Introduction
Aging is a normal physiological process that not
only increases susceptibility to diseases, but also gradually
reduces physiological functions (1). Age-related hearing loss (AHL), also
referred to as presbycusis, is a type of sensorineural deafness
with bilateral high-frequency hearing loss that progressively
declines with age (2).
Aging-related changes in the central auditory system have a
negative impact on auditory perception and verbal communication, or
both (3). However, the mechanisms
involved in central presbycusis remain unclear.
A number of aging pathways are closely related to
cellular protein homeostasis, suggesting that maintaining a healthy
protein homeostasis is crucial for longevity (4). Accumulating lines of evidence have
indicated that the unfolded protein response (UPR) is the link
between aging and proteostasis in a variety of vital organisms
(4,5). Hearing loss caused by both
environmental and genetic factors is improved by regulating the UPR
(6). The continuous aggravation
of the endoplasmic reticulum (ER) and mitochondrial (MT) stress
participates in the pathogenesis of AHL. Heat shock protein (HSP)-
or stress-related chaperones have been identified as markers of the
UPR, and a number of studies have been conducted mainly on the UPR
signaling pathways (7-9). However, the specific mechanisms and
proteostatic UPR signaling pathways involved in age-related
diseases remain unknown.
Metformin, an AMP-activated protein kinase (AMPK)
activator, is a medication used in the first-line treatment of type
2 diabetes. Metformin readily crosses the blood-brain barrier and
is distributed to various regions in the brain (10), and recently, it has been reported
to promote neurogenesis, thereby providing potential
neuroprotection (11). However,
whether metformin interacts with the UPR and the exact role of
metformin in the UPR remain unclear. It is well known that
AMPK/ERK1/2 are growth factor signals and important metabolic
regulators; however, a link between changes in the auditory system
and the UPR has not yet been reported, at least to the best of our
knowledge. Similarly, few studies have reported the influence of
metformin on ERK1/2 (12,13), which directs various metabolic
processes.
In this study, we investigated the molecular
mechanisms of the UPR and AMPK/ERK1/2 pathway in the aging process
and the effects of metformin on the process of age-related
neurodegeneration. The findings of this study may provide a novel
strategy for the protection of the brain against aging,
specifically by treatment with metformin.
Materials and methods
Animal procedures
A total of 200 male Sprague-Dawley (SD) rats
(weighing 80-100 g, 3 weeks old) were purchased from the
Experimental Animal Center of Tongji Medical College, Huazhong
University of Science and Technology. The rats were provided with
free access to standard food and water and were housed in cages
with an ambient temperature of 25°C, a controlled humidity of 50%
and a 12/12-h light/dark cycle. Prior to treatment, all rats were
allowed to acclimatize to their environment for 1 week and were
then randomly divided into 3 groups as follows (n=65-67 per group):
The control group, the D-galactose (D-gal) group and the D-gal +
metformin group (Met group). The rats in the D-gal and Met groups
were treated with D-gal (dissolved in 0.9% saline, 500 mg/kg,
Sigma) via subcutaneous injection for 8 weeks, while the rats in
the control group were administered an equal volume of normal
saline on the same schedule. The rats in the Met group were
subjected to the daily administration of D-gal (subcutaneously) and
metformin (intragastrically; metformin was dissolved in drinking
water, 150 mg/kg/day, Dongrui Chemical Co.) for the same 8 weeks.
After the 8-week establishment of the model, the rats in the 3
groups were further randomly divided into the 3-month-old,
9-month-old (6 months post-injection) and 15-month-old (12 months
post-injection) subgroups. Metformin was intragastrically
administered to the rats in the Met groups (the 9-month-old Met
group and the 15-month-old Met group) for 8 weeks before each
sacrifice time point. The rats in the control and D-gal groups
received the same dose of water without metformin on the same
schedule. More detailed information on the animal procedures is
provided in Fig. S1. All
experimental procedures involving the care of animals were
performed in strict accordance with the recommendations of the
Guide for the Care and Use of Laboratory Animals of the National
Institutes of Health (14). The
protocol was under the supervision of the Committee on the Ethics
of Animal Experiments of Huazhong University of Science and
Technology (Wuhan, China; permit no. IACUC S2219).
Auditory functions were examined by the auditory
brain-stem response (ABR) just before each sacrifice time point.
When the rats were sacrificed, blood was harvested, and the brain
tissues were immediately used in experiments or stored at −80°C for
later use.
Cell culture and treatment
Highly differentiated rat pheochromocytoma cells
(PC12 cells) were obtained from the Ministry of Education Key
Laboratory for Environment and Health of Tongji Medical College
(HUST). The cells were cultured in RPMI-1640 medium with 10% fetal
bovine serum (Gibco, Thermo Fisher Scientific) and 100 units/ml
penicillin at 37°C in an incubator containing 5% CO2 and
humidified air. At 24 h after seeding, the cells were pre-treated
with the ERK1/2 inhibitor, U0126 (Sigma), and the AMPK inhibitor,
compound C (Sigma), for 30 min. The cells were then treated with or
without metformin (1,1-dimethylbiguanide hydrochloride, 100
µmol/ml, Sigma) and cultured for 24 h as previously
described (15), followed by
incubation with D-gal (15 mg/ml) at 37°C for an additional 48 h.
Detailed information on the treatment of each group of PC12 cells
is described in Fig. S2.
PC12 senescence model
PC12 cells were treated with increasing
concentrations of D-gal (3, 6, 9, 12, 15, 18, 21 and 24 mg/ml) for
48 h; there was a significant decrease in cell number from 18 mg/ml
(Fig. S3). The Cell Counting
kit-8 (CCK-8) assay (Dojindo Laboratories) was used to determine
the cell viability and cell proliferation according to the
manufacturer's instructions and as previously described (16). D-gal was used to induce senescence
in the PC12 cells, which were pre-treated with compound C (40
µmol/l), U0126 (10 µmol/l) or metformin (100
µmol/l) for 24 h for further experiments. The selected
concentrations of the chemical inhibitors, D-gal and metformin did
not affect cell viability (Fig.
S3). The β-galactosidase (β-Gal) assay, which measures the
activity of β-Gal by immunohistochemistry, is a simple and
effective method for measuring age-related changes in vitro
and in vivo (17). The
PC12 cells were stained using the β-Galactosidase Staining Kit
(C0602, Beyotime) according to the manufacturer's instructions.
Flow cytometry
Annexin V/PI analysis
The Annexin V-FITC Apoptosis Detection kit (KeyGen
Biotech) was used to detect cell apoptosis. The PC12 cells were
harvested with EDTA-free 0.25% trypsin at 37°C in a cell incubator,
washed twice with phosphate-buffered saline (PBS) and resuspended
in 500 µl of binding buffer. Subsequently, 5 µl of
Annexin V-FITC and PI were added to the PC12 cells for 15 min in
the dark at room temperature and analyzed using a flow cytometer
(Olympus).
Measurement of mitochondrial membrane
potential (ΔΨm)
A Mitochondrial Membrane Potential Assay kit with
JC-1 (Beyotime) was used to detect changes in ΔΨm according to the
manufacturer's instructoins. After each treatment, changes in the
ΔΨm of the PC12 cells were detected using the JC-1 assay.
Measurement of ATP production
The ATP content in the cultured PC12 cells was
determined using an adenosine 5′-triphosphate assay kit (A095,
Nanjing) according to the manufacturer's instructions.
Hearing test: ABR
The auditory threshold was examined by evoking the
ABR 4 times during the entire experiment (at 1, 3, 9 and 15 months
of age). The rats (n=10 per group) were anesthetized with ketamine
(100 mg/kg) and chlorpromazine (5 mg/kg) via intraperitoneal
injection (18), placed in a
double-walled radiofrequency and electrically shielded booth, and
an insert earphone was set. Stainless steel needle electrodes were
fixed subcutaneously ventrolateral to the right and left ears and
the vertex for recording. The detailed procedures of the ABR test
have been previously reported (1).
Morphological observation: Nissl staining
analysis
The rat brains were fixed in 10% neutral
formaldehyde solution, embedded in paraffin, and then sectioned at
5 µm. The sections were deparaffinized following standard
procedures. The brain slices were soaked in 0.3% toluidine blue
solution for 40 min at room temperature. The sections were then
dehydrated using 95% ethanol for 5 sec, slightly counterstained
with 0.5% eosin for 5 sec, cleared using xylene, placed under cover
slips and analyzed under a light microscope (Leica).
Measurement of antioxidant enzyme
activity
The levels of malondialdehyde (MDA), which results
from lipid peroxidation and the activities of glutathione (GSH),
superoxide dismutase (SOD) and catalase (CAT), were measured in
vitro and in vivo using the appropriate kits (Nanjing
Jiancheng Institute of Biological Engineering) according to the
manufacturer's instructions and as previously described (19).
Western blot analysis
Tissues from both sides of the auditory cortex were
dissected carefully on ice, and total protein was then extracted
immediately from the tissues and from the PC12 cells with an
ice-cold radioimmunoprecipitation assay (RIPA) lysis solution
(Beyotime). The protein concentration of the samples from both the
PC12 cells and the SD rats was quantified using a BCA Protein Assay
kit (Beyotime). Equal amounts of protein from each sample were
separated by 10-12% sodium dodecylsulfate polyacrylamide gel
electrophoresis (SDS-PAGE) and electrotransferred onto
poly-vinylidene difluoride (PVDF) membranes. The membranes were
blocked with 5% BSA for 1 h and incubated overnight at 4°C with an
appropriate dilution of specific primary antibodies. The following
antibodies were used: Anti-extracellular signal-regulated kinase
(ERK)1/2 and anti-p-ERK1/2 (diluted 1:1,000, 4695P and 4370P, Cell
Signaling Technology); anti-forkhead transcription factor (Foxo)3
(diluted 1:1,000, NBP2-24579, Novus Biologicals); anti-p-Foxo3
(diluted 1:1,000, ARG51655, Arigo Biolaboratories); anti-HSP90α
(diluted 1:2,000, GTX109753, GeneTex); anti-HSP60 (diluted 1:1,000,
4870, Sigma); anti-AMPKα1/2 (diluted 1:1,000, 21191, Signalway);
anti-p-AMPKα1/2 (diluted 1:600, sc-33524, Santa Cruz
Biotechnology); anti-GRP78 (diluted 1:2,000, GTX102580, GeneTex);
anti-GADD153/CHOP (diluted 1:3,000, NBP2-13172, Novus Biologicals);
anti-caspase-3 and anti-cleaved caspase-3 (diluted 1:1,000, 9662
and 9661, Cell Signaling Technology); anti-p53 (1:200, sc-98, Santa
Cruz Biotechnology); anti-α-tubulin (diluted 1:1,000, ab18251,
Abcam) and anti-β-actin (diluted 1:3,000, Mab1445, Lianke). The
membranes were washed 4 times before being incubated with
appropriate horseradish peroxidase (HRP)-conjugated secondary
antibodies (diluted 1:4,000, ANT019 and ANT020, AntGene) for 1 h at
room temperature and developed using ECL Plus reagents (Beyotime)
to visualize the membranes. Protein quantification was performed
using Quantity One 4.6.2 Software (Bio-Rad).
