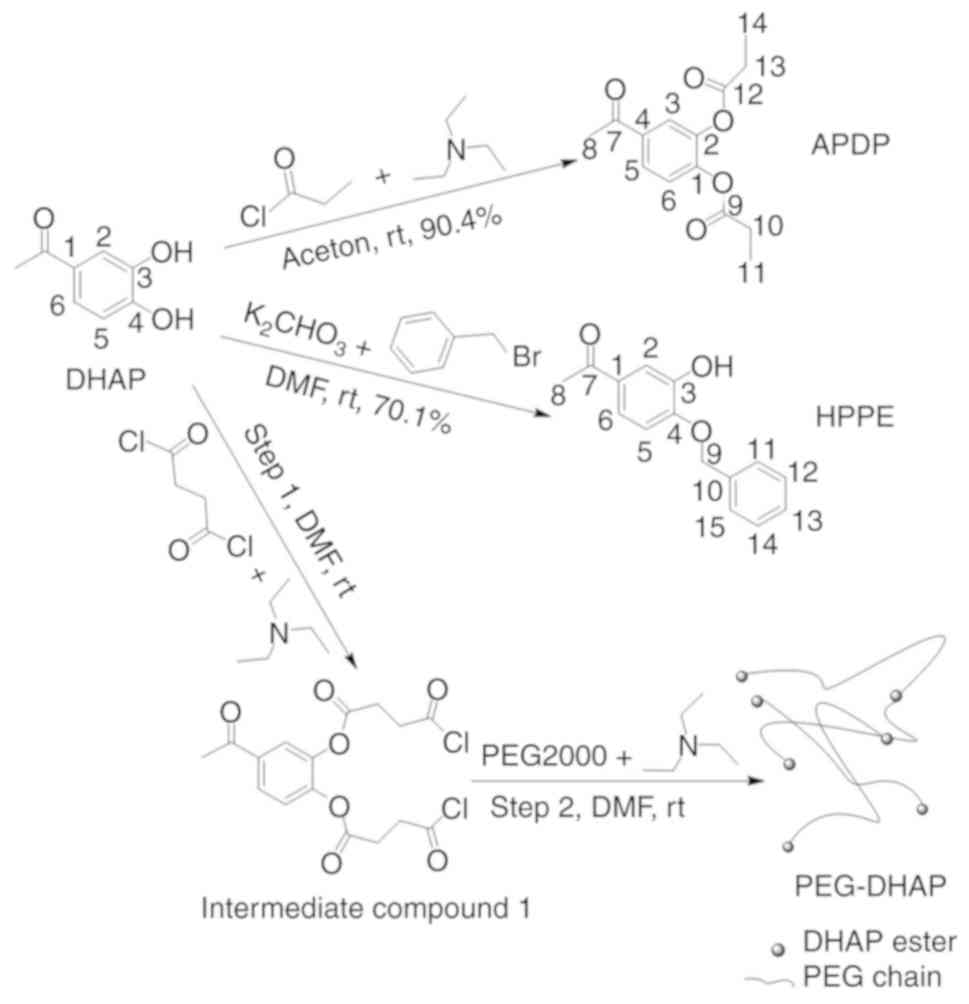
Introduction
In 1971, 3,4-dihydroxyacetophenone (DHAP; also
referred to as Qingxintong) was isolated from the leaves of Ilex
Pubescens Hook et Arnvar glaber Chang, which is a
traditional Chinese herb that improves blood circulation (1). Since then, several studies have
demonstrated that DHAP effectively improves the clinical symptoms
of patients with angina pectoris, pulmonary heart disease and
pregnancy-induced hypertension (2-12).
Clinical and animal pharmacological studies have revealed that DHAP
exerts antiplatelet aggregation effects (13-18), inhibits cyclooxygenase (18), reduces the fluidity of platelet
membranes (19) and inhibits
thrombus formation by regulating the ratio of thromboplastin to
thromboplastin inhibitor in the human umbilical vein endothelium
(20). DHAP also dredges
microcirculation (6,7,21-23). It has been demonstrated that DHAP
significantly enhances the delayed K+ current of
vascular smooth muscle and promotes the opening of K+
channels, thereby relaxing smooth muscle, relaxing the coronary
arteries and improving myocardial function (21,24). Additionally, DHAP has been shown
to correct the imbalance of thromboxane
TAX2/prostacyclin PGI2 and the imbalance of
NO/endothelin in maternal and uterine placental vascular beds,
thereby improving uterine and placental blood circulation in
patients with pregnancy-induced hypertension (9,25,26). DHAP also exerts anti-inflammatory
effects and affects the occurrence and development of
atherosclerotic inflammatory responses through various mechanisms
(27-32).
Although clinical studies have demonstrated that
DHAP exerts therapeutic effects on cardiovascular and pulmonary
heart disease, it is not administered as a clinical drug due to it
being a short-acting compound that is eliminated from the body
relatively quickly. Pharmacokinetic studies in animal experiments
have revealed that DHAP is absorbed rapidly and eliminated quickly,
that the absorption half-life and the elimination half-life of
intramuscular (i.m.) injection and intragastric (i.g.)
administration are in the range of 0.05-0.07 and 0.14-0.29 h,
respectively, and that it exhibits low oral bioavailability in the
range of 9.16-22.05% due to severe first-pass metabolism (33,34). Since the in vivo acting
time of DHAP is very short, frequent administration is required to
maintain an acceptable blood concentration to achieve its
clinically therapeutic purpose. These shortages significantly
impede the clinical application of DHAP.
The chemical structure of DHAP is presented in
Fig. 1. DHAP is soluble in water
and has a low molecular weight (152 g/mol), meaning its absorption
and distribution is rapid and primarily occurs via passive
diffusion across intestinal epithelial cells. Similar to most
phenolic and catechol containing drugs, DHAP tends to be rapidly
cleared by sulfation, glucuronidation and 3-O-methylation in
vivo (35). Modifying the
sites of metabolism and phenolic hydroxyls of DHAP, as is the case
for esterification and etherification, affects certain
pharmacokinetic processes and oral bioavailability. In the present
study, three derivatives of DHAP were prepared via modification of
C-3 or C-4 phenol hydroxyls. 4-Acetyl-1,2-phenylene dipropionate
(APDP) was prepared by esterification of C-3 and C-4 phenol
hydroxyls, 1-(3-hydroxy-4-phenoxy-phenyl)-ethanone (HPPE) was
prepared via etherification of the C-4 phenol hydroxyl, and the
polymer derivative (PEG-DHAP) was prepared by attaching DHAP to
polyethylene glycol 2000 (PEG 2000) via ester bonds. Ester and
ether bonds were selected as they can be cleavage-catalyzed by
enzymes in vivo, after which the molecular structure of DHAP
may be recovered. Compared with DHAP, APDP and HPPE contain
hydrophobic hydrocarbon groups, which confer lower water solubility
and higher oil-water distribution coefficients. For the polymer
derivative, PEG-DHAP, the PEG barriers protect DHAP against rapid
clearance in vivo, as only free DHAP diffusing out of the
PEG barriers may be cleared. The pharmacokinetic processes of these
derivatives differ from those of DHAP, which is expected to a
certain extent.
In the present study, certain physicochemical
properties closely associated with absorption and distribution were
assessed. These included the water solubility and oil-water
partition coefficients of DHAP, APDP and HPPE, the mean particle
size of PEG-DHAP in aqueous solution and the release of three DHAP
derivatives in vitro and in vivo. The concentrations
of the three derivatives in rat blood following a single oral
administration were measured. The pharmacokinetic parameters were
compared with those of DHAP to assess whether these derivatives may
prolong in vivo acting time and improve oral
bioavailability. The antiplatelet aggregation was then selected as
an indicator of activity to discuss the bioactivity of these
derivatives.
Materials and methods
Chemicals
DHAP (≥98%) was obtained from Jinan Luxin Chemical
Technology Co., Ltd. Propionyl chloride and succinyl chloride, each
of analytical grade, were purchased from Aladdin Reagents Co., Ltd.
