DEGs are presented in Tables SII and SIII. The enrichment
results demonstrated that LADEGs were significantly associated with
a number of functions and pathways, including coagulation,
inflammation and remodeling amongst others, suggesting a fairly
complicated regulatory network in AF. Enrichment terms of RADEGs
and DADEGs were both similar with LADEGs to a certain extent, which
may explain their respective associations.
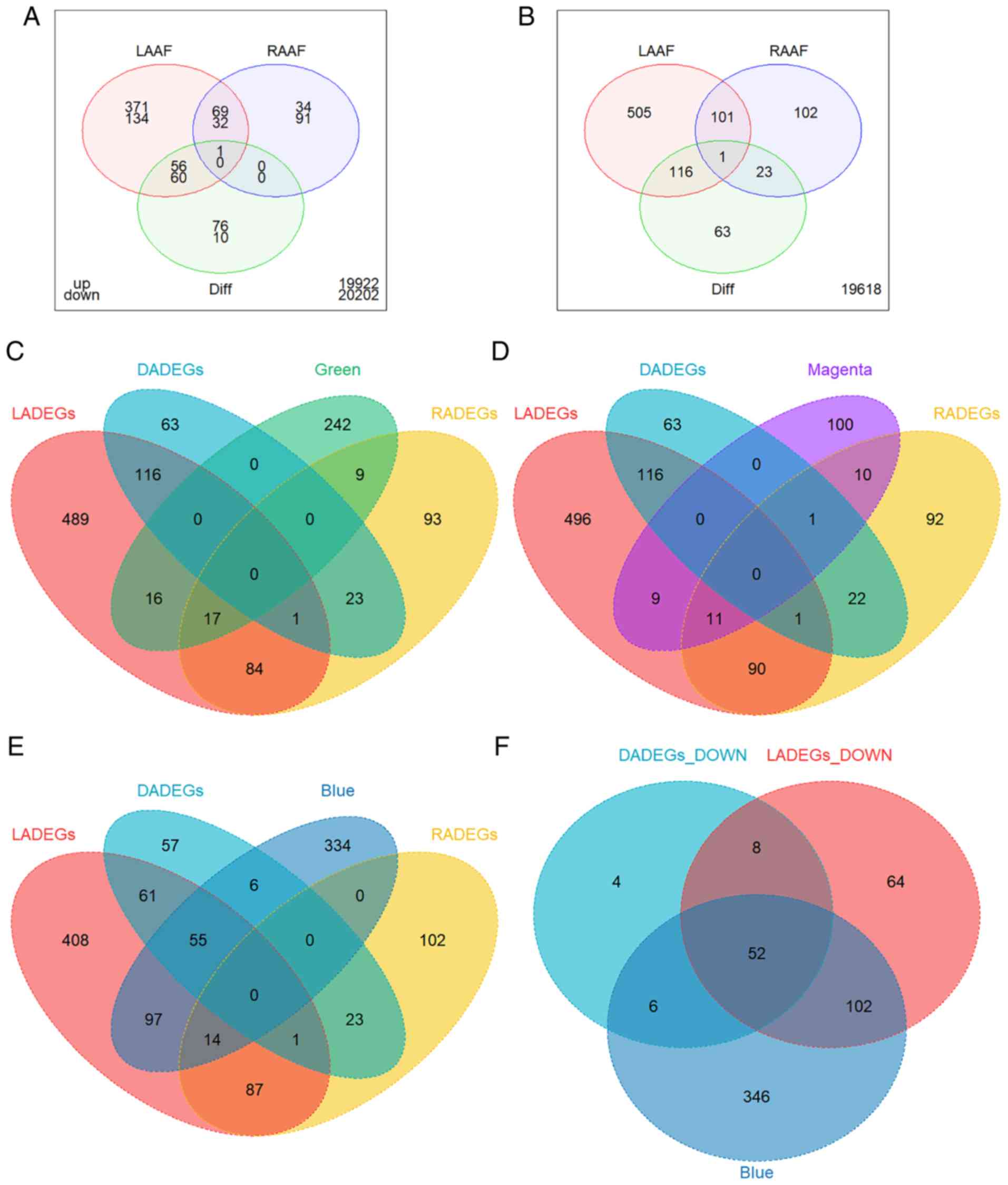 | Figure 6Overlap of DEGs and modules of
interest. (A) Venn diagram of LADEGs, RADEGs and DADEGs in UP DEGs
(above) and DOWN DEGs (below). The numbers outside the diagram
represent the sum of genes which did not belong to any category of
DEG. Venn diagram of LADEGs, RADEGs and DADEGs and the (B) total
DEGs, (C) genes of the green module, (D) genes of the magenta
module, and (E) genes of the blue module. (F) Venn diagram of DOWN
LADEGs, DOWN DADEGs and genes of the blue module. DEGs,
differentially expressed genes; LA, left atria; RA, right atria;
LADEGs, differentially expressed genes in the LA; RA, right atria;
RADEGs, differentially expressed genes in the RA; DADEGs,
differentially expressed genes in LA/RA ratio; UP, upregulated;
DOWN, downregulated. |
Association between DEGs and the modules
of interest
Complex interactions and enrichment patterns of the
aforementioned DEGs analysis highlighted the potential of
integrated methods such as WGCNA to explore the gene co-expression
network in AF. The green, magenta and blue modules were overlapped
with the DEGs, in order to validate the results. The genes of both
the green and magenta modules exhibited a degree of overlap with
LADEGs or RADEGs, but almost no overlap with DADEGs (Fig. 6C and D). As these two modules were
characteristically enriched in specific functions, some of which
were also parts of enriched pathways in the LADEGs or RADEGs, they
may be used to validate the important functional roles of these two
modules in AF. The genes of the blue module exhibited the largest
amount of overlap with the downregulated genes of LADEGs and
DADEGs, confirming the association between the blue module with AF
and LA. Genes in the blue module also showed notably less overlap
with the RADEGs (Fig. 6E and F),
similar to the overlapping patterns of DADEGs. Enrichment results
also demonstrated that several significant pathways associated with
the blue module were similar to pathways associated with the
down-regulated DADEGs or LADEGs. Overall, the comparisons between
DEGs and modules of interest verified the reliability of the
WGCNA.