Quantitative polymerase chain reaction
(qPCR)
Total DNA was extracted from the tissues from both
sides of the auditory cortex and from 107 cultured
neurons using a Genomic DNA Purification kit (Tiangen Biotech Co.,
Ltd.). The purification and concentration of the DNA was evaluated
with a Gene Quant Pro DNA/RNA Calculator (BioChrom). Primers and
probes specifically designed for mitochondrial DNA (mtDNA) CD and
mtDNA D-loop have been described previously (20). The LC-480 Real-time PCR system
(Roche Diagnostics Ltd.) was used for performing the PCR
amplification. TaqMan PCR mix (2X, 10 µl, Takara), a probe
(0.2 µl for each, 10 mM), primers (0.4 µl of each
reverse and forward, 10 mM), distilled water (5 µl), and
sample DNA (4 µl, 10 ng/ml) comprised the 20-µl
reaction mixture. The cycling conditions were as follows: An
initial phase at 95°C for 30 sec, then 40 cycles at 95°C for 5 and
30 sec at 60°C. ΔCq (Cqdeletion−CqD-loop) was
used to calculate the abundance of the mtDNA 4,834 bp deletion. The
relative expression indicates the factorial difference in the
deletions between the subgroups, and it was calculated using the
2−ΔΔCq method (21),
where ΔΔCq=ΔCqmtDNA deletion in D-gal
group−ΔCqmtDNA deletion in control group or
ΔΔCq=ΔCqmtDNA deletion in Met group−ΔCqmtDNA
deletion in D-gal group. qPCR and the detailed method of
calculation were performed as reported previously by our laboratory
(22).
Electron microscopy
Transmission electron microscopy (TEM) was used to
examine the ultrastructure of the auditory cortex and the cultured
PC12 cells. After the PC12 cells were treated with the appropriate
drugs, the cells were fixed in 2.5% glutaraldehyde (Sigma) at 4°C
for 24 h. A total of 27 (n=3 from each subgroup) were used for the
TEM assay. The extraction of the specimens and the preparation,
conditions, and procedures were performed as previously described
(22,23).
TUNEL assay
A TUNEL Apoptosis Detection kit (FITC; Yeasen) was
used to detect PC12 cell apoptosis. At the same time, the nuclei
were counterstained with a DAPI staining solution (1 µg/ml;
Beyotime) at room temperature for 3-5 min. A laser-scanning
confocal microscope (Nikon) was used to examine the cells.
Statistical analysis
The data are expressed as the means ± standard
deviation (SD). Statistical analyses were conducted using GraphPad
Prism 5.0 software and Microsoft Excel. A two-tailed, unpaired
Student's t-test were used to determine statistical significance
when comparing 2 groups, and one-way analysis of variance (ANOVA)
followed by a Dunnett's multiple comparisons test was used to
compare differences between >2 groups. Differences with a
P-value <0.05 were considered to indicate statistically
significant differences. All experiments were repeated
independently at least 3 times.
Results
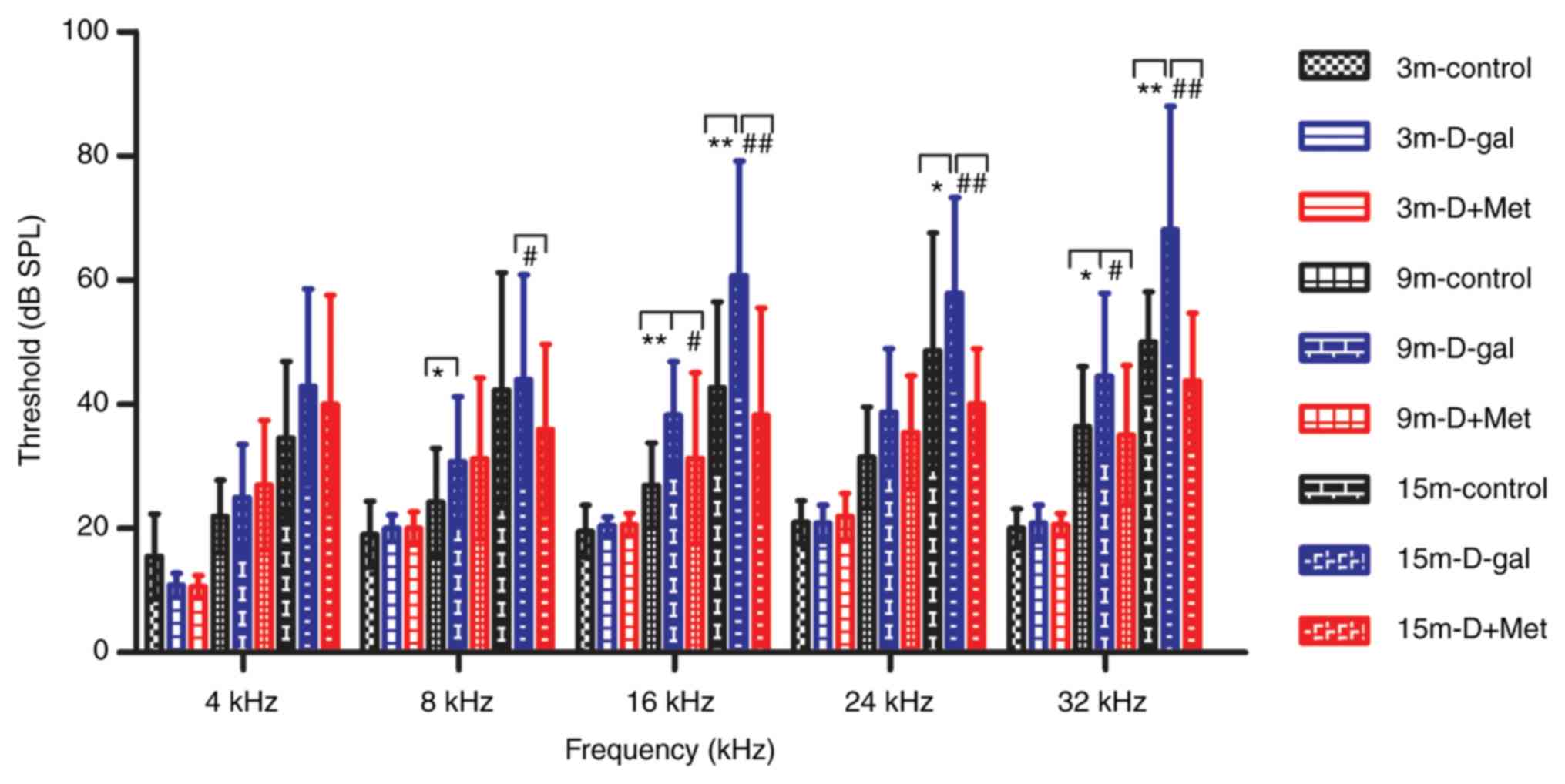
Metformin treatment alleviates
age-related hearing loss
The ABR threshold was significantly elevated in the
15-month-old rats compared with the 3-month-old rats. There was a
significant increase in the average ABR threshold in the D-gal
group compared with the control group, while metformin
significantly suppressed this elevation, particularly in the
15-month-old rats (Fig. 1). This
finding indicated that D-gal accelerated AHL and that metformin
exerted a protective effect on D-gal-induced aging in rats.
Metformin attenuates the
neurodegeneration induced by D-gal in vivo and in vitro
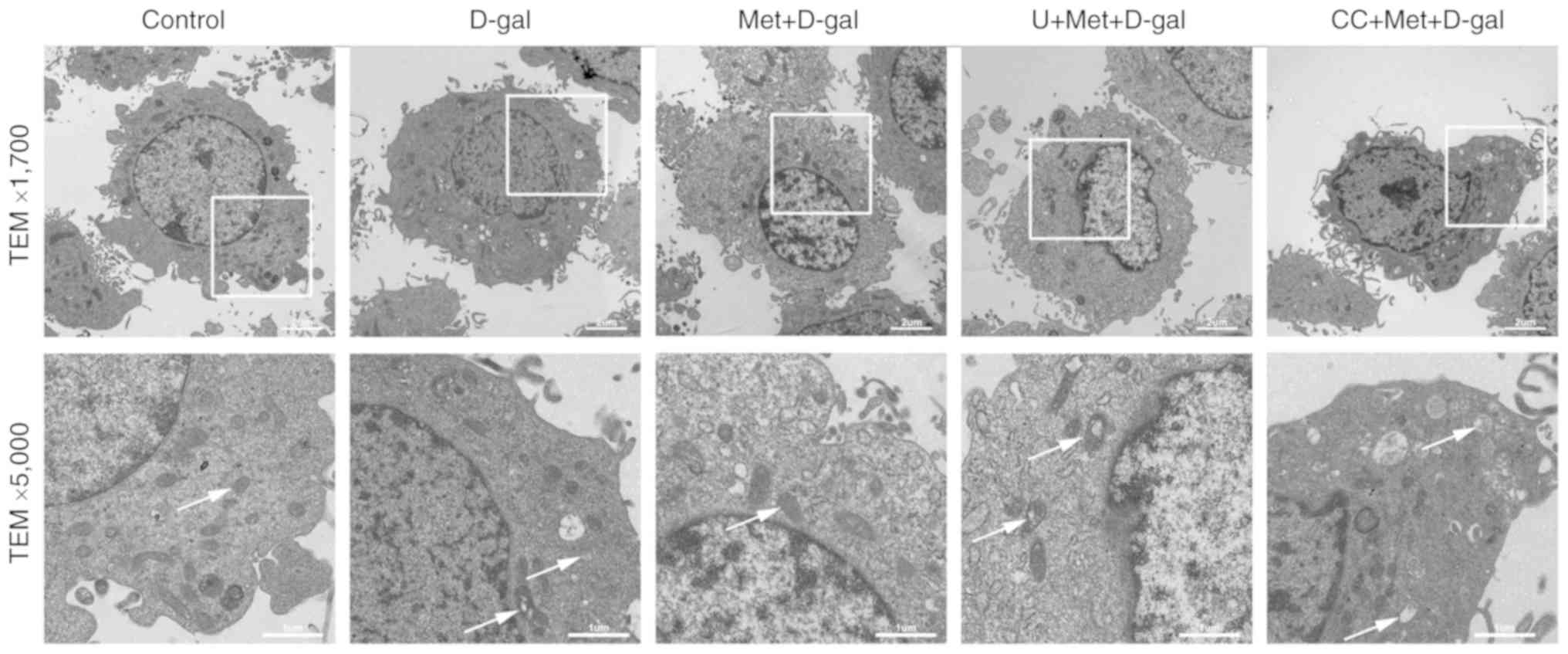
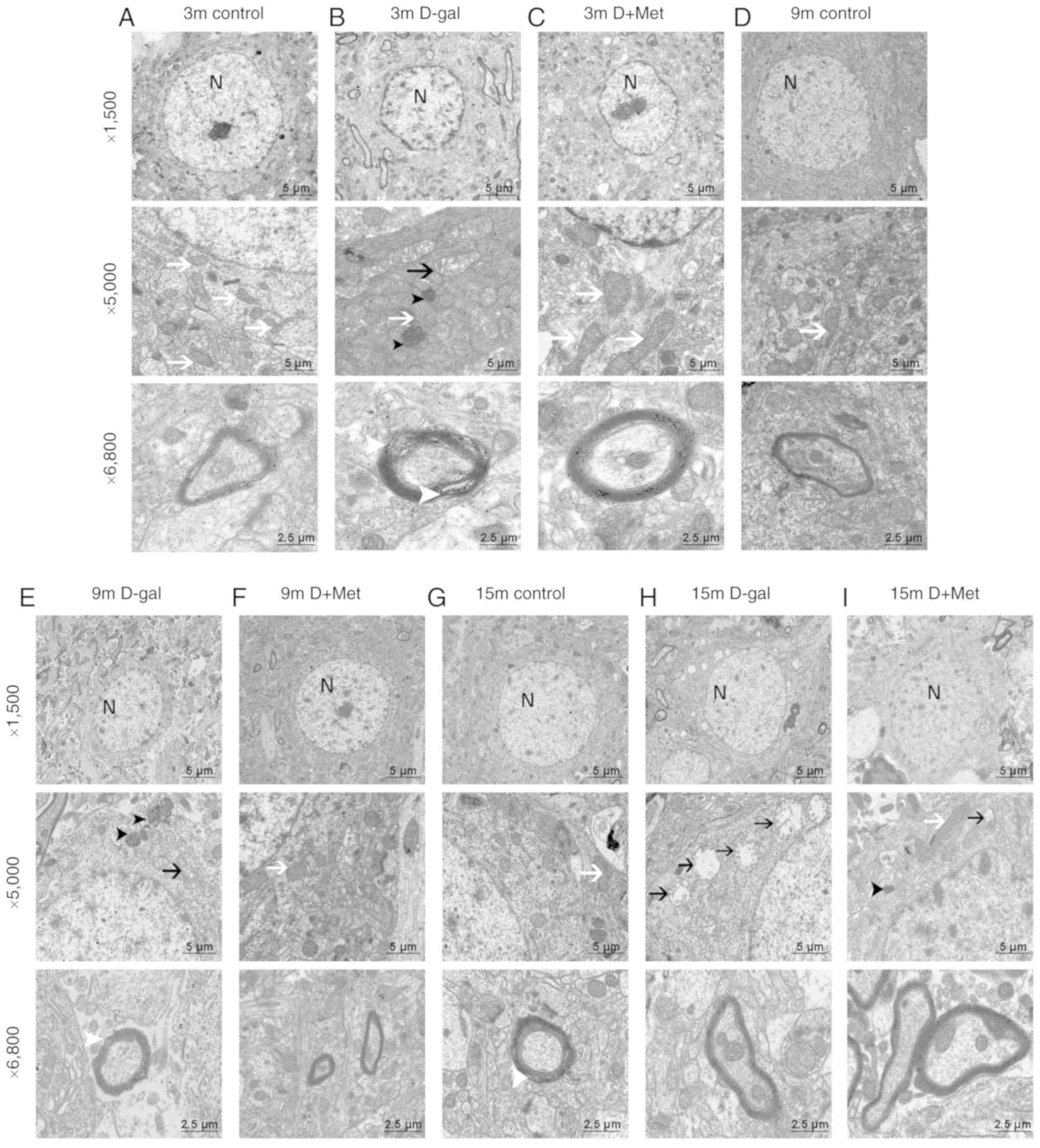
TEM was used to examine the effects of metformin on
neurodegeneration at the ultrastructural level in the auditory
cortex and in cultured PC12 cells. The sections from the PC12 cells
were analyzed at ×1,700 and ×5,000 magnification (Fig. 4). The sections from the auditory
cortex were analyzed at ×1,500, ×5,000, and ×6,800 magnification,
respectively focusing on the nuclei, organelles and the myelin. In
all rats in the 3-month-old groups (Fig. 2A-C), the nuclear membrane was
intact, and the nuclei (N) were round in shape. The chromatin in
the 9-month-old control group (Fig.