Benzyl bromide and N,N-dimethylformamide (DMF), each of analytical
grade, were purchased from Sinopharm Chemical Reagent Co., Ltd. and
re-evaporated prior to use. Triethylamine (TEA) and acetone, both
of analytical grade, were purchased from Tianjin Kemiou Chemical
Reagent Co., Ltd. and dehydrated prior to use. PEG 2000, an
experimental reagent, was purchased from Tianjin Damao Chemical
Reagent Factory. Adenosine diphosphate (ADP), a biochemical
reagent, was purchased from Sigma-Aldrich; Merck KGaA and dissolved
in physiological saline prior to use. Other chemicals were
purchased from local chemical reagent suppliers.
Animals
Male rabbits and male/female Wistar rats (weight,
250-300 g) were purchased form the Experimental Animal Center of
Shandong Lukang Pharmaceutical Co. (license nos. 0017075 and
0010061, respectively). The experimental procedures were approved
by the Animal Experimentation Ethics Committee of Weifang Medical
College and were conducted in accordance with the guidelines of the
National Health and Medical Research Council of China for the care
and use of animals. Pior to the experiments, the rabbits and rats
were reared for 1 week in an environment with a temperature of 25°C
and a humidity of 40-70%. They were fasted only from food but were
still provided with water for 12 h prior to the experiment. For the
determination of the behaviors of derivatives releasing DHAP in the
in vitro and in vivo experiments, 21 rats were used,
and blood was obtained from the fundus venous plexus after the rats
inhaled isoflurane for anesthesia. The dose of isoflurane used was
3-4% for induction and 2-2.5% for maintenance, and the oxygen flow
rate was 500-700 ml/min. These rats continued to be raised for 2
weeks for other uses. In the pharmacokinetic experiments, 36 rats
were used, and blood was obtained from the fundus venous plexus
after the rats inhaled isoflurane for anesthesia. The dose of
isoflurane used was the same as the one described above for the
determination of derivatives releasing DHAP. These rats were
sacrificed by cervical dislocation after the end of the in
vivo experiment. For the in vitro antiplatelet
aggregation activity experiments, 3 rabbits were used. The rabbit
ear vein was injected with a concentration of 3% sodium
pentobarbital solution for anesthesia at a dose of 30 mg/kg.
Following the collection of approximately 80-100 ml of blood from
the carotid artery, the rabbit ear veins were injected with air to
terminate life. For the in vivo antiplatelet aggregation
activity experiments, 54 rats were used and were anesthetized with
sodium pentobarbital (3%) via an intraperitoneal injection at a
dose of 40 mg/kg. The chest of the animals was then opened and 5 ml
blood were obtained from the heart, and the rats were subsequently
decapitated.
Synthesis of APDP
DHAP (2.01 g; 13.25 mmol) was dissolved in 140 ml
acetone, followed by the addition of TEA (4.60 ml; 33.00 mmol) and
stirring at room temperature for 1 h. Propionyl chloride (3.00 ml;
34.37 mmol) was dissolved in 10 ml acetone and slowly dropped into
the DHAP solution on ice. The solution was then stirred at room
temperature for 8 h. The reaction solution was evaporated to
dryness under reduced pressure, and the remaining residue was
dissolved in ethyl acetate. The solution was washed successively
with water and saturated sodium chloride aqueous solution. The
resultant ethyl acetate layer was dried over anhydrous magnesium
sulfate overnight. The solution was filtered and the filtrate was
evaporated to dryness under reduced pressure to create a colorless
powder.
Synthesis of HPPE
DHAP (2.54 g; 16.70 mmol) and anhydrous potassium
carbonate (2.31 g; 16.72 mmol) were sequentially added into 42 ml
DMF under a nitrogen atmosphere and stirred at room temperature for
1 h. Benzyl bromide (1.99 ml; 16.74 mmol) was slowly dropped into
the aforementioned mixture in an ice bath. The mixture was
subsequently stirred at room temperature for 90 min. The mixture
was filtered and the filtrate was diluted with 5-fold volumes of
ethyl acetate, after which time the solution was washed
sequentially with water and saturated sodium chloride aqueous
solution. The ethyl acetate phase was dried over anhydrous
magnesium sulfate overnight. The solution was filtered, and the
filtrate was evaporated to dryness under reduced pressure to create
a light-yellow powder.
Synthesis of PEG-DHAP
DHAP (1.00 g; 6.60 mmol) and TEA (2.10 ml; 15.12
mmol) were dissolved in 50 ml DMF and the solution was stirred at
room temperature for 1 h. Succinyl chloride (1.7 ml; 14.53 mmol)
was dropped into the solution slowly in an ice bath. The solution
was subsequently stirred at room temperature for 8 h to yield an
intermediate (product 1) solution. PEG 2000 (6.63 g; 3.32 mmol) and
TEA (1.06 ml; 7.64 mmol) were dissolved in 50 ml DMF, after which
time the solution was stirred at room temperature for 1 h.
Intermediate (product 1) solution was dropped into the second
solution slowly. The resultant solution was then stirred at room
temperature for 24 h. After the reaction had taken place, the
solution was concentrated under reduced pressure and dialyzed in a
dialysis bag with a molecular weight of 1,500 against deionized
water for 3 days. The samples were then freeze-dried to create a
light brown powder.
Water solubility determination
Water solubility was determined according to the
method previously described by Montenegro et al (36). An excess quantity of compound was
weighed into a glass tube containing 2 ml of water, and the tube
was sealed with a Teflon-lined cap. The mixture was stirred with a
magnetic stirrer for 24 h at room temperature and then filtered.
The quality of the compound in its saturated solution was
determined via HPLC.
Oil-water partition coefficient
determination
The oil-water partition coefficient was determined
using the classical shake flask method (37). The quality of the compound in
solution was determined via HPLC.
Content of DHAP in PEG-DHAP
PEG-DHAP (30 mg) was dissolved in 2 ml distilled
water and sealed in a dialysis bag (Spectrumlabs, MD31, MW
500-1,000). The dialysis bag was incubated in 50 ml aqueous
hydrochloric acid solution (pH 2) at room temperature. Hydrochloric
acid solution (0.5 ml) was sampled and the quality of DHAP was
determined via HPLC. After each sampling, 0.5 ml hydrochloric acid
solution (pH 2) were added. The measurement continued until the
quality of DHAP no longer changed.
HPLC conditions
A 1260 Infinity chromatography system (Agilent
Technologies, Inc.) was used. It was equipped with a DAD detector
(G1315D VL; Agilent Technologies, Inc.), a column (Hypersil ODS2;
250×4.60 mm; 5 µm; Illit) and an injector (2PS/6PT; Mon Inj;
600 bar; Agilent Technologies, Inc.). The sample volume was 20
µl and the flow rate was 1 ml/min. The eluent was composed
of methanol and water, with a volume ratio of 60:40. The elution
was performed at room temperature and was detected at a wavelength
of 275 nm.
Particle size determination of PEG-DHAP
in aqueous solution
PEG-DHAP powder was dissolved in distilled water by
ultrasonication to obtain a 0.5 mg/l PEG-DHAP solution. Particle
size and distribution were determined using the Malvern Zetasizer
Nano Series 90 (Malvern Instruments, Ltd.
Determination of the behaviors of
derivatives releasing DHAP in vitro and preparation of plasma
samples
Rat blood (0.5 ml) was collected into a centrifuge
tube containing 55 µl 3.8% (w/v) sodium citrate solution via
the fundus venous plexus and centrifuged at 1,425 × g for 10 min at
room temperature. The supernatant plasma was then separated.
Subsequently, 100 µl plasma were precisely obtained and 10
µl methanol solution of phenol (internal standard) with 10
µl methanol solution of derivative were added. The mixture
was then vortexed and 400 µl ethyl acetate were added.
Following a second vortex and centrifugation at 1,425 × g for 10
min at room temperature, the supernatant was collected. Ethyl
acetate (200 µl) was further added to the precipitate, which
was vortexed and centrifuged at 1,425 × g for 10 min at room
temperature. The supernatant was collected and combined with that
obtained previously. The combined supernatants were then dried with
nitrogen and dissolved in 100 µl mobile phase for HPLC
analysis. For comparison, the methanol solutions of the derivatives
were also analyzed by HPLC.