Validation of hub genes in public
database
Although none of the green module hub genes were
present in the clusters of LADEGs, RADEGs or DADEGs, they were all
significantly overexpressed in the LA or RA in AF compared with SR.
However, no statistical difference in the LA/RA ratios was observed
between the AF and SR groups (Fig.
7A-C). Similarly, only 1 magenta hub gene, TCEAL2, was present
in the LADEGs and RADEGs; FTX was present in the LADEGs and MCM3AP
was not present in either. Nevertheless, the expression levels of
all 3 genes were significantly down-regulated in both LA and RA,
and were not differentially expressed in LA/RA ratio (Fig. 7D-F). These results demonstrated
the validity of the hub genes identified the in green and magenta
modules. Through further validation of the hub genes in other
datasets (Table SIV), it was
identified that CRADD was significantly upregulated in AF compared
with SR in the RA in GSE115574. The expression levels of ACAT1 and
FTX in the RA in GSE115574 were in agreement with the results of
analysis of the GSE79768 dataset (ACAT1, P=0.06; FTX, P=0.05). FTX
and TCEAL2 expression was also significantly decreased in AF in the
GSE2240 dataset. As the datasets varied in sample size and had
different criteria for inclusion/exclusion, not all of the hub
genes identified in the GSE79768 dataset were verified in the other
two datasets. In addition, as patients had different cardiovascular
diseases between the datasets, the potential differences in the
comorbidities and demographics of the patients may represent
confounding variables. As such, additional studies and datasets are
required to verify the value of these hub genes.
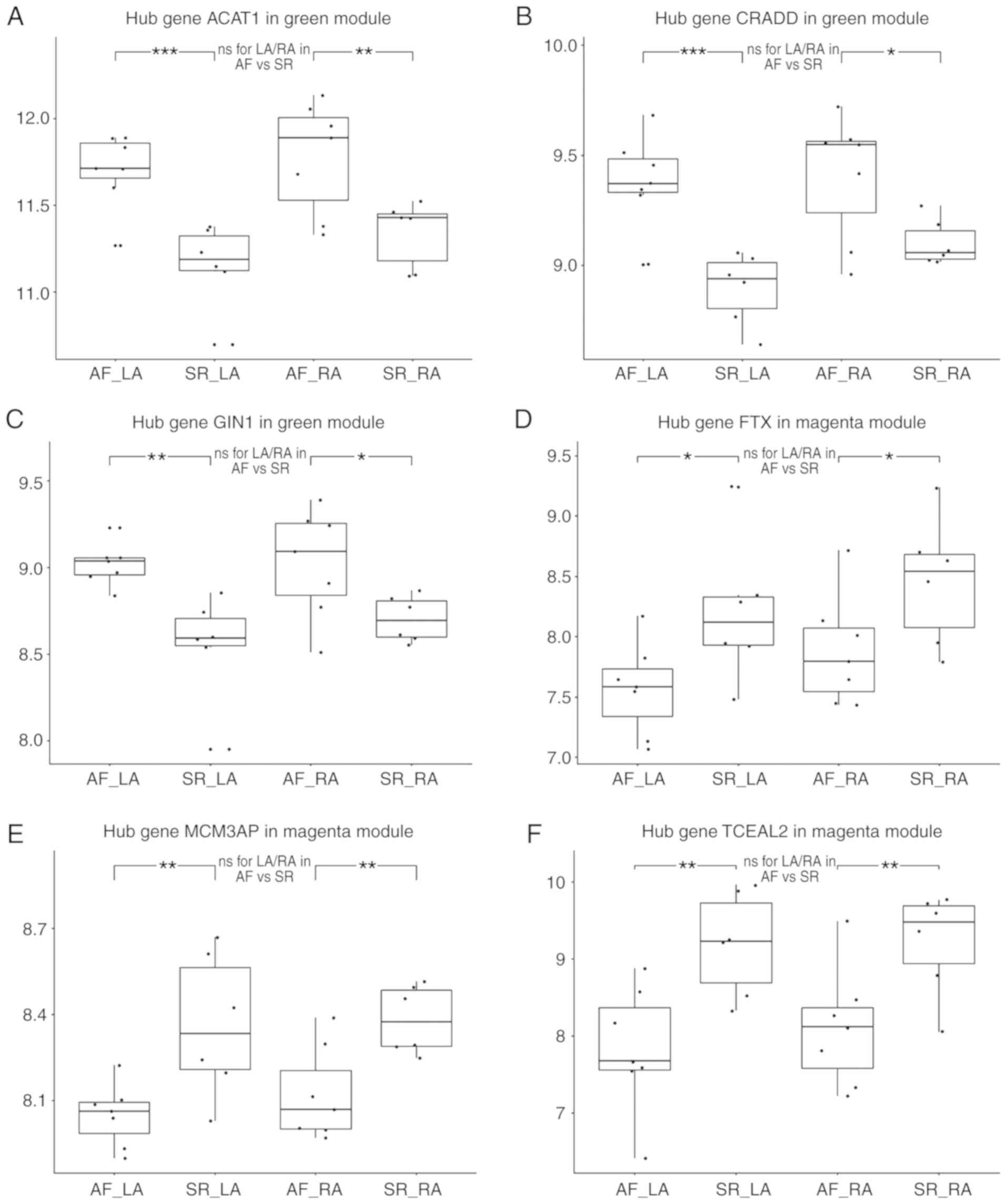 | Figure 7Validation of hub genes at the
expressional level. Validation of hub genes (A) ACAT1, (B) CRADD
and (C) GIN1 in the green module. Expression values of each hub
gene in the LA of AF, LA of SR, RA of AF, RA of SR, from left to
right. These hub genes were significantly overexpressed in both LA
and RA compared with SR, while no statistical difference was
observed the in LA/RA ratio. Validation of the hub genes (D) FTX,