2D) was uniform, and the mitochondria (white arrows) were
normal. In the 9-month-old D-gal group (Fig. 2E), some irregular nuclei and
condensed chromatin were noted; moreover, these changes were more
significant in the rats in the 15-month-old D-gal group (Fig. 2H). Visible ultrastructural
degeneration, such as condensed chromatin, vacuolated mitochondria
(black arrows), and irregular nuclei, were found in the
15-month-old rats (Fig. 2G-I).
Additionally, the myelin was swollen and disrupted (white
arrowhead), and lipofuscin was abundant (black arrowhead).
Metformin treatment alleviated these changes induced by D-gal, as
the ultrastructure of the organelles appeared healthier (Fig. 2C, F and I).
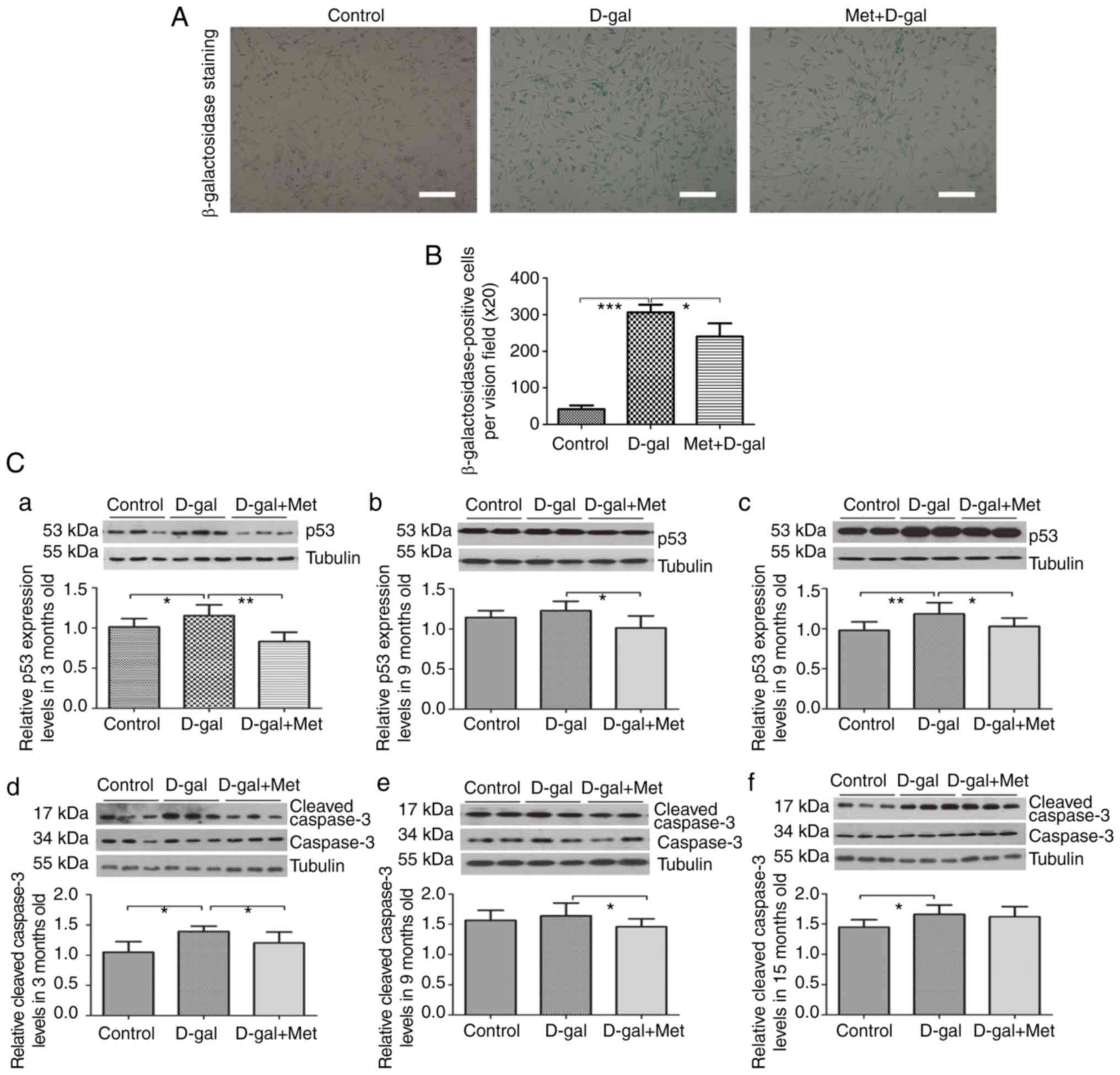
In addition, β-galactosidase staining was performed
to detect the apoptosis of PC12 cells. A significant accumulation
of positively stained cells was observed in the D-gal (15 mg/ml)
group, while pre-treatment with metformin reduced the number of
positive cells (Fig. 3A and B).
Considering these results, our model of senescent PC12 cells, which
was induced by 15 mg/ml D-gal, was successful and was used in our
subsequent experiments.
In our in vitro experiments, abundant normal
mitochondria (white arrows), an intact nuclear membrane and
normal-shaped nuclei were observed in the PC12 cells. However, in
the D-gal group, the mitochondria were swollen and vacuolated
(white arrows); however, pre-treatment with metformin alleviated
the mitochondrial damage. However, the inhibition of ERK1/2 and
AMPK aggravated these changes (Fig.
4).
Toluidine blue staining was used to determine
neuronal loss in the auditory cortex region, the sections were
analyzed at ×200 and ×400 magnification (Fig. 5). The results demonstrated that
the number of neurons in the auditory cortex regions was
significantly reduced in the D-gal group compared with the control
group, and these alterations were significantly attenuated by
metformin treatment. In the rats in the same age groups, the rats
in the control group and Met group had numerous neurons compared
with those in the D-gal group, and the neurons in the rats in the
control group and Met group were arranged tightly and were
morphologically intact. An irregular structure, a decreased number
of pyramidal cells, and degenerated pyramidal cells were visible in
the auditory cortex region of the brains of 15-month-old rats
(Fig. 5A and B).
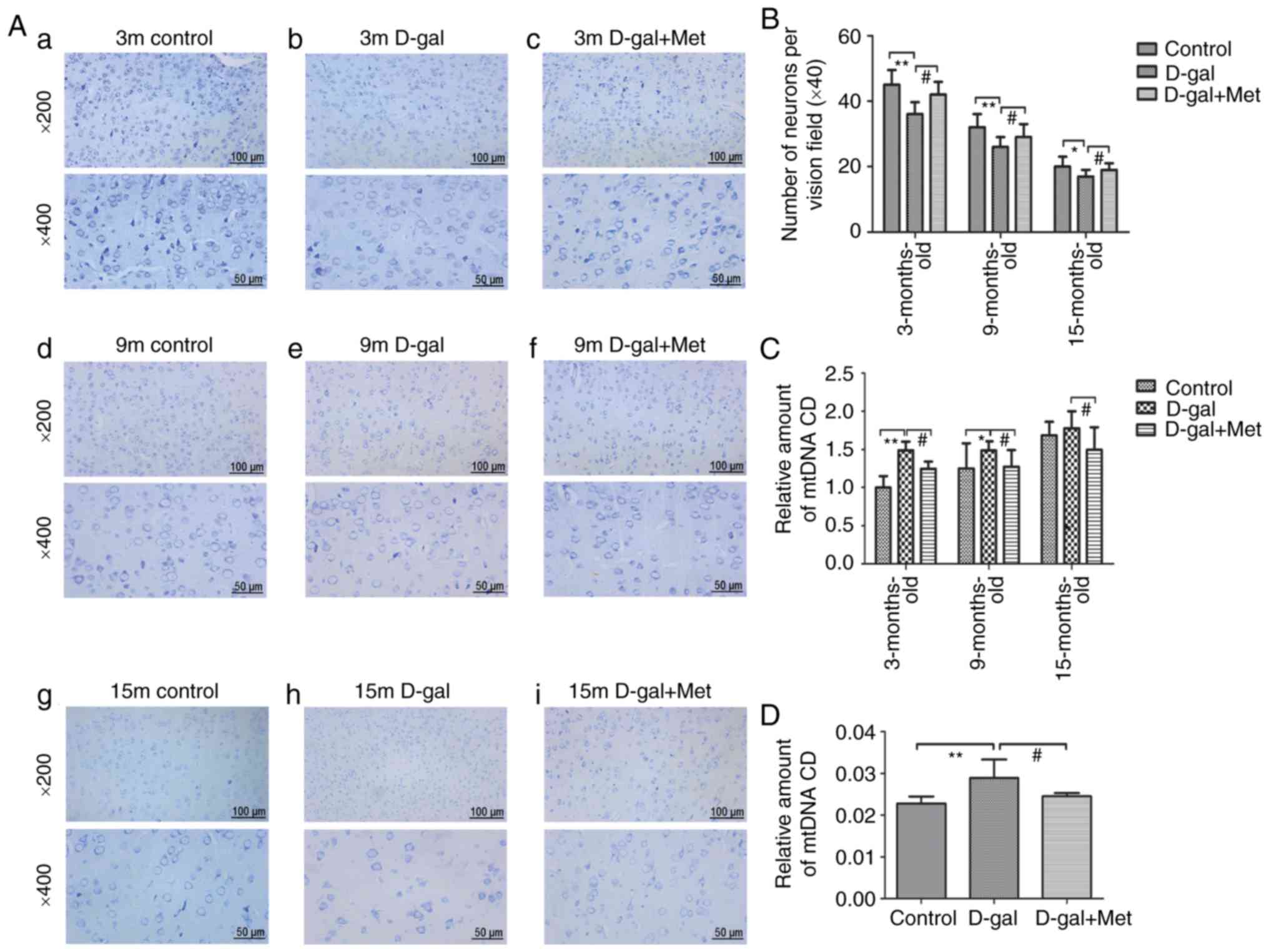 | Figure 5(A) Sections from the auditory cortex
of rats were stained with toluidine blue and used to count
surviving neurons. The sections from the auditory cortex were
analyzed at ×200 and ×400 magnification, (a-c) 3-Month-old rats;
(d-f) 9-month-old rats; (g-i) 15-month-old rats. The top images
were analyzed at ×200 magnification, and the bottom images were
analyzed at ×400 magnification (n=6 for each subgroup); each
paraffin-embedded rat brain sample was cut into several slices; the
images in the top and bottom panels are the same mouse brain, but
from different slices. (B) The corresponding statistical chart is
displayed. (C) qPCR analysis of CD 4,834 levels in the 3-, 9-, and
15-month-old control, D-gal and Met groups. (D) qPCR analysis of CD
4,834 levels in PC12 cells. *P<0.05,
**P<0.01; #P<0.05. The data are
displayed as the means ± SD; n=6 for each subgroup. m, months;
D-gal, D-galactose; Met, metformin; CD, common deletion. |
Metformin alleviates the diminished
function of the antioxidant system via the AMPK/ERK1/2 signaling
pathways
We examined the activity of antioxidants and
investigated the mechanisms underlying the effects of metformin on
antioxidants. The activities of the antioxidants, GSH, SOD and CAT,
were decreased in the blood serum of the rats in the 3-, 9- and
15-month-old D-gal groups, respectively, compared with those in the
age-matched control groups, while concurrent metformin treatment
significantly attenuated this decrease (Fig. 6A-C). In addition, we also found
that the levels of CAT, GSH and SOD were reduced with age. These
alterations in enzyme activity were also detected in the PC12
cells, suggesting that GSH, SOD and CAT enzyme activities were
markedly decreased in the senescent aging model and increased upon
treatment with metformin (Fig.