The percentage of derivative converted to DHAP was
calculated using internal standard correction factor method as the
following formula.
where ADHAP¯ is the average
value of the peak area of DHAP, Aphenol¯ is the average value of the peak
area of internal standard phenol and Aderivative¯ is the average value of the
peak area of derivatives.
Determination of the behaviors of
derivatives releasing DHAP in vivo
The rats were randomly divided into 3 groups (APDP,
HPPE and PEG-DHAP) with 6 rats in each. The derivatives were
dissolved in physiological saline to prepare a solution or
suspension. The doses used for the rats (i.g.), as recommended by
previous studies (33,34), were 0.79 mmol/kg APDP and HPPE,
and 0.55 g/kg PEG-DHAP (containing 0.45 mmol DHAP). At 10 min after
the administration, 0.5 ml blood were collected via the fundus
venous plexus. The preparation of the plasma sample was the same as
the aforementioned, apart from the fact that a methanol solution of
derivative was added.
HPLC conditions
The aforementioned HPLC conditions were used, apart
from the elution and detection wavelength. The elution was a linear
gradient of methanol and water: 0 min (40:60 v/v), 5.5 min (60:40
v/v) and 15 min (40:60 v/v). The detection wavelength was 260
nm.
Preparation of plasma for antiplatelet
aggregation activity
Blood was collected from the carotid artery of
rabbits or the heart of rats. Sodium citrate solution (3.8% w/v)
was used as the anticoagulant and was mixed with blood at a volume
ratio of 1:9. Platelet-rich plasma (PRP) was obtained by
centrifugation at 285 × g for 10 min at room temperature.
Platelet-poor plasma (PPP) was obtained by centrifugation of the
remaining blood at 1,425 × g for 15 min at room temperature. The
number of platelets in PRP was adjusted using PPP to ~5×108
cells/ml.
Antiplatelet aggregation activity of
rabbits in vitro
PRP (300 µl) was pre-incubated for 3 min at
37°C with various concentrations of compound that were
pre-dissolved in dimethyl sulfoxide (DMSO). The final concentration
of DMSO in PRP was <1.0% (v/v) to eliminate a false positive
result. Platelet aggregation was subsequently stimulated by ADP
(final concentration in PRP, 10 µmol/l). Aggregation was
monitored using a platelet aggregometer (LBY-NJ4A; Precill) at a
constant stirring speed of 1,200 rpm, and aggregation rates were
recorded for 5 min to determine the percentage aggregation. The
aggregation inhibition rate (AIR) was calculated using the
following formula:
where AR stands for the aggregation rate.
Antiplatelet aggregation activity in vivo
in rats
Wistar rats were randomly divided into 9 groups (the
vehicle, DHAP10, APDP10, HPPE10, PEG-DHAP10, DHAP40, APDP40, HPPE40
and PEG-DHAP40), 6 in each group. The compounds were dissolved in
physiological saline to prepare a solution or suspension. The doses
used for the rats (i.g.) were 0.79 mmol/kg DHAP, APDP and HPPE, and
0.55 g/kg PEG-DHAP (containing 0.45 mmol DHAP). The rats in the
vehicle group were administered the same volume of i.g.
physiological saline. After 10 and 40 min of administration, 5 ml
blood were collected.
PRP was pre-incubated for 3 min at 37°C and then
stimulated by ADP. The aforementioned procedures were used to
assess the concentration of ADP and for the measurement of platelet
aggregation.
Statistical analysis for antiplatelet
aggregation activity
Data are presented as the means ± SD. Statistical
significance was determined by an unpaired t-test or two-way ANOVA
followed by Tukey's multiple comparisons test (SPSS 9.0 for
Windows; SPSS, Inc.). A value of P<0.05 was considered to
indicate a statistically significant difference with α=0.05%. In
the in vitro experiments, each drug concentration group was
independently determined 4 times. In the in vivo
experiments, there were 6 rats in each group, and the blood of each
rat was independently determined 4 times.
HPLC conditions for the measurement of
blood concentration
The HPLC conditions were same as the aforementioned,
apart from the detection wavelength, which was set to 275 nm for
DHAP and 246 nm for HPPE.
Specific investigation
The internal standard and DHAP were added to both
blank plasma and plasma samples, and these plasma samples were
measured using HPLC to assess the specificity of chromatographic
conditions.
Preparation of DHAP and HPPE standard
curves in plasma
Serial concentrations of DHAP or HPPE standard
solution were prepared using methanol as a solvent. Standard
solution (10 µl) and methanol solution of phenol (internal
standard; 10 µl) were added to 100 µl blank plasma.
Each mixture was processed as described above in the paragraph
entitled 'Determination of the behaviors of derivatives releasing
DHAP in vitro and preparation of plasma samples'. The peak
area ratio (DHAP or HPPE vs. internal standard) of each sample was
determined by HPLC and plotted against the concentration. Linear
regression was performed to produce the standard curve.
Recovery and precision control
High, medium and low concentrations of DHAP or HPPE
standard solutions were added to the blank plasma, creating final
concentrations of 0.003, 0.013 and 0.053 mmol/l, respectively. Each
sample was processed as described in the paragraph entitled
'Determination of the behaviors of derivatives releasing DHAP in
vitro and preparation of plasma samples', apart from the fact
that the derivative methanol solution was added and then determined
by HPLC. Recovery rates were calculated according to the standard
curve of DHAP or HPPE in plasma.
Three concentrations of the plasma solutions, namely
0.003, 0.013 and 0.053 mmol/l, were prepared in triplicate and
determined by two HPLC experiments performed in 1 day and then
continuously for 5 days. The relative standard deviation (RSD)
within-day and between days was subsequently calculated.
Method of administration, blood
collection and processing
The rat i.g. DHAP and all 3 derivatives were the
same as those aforementioned. The i.m. and intravenous (i.v.) doses
of DHAP administered to the rats were 0.39 and 0.20 mmol/kg,
respectively. Following administration, 0.5 ml blood were collected
via the fundus venous plexus into a centrifuge tube containing 55
µl of 3.8% (w/v) sodium citrate solution at a series of time
points (DHAP i.v.: 5.0, 17.0, 20.0, 25.0, 35.0 45.0, 60.0, 75.0, 90
and 120.0 min; DHAP i.m.: 1.0, 5.0, 10.0, 15.0, 20.0, 25.0, 30.0,
45.0, 60.0, 75.0, 90.0 and 120.0 min; DHAP i.g.: 3.0, 5.0, 10.0,
18.0, 25.0, 29.0, 44.0, 60.0, 75.0 and 120.0 min; APDP i.g., HPPE
i.g. and PEG-DHAP i.g.: 0.05, 0.083, 0.17, 0.25, 0.5, 1.0, 2.0,
4.0, 6.0, 12.0, 24.0 and 48.0 h). Samples were subsequently
processed according to the aforementioned blood processing
procedure, except for the addition of methanol solution of
derivative.
Data processing
Drug and statistics software (DAS; version 2.1.1)
provided by Shanghai Bojia Pharmaceutical Technology Co., Ltd. was
used to process the data. Additionally, the pharmacokinetic
parameters were calculated. The relative oral bioavailability of
the derivatives (FR%) was calculated using the following
formula:
where D stands for the dose for i.g. administration.
Results
Synthesis of derivatives
APDP was prepared through the acylation of
3-hydroxyl and 4-hydroxyl via propionyl chloride. HPPE was prepared
through the etherification of 4-hydroxyl via benzyl bromide. The
polymer derivative, PEG-DHAP, was prepared by bonding DHAP to
polyethylene glycol 2000 via ester bonds. The preparation process
and conditions were simple and are presented in Fig. 1. The structural identification
spectra of DHAP and the 3 derivatives are presented in Figs. S1-S4. The synthesis of the
derivates was as follows.