(E) MCM3AP and (F) TCEAL2 in the magenta module. These hub genes
were downregulated, and no statistically significant difference was
observed in the LA/RA ratio. AF, atrial fibrillation; LA, left
atria; SR, sinus rhythm; RA, right atria; ns, no statistical
difference; *P<0.05, **P<0.01 and
***P<0.001. ACAT1, acetyl-CoA Acetyltransferase 1;
CRADD, death domain-containing protein CRADD; GIN1, gypsy
retrotransposon integrase 1; LA, left atria; AF, atrial
fibrillation; SR, sinus rhythm; RA, right atria; FTX, FTX
transcript, XIST regulator; MCM3AP, minichromosome maintenance
complex component 3 associated protein; TCEAL2, transcription
elongation factor A like 2. |
Validation of hub genes in AF rat
models
As TCEAL2 is not present in the rat genome, the
expression levels of the other 5 hub genes were measured for
validation in the AF rat model generated in the present study. As
demonstrated in Fig. 8B-D, the
expression levels of hub genes ACAT1, CRADD and GIN1 in RA were all
significantly increased in AF group compared with those in control
(SR) group (P<0.05). In LA, the expression level of GIN1 was
significantly increased in the AF group (P<0.001; Fig. 8D), and the expression levels of
lncRNA FTX was significantly decreased compared with those in
control group (P<0.05; Fig.
8E). Besides, the expression levels of ACAT1 and CRADD in LA
were observed to be increased in AF group, and the expression
levels of FTX in RA were decreased in AF group, but these
differences were not statistically significant. In addition, there
was no significant difference in the LA/RA ratios between the AF
and control groups in any of these hub genes. These results were in
concordance with the results of the primary dataset (Fig. 7). However, no statistically
significant expression difference in MCM3AP was identified between
the AF and control groups in either the LA or RA.
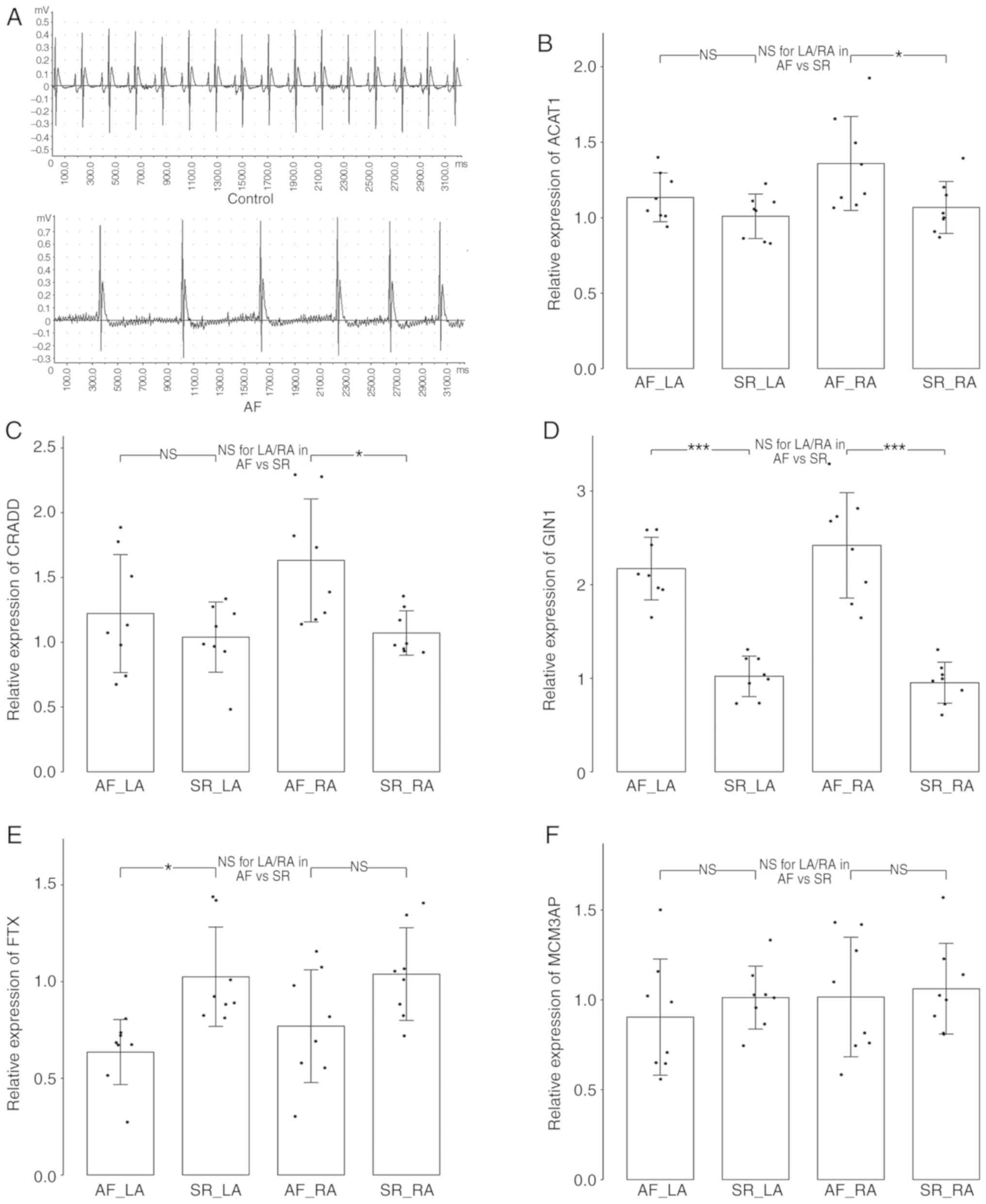 | Figure 8Validation of hub genes in AF rat
models. (A) The electrocardiogram manifestations of rats in control
group (SR; upper panel) and AF group (induced AF; lower panel).