6E-G). MDA is a product of lipid peroxidation mediated by
oxygen free radicals and is widely known as a marker of oxidative
stress (24). The data indicated
that the MDA levels in the D-gal group increased significantly
compared with those in the age-matched control group. In the
metformin group, the MDA level was decreased compared with the
age-matched D-gal group. In addition, the levels of MDA increased
with aging (Fig. 6D and H). All
these changes caused by D-gal were attenuated by metformin
treatment, while the effect was partially suppressed when the PC12
cells were treated with the AMPK and ERK1/2 inhibitors (Fig. 6E-H). The results also implied that
the improved antioxidant capacity induced by metformin occurred
through the AMPK and ERK1/2 signaling pathways to a certain
extent.
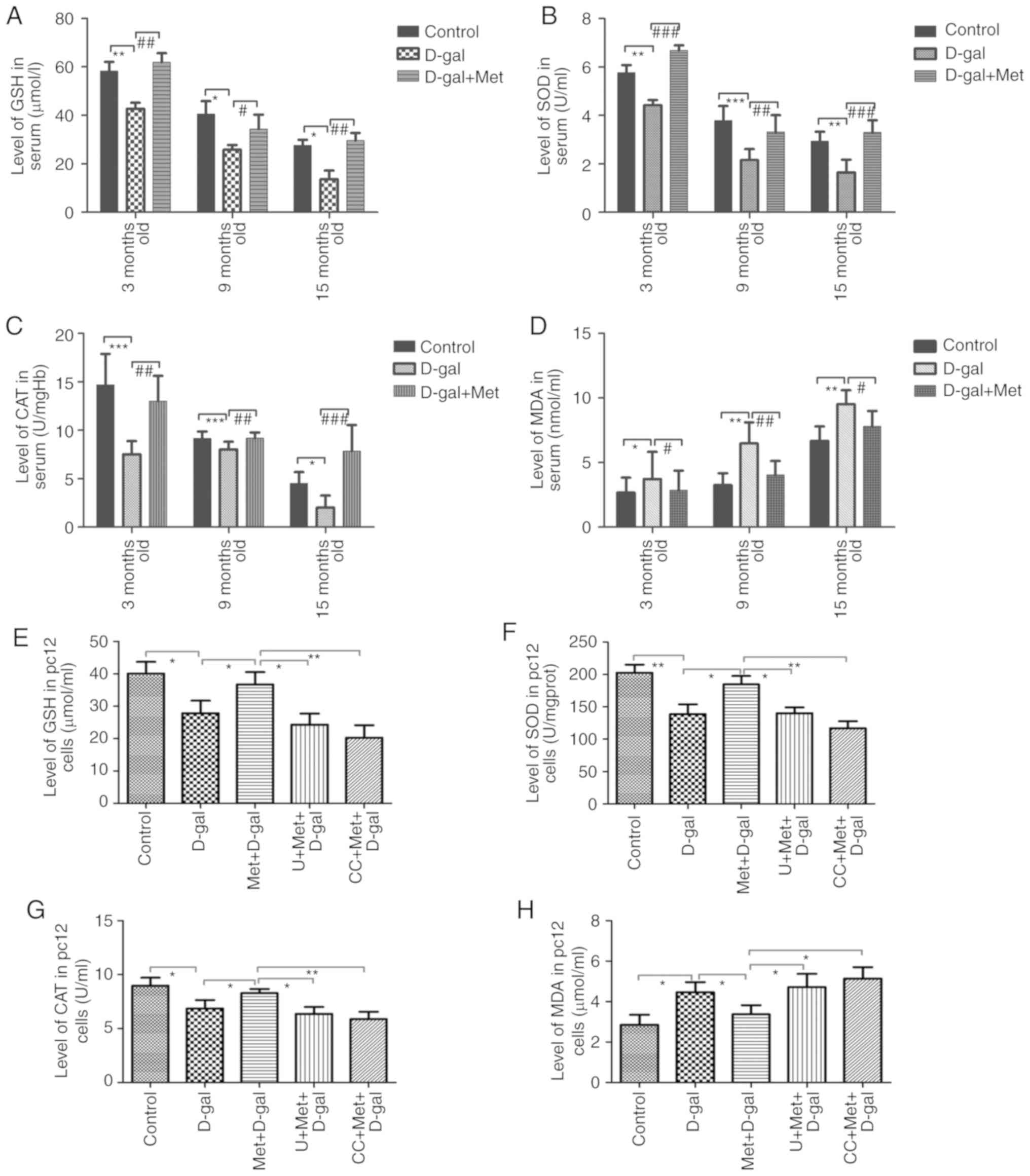 | Figure 6Analysis of antioxidants in
vitro and in vivo. Metformin treatment significantly
increased the (A) GSH, (B) SOD and (C) CAT levels in the auditory
cortex of D-gal-injected rats. Metformin treatment significantly
decreased the (D) MDA level compared with that in the respective
D-gal groups. The activities of these enzymes were detected in PC12
cells, suggesting that (E) GSH, (F) SOD, (G) CAT and (H) MDA enzyme
activities were markedly decreased in the senescent aging model and
increased upon treatment with metformin; however, this upregulation
was prevented significantly by inhibitors of AMPK and ERK1/2. The
data are presented as the means ± SD (n=6). *P<0.05,
**P<0.01, ***P<0.001,
#P<0.05, ##P<0.01,
###P<0.001. D-gal, D-galactose; Met, metformin; SOD,
superoxide dismutase; GSH, glutathione; MDA, malondialdehyde. |
Metformin protects the mitochondria
through the AMPK/ERK1/2 pathways
The function of the mitochondria was determined
through the evaluation of mitochondrial membrane permeabilization
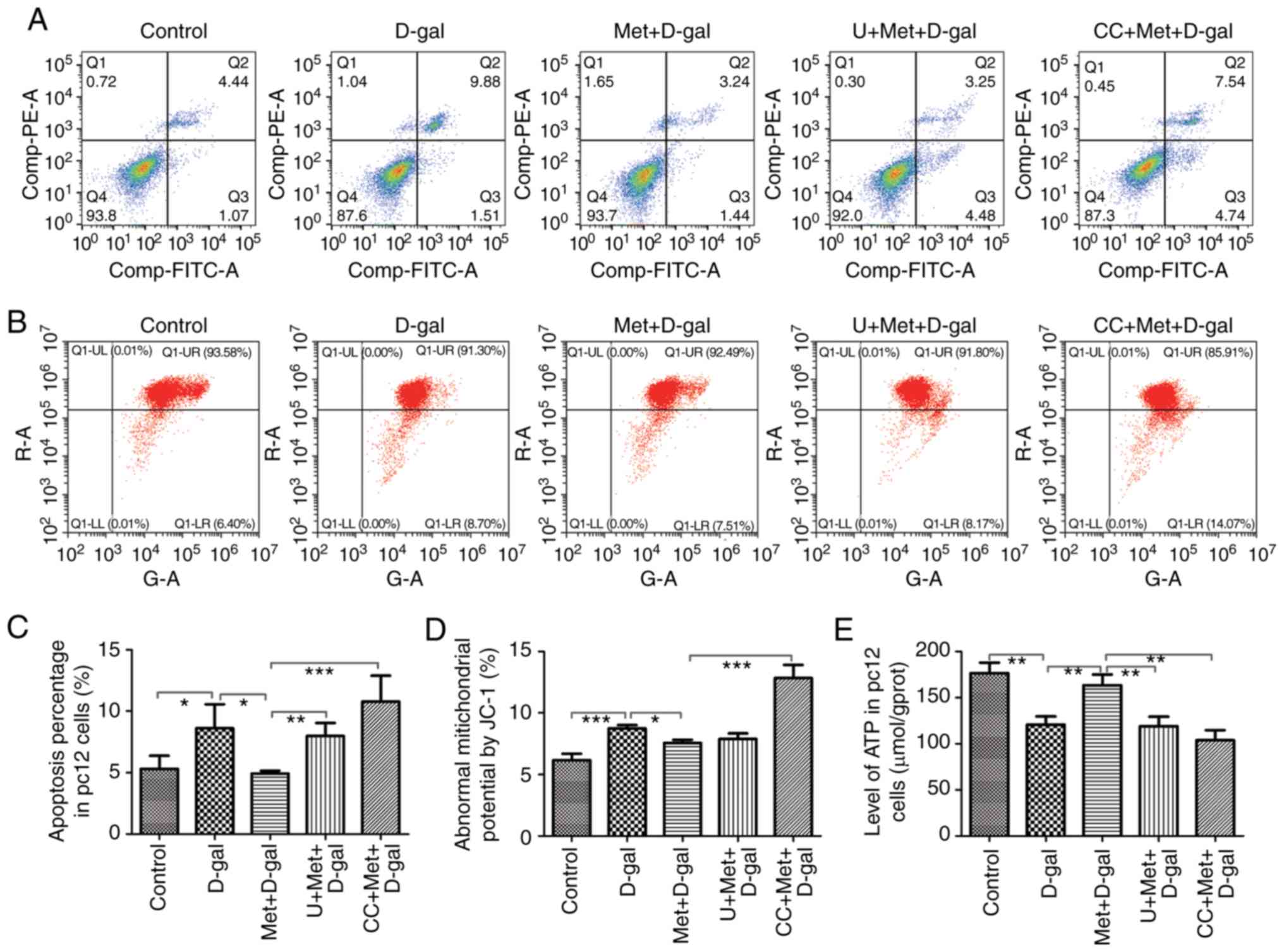
and ATP production. The flow cytometry data revealed a decreased
mitochondrial membrane permeabilization in the D-gal group, which
indicated that the number of early apoptotic mitochondria was
increased (Fig. 7B and D).