APDP (yield, 90.3%; purity, 97.6%)
1H NMR (DMSO-d6, 600 MHz): δ7.86
(1H, dd, H-6), 7.8 (1H, d, H-2), 7.3 (1H, d, H-5), 2.60 (7H, br,
O=C-CH3, -CH2-), 1.27 (6H,t,
-CH3); 13C NMR (DMSO-d6, 150 MHz):
δ196.05(C-7), 171.57(C-12), 171.21 (C-9), 146.1 (C-4), 142.33
(C-3), 135.47 (C-1), 126.75 (C-6), 123.68 (C-2), 123.62 (C-5),
27.50 (C-13), 27.41 (C-10), 26.55 (C-8), 9.09 (C-11), 9.03 (C-14);
FTIR (KBr): 3,100.83-3,060.00 (ν=C-H), 2,989.03-2,884.24
(νC-H), 1,764.53 (νC=O, ester), 1,683.37
(νC=O, ketone), 1,601.74-1,462.28 (νC=C, aromatic
ring), 1,421.65-1,318.23 (δC-H), 1,279.26-1,174.37
(νC-O-C, ester). EI-MS, 265.1072 m/z,
(M+H)+, C14H17O5. mp
72.0-73.7°C.
HPPE (yield, 70.1%; purity, 99.1%)
1H NMR (DMSO-d6, 600 MHz): δ9.42
(1H, s, 3-OH), 7.49 (1H, dd, H-6), 7.42 (5H, br, H-11, H-12, H-13,
H-14, H-15), 7.34 (1H, d, H-2), 7.09 (1H, d, H-5), 5.21 (2H, s,
-CH2-), 2.47 (3H, s, O=C-CH3); 13C NMR
(DMSO-d6, 150 MHz): δ196.83 (C-7), 151.46 (C-4),147.17 (C-3),
137.23 (C-10), 130.83 (C-1), 128.87 (C-12, C14), 128.34 (C-13),
128.14 (C-11, C-15), 121.65 (C-6), 115.22 (C-2), 70.23 (C-9), 26.75
(C-8); FTIR (KBr): 3,173.39 (ν=C-H), 1,653.42
(νC=O, ketone), 1,605.93-1,428.72 (νC=C, aromatic
ring), 1,276.96, 1,211.98 (δC-H), 1,141.52
(νC-O-C). EI-MS, 243.1016 m/z, (M+H)+,
C15H15O3. mp 122.0-123.4°C.
PEG-DHAP (yield, 52.7%)
1H NMR (DMSO-d6, 600 MHz):
δ6.57-8.24 (3-OH and protons of aromatic ring), 3.52
(-OCH2CH2O-); FTIR (KBr): 3,426.59
(νOH), 2,880.85 (νC-H), 1,731.72
(νC=O), 1,588.12 (νC=C, aromatic ring),
1,467.53~1,242.20 (δC-H), 1,113.34 (νC-O-C,
ester). DHAP content, 12.47%.
Physicochemical properties of DHAP and
its derivatives
The water solubility and oil-water partition
coefficient (presented using LogP) of compounds are presented in
Table I. DHAP was found to be
highly soluble in water, with a solubility of 176.68 mg/ml. It also
exhibited a low oil-water partition coefficient (LogP=0.90),
suggesting that it can be absorbed rapidly through epithelial
cells, similarly to water-soluble propofol. APDP and HPPE exhibited
markedly lower water solubilities and higher oil-water distribution
coefficients compared with DHAP, due to possessing hydrophobic
hydrocarbyl chains (Table I).
Their LogP-values were in the range of 0-3, which is optimal for
the penetration of orally administered drugs through the
phospholipid biomembrane via passive diffusion (38). PEG-DHAP is highly water-soluble.
The mean particle size of PEG-DHAP in its 0.5 mg/l aqueous solution
was 260.90 nm, suggesting that PEG-DHAP nanoparticles can be
absorbed via epithelial endocytosis (39).
 | Table IValues of water solubility and
LogP of compoundsa. |
Table I
Values of water solubility and
LogP of compoundsa.
|
Solubility/LogP | DHAP | APDP | HPPE | PEG-DHAP |
|---|
| Water solubility
(mg/ml) | 176.68±12.33 | 0.23±0.02 | 0.15±0.007 | 48.33±3.73 |
| LogP | 0.904±0.022 | 1.538±0.063 | 2.219±0.097 | - |
Derivative release of DHAP in vitro
Selecting an appropriate UV detection wavelength is
critical for the simultaneous quantitative determination of
derivatives and DHAP in plasma by HPLC. As presented in Fig. S5, the maximum absorption
wavelengths of all 3 derivatives were at 246 nm, with that of DHAP
being 275 nm. The absorption curves intersected at a wavelength of
260 nm, they exhibited the same degree of absorption at 260 nm.
Therefore, 260 nm was selected as the detection wavelength for the
simultaneous quantitative determination of the 3 DHAP
derivatives.
As presented in the HPLC chromatograms depicted in
Fig. 2, the retention times
(tR) of DHAP, phenol (internal standard), APDP,
HPPE and PEG-DHAP were ~4.36, 6.05, 11.48, 11.57 and 2.74 min,
respectively. The peak area values of the main chromatographic
peaks are shown in Table II. The
3 derivatives existed in different forms within in vitro
plasma, as presented in Fig.
2E-G. A total of 89.40% of APDP transformed into DHAP (Fig. 2E), 7.90% of PEG-DHAP released DHAP
(Fig. 2G), and almost all HPPE
existed as the prototype in plasma (Fig. 2F). The release of DHAP from APDP
and PEG-DHAP may be due to the hydrolysis of ester bonds catalyzed
by carboxylesterases in plasma. The hydrolysis of ether bonds in
HPPE requires CYP-450, which is insufficient in plasma (40). Therefore, HPPE does not transform
into DHAP in plasma in vitro.
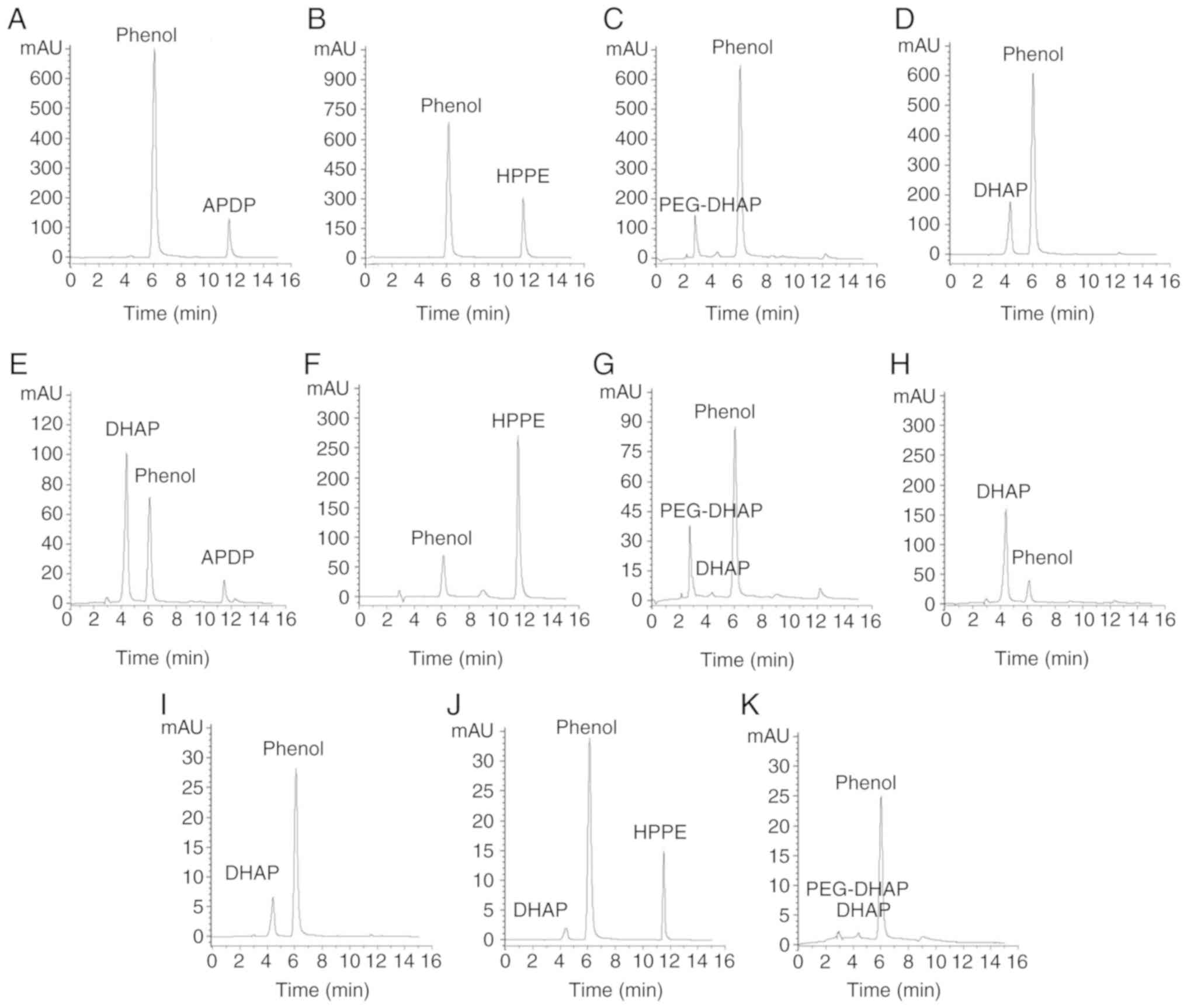 | Figure 2Behaviors of derivatives releasing
DHAP in vitro and in vivo. HPLC chromatograms of APDP
(A) in methanol, (E) in plasma in vitro and (I) in plasma
in vivo; HPLC chromatograms of HPPE (B) in methanol, (F) in
plasma in vitro and (J) in plasma in vivo; HPLC
chromatograms of PEG-DHAP (C) in methanol, (G) in plasma in
vitro and (K) in plasma in vivo; HPLC chromatograms of
DHAP (D) in methanol, (H) in plasma in vitro. In in
vitro experiments, n=3; in in vivo experiments, n=6.