Validation of the expression levels of hub genes (B) ACAT1, (C)
CRADD, (D) GIN1, (E) FTX and (F) MCM3AP by reverse transcription
quantitative polymerase chain reaction. The relative expression
levels were presented with mean ± standard deviation.
*P<0.05, **P<0.01 and
***P<0.001. AF, atrial fibrillation; LA, left atria;
SR, sinus rhythm; RA, right atria; ns, no statistical difference;
ACAT1, acetyl-CoA Acetyltransferase 1; CRADD, death
domain-containing protein CRADD; GIN1, gypsy retrotransposon
integrase 1; FTX, FTX transcript, XIST regulator; MCM3AP,
minichromosome maintenance complex component 3 associated
protein. |
Discussion
AF is the most common type of cardiac arrhythmia and
is associated with a poor prognosis in patients with cardiac
diseases (1); however, treatments
for patients with AF remain unsatisfactory (35). The underlying pathophysiological
mechanisms of AF are extremely complicated, thus WGCNA may be an
effective method of mining valuable data to analyze complicated
genetic networks. In the present study, WGCNA was performed using R
with an AF dataset, which contained gene expression data from
paired left and right atrial tissues from cardiac patients with AF
or SR. The green and magenta modules were identified as the key
modules of AF, with the highest level of significant associations
with AF. The top 3 genes of each key module with the highest
intramodular connectivity were identified as hub genes for AF. In
addition, another module of interest was selected that was
associated with both AF and LA, to examine the potential
association between AF and the genetic differences between the LA
and RA. The results of the enrichment analysis suggested that the
modules of interest used in the present study were associated with
the pathological processes of energy metabolism, inflammation and
apoptosis, coagulation, complement or ECM formation. These results
provided a partial description of the comprehensive regulatory
network in AF, which may improve the current understanding of the
mechanisms underlying AF, and may assist in identifying potential
therapeutic targets.
The present study used the GEO dataset GSE79768 to
perform WGCNA. The original study involving this dataset, conducted
by Tsai et al (14),
revealed the differences between LA-to-RA transcriptional profiles
between AF and SR, and suggested that AF was associated with
differential LA-to-RA gene expression, which was related to
specific ion channels and pathways, as well as upregulation of
thrombogenesis-associated genes in the LA appendage. The major
differences in the genes or modules obtained in the present study,
compared with the results from the study by Tsai et al
(14) was that the present study
used a more comprehensive method, WGCNA, not only identifying the
blue module as an important regulatory module of AF with increased
specificity in the LA associated with complement, coagulation and
extracellular matrix formation, expanding upon their results in
LA-to-RA DEGs associated-pathways, but also identifying 2 critical
modules for AF: The green module, which was associated with energy
metabolism; and the magenta module, which was associated with
multiple interactive pathways associated with apoptosis and
inflammation. The in-depth analyses performed in the present and
the results obtained cannot be obtained from conventional
microarray DEGs analyses.
Among the 20 modules identified by WGCNA, the green
module exhibited the greatest positive correlation with AF.
Enrichment analysis indicated that the genes in the green module
were primarily associated with pathways related to energy substance
metabolism and potentially located in the mitochondria. Several
studies have suggested that altered atrial metabolism may serve an
important role in the pathophysiology of AF (36-38). Previous genomic, metabolomic and
proteomic studies have also demonstrated that metabolic genes and
products were notably altered in patients with AF compared to
patients with SR (39,40). As a result of irregular
high-frequency excitation and contraction, AF-associated metabolic
stress occurs when there is a decreased capacity of energy to
supplement the demands of atrial tissues, combined with an increase
in metabolism of ketone bodies, AMPK activation, mitochondria
dysfunction and reactive oxygen species generation, which
accelerates gradual atrial remodeling (41). KEGG pathway analysis of the
magenta module indicated that it was primarily associated with the
Hippo signaling pathway. The Hippo pathway is a highly conserved
mammalian pathway, involved in regulation of cell proliferation,
apoptosis and other cellular functions, and serves an important
role in the development of the heart, regeneration and cardiac
diseases (42-44). Although, to the best of our
knowledge, there are currently no studies investigating the role of
the Hippo pathway in AF, several recent studies have demonstrated
its role in myocardial infarction, hypertrophy and heart failure
(43). Del Re et al
(45) revealed that depletion of
Yes-associated protein isoform 1 (Yap1), a downstream protein in
the Hippo pathway, augmented apoptosis and fibrosis and decreased
compensatory cardiomyocyte hypertrophy, thereby exacerbating injury
following chronic myocardial infarction, suggesting that Yap1 is
critical in the homeostasis of the physiological processes of the
heart. Leach et al (46)
demonstrated that deletion of the Hippo pathway component Salvador
(Salv) in mouse hearts with ischemic heart failure following
myocardial infarction decreased fibrosis, increased scar border
vascularity and recovery of the pumping function, thus reversing
heart failure; whereas the Yap target gene Parkin RBR E3 ubiquitin
protein ligase was essential for heart repair in Salv knock-out
mice. The Hippo pathway interacts with multiple transcription
factors that affects a variety of genes and pathways including
proinflammatory genes and the Wnt pathway (43,44), and these interactions were also
predicted by GO enrichment of the magenta module in the present
study. Furthermore, apoptosis and fibrotic activation mediated by
inflammation may contribute to structural remodeling of the atria
(35), and the Hippo pathway is
involved in these pathological mechanisms in cardiovascular systems
(43). Therefore, the magenta
module may be an integrated functional cluster that regulates
myocardial apoptosis, inflammation and remodeling, and may be
mediated by the Hippo pathway. Further exploration of this module
is required to identify the precise mechanisms associated with the
development of AF.