Annexin V/PI staining and flow cytometry analysis of cell death
were repeated 4 times and similar results were obtained (Fig. 7A). The average values of the
percentages of apoptotic cells from repeated experiments are shown
in Fig. 7C. The number of
apoptotic mitochondria significantly decreased with metformin
treatment, while the protective effect was suppressed when the PC12
cells were treated with AMPK and ERK1/2 inhibitors (Fig. 7). In addition, we detected the ATP
levels in the PC12 cells. ATP, which is produced by mitochondria,
is the most intuitive indicator of MT function. The levels of ATP
decreased in the D-gal group, but increased with metformin
treatment; however, when the PC12 cells were simultaneously treated
with AMPK or ERK1/2 inhibitors, metformin could not increase the
ATP levels (Fig. 7E). Taken
together, these results indicated that metformin protected the
mitochondria through the AMPK and ERK1/2 signaling pathways to a
certain extent.
Metformin treatment decreased the accumulation of
the mtDNA common deletion (CD) in vitro and in vivo
(Fig. 5C and D). Previous studies
have confirmed that the mitochondrial 4,834-bp deletion is a
biomarker of AHL, and the 4,834-bp deletion gradually deteriorates
with age (25,26). In this study, compared with
age-matched control groups, the D-gal groups exhibited a markedly
elevated CD level (Fig. 5C); CD
accumulated with age, and metformin decreased the CD level
throughout the process. The same phenomenon was observed in the
PC12 cells (Fig. 5D).
Metformin prevents apoptosis via the
AMPK/ERK1/2 signaling pathways
To investigate the stress-related apoptotic factors,
caspase-3 and p53, and to examine the effects of metformin on these
proteins, changes in protein expression were examined by western
blot analysis. In the auditory cortex tissues, significant
increases in p53 and cleaved caspase-3 expression were noted after
the injection of D-gal. However, we found a significantly decreased
expression of p53 and cleaved caspase-3 following treatment with
metformin (Fig. 3C). We also
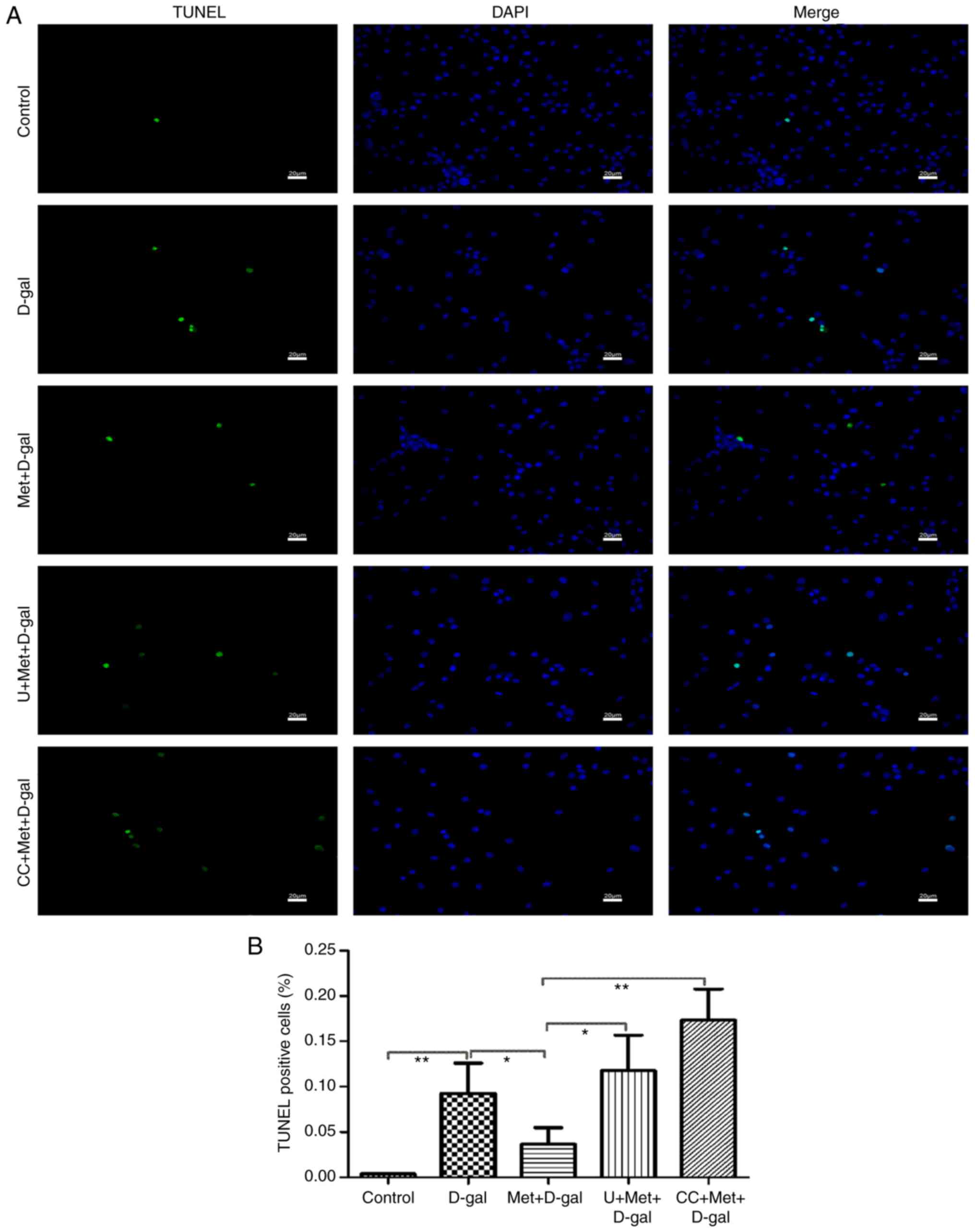
analyzed apoptosis by counting the number of cells stained by TUNEL
assay. Apoptosis Detection kit (FITC) staining and Annexin V/PI
staining were performed on the PC12 cells. The right upper quadrant
and right lower quadrant represent the late and early apoptotic
cells, respectively. The number of apoptotic cells in the Q2 and Q3
panels (upper and lower right) was calculated, and the percentages
of cells undergoing apoptosis was determined (Fig. 7A). Significant increases in the
number of TUNEL-positive cells were noted after the injection of
D-gal; however, treatment with metformin decreased the percentage
of apoptotic cells (Fig. 8).
Moreover, treatment with AMPK and ERK1/2 inhibitors partially
reversed the protective effects of metformin. Thus, metformin
alleviated the apoptosis of the PC12 cells through the AMPK and
ERK1/2 pathways.
Metformin regulates the UPR to maintain
protein homeostasis in vivo and in vitro
Protein misfolding and aggregation are common
pathological features of a number of neurodegenerative diseases
(27). In this study, significant
increases in the levels of UPR-related proteins were observed after
the injection of D-gal, which is consistent with the findings of
previous studies (28,29). The ER stress markers, GRP78 and
CHOP, are also known as major modulators of the ER-UPR. HSP90 and
HSP60 are chaperone proteins that assist other proteins in folding
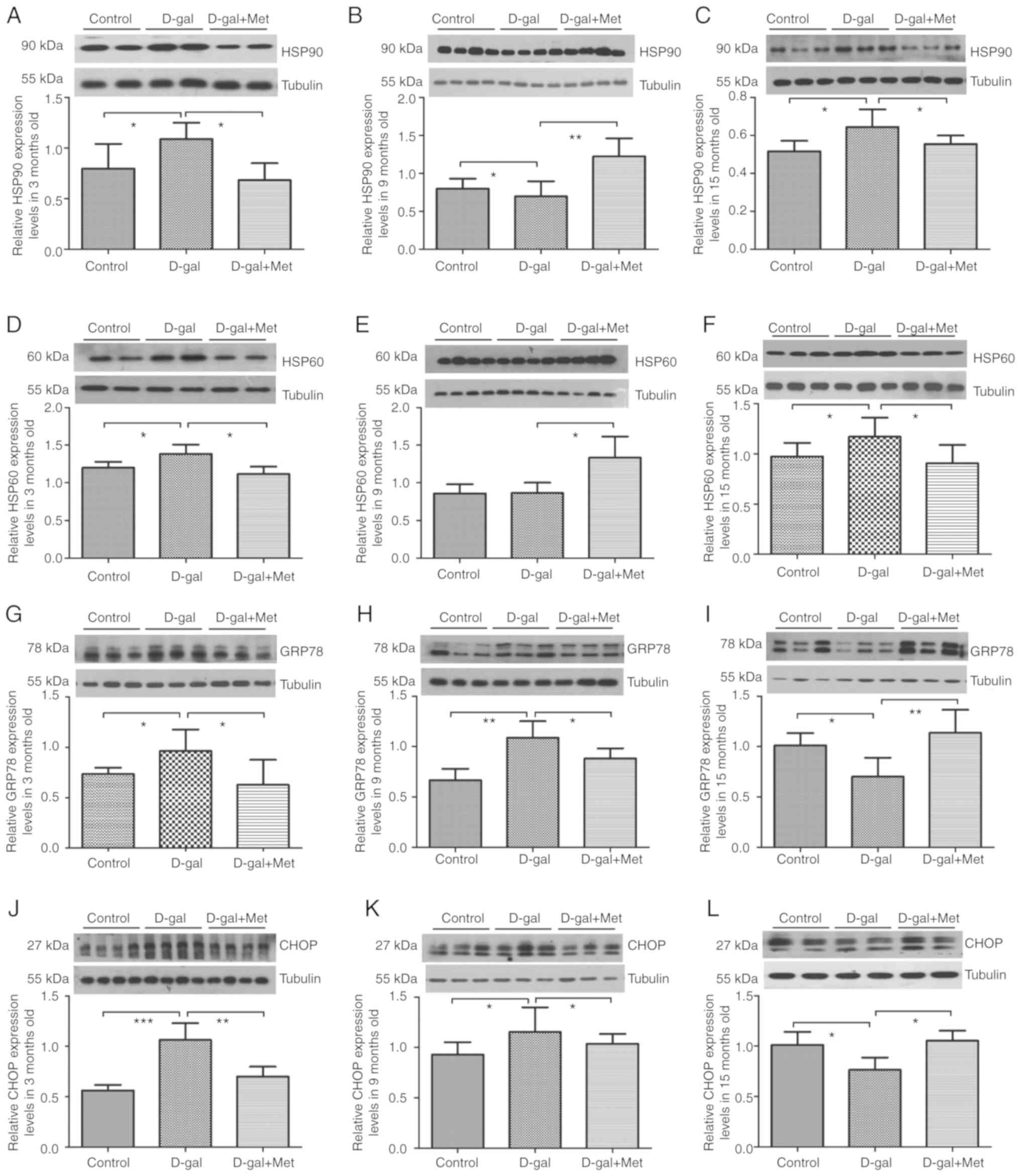
properly and are also modulators of the MT-UPR. In our in
vivo experiments, we found that the expression levels of HSP90,
HSP60, GRP78 and CHOP were upregulated in the 3-month-old D-gal
groups; however, metformin significantly decreased these expression
levels. The levels of the MT-UPR chaperone proteins, HSP90 and
HSP60, were still upregulated in the 15-month-old D-gal groups, and
metformin significantly decreased these expression levels. However,
the expression levels of the ER-UPR chaperone proteins, GRP78 and
CHOP, decreased in the 15-month-old D-gal group compared with the
control group, but increased in the 15-month-old Met group compared
with the D-gal group (Fig. 9). In
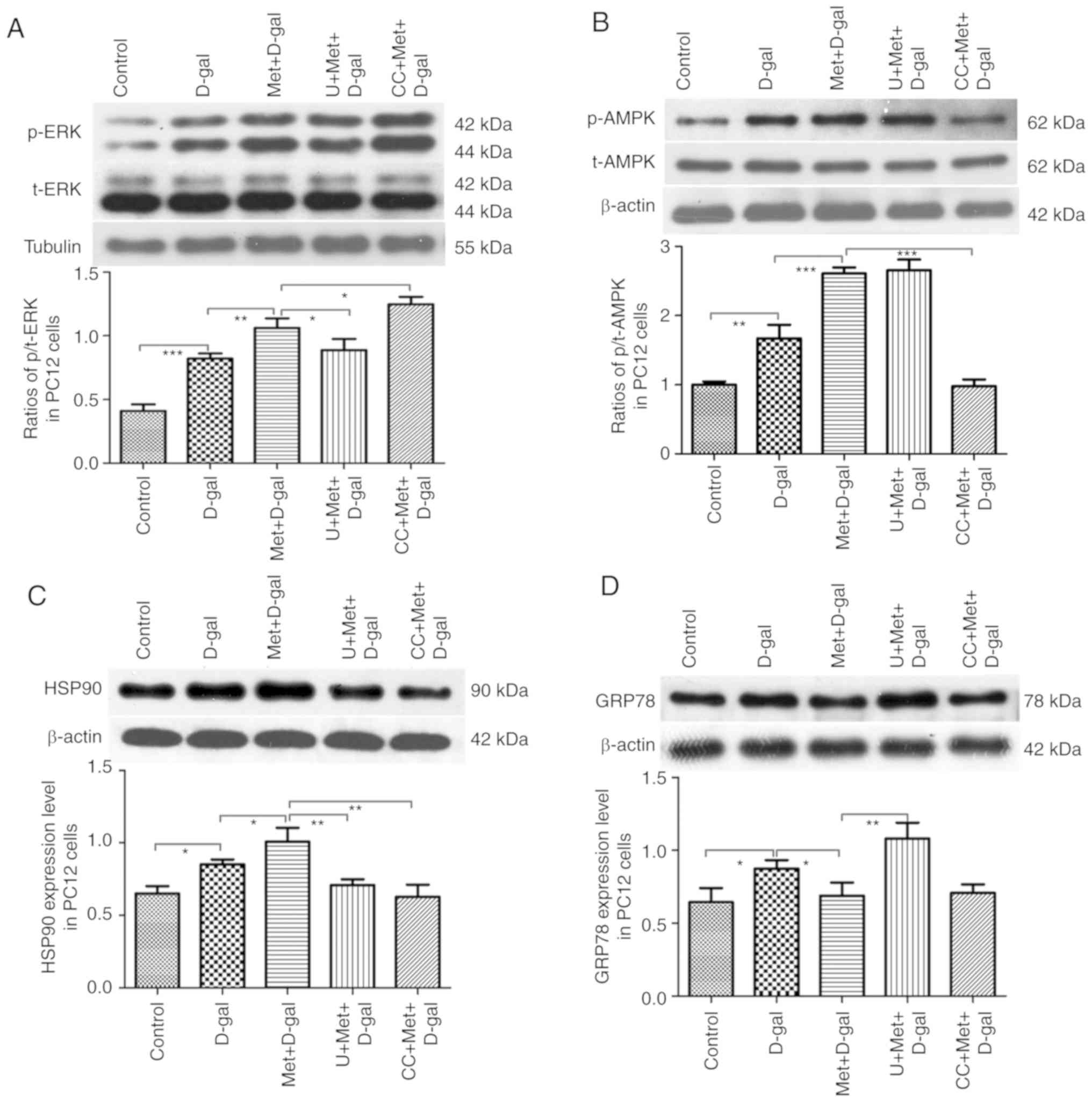
our in vitro experiments, HSP90 expression was upregulated
following treatment with D-gal, and its expression increased
further following additional treatment with metformin. Treatment
with AMPK and ERK1/2 inhibitors significantly reversed the effects
of metformin (Fig. 10C). A