DHAP, 3,4-dihydroxyacetophenone; APDP, 4-acetyl-1,2-phenylene
dipropionate; HPPE, 1-(3-hydroxy-4-phenoxy-phenyl)-ethanone. |
| Table IIValues of the HPLC chromatography
peak area in study of the behaviors of derivatives releasing DHAP
in vitro and in vivoa. |
Table II
Values of the HPLC chromatography
peak area in study of the behaviors of derivatives releasing DHAP
in vitro and in vivoa.
| Phenol | DHAP | APDP | HPPE | PEG-DHAP |
|---|
| APDP in
methanolb | 12,561.5±345.6 | - | 1,897.2±117.9 | - | - |
| HPPE in
methanolb | 11,862.3±218.7 | - | - | 2,837.8±163.6 | - |
| PEG-DHAP in
methanolb | 11,511.2±316.2 | - | - | - | 1,764.8±110.1 |
| DHAP in
methanolb | 10,636.7±228.4 | 2,023.3±89.2 | - | - | - |
| APDP in plasma
in vitrob | 1,594.2±115.2 | 2,225.1±179.3 | 263.7±18.3 | - | - |
| HPPE in plasma
in vitrob | 1,562.5±127.5 | - | - | 5,254.3±213.6 | - |
| PEG-DHAP in plasma
in vitrob | 1,448.0±137.4 | 36± | - | - | 419.6±40.8 |
| DHAP in plasma
in vitrob | 1,525.7±72.7 | 7,039.0±229.5 | - | - | - |
| APDP in plasma
in vivoc | 648.6±47.5 | 177.0±8.4 | - | - | - |
| HPPE in plasma
in vivoc | 718.0±50.5 | 43.3±2.1 | - | 269.8±19.4 | - |
| PEG-DHAP in plasma
in vivoc | 542.8±32.7 | 15.9±1.3 | - | - | 22.1±1.8 |
Derivatives releasing DHAP in vivo
The present study assessed whether the derivatives
released DHAP following i.g. administration to rats to determine
which form was active in vivo and which form may be a target
for blood concentration determination. When comparing the
derivative HPLC chromatograms in methanol (Fig. 2A-C) and in plasma following i.g.
administration (Fig. 2I-K), all
APDP and a quantity of HPPE and PEG-DHAP transformed into or
released DHAP ~10 min after the administration. For PEG-DHAP, only
the free DHAP transported across the PEG barriers was active.
Therefore, DHAP is the active form of both APDP and PEG-DHAP in
plasma after absorption and distribution. For HPPE, the original
form and DHAP produced by metabolism were present in the blood,
synchronously, 10 min after oral administration. Whether the
remaining HPPE may be used as the active substance for determining
blood concentrations depends on whether it is active or not.
Antiplatelet aggregation activity
If HPPE itself exerts anti-platelet aggregation
activity, it is necessary to determine the blood concentration of
HPPE. Therefore, the in vitro anti-platelet aggregation
activity of DHAP and its derivatives was determined.
DHAP exhibited antiplatelet aggregation activity.
Its in vitro activity was concentration-dependent, as
presented in Fig. 3A, and the
half-maximal inhibitory concentration (IC50) was 99.22
µmol/l. The IC50 of APDP was 104.59
µmol/l, which was almost equal to that of DHAP. This result
may be due to the transformation of almost all APDP into DHAP
following incubation in plasma for 5 min. The IC50 of
HPPE was 92.35 µmol/l, which was also similar to DHAP. Since
HPPE cannot be cleaved into DHAP in in vitro plasma
(Fig. 2F), the results indicated
that HPPE itself exhibited antiplatelet aggregation activity,
similar to DHAP. HPPE may therefore be used as one of the active
substances for the determination of blood concentration. The
IC50 of PEG-DHAP was 0.25 mg/ml, which was equivalent to
173.20 µmol/l of DHAP (Fig.
3B). The higher IC50 value demonstrated herein may
be due to the slow diffusion of DHAP across PEG barriers, a notion
that implies the possibility of slow release DHAP.
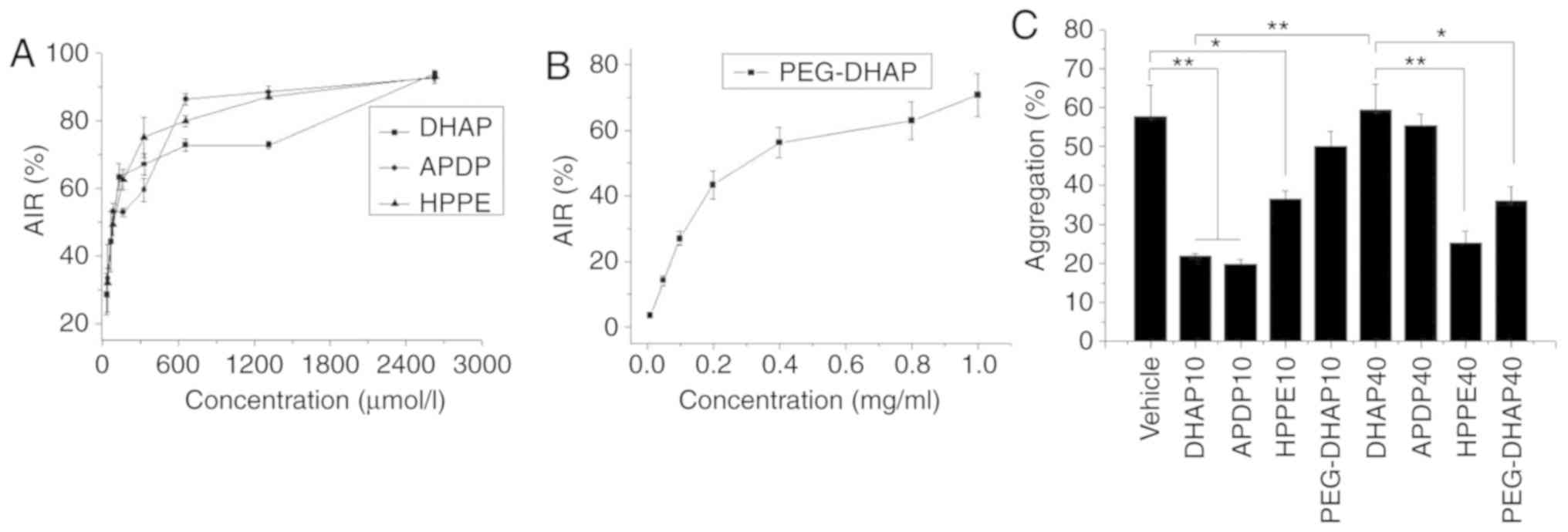 | Figure 3Antiplatelet aggregation activity (A)