As the green and magenta modules exhibited the
highest levels of positive and negative correlation with AF, the
hub genes in these modules were screened. The top 3 genes in each
module were identified as the intramodular hub genes of AF
following confirmation of their close association with AF. A total
of 3 protein coding genes, ACAT1, CRADD and GIN1 in the green
module, and 1 lncRNA, FTX, and 2 protein coding genes TCEAL2 and
MCM3AP in the magenta module were identified. ACAT1 is expressed in
macrophages, and it has been demonstrated that it generates
cholesterol ester of foam cells, thereby serving an important role
in early atherosclerotic lesions (47,48). Although, to the best of our
knowledge, there are no studies providing evidence of a
relationship between ACAT1 and AF, a close association between AF
and atherosclerosis has been described (49). Therefore, ACAT1 may be associated
with an increased risk of atherosclerosis in patients with AF.
CRADD was demonstrated to serve a role in the regulation of
apoptosis in a number of different types of tissues (50). Therefore, it may be possible that
CRADD modulates apoptosis in AF, although to date this has not been
conclusively demonstrated. FTX is a lncRNA that regulates
cardiomyocyte apoptosis by targeting miR-29b-1-5p and Bcl2l2
(51), therefore, it may also
mediate apoptosis in AF as well. In addition, several hub genes in
the magenta module were all associated with apoptosis. Apoptosis in
cardiomyocytes is accompanied by fibroblast recruitment and ECM
deposition in AF (52), which
contributes to atrial remodeling. The data from these studies
support the results of the present study that suggest the
involvement of apoptosis in the pathophysiology of AF. MCM3AP is a
potent natural inhibitor of initiation of DNA replication (53). TCEAL2 is a member of transcription
elongation factor A (SII)-like gene family of proteins, which
modulates transcription in a promoter context-dependent manner and
is considered an important nuclear target for intracellular signal
transduction (54). GIN1 is
ubiquitously expressed in various different types of tissues, and
may therefore possess an essential function (55), although the exact function remains
to be determined. To the best of our knowledge, MCM3AP, TCEAL2 and
GIN1 have not been studied in cardiac pathophysiology.
Nevertheless, they may serve an important role in AF, as they were
identified to be crucial in the AF-associated co-expression key
modules and were differentially expressed between AF and SR in both
atria in the present study. Therefore, future studies on the
function of these hub genes in AF is required. In addition to these
hub genes, a number of other genes in the green and magenta modules
were also associated with pathophysiological processes of AF. For
example, heat shock protein B3 and stress-70 protein,
mitochondrial, present in the green module, have been demonstrated
to be associated with AF (56,57), and platelet basic protein in the
magenta module was associated with platelet activity and is a
marker of thrombogenesis and platelet activation in AF (58).
The LA is more likely to be a driver in AF and is
more likely to be associated with stroke compared with RA (15-17). The blue module was selected as
another module of interest that was significantly correlated with
both AF and the LA. The majority of the downregulated DADEGs were
overlapped with the genes of the blue module, primarily due to the
down-regulated expression of LADEGs, suggesting that the genes of
the blue module may be an important regulatory module of AF with an
increased specificity in the LA. Enrichment analysis indicated that
the blue module was primarily associated with complement,
coagulation activity and ECM formation. AF is associated with
elevated levels of inflammation, which contributes to
thrombo-embolic complications (35,59). However, different studies have
described contrasting results regarding the effects of the
complement system in AF (60). A
recent proteomics study demonstrated that left atrial low voltage
areas (LVA) in patients with AF, which corresponded to areas of
fibrotic and electrically silent myocardial tissue, were associated
with an imbalance in coagulation and complement pathways, and
complement and coagulation molecules were differentially expressed,
including downregulation of C1q-B, C1q-C, CFH and plasminogen
compared with patients without LVA (61). ECM dysregulation caused atrial
fibrotic remodeling in AF through inflammation and oxidative stress
(62), and studies have
identified that ECM-associated genes are differentially expressed,
with notable differences in gene expression between LA and RA in AF
(14,63). The homeostasis between ECM
composition and complement cascade effectors modulates the net
effect of ECM macromolecules on the complement cascade (64). Therefore, the results of the
present study suggest a complex interaction network of the
complement, coagulation and ECM systems. Furthermore, the blue
module was positively associated with the LA, suggesting that the
blue module may be associated with the differences in
hypercoagulation and structure remodeling in the different atria.
Based on the results of the present and previous studies, the genes
of the blue module may serve an important role in inflammation,
coagulation and fibrosis, particularly in the LA, which may
contribute to the pathogenesis or maladaptation of AF.
The present study has certain limitations. First, as
the original article of the dataset discussed, there were no
healthy controls and multiple factors including other underlying
clinical traits that were not described in the dataset (14), which may have affected the
reliability of the results of the present study. The analysis was
focused on only 1 dataset due to the limited access to gene
expression data that were collected from the paired LA and RA in
patients with AF or SR. Therefore, additional datasets should be
analyzed, if available, to obtain more representative results.
Additionally, the pathophysiology of AF in humans is complex, and
animal models could only partially reproduce the pathophysiologic
spectrum of clinical AF. Further studies are required to validate
these data.