significant elevation in GRP78 expression was also observed in the
PC12 cells following treatment with D-gal; however, treatment with
metformin led to a significant decline in GRP78 expression.
Conversely, in the groups pre-treated with U0126, metformin did not
decrease GRP78 expression, but when the groups were pre-treated
with compound C, changes were not evident (Fig. 10D). These findings focus on the
UPR pathways, suggesting the role of metformin in the maintenance
of cellular homeostasis.
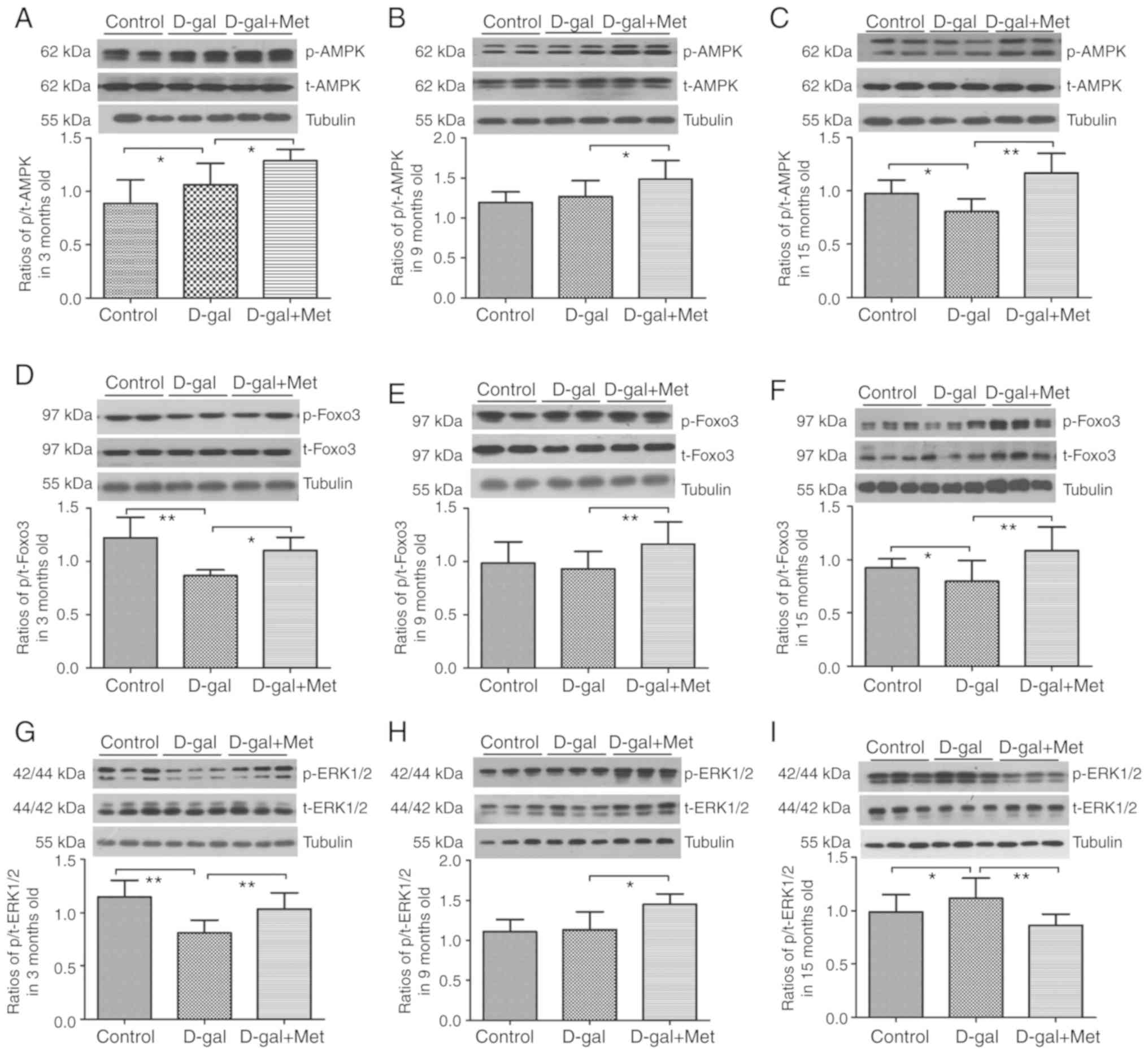
Metformin regulates the AMPK/ERK1/2
signaling pathways in vivo and in vitro during aging
AMPK and ERK1/2 act independently, and the crosstalk
between them is frequent. Notably, the crosstalk is highly
variable; both cross-inhibition and cross-activation can occur
depending on the cell type (30).
In this study, metformin treatment significantly activated p-AMPK
and p-Foxo3α in the 3-, 9-, and 15-month-old rats (Fig. 11A and B). A significant decrease
in the phosphorylation levels of Foxo3α and ERK1/2 was noted after
the injection of D-gal. By contrast, we observed an increase in the
Foxo3α and ERK1/2 phosphorylation levels in the 3-month-old
metformin group (Fig. 11B and
C); however, the ERK1/2 phosphorylation levels were decreased
in the 15-month-old metformin group (Fig. 11C). In our in vitro
experiment, a significant elevation in the phosphorylation levels
of ERK1/2 and AMPK was noted following treatment with D-gal, and a
further elevation in the levels of these proteins was found
following co-treatment with metformin. In addition, the inhibition
of ERK1/2 blocked the effects mediated by metformin. However,
surprisingly, the inhibition of AMPK slightly increased the
phosphorylation level of ERK1/2. Thus, there was some association
between ERK1/2 and AMPK, indicating that the AMPK pathway can
inhibit the ERK1/2 pathway in PC12 cells to a certain extent
(Fig. 10A and B).
Discussion
D-gal accelerates the process of
age-related neurodegeneration
Mice treated with D-gal exhibited oxidant damage in
the blood, brain and liver, and D-gal treatment in mice was
confirmed as a successful mimetic aging model (31). Our previous study confirmed that
D-gal injection induced a mimetic aging effect in the inner ear of
Wistar rats (32). Eight weeks of
D-gal treatment successfully established an ideal mimetic SD rat
model of presbycusis in our previous study (33).
In this study, a significant decline in the levels
of antioxidants was noted after the injection of D-gal, as observed
in several neurodegenerative disorders. ROS production increases
with age, and it is known that a lack of proteostasis, oxidative
stress and the dysfunction of organelles are critical factors
leading to the onset of age-related diseases (34-36). The mitochondria play a central
role in a number of metabolic tasks, such as the regulation of
cellular metabolism, cellular proliferation regulation, calcium
signaling and hormonal signaling; however, the function of the
mitochondria decreases with age (37-39). Studies have suggested that the
MT-UPR is triggered when mitochondrial DNA and nuclear DNA are not
balanced (40,41). Multiple studies have indicated
that there is a strong associatoin between the UPR and longevity,
physiology, and age-related diseases (40,42,43). The ROS signal is a major modulator
in the process of aging (44);
however, the effect of the MT-UPR on longevity does not depend on
the ROS signal (40). The
4,834-bp mtDNA deletion in rats is similar to the 3,867-bp deletion
in mice and the 4,977-bp deletion in humans; these deletions are
also known as CDs, and they have been reported to be the most
common age-related mtDNA damage and are used as biomarkers of aging
(45-47). Animal models of aging established
by our laboratory can trigger oxidative stress and increase the
accumulation of the CD in the peripheral and central auditory
system (19,48). In this study, we also found the
same results in PC12 cells, and the CD accumulated more readily in
aged rats. The mitochondrial ultrastructure was damaged, and flow
cytometry data revealed that the decreased mitochondrial membrane
permeabilization after the injection of D-gal, which indicated that
the number of early apoptotic mitochondria was increased. Both
mtDNA mutations and mitochondrial ultrastructural damage in
vitro and in vivo weaken mitochondrial function. Herein,
significant decreases in the number of neurons and severe apoptosis
were observed after the injection of D-gal. At the same time, the
p53- and caspase-3-dependent apoptosis pathways were activated by
D-gal.