AIR of DHAP, APDP and HPPE in vitro (B) AIR of PEG-DHAP
in vitro (C) AR of DHAP and derivatives in vivo. The
data are presented as the means ± SD. *P<0.05 and
**P<0.01, with comparisons indicated by lines.
DHAP10, APDP10, HPPE10 and PEG-DHAP10, indicate 10 min after the
administration of the compound; DHAP40, APDP40, HPPE40 and
PEG-DHAP40, indicate 40 min after the administration of the
compound. In in vitro experiments each drug concentration
group was independently determined 4 times. In in vivo
experiments used 6 rats in each group, and the blood of each rat
was independently determined 4 times. DHAP,
3,4-dihydroxyacetophenone; APDP, 4-acetyl-1,2-phenylene
dipropionate; HPPE, 1-(3-hydroxy-4-phenoxy-phenyl)-ethanone. |
In vivo antiplatelet aggregation measurements
were performed in the current study to further assess the in
vivo processes and the activity of the 3 derivatives. As
presented in Fig. 3C, DHAP
(P=0.003) and APDP (P=0.002) significantly inhibit platelet
aggregation after 10 min of i.g. administration compared with the
vehicle, indicating that they functioned rapidly in vivo.
Following 10 min of i.g. administration, HPPE and PEG-DHAP
exhibited a lower antiplatelet aggregation activity and a slower
action time compared to DHAP. After 40 min of i.g. administration,
the antiplatelet aggregation activity of DHAP was significantly
reduced compared with 10 min of administration (P=0.003), and APDP
exhibited very weak activities, while HPPE (P=0.006) and PEG-DHAP
(P=0.021) demonstrated significant activity compared with 40 min of
administration of DHAP. The results confirmed that DHAP has a short
acting time in vivo and is rapidly adsorbed and eliminated,
which is in congruence with previously reported data (13,16). APDP rapidly transformed into DHAP
in vivo, thereby allowing for rapid elimination. The
long-lasting activity of HPPE and PEG-DHAP imply that the rapid
metabolism of DHAP may be delayed by HPPE and PEG-DHAP, indicating
their ability to prolong acting time in vivo and improve the
oral bioavailability of DHAP.
Assessments of HPLC analytical
method
Phenol was selected as the internal standard. Under
the HPLC conditions selected for the determination of blood
concentration, the target substances were well separated from
phenol and were not interfered with by the endogenous substances of
plasma (Figs. 2 and 4A). DHAP was the active substance for
the determination of APDP and PEG-DHAP blood concentrations, and
HPPE and DHAP were the active substances for the determination of
HPPE blood concentrations. Therefore, the standard curves of DHAP
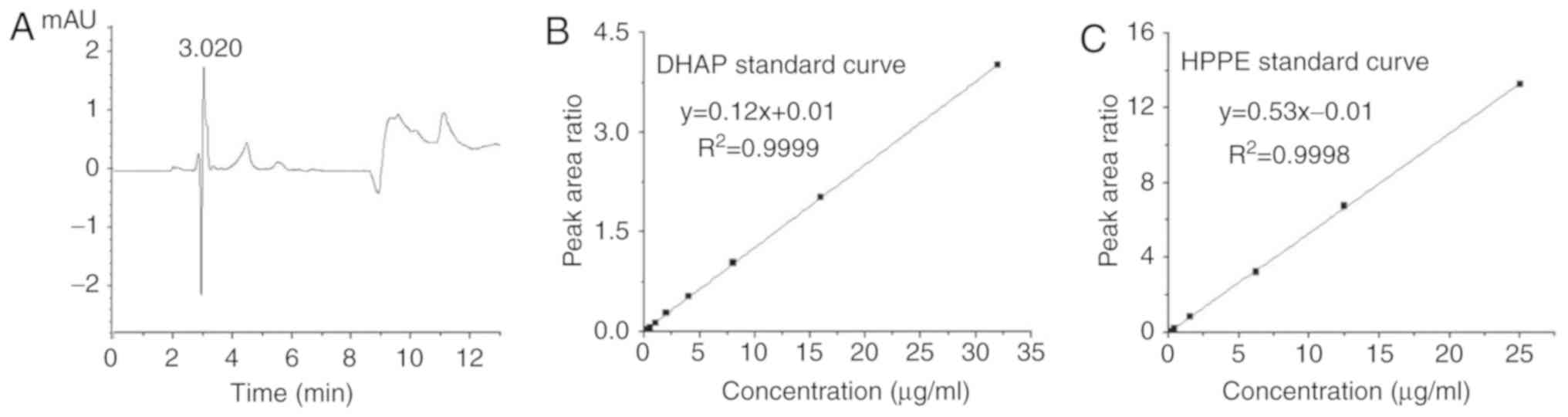
and HPPE were determined. Within the range of 0.10-2.00
µg/ml, the association between the plasma concentration of
DHAP, x, and the peak area ratio (DHAP vs. internal
standard), y, was linear. The linear regression equation
was, R2=0.9999 (Fig. 4B). In the range of 0.10-25.00
µg/ml, the association between the plasma concentration of
HPPE, x, and the peak area ratio (HPPE vs. internal
standard), y, was linear. The linear regression equation
was, R2 = 0.9998 (Fig. 4C). As presented in Table III, the values of recovery of
the high, medium and low concentrations of plasma DHAP and HPPE
were in the range of 86.00-105.67 and 92.24-104.62%, respectively.
Within-day precision was presented using RSD and was in the range
of 1.42-3.84 and 1.12-4.27%, respectively. The between-day
precision of RSD was in the range of 5.36-15.29 and 1.15-5.99%,
respectively. Additionally, solution storage stability was within
the range of 84.12-95.00 and 96.76-98.47%, respectively. The
results revealed that the analytical method used for the
determination of blood concentrations met the requirements of
pharmacokinetic studies.
| Table IIIParameters used to evaluate the
method of blood concentration determinationa. |
Table III
Parameters used to evaluate the
method of blood concentration determinationa.
| Compound | Concentration
(mmol/l) | Recoveryb (%) | Within-day RSD
(%) | Between-days RSD
(%) | Storage
stabilityb (%) |
|---|
| DHAP | 0.003 | 86.00±26.21 | 3.84 | 14.07 | 84.12±12.25 |
| 0.013 | 105.67±3.93 | 1.42 | 15.29 | 88.85±8.42 |
| 0.053 | 101.52±1.10 | 2.64 | 5.36 | 95.00±3.61 |
| HPPE | 0.003 | 92.24±17.02 | 4.27 | 5.99 | 96.76±4.47 |
| 0.013 | 98.77±4.41 | 2.58 | 3.17 | 96.81±6.23 |
| 0.053 | 104.62±2.29 | 1.12 | 1.15 | 98.47±3.20 |
Blood concentration and
bioavailability
In the present study, the pharmacokinetic parameters
of DHAP administered i.v. to rats were consistent with a
two-compartment model, which was same as reported previously
(33,34). The values of distribution
half-life t1/2(α) and elimination half-life
t1/2(β) were 0.045 and 0.282 h, respectively. Due to the
rapid distribution and elimination of DHAP when administered i.v.,
according to a previous study (41), a single-compartment model
simulation was used to compare with other groups. The
pharmacokinetic parameters of the other drug-administered groups
were consistent with the single-compartment model, as presented in
Table IV. The mean blood
concentration-time curves are presented in Fig. 5.
| Table IVPharmacokinetic parameters of the
tested compoundsa. |
Table IV
Pharmacokinetic parameters of the
tested compoundsa.