In summary, WGCNA was performed on an AF dataset.
Among the 20 modules, the green and magenta modules were identified
as the most critical modules for AF, from which 6 hub genes, ACAT1,
CRADD, GIN1, FTX, TCEAL2 and MCM3AP, were screened out, and
postulated to serve key roles in pathophysiological mechanisms of
AF. The green module was identified to be associated with energy
metabolism, and the magenta module was associated with multiple
complex interactive pathways of apoptosis and inflammation. In
addition, the blue module was identified to be an important
regulatory module of AF with higher specificity for the LA, and was
primarily associated with complement, coagulation and ECM. These
data may promote future experimental studies investigating the
roles of the genes in cardiac function and pathophysiology, a
number of which have not been previously described. Additionally,
the genes identified may serve as novel therapeutic targets for
treating patients, and assist in attaining an improved
understanding of the underlying mechanisms of AF.
Supplementary Data
Acknowledgments
Not applicable.
Funding
This study was supported by the National Natural
Science Foundation of China (grant nos. 91639301 and 81770458), the
Key Project of Research and Development Plan of Shaanxi Province
(grant no. 2017ZDCXL-SF-02-04-01), the Clinical Research Award of
the First Affiliated Hospital of Xi'an Jiaotong University (grant
no. XJTU1AF-CRF-2016-004), and the Natural Science Foundation of
Shaanxi Province (grant no. 2018JM7063).
Availability of data and materials
The main dataset analyzed during the present study
is available from the Gene Expression Omnibus repository
(https://www.ncbi.nlm.nih.gov/geo) with
GSE accession no., GSE79768. The DOI of the original article which
generated this dataset is 10.1016/j.ijcard.2016.07.103. The two
external datasets for validation are also available from GEO with
GSE accession nos., GSE2240 and GSE115574.
Authors' contributions
WL performed the weighted gene co-expression network
analysis and was a major contributor in writing the manuscript. LW
and YW annotated the results of the network analysis and made
important modifications to the manuscript. ZY and JZ designed the
research project and created the final revision of the manuscript.
WL and JZ performed the animal experiments in this study. All
authors read and approved the final version of the manuscript.
Ethics approval and consent to
participate
The animal study was approved by the Institutional
Ethics Committee for Animal Experiments of Xi'an Jiaotong
University. All procedures conformed to the Guide for the Care and
Use of Laboratory Animals published by the National Institutes of
Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Zimetbaum P: Atrial Fibrillation. Ann
Intern Med. 166:ITC33–ITC48. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Odutayo A, Wong CX, Hsiao AJ, Hopewell S,
Altman DG and Emdin CA: Atrial fibrillation and risks of
cardiovascular disease, renal disease, and death: Systematic review
and meta-analysis. BMJ. 354:i44822016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ferrari R, Bertini M, Blomstrom-Lundqvist
C, Dobrev D, Kirchhof P, Pappone C, Ravens U, Tamargo J, Tavazzi L
and Vicedomini GG: An update on atrial fibrillation in 2014: From
pathophysiology to treatment. Int J Cardiol. 203:22–29. 2016.
View Article : Google Scholar
|
|
4
|
Liu Y, Shi Q, Ma Y and Liu Q: The role of
immune cells in atrial fibrillation. J Mol Cell Cardiol.
123:198–208. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nattel S: Molecular and cellular
mechanisms of atrial fibrosis in atrial fibrillation. JACC Clin
Electrophysiol. 3:425–435. 2017. View Article : Google Scholar
|
|
6
|
Qian C, Li H, Chang D, Wei B and Wang Y:
Identification of functional lncRNAs in atrial fibrillation by
integrative analysis of the lncRNA-mRNA network based on competing
endogenous RNAs hypothesis. J Cell Physiol. 234:11620–11630. 2019.
View Article : Google Scholar
|
|
7
|
Prystowsky EN, Padanilam BJ and Fogel RI:
Treatment of atrial fibrillation. JAMA. 314:278–288. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang B and Horvath S: A general framework
for weighted gene co-expression network analysis. Stat Appl Genet
Mol Biol. 4:Article172005. View Article : Google Scholar
|
|
9
|
Liu S, Wang Z, Chen D, Zhang B, Tian RR,
Wu J, Zhang Y, Xu K, Yang LM, Cheng C, et al: Annotation and
cluster analysis of spatiotemporal- and sex-related lncRNA
expression in rhesus macaque brain. Genome Res. 27:1608–1620. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yin L, Cai Z, Zhu B and Xu C:
Identification of key pathways and genes in the dynamic progression
of HCC based on WGCNA. Genes (Basel). 9:E922018. View Article : Google Scholar
|
|
11
|
Zhang X, Feng H, Li Z, Li D, Liu S, Huang
H and Li M: Application of weighted gene co-expression network
analysis to identify key modules and hub genes in oral squamous
cell carcinoma tumorigenesis. OncoTargets Ther. 11:6001–6021. 2018.
View Article : Google Scholar
|
|
12
|
Ahmed M, Lai TH, Zada S, Hwang JS, Pham
TM, Yun M and Kim DR: Functional linkage of RKIP to the epithelial
to mesenchymal transition and autophagy during the development of
prostate cancer. Cancers (Basel). 10:E2732018. View Article : Google Scholar
|
|
13
|
Radulescu E, Jaffe AE, Straub RE, Chen Q,
Shin JH, Hyde TM, Kleinman JE and Weinberger DR: Identification and
prioritization of gene sets associated with schizophrenia risk by
co-expression network analysis in human brain. Mol Psychiatry. Nov
26–2018.Epub ahead of print. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsai FC, Lin YC, Chang SH, Chang GJ, Hsu
YJ, Lin YM, Lee YS, Wang CL and Yeh YH: Differential left-to-right
atria gene expression ratio in human sinus rhythm and atrial
fibrillation: Implications for arrhythmogenesis and thrombogenesis.