The UPR protects organelle protein
homeostasis in the aging brain
Recently, a number of researchers have attempted to
clarify the mechanism sof the UPR in maintaining homeostasis
(49). Although it has been
reported that the UPR is involved in age-related neurodegenerative
diseases (27); however, the
specific role and function of the UPR in various diseases is fairly
complex and controversial.
This study indicated an increase in UPR in the D-gal
groups. The accumulation of unfolded or misfolded proteins induced
by chronic D-gal injection resulted in the elevation in the levels
of the stress response-related proteins, HSP90, HSP60, GRP78 and
CHOP. In some cases, the aberrant accumulation of UPR-related
proteins can be a dangerous signal to activate the inflammatory
response (50) and shorten
lifespan (40). In this study,
hearing ability, the morphology of organelles and multiple
functions were improved by achieving a balance of the UPR by
metformin treatment. Notably, in the 15-month-old D-gal group,
GRP78 and CHOP expression decreased significantly, and greater
apoptosis and significant neuronal death were observed. This
finding may illustrate that aged rats cannot deal with chronic and
excessive stress from D-gal and that ER cannot trigger a sufficient
UPR to remove or degrade the unfolded or misfolded proteins. Our
results are in accordance with those of other studies that the
proper activation and mild stimulation of the UPR can ameliorate
metabolic and age-related diseases, yet the improper or excessive
activation of the UPR can release harmful signals (50-52). These results indicate that ER-UPR
in aged rats cannot cope with long-term persistent oxidative stress
or trigger adequate UPR signals to combat the stress of unfolded
proteins. Our data also indicate that the UPR is time-dependent and
that metformin suppresses the overexpressed MT-UPR in rats
following exposure to D-gal, whereas metformin activates the
ER-UPR, which is beneficial for healthy brain metabolism, in aged
rat brains.
Metformin protects neurons via the UPR
through the AMPK/ERK1/2 signaling pathways
In this study, metformin significantly increased
antioxidant capability in vitro and in vivo. Other
researchers also reported that metformin exerts antioxidant effects
on the brain during oxidative stress (53). In this study, a significant
decrease in the occurrence of CD was observed in rats treated with
metformin and in senescent PC12 cells. Additionally, metformin
alleviated the level of apoptosis and changes in ultrastructural
morphology through the AMPK and ERK1/2 pathways. Metformin
significantly decreased neuronal apoptosis and reversed abnormal
neuronal damage; thus, this study indicated that metformin afforded
a useful benefit, maintaining a healthy state and delaying the
aging process of the auditory system.
Our data not only confirmed that UPR pathways are
potential regulating points for the effects of metformin on
proteostasis, but also indicated that the inhibition of AMPK or
ERK1/2 can influence the UPR. Our investigations revealed long-term
changes in AMPK and ERK1/2 signaling in the auditory cortex and in
a cellular model of senescence.
ERK1/2 is a member of the MAPK family and integrates
external signals into signaling events, promoting a large amount of
cell growth and proliferation (54). In many cases, the phosphorylation
of ERK1/2 results in the activation of its kinase activity and
leads to prosurvival factors (55,56). By contrast, others have reported
that ERK1/2 is often related to cell death and apoptosis (57-60). The level of phosphorylated ERK1/2
and its specific function vary with different stimuli, stimulus
methods and stimulus duration. In fact, the role of ERK1/2 in the
aging process pf the brain remains unknown. A few studies have
indicated that the phosphorylation of ERK1/2 in brain tissue
decreases with age (61,62). This study found that oxidative
stress or unfolded protein stress triggered by D-gal deactivated
ERK1/2 in 3-month-old rats, yet metformin recovered the
phosphorylation levels to regulate cell proliferation and
differentiation in rat brains. However, in 15-month-old rats, the
results revealed the overexpression of p-ERK1/2 in aged
D-gal-treated rats. This finding was consistent with the changes
observed in the morphological and functional experiments. The
persistent activation of ERK1/2 contributes to oxidative toxicity
and induces cell death (63), and
metformin markedly reverses phosphorylation to protect neurons from
apoptosis or death. In our in vitro experimetns, D-gal and
metformin activated ERK1/2 and AMPK phosphorylation. To our
surprise, AMPK inhibition slightly increased the phosphorylation
level of ERK1/2, indicating that the AMPK pathway can partially
inhibit the ERK1/2 pathway in PC12 cells.
The crosstalk between the AKT signaling pathway and
the ERK1/2 signaling pathway is dynamic. It has been reported that
ERK1/2 activation is accompanied by a gradual decrease in both AKT
levels and apoptosis (64), and
phosphorylated AKT inhibits the activation of ERK1/2 (65). The findings of this study that
increased levels of AMPK in rat brains were accompanied by a
decrease in ERK1/2 and that an increase in ERK1/2 was accompanied
by a decrease in AMPK. However, whether metformin suppresses ERK1/2
activity in aged rats through the regulation of AMPK or through UPR
signaling requires further investigation. Our findings also
indicated that Foxo3α may be a regulator of the protection of
neuronal survival against D-gal by metformin.
In conclusion, the present data suggest that D-gal
injection induces multiple molecular and functional changes,
oxidative stress, mitochondrial dysfunction and abnormal apoptosis
in the brain similar to natural aging. Metformin regulates the UPR
to promote neuronal survival and cellular homeostasis, ultimately
resulting in the enhanced hearing ability of D-gal-treated rats.
The protective effect of metformin involves the regulation of the
UPR through the AMPK/ERK1/2 pathways. Our studies demonstrated a
crosstalk between the AMPK/ERK1/2 and UPR signaling pathways.
Metformin-induced cell protection in vitro and in
vivo may provide novel therapeutic targets for postponing the
aging process of the brain and suggest that metformin may be a
potential therapeutic treatment for age-related diseases.
Supplementary Data
Funding
This study was supported by research grants from the
National Natural Science Foundations of China (nos. 81873700 and
81230021).
Availability of data and materials
The analyzed datasets generated during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
HC, WK and WJK conceived and designed the
experiments; HC, BH, YH, XZ, ZH, XC, HS, JY, YL and XY performed
the experiments; HC, WK and WJK analyzed the data; HC, BH, YH, WK
and WJK wrote the manuscript. All the authors have read and
approved the final version of this manuscript.
Ethics approval and consent to
participate
All experimental procedures involving the care of
animals were performed in strict accordance with the
recommendations of the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health (14). The protocol was under the
supervision of the Committee on the Ethics of Animal Experiments of
Huazhong University of Science and Technology (Wuhan, China; permit
no. IACUC S2219).
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
D-gal
|
D-galactose
|
|
ABR
|
auditory brainstem response
|
|
mtDNA
|
mitochondrial DNA
|
|
ROS
|
reactive oxygen species
|
|
CD
|
common deletion
|
|
SOD
|
superoxide dismutase
|
|
GSH
|
glutathione
|
|
MDA
|
malondialdehyde
|
|
TEM
|
transmission electron microscopy
|
|
qPCR
|
quantitative polymerase chain
reaction
|
|
MT
|
mitochondrial
|
|
ER
|
endoplasmic reticulum
|
|
UPR
|
unfolded protein response
|
|
Foxo
|
forkhead transcription factor, class O
transcription
|
|
AMPK
|
AMP-activated protein kinase
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
HSP90
|
heat shock protein 90
|
|
HSP60
|
heat shock protein 60
|
|
CHOP
|
C/EBP homologous protein
|
|
GRP78
|
glucose-regulated protein 78
|
|
TUNEL
|
terminal deoxynucleotidyl
transferase-mediated deoxyuridine 5′-triphosphate nick-end
labeling
|
|
ANOVA
|
analysis of variance
|
Acknowledgments
Not applicable.
References
|
1
|
Du Z, Yang Y, Hu Y, Sun Y, Zhang S, Peng
W, Zhong Y, Huang X and Kong W: A long-term high-fat diet increases
oxidative stress, mitochondrial damage and apoptosis in the inner
ear of D-galactose-induced aging rats. Hear Res. 287:15–24. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gates GA and Mills JH: Presbycusis.
Lancet. 366:1111–1120. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Humes LE, Dubno JR, Gordon-Salant S,
Lister JJ, Cacace AT, Cruickshanks KJ, Gates GA, Wilson RH and
Wingfield A: Central presbycusis: A review and evaluation of the
evidence. J Am Acad Audiol. 23:635–666. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Taylor RC and Dillin A: Aging as an event
of proteostasis collapse. Cold Spring Harb Perspect Biol.
3:a0044402011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Jensen MB and Jasper H: Mitochondrial
proteostasis in the control of aging and longevity. Cell Metab.
20:214–225. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li J, Akil O, Rouse SL, McLaughlin CW,
Matthews IR, Lustig LR, Chan DK and Sherr EH: Deletion of Tmtc4
activates the unfolded protein response and causes postnatal
hearing loss. J Clin Invest. 128:5150–5162. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chaube R: Can UPR integrate fasting and
stem cell regeneration? FRONT CHEM. 3:52015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Duran-Aniotz C, Martínez G and Hetz C:
Memory loss in Alzheimer's disease: Are the alterations in the UPR
network involved in the cognitive impairment? Front Aging Neurosci.
6:82014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Woehlbier U and Hetz C: Modulating stress
responses by the UPRosome: A matter of life and death. Trends
Biochem Sci. 36:329–337. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhao RR, Xu XC, Xu F, Zhang WL, Zhang WL,
Liu LM and Wang WP: Metformin protects against seizures, learning
and memory impairments and oxidative damage induced by
pentylenetetrazole-induced kindling in mice. Biochem Biophys Res
Commun. 448:414–417. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Patil SP, Jain PD, Ghumatkar PJ, Tambe R
and Sathaye S: Neuroprotective effect of metformin in MPTP-induced
Parkinson's disease in mice. Neuroscience. 277:747–754. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liang D, Song Z, Liang W, Li Y and Liu S:
Metformin inhibits TGF-beta 1-induced MCP-1 expression through
BAMBI-mediated suppression of MEK/ERK1/2 signalling. Nephrology
(Carlton). 24:481–488. 2019. View Article : Google Scholar
|
|
13
|
Tao L, Li D, Liu H, Jiang F, Xu Y, Cao Y,
Gao R and Chen G: Neuroprotective effects of metformin on traumatic
brain injury in rats associated with NF-κB and MAPK signaling
pathway. Brain Res Bull. 140:154–161. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. National
Academies Press; Washington, DC: 2011
|
|
15
|
Khallaghi B, Safarian F, Nasoohi S,
Ahmadiani A and Dargahi L: Metformin-induced protection against
oxidative stress is associated with AKT/mTOR restoration in PC12
cells. Life Sci. 148:286–292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Jiang HY, Yang Y, Zhang YY, Xie Z, Zhao
XY, Sun Y and Kong WJ: The dual role of poly(ADP-ribose)
polymerase-1 in modulating parthanatos and autophagy under
oxidative stress in rat cochlear marginal cells of the stria
vascularis. Redox Biol. 14:361–370. 2018. View Article : Google Scholar
|
|
17
|
Itahana K, Campisi J and Dimri GP: Methods
to detect biomarkers of cellular senescence: The
senescence-associated beta-galactosidase assay. Methods Mol Biol.