| Parameter | Unit | DHAP i.v. | DHAP i.m. | DHAP i.g. | APDP | HPPE | PEG-DHAP |
|---|
|
t1/2d | h | 0.15±0.02 | 0.16±0.015 | 0.16±0.02 | 0.13±0.01 | 1.06±0.14 | 1.80±0.27 |
|
Ked | 1/h | 4.81±0.73 | 4.36±0.383 | 4.27±0.41 | 5.35±0.34 | 0.66±0.08 | 0.39±0.05 |
|
V1/Fd | l/kg | 0.48±0.05 | 0.71±0.07 | 4.03±0.83 | 2.49±0.63 | 6.97±2.17 | 13.48±4.08 |
| CL/Fd | l/h/kg | 2.30±0.38 | 3.121±0.56 | 17.39±4.81 | 13.35±3.56 | 4.70±1.78 | 5.36±2.05 |
| AUC
(0-t)d | µmol/l x
h | 66.32±1.67 | 124.22±3.50 | 47.78±2.11 | 34.59±2.29 | 118.06±15.04 | 6.69±4.35 |
| AUC (0-∞)d | µmol/l x
h | 87.48±2.02 | 129.94±3.66 | 48.64±2.18 | 35.94±2.48 | 123.26±15.54 | 7.03±4.79 |
|
Kad | 1/h | | 5.13±0.64 | 11.77±1.40 | 6.43±0.56 | 12.01±2.43 | 0.44±0.06 |
|
t1/2Kad | h | | 0.14±0.02 | 0.06±0.01 | 0.11±0.01 | 0.06±0.01 | 1.62±0.25 |
| Tlagd | h | | 0.04±0.01 | 0.066±0.00 | 0.04±0.00 | 0.04±0.00 | 0.03±0.03 |
| MRT (0-t)e | h | 0.22±0.03 | 0.46±0.01 | 0.38±0.02 | 0.36±0.01 | 1.35±0.24 | 4.36±0.28 |
| F % | | | 93.81b | 18.01b | 126.14c | 394.78c | 425.44c |
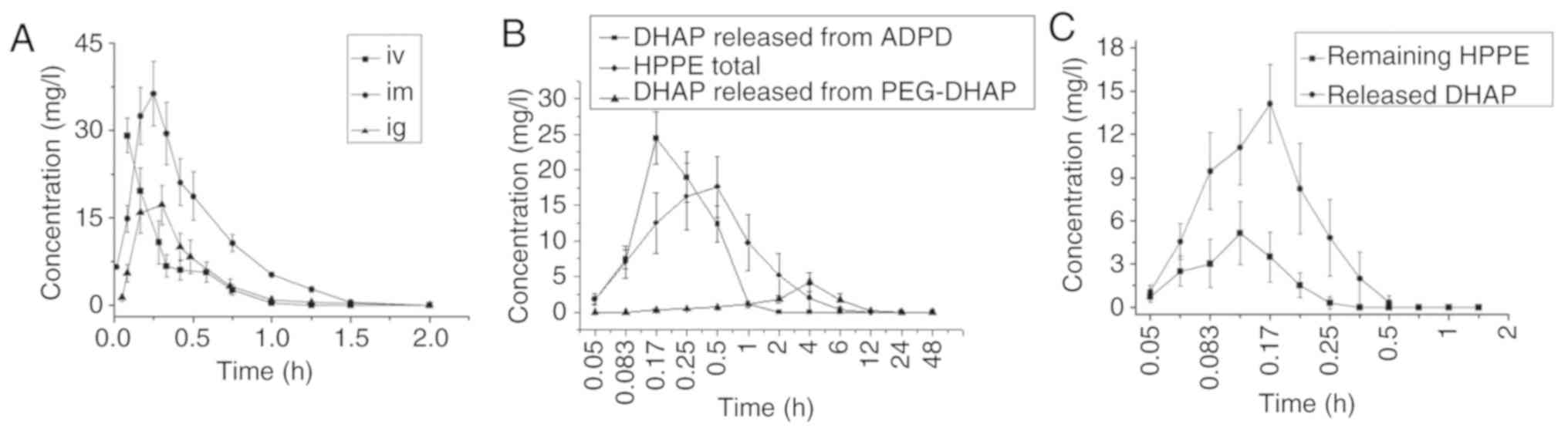
As presented in Fig.
5A, DHAP exhibited rapid absorption and rapid elimination rates
after i.v., i.m. and i.g. administration in rats, as reported in
previous studies involving monkeys and rabbits (33,34). It can be detected in plasma within
3 min after i.m. and i.g. administration. The mean residence time
in the body (MRT (0-t)) was within the range of
0.22-0.46 h, and the elimination half-life (t1/2), was
in the range of 0.15-0.16 h. Its very short in vivo acting
time and rapid elimination rate may be due to the severe first pass
effect that was confirmed by the markedly higher bioavailability of
i.m. administration relative to i.g. administration (Table IV). The severe first pass effect
may also be confirmed via the i.v.-clearance value listed in
Table III of 2.30 l/h/kg, which
was greater than the liver blood flow rate of the rats. This
indicated that DHAP exhibited both hepatic and extrahepatic
metabolisms.
Compared with DHAP, APDP exhibited a much lower
water solubility and a higher oil-water partition coefficient and
was therefore easier to pass through the biofilm via passive
diffusion, making its absorption rate faster. As presented in
Table IV, its MRT
(0-t), t1/2 and FR% were
0.36, 0.13 h and 126.14%, respectively. However, no marked
difference from the DHAP i.g. group was observed. For HPPE, the
average blood concentration-time curves of its original form and
DHAP form produced by metabolism are presented in Fig. 5C. Its pharmacokinetic parameters
were calculated using total the concentration of HPPE and DHAP. Its
MRT (0-t), t1/2 and FR was
1.35, 1.06 h and 394.78%, respectively. The results were different
to those of the DHAP i.g. group. HPPE therefore prolongs the in
vivo acting time and half-life of DHAP and improves its oral
bioavailability. For PEG-DHAP, as presented in Table IV, its value of
MRT(0-t) was 4.36 h, which was 11.47-fold higher than
that of DHAP. Its value of t1/2 was also 1.80 h, which
was 11.88-fold higher than that of DHAP. Finally, its value of
FR% was 425.44%. A prolonged in vivo
acting time and an improved oral bioavailability of PEG-DHAP were
obtained.
Discussion
The phenolic hydroxyl groups of catechol compounds
are essential for biological activity. They are also the main sites
of metabolic inactivation (35).
Modifying the phenolic hydroxyls of DHAP may impact its associated
pharmacokinetic processes and oral bioavailability. Fortunately,
these phenolic hydroxyls can be modified easily and there are many
simple and classic chemical reactions available for reference and
selection. In the present study, the esterification or
etherification of the phenolic hydroxyl groups located at C3 or C4
were selected as the generated derivatives as they may be cleavage
catalyzed by enzymes in vivo to recover DHAP. Furthermore,
the synthetic methods that obtained high yields utilized in the
present study can be implemented in any laboratory.
DHAP is a water-soluble small molecule that is
absorbed rapidly through epithelial cells in the present study,
similarly to the water soluble propofol. DHAP was also eliminated
rapidly regardless of the route of administration and exhibited a
severe first pass effect, which was in congruence with other
studies on monkeys and rabbits (33,34). The derivatives produced in the
current study exhibited different physicochemical properties,
biological activities or in vivo processes compared with
DHAP. APDP was produced as a derivative with two propionate groups
at C1 and C2. These hydrophobic carbon chains caused APDP to
possess a much lower water solubility and much higher values of
LogP, resulting in a more rapid absorption rate compared with DHAP.
However, APDP exhibited no signifi-cant differences in antiplatelet
activity, in vivo process and oral bioavailability when
compared with DHAP. Although APDP was a derivative obtained by
modifying the main metabolic sites of DHAP, its modified ester
bonds were rapidly hydrolyzed in intestinal cells and the liver
during absorption, which recovered the hydroxyls and lost the
protection of the metabolic sites. This was also confirmed by the
results of the derivative-releasing DHAP experiments, which
revealed that plasma APDP recovers to DHAP rapidly, whether in
vivo or in vitro. Therefore, this did not prolong the
in vivo acting time and half-life of DHAP or improve its
oral bioavailability. Although HPPE was also a derivative obtained
via the derivatization of the phenolic hydroxyl group of DHAP, the
introduced hydrophobic carbon chain was attached to the phenyl ring
via an ether bond and thus it exerted different antiplatelet
aggregation activities and in vivo processes from APDP. HPPE
is an aryl ether compound, and its metabolic pathway involves the
O-dealkylation reaction, which transpires within the liver. In
this, the carbon-oxygen bond is cleaved to produce a phenol group.