Int J Cardiol. 222:104–112. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gaborit N, Steenman M, Lamirault G, Le
Meur N, Le Bouter S, Lande G, Léger J, Charpentier F, Christ T,
Dobrev D, et al: Human atrial ion channel and transporter subunit
gene-expression remodeling associated with valvular heart disease
and atrial fibrillation. Circulation. 112:471–481. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kamel H, Okin PM, Elkind MS and Iadecola
C: Atrial fibrillation and mechanisms of stroke: Time for a new
model. Stroke. 47:895–900. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Voigt N, Trausch A, Knaut M, Matschke K,
Varró A, Van Wagoner DR, Nattel S, Ravens U and Dobrev D:
Left-to-right atrial inward rectifier potassium current gradients
in patients with paroxysmal versus chronic atrial fibrillation.
Circ Arrhythm Electrophysiol. 3:472–480. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liao Q, Liu C, Yuan X, Kang S, Miao R,
Xiao H, Zhao G, Luo H, Bu D, Zhao H, et al: Large-scale prediction
of long non-coding RNA functions in a coding-non-coding gene
co-expression network. Nucleic Acids Res. 39:3864–3878. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Du Z, Fei T, Verhaak RG, Su Z, Zhang Y,
Brown M, Chen Y and Liu XS: Integrative genomic analyses reveal
clinically relevant long noncoding RNAs in human cancer. Nat Struct
Mol Biol. 20:908–913. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang G, Sun H, Zhang Y, Zhao H, Fan W, Li
J, Lv Y, Song Q, Li J, Zhang M and Shi H: Characterization of
dysregulated lncRNA-mRNA network based on ceRNA hypothesis to
reveal the occurrence and recurrence of myocardial infarction. Cell
Death Discov. 4:352018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liao Y, Smyth GK and Shi W: The Subread
aligner: Fast, accurate and scalable read mapping by seed-and-vote.
Nucleic Acids Res. 41:e1082013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Langfelder P and Horvath S: WGCNA: An R
package for weighted correlation network analysis. BMC
Bioinformatics. 9:5592008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yu G, Wang LG, Han Y and He QY:
clusterProfiler: An R package for comparing biological themes among
gene clusters. OMICS. 16:284–287. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ashburner M, Ball CA, Blake JA, Botstein
D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT,
et al: Gene ontology: Tool for the unification of biology. The Gene
Ontology Consortium Nat Genet. 25:25–29. 2000.
|
|
25
|
The Gene Ontology Consortium: The Gene
Ontology Resource: 20 years and still GOing strong. Nucleic Acids
Res. 47:D330–D338. 2019. View Article : Google Scholar :
|
|
26
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar
|
|
27
|
Kanehisa M, Sato Y, Furumichi M, Morishima
K and Tanabe M: New approach for understanding genome variations in
KEGG. Nucleic Acids Res. 47:D590–D595. 2019. View Article : Google Scholar :
|
|
28
|
Kanehisa M: Toward understanding the
origin and evolution of cellular organisms. Protein Sci.
28:1947–1951. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shannon P, Markiel A, Ozier O, Baliga NS,
Wang JT, Ramage D, Amin N, Schwikowski B and Ideker T: Cytoscape: A
software environment for integrated models of biomolecular
interaction networks. Genome Res. 13:2498–2504. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Horvath S and Dong J: Geometric
interpretation of gene coexpression network analysis. PLoS Comput
Biol. 4:e10001172008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Phipson B, Lee S, Majewski IJ, Alexander
WS and Smyth GK: Robust hyperparameter estimation protects against
hypervariable genes and improves power to detect differential
expression. Ann Appl Stat. 10:946–963. 2016. View Article : Google Scholar
|
|
32
|
Ritchie ME, Phipson B, Wu D, Hu Y, Law CW,
Shi W and Smyth GK: limma powers differential expression analyses
for RNA-sequencing and microarray studies. Nucleic Acids Res.
43:e472015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lv X, Li J, Hu Y, Wang S, Yang C, Li C and
Zhong G: Overexpression of miR-27b-3p targeting Wnt3a regulates the
signaling pathway of Wnt/β-catenin and attenuates atrial fibrosis
in rats with atrial fibrillation. Oxid Med Cell Longev.
2019:57037642019. View Article : Google Scholar
|
|
34
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
35
|
Hu YF, Chen YJ, Lin YJ and Chen SA:
Inflammation and the pathogenesis of atrial fibrillation. Nat Rev
Cardiol. 12:230–243. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ghezelbash S, Molina CE and Dobrev D:
Altered atrial metabolism: An underappreciated contributor to the
initiation and progression of atrial fibrillation. J Am Heart
Assoc. 4:e0018082015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Lenski M, Schleider G, Kohlhaas M, Adrian
L, Adam O, Tian Q, Kaestner L, Lipp P, Lehrke M, Maack C, et al:
Arrhythmia causes lipid accumulation and reduced glucose uptake.
Basic Res Cardiol. 110:402015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Heijman J and Dobrev D: Irregular rhythm
and atrial metabolism are key for the evolution of proarrhythmic
atrial remodeling in atrial fibrillation. Basic Res Cardiol.
110:412015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Mayr M, Yusuf S, Weir G, Chung YL, Mayr U,
Yin X, Ladroue C, Madhu B, Roberts N, De Souza A, et al: Combined
metabolomic and proteomic analysis of human atrial fibrillation. J
Am Coll Cardiol. 51:585–594. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chiang DY, Zhang M, Voigt N, Alsina KM,
Jakob H, Martin JF, Dobrev D, Wehrens XH and Li N: Identification
of microRNA-mRNA dysregulations in paroxysmal atrial fibrillation.