371:21–31. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu X, Wang Y, Sun Y, Chen S, Zhang S, Shen
L, Huang X, Lin X and Kong W: Reduced expression of Connexin26 and
its DNA promoter hypermethylation in the inner ear of mimetic aging
rats induced by d-galactose. Biochem Biophys Res Commun.
452:340–346. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kong WJ, Hu YJ, Wang Q, Wang Y, Han YC,
Cheng HM, Kong W and Guan MX: The effect of the mtDNA4834 deletion
on hearing. Biochem Biophys Res Commun. 344:425–430. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nicklas JA, Brooks EM, Hunter TC, Single R
and Branda RF: Development of a quantitative PCR (TaqMan) assay for
relative mitochondrial DNA copy number and the common mitochondrial
DNA deletion in the rat. Environ Mol Mutagen. 44:313–320. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
22
|
Sun HY, Hu YJ, Zhao XY, Zhong Y, Zeng LL,
Chen XB, Yuan J, Wu J, Sun Y, Kong W and Kong WJ: Age-related
changes in mitochondrial antioxidant enzyme Trx2 and
TXNIP-Trx2-ASK1 signal pathways in the auditory cortex of a mimetic
aging rat model: Changes to Trx2 in the auditory cortex. FEBS J.
282:2758–2774. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xia MY, Zhao XY, Huang QL, Sun HY, Sun C,
Yuan J, He C, Sun Y, Huang X, Kong W and Kong WJ: Activation of
Wnt/β-catenin signaling by lithium chloride attenuates
d-galactose-induced neurodegeneration in the auditory cortex of a
rat model of aging. FEBS Open Bio. 7:759–776. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tian YY, Jiang B, An LJ and Bao YM:
Neuroprotective effect of catalpol against MPP(+)-induced oxidative
stress in mesencephalic neurons. Eur J Pharmacol. 568:142–148.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kong WJ, Wang Y, Wang Q, Hu YJ, Han YC and
Liu J: The relation between D-galactose injection and mitochondrial
DNA 4834 bp deletion mutation. Exp Gerontol. 41:628–634. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zeng L, Yang Y, Hu Y, Sun Y, Du Z, Xie Z,
Zhou T and Kong W: Age-related decrease in the mitochondrial
sirtuin deacetylase Sirt3 expression associated with ROS
accumulation in the auditory cortex of the mimetic aging rat model.
PLoS One. 9:e880192014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bernales S, Soto MM and McCullagh E:
Unfolded protein stress in the endoplasmic reticulum and
mitochondria: A role in neuro-degeneration. Front Aging Neurosci.
4:52012. View Article : Google Scholar
|
|
28
|
Wang W, Sun Y, Chen S, Zhou X, Wu X and
Kong W and Kong W: Impaired unfolded protein response in the
degeneration of cochlea cells in a mouse model of age-related
hearing loss. Exp Gerontol. 70:61–70. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen J, Huang Z, Wu X, Kang J, Ren Y, Gao
W, Lu X, Wang J, Ding W, Nakabeppu Y, et al: Oxidative stress
induces different tissue dependent effects on Mutyh-deficient mice.
Free Radic Biol Med. 143:482–493. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mendoza MC, Er EE and Blenis J: The
Ras-ERK and PI3K-mTOR pathways: Cross-talk and compensation. Trends
Biochem Sci. 36:320–328. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ho SC, Liu JH and Wu RY: Establishment of
the mimetic aging effect in mice caused by D-galactose.
Biogerontology. 4:15–18. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kong W, Hu Y, Wang Q, Xu L, Wang Y, Han Y,
Li J, Liu B and Kong W: Establishment of model with inner ear
mimetic aging and mtDNA 4834 bp deletion in rats. Lin Chuang Er Bi
Yan Hou Ke Za Zhi. 20:888–890. 8932006.In Chinese.
|
|
33
|
Zhong Y, Hu Y, Peng W, Sun Y, Yang Y, Zhao
X, Huang X, Zhang H and Kong W: Age-related decline of the
cytochrome c oxidase subunit expression in the auditory cortex of
the mimetic aging rat model associated with the common deletion.
Hear Res. 294:40–48. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Balaban RS, Nemoto S and Finkel T:
Mitochondria, oxidants, and aging. Cell. 120:483–495. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lin MT and Beal MF: Mitochondrial
dysfunction and oxidative stress in neurodegenerative diseases.
Nature. 443:787–795. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sands WA, Page MM and Selman C:
Proteostasis and ageing: Insights from long-lived mutant mice. J
Physiol. 595:6383–6390. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McBride HM, Neuspiel M and Wasiak S:
Mitochondria: More than just a powerhouse. Curr Biol. 16:R551–R560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Klinge CM: Estrogenic control of
mitochondrial function and biogenesis. J Cell Biochem.
105:1342–1351. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Weinberg F and Chandel NS: Mitochondrial
metabolism and cancer. Ann N Y Acad Sci. 1177:66–73. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Houtkooper RH, Mouchiroud L, Ryu D,
Moullan N, Katsyuba E, Knott G, Williams RW and Auwerx J:
Mitonuclear protein imbalance as a conserved longevity mechanism.
Nature. 497:451–457. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Yoneda T, Benedetti C, Urano F, Clark SG,
Harding HP and Ron D: Compartment-specific perturbation of protein
handling activates genes encoding mitochondrial chaperones. J Cell
Sci. 117:4055–4066. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Shpilka T and Haynes CM: The mitochondrial
UPR: Mechanisms, physiological functions and implications in
ageing. Nat Rev Mol Cell Biol. 19:109–120. 2018. View Article : Google Scholar
|
|
43
|
Beck JS, Mufson EJ and Counts SE: Evidence
for mitochondrial UPR gene activation in familial and sporadic
Alzheimer's disease. Curr Alzheimer Res. 13:610–614. 2016.
View Article : Google Scholar
|
|
44
|
Zarse K, Schmeisser S, Groth M, Priebe S,
Beuster G, Kuhlow D, Guthke R, Platzer M, Kahn CR and Ristow M:
Impaired insulin/IGF1 signaling extends life span by promoting
mitochondrial L-proline catabolism to induce a transient ROS
signal. Cell Metab. 15:451–465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Mohamed SA, Hanke T, Erasmi AW, Bechtel
MJ, Scharfschwerdt M, Meissner C, Sievers HH and Gosslau A:
Mitochondrial DNA deletions and the aging heart. Exp Gerontol.
41:508–517. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Chen B, Zhong Y, Peng W, Sun Y, Hu YJ,
Yang Y and Kong WJ: Increased mitochondrial DNA damage and
decreased base excision repair in the auditory cortex of
D-galactose-induced aging rats. Mol Biol Rep. 38:3635–3642. 2011.
View Article : Google Scholar
|
|
47
|
Zeng Z, Zhang Z, Yu H, Corbley MJ, Tang Z
and Tong T: Mitochondrial DNA deletions are associated with
ischemia and aging in Balb/c mouse brain. J Cell Biochem.
73:545–553. 1999. View Article : Google Scholar
|
|
48
|
Chen B, Zhong Y, Peng W, Sun Y and Kong
WJ: Age-related changes in the central auditory system: Comparison
of D-galactose-induced aging rats and naturally aging rats. Brain
Res. 1344:43–53. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Eletto D, Chevet E, Argon Y and
Appenzeller-Herzog C: Redox controls UPR to control redox. J Cell
Sci. 127:3649–3658. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Hill S and Van Remmen H: Mitochondrial
stress signaling in longevity: A new role for mitochondrial
function in aging. Redox Biol. 2:936–944. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Cohen E, Bieschke J, Perciavalle RM, Kelly
JW and Dillin A: Opposing activities protect against age-onset
proteotoxicity. Science. 313:1604–1610. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Labunskyy VM, Gerashchenko MV, Delaney JR,
Kaya A, Kennedy BK, Kaeberlein M and Gladyshev VN: Lifespan
extension conferred by endoplasmic reticulum secretory pathway
deficiency requires induction of the unfolded protein response.
PLoS Genet. 10:e10040192014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Correia S, Carvalho C, Santos MS, Proença
T, Nunes E, Duarte AI, Monteiro P, Seiça R, Oliveira CR and Moreira
PI: Metformin protects the brain against the oxidative imbalance
promoted by type 2 diabetes. Med Chem. 4:358–364. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kurioka T, Matsunobu T, Satoh Y, Niwa K,
Endo S, Fujioka M and Shiotani A: ERK2 mediates inner hair cell
survival and decreases susceptibility to noise-induced hearing
loss. Sci Rep. 5:168392015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Arthur DB, Georgi S, Akassoglou K and
Insel PA: Inhibition of apoptosis by P2Y2 receptor activation:
Novel pathways for neuronal survival. J Neurosci. 26:3798–3804.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Stanciu M, Wang Y, Kentor R, Burke N,
Watkins S, Kress G, Reynolds I, Klann E, Angiolieri MR, Johnson JW
and DeFranco DB: Persistent activation of ERK contributes to
glutamate-induced oxidative toxicity in a neuronal cell line and
primary cortical neuron cultures. J Biol Chem. 275:12200–12206.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Liu L, Cao Y, Chen C, Zhang X, McNabola A,
Wilkie D, Wilhelm S, Lynch M and Carter C: Sorafenib blocks the
RAF/MEK/ERK pathway, inhibits tumor angiogenesis, and induces tumor
cell apoptosis in hepatocellular carcinoma model PLC/PRF/5. Cancer
Res. 66:11851–11858. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Tavares R and Pathak SK: Helicobacter
pylori Secreted protein HP1286 triggers apoptosis in macrophages
via TNF-independent and ERK MAPK-dependent pathways. Front Cell
Infect Microbiol. 7:582017. View Article : Google Scholar :
|
|
60
|
Feng X, Sun T, Bei Y, Ding S, Zheng W, Lu
Y and Shen P: S-nitrosylation of ERK inhibits ERK phosphorylation
and induces apoptosis. Sci Rep. 3:18142013. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Allen EN, Potdar S, Tapias V, Parmar M,
Mizuno CS, Rimando A and Cavanaugh JE: Resveratrol and pinostilbene
confer neuroprotection against aging-related deficits through an
ERK1/2-dependent mechanism. J Nutr Biochem. 54:77–86. 2018.
View Article : Google Scholar
|
|
62
|
Zhen X, Uryu K, Cai G, Johnson GP and
Friedman E: Age-associated impairment in brain MAPK signal pathways
and the effect of caloric restriction in Fischer 344 rats. J
Gerontol A Biol Sci Med Sci. 54:B539–B548. 1999. View Article : Google Scholar
|
|
63
|
Zhuang S and Schnellmann RG: A
death-promoting role for extracellular signal-regulated kinase. J
Pharmacol Exp Ther. 319:991–997. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sinha D, Bannergee S, Schwartz JH,
Lieberthal W and Levine JS: Inhibition of ligand-independent ERK1/2
activity in kidney proximal tubular cells deprived of soluble
survival factors up-regulates Akt and prevents apoptosis. J Biol
Chem. 279:10962–10972. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Moelling K, Schad K, Bosse M, Zimmermann S
and Schweneker M: Regulation of Raf-Akt cross-talk. J Biol Chem.
277:31099–31106. 2002. View Article : Google Scholar : PubMed/NCBI
|