Although the rate of this metabolism is very rapid, HPPE tends to
accumulate in the lipid layer of biofilms, releasing slowly into
bodily fluids due to its low water solubility and high oil-water
partition coefficient. Therefore, HPPE transformed into DHAP at a
slower rate relative to ADPD. The average blood concentration-time
curves of HPPE (Fig. 5C) and the
derivative-releasing DHAP experiments revealed that a portion of
HPPE transformed into DHAP in vivo, a short time after
absorption. However, some still remained within the blood and DHAP
was gradually released with an extended administration time. HPPE
itself also exhibited significant antiplatelet aggregation
activity. During a certain administration time, the activity was
contributed to by both the released DHAP and the remaining HPPE.
Therefore, the kinetic parameters of HPPE were calculated in the
present study based on the total concentrations of DHAP and HPPE in
the blood. The significantly longer in vivo time, higher
relative oral bioavailability and longer-lasting antiplatelet
aggregation activity after oral absorption confirmed that HPPE
improves the clinical application defects of DHAP. PEG-DHAP is a
polymer derivative obtained by linking DHAP to crosslinked PEG 2000
via ester bonds. Its dose and the unit of measurement were not same
as those of DHAP, APDP and HPPE. PEG-DHAP is a high molecular drug,
and the molecular weight is not very accurate. Therefore, the dose
unit was expressed by the mass of the weighing. In order to
facilitate a comparison with DHAP and two other small molecular
prodrugs, we calculated the moles of DHAP contained in the dosage
of PEG-DHAP. The lower dose of PEG-DHAP was selected as the same
dose of PEG-DHAP was dissolved in 1.5 ml of normal saline to form a
very viscous solution, which could not guarantee the normal
administration of intragastric administration. The effective
results of several animal experiments were obtained at these doses
i) in vivo release after administration only to determine
out whether the drug can release DHAP; ii) determination of IC50 of
in vivo antiplatelet aggregation; and iii) pharmacokinetic
experiments). PEG-DHAP disperses into nanoparticles within bodily
fluids and is adsorbed via the phagocytosis of epithelial cells
(39). Although ester bonds can
be rapidly hydrolyzed and catalyzed by carboxylesterases present in
the blood, cytosol and endoplasmic reticulum, it takes some time
for esterases and associated metabolizing enzymes to cross the PEG
barrier and come into contact with DHAP. Additionally, it takes
some time for free DHAP to migrate out of the PEG barrier to reach
sites at which it may exhibit pharmacological activity. Therefore,
for PEG-DHAP, a prolonged in vivo acting time and improved
oral bioavailability were obtained.
In the present study, other metabolites of DHAP,
including sulfate, gluconate metabolites were not considered.
Previous pharmacodynamic studies have demonstrated that DHAP
exhibits the strongest antiplatelet aggregation activity following
10 min of i.g. administration, after which the activity continues
to decrease, and disappears completely after 40 min (13,16). Pharmacokinetics also revealed that
DHAP reaches a peak plasma concentration at 5-10 min after i.g.
administration, after which rapid metabolism is observed, and
plasma DHAP is absent after 30-40 min (33,34). DHAP has a catechol structure and
is rapidly metabolized in vivo to gluconate and sulfate
metabolites. Combined with the pharmacokinetic and pharmacodynamics
results of previous studies, it can be inferred that these
gluconate and sulfate metabolites have no antiplatelet aggregation
activity. Therefore, only the concentrations of possible active
substances in blood were determined in the present study after ig
administration in rats. DHAP in blood was measured during the blood
concentration determination of APDP and PEG-DHAP, and both
concentrations of blood HPPE and DHAP were measured in the
pharmacokinetic study of HPPE.
DHAP is a water-soluble small molecule that
exhibits severe first pass metabolism, which is the reason for its
short acting time in vivo and relatively low oral
bioavailability. By modifying the metabolic sites of DHAP, three
derivatives, including APDP, HPPE and PEG-DHAP, were prepared in
the current study to alter the pharmacokinetics of DHAP. These
derivatives transformed into or released DHAP in vivo. Among
these, HPPE and PEG-DHAP significantly increased the acting time
in vivo and the oral bioavailability of DHAP. The results of
the current study may provide a possible reference point for the
development of DHAP as an oral drug. Furthermore, these results may
be useful for researchers to address the poor bioavailability of
phenolic drugs.
Supplementary Data
Funding
The study was supported by the National Natural
Science Foundation of China (grant no. 31600386), the Natural
Science Foundation of Shandong Province (grant no. ZR2016HM47) and
the Medical and Health Science and Technology Development Project
of Shandong Province (grant no. 2014WSB27002).
Availability of data and materials
All data generated or analyzed during this study
are included in this published article.
Authors' contributions
NS and YD were responsible for the synthesis of
derivatives and pharmacokinetic experiments. MQ and YL were
responsible for the determination of physicochemical properties and
platelet aggregation experiments. MW and LW were responsible for
raising the animals and treating the animals used in the
experiments. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All procedures performed in experiments involving
animals were approved by the Animal Experimentation Ethics
Committee of Weifang Medical College and in accordance with the
guidelines of the National Health and Medical Research Council of
China for the care and use of animals for scientific purposes.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Dr Bo Zhang and Dr
Xuejian Wang from the School of Pharmacy, Weifang Medical College
for their helpful assistance with the calculation of
pharmacokinetic parameters.
References
|
1
|
Beijing Pharmaceutical Industry Research
Institute, Chinese People's Liberation Army 157 Hospital: Study on
the active constituents of leaves of Ilex pubescens Hook et Arn var
glaber Chang. Chin Trad and Herb Drugs Commun. 8:7–10. 1977.In
Chinese.
|
|
2
|
Research Group of the Therapeutic Effects
of Qingxintong: Summary of the treatment effects of Qingxintong on
angina pectoris of coronary heart disease. Beijing Pharm Industry.
1:23–27. 1983.In Chinese.
|
|
3
|
Chinese People's Liberation Army 157
Hospital: Clinical observation of 50 cases of coronary heart
disease treated with Qingxintong injection. Beijing Pharm Industry.
1:27–29. 1983.In Chinese.
|
|
4
|
Beijing pharmaceutical industry research
institute: Effect of Qingxintong on thrombosis in patients with
coronary heart disease. Beijing Pharm Industry. 1:16–17. 1983.In
Chinese.
|
|
5
|
Beijing Pharmaceutical Research Institute:
Research on the pharmacologic effect of leaves of Qingxintong on
coronary heart disease. Chin Trad and Herb Drugs. 11:358–366.
1980.In Chinese.
|
|
6
|
Lin CL, Zhang ZX and Xu YJ: Therapeutic
mechanism of qingxintong on chronic obstructive pulmonary disease.
Chin J Tuberculosis and Respirat. 18:97–98. 1995.In Chinese.
|
|
7
|
Lin CL, Zhang ZX, Xu YJ and Ni W: Effects
of qingxintong on hemodynamics and atrial natriuretic peptide and
cyclic glucosides in patients with chronic obstructive pulmonary
disease. Chin J Integrated Trad Chin Western Med. 15:131–133.
1995.In Chinese.
|
|
8
|
Lin CL, Zhang ZX and Xu YJ: Effects of
3.4-dihydroxyaceto-phenine on hemorheology and plasma levels of
TXA2, CD62P in patients with chronic pulmonary heart disease. J
Clin Internal Med. 19:196–198. 2002.In Chinese.
|
|
9
|
Sun YP, Ma TY and Wu XR: Clinical analysis
and mechanism of treatment of pregnancy-induced hypertension with
Qingxintong. Practical J Obstet Gynecol. 7:141–143. 1991.In
Chinese.
|
|
10
|
Huang YP and Ma TY: Treatment of
intrauterine growth retardation with qingxintong. Chin J Obstet
Gynecol. 28:333–336. 1993.In Chinese.
|
|
11
|
Wu XR, Li Y, Yang DS, Han BJ, Si YZ, Shan
ZZ, Ma TY, Wen LZ, Sun YP and Huang YP: The utero-placental
circulation, eugenics and the prevention and treatment of high risk
pregnancies. J Tongji