Int J Cardiol. 184:190–197. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Harada M, Melka J, Sobue Y and Nattel S:
Metabolic considerations in atrial fibrillation-mechanistic
insights and therapeutic opportunities. Circ J. 81:1749–1757. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhou Q, Li L, Zhao B and Guan KL: The
hippo pathway in heart development, regeneration, and diseases.
Circ Res. 116:1431–1447. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Ikeda S and Sadoshima J: Regulation of
myocardial cell growth and death by the hippo pathway. Circ J.
80:1511–1519. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang J, Liu S, Heallen T and Martin JF:
The Hippo pathway in the heart: Pivotal roles in development,
disease, and regeneration. Nat Rev Cardiol. 15:672–684. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Del Re DP, Yang Y, Nakano N, Cho J, Zhai
P, Yamamoto T, Zhang N, Yabuta N, Nojima H, Pan D and Sadoshima J:
Yes-associated protein isoform 1 (Yap1) promotes cardiomyocyte
survival and growth to protect against myocardial ischemic injury.
J Biol Chem. 288:3977–3988. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Leach JP, Heallen T, Zhang M, Rahmani M,
Morikawa Y, Hill MC, Segura A, Willerson JT and Martin JF: Hippo
pathway deficiency reverses systolic heart failure after
infarction. Nature. 550:260–264. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Rudel LL, Lee RG and Parini P: ACAT2 is a
target for treatment of coronary heart disease associated with
hypercholesterolemia. Arterioscler Thromb Vasc Biol. 25:1112–1118.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Fazio S, Dove DE and Linton MF: ACAT
inhibition: Bad for macrophages, good for smooth muscle cells?
Arterioscler Thromb Vasc Biol. 25:7–9. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
da Silva RM: Influence of inflammation and
atherosclerosis in atrial fibrillation. Curr Atheroscler Rep.
19:22017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Ahmad M, Srinivasula SM, Wang L, Talanian
RV, Litwack G, Fernandes-Alnemri T and Alnemri ES: CRADD, a novel
human apoptotic adaptor molecule for caspase-2, and FasL/tumor
necrosis factor receptor-interacting protein RIP. Cancer Res.
57:615–619. 1997.PubMed/NCBI
|
|
51
|
Long B, Li N, Xu XX, Li XX, Xu XJ, Guo D,
Zhang D, Wu ZH and Zhang SY: Long noncoding RNA FTX regulates
cardiomyocyte apoptosis by targeting miR-29b-1-5p and Bcl2l2.
Biochem Biophys Res Commun. 495:312–318. 2018. View Article : Google Scholar
|
|
52
|
Harada M, Van Wagoner DR and Nattel S:
Role of inflammation in atrial fibrillation pathophysiology and
management. Circ J. 79:495–502. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Takei Y, Assenberg M, Tsujimoto G and
Laskey R: The MCM3 acetylase MCM3AP inhibits initiation, but not
elongation, of DNA replication via interaction with MCM3. J Biol
Chem. 277:43121–43125. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kim YS, Hwan JD, Bae S, Bae DH and Shick
WA: Identification of differentially expressed genes using an
annealing control primer system in stage III serous ovarian
carcinoma. BMC Cancer. 10:5762010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Lloréns C and Marín I: A mammalian gene
evolved from the integrase domain of an LTR retrotransposon. Mol
Biol Evol. 18:1597–1600. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Kirmanoglou K, Hannekum A and Schafler AE:
Expression of mortalin in patients with chronic atrial
fibrillation. Basic Res Cardiol. 99:404–408. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
García A, Eiras S, Parguiña AF, Alonso J,
Rosa I, Salgado- Somoza A, Rico TY, Teijeira-Fernández E and
González-Juanatey JR: High-resolution two-dimensional gel
electrophoresis analysis of atrial tissue proteome reveals
down-regulation of fibulin-1 in atrial fibrillation. Int J Cardiol.
150:283–290. 2011. View Article : Google Scholar
|
|
58
|
Lip GY, Lip PL, Zarifis J, Watson RD,
Bareford D, Lowe GD and Beevers DG: Fibrin D-dimer and
beta-thromboglobulin as markers of thrombogenesis and platelet
activation in atrial fibrillation. Effects of introducing
ultra-low-dose warfarin and aspirin. Circulation. 94:425–431. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hijazi Z, Oldgren J, Siegbahn A, Granger
CB and Wallentin L: Biomarkers in atrial fibrillation: A clinical
review. Eur Heart J. 34:1475–1480. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Lappegård KT, Garred P, Jonasson L,
Espevik T, Aukrust P, Yndestad A, Mollnes TE and Hovland A: A vital
role for complement in heart disease. Mol Immunol. 61:126–134.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Kornej J, Büttner P, Hammer E, Engelmann
B, Dinov B, Sommer P, Husser D, Hindricks G, Völker U and Bollmann
A: Circulating proteomic patterns in AF related left atrial
remod-eling indicate involvement of coagulation and complement
cascade. PLoS One. 13:e01984612018. View Article : Google Scholar
|
|
62
|
Lin CS and Pan CH: Regulatory mechanisms
of atrial fibrotic remodeling in atrial fibrillation. Cell Mol Life
Sci. 65:1489–1508. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhu H, Zhang W, Zhong M, Zhang G and Zhang
Y: Differential gene expression during atrial structural remodeling
in human left and right atrial appendages in atrial fibrillation.
Acta Biochim Biophys Sin (Shanghai). 43:535–541. 2011. View Article : Google Scholar
|
|
64
|
Gialeli C, Gungor B and Blom AM: Novel
potential inhibitors of complement system and their roles in
complement regulation and beyond. Mol Immunol. 102:73–83. 2018.
View Article : Google Scholar : PubMed/NCBI
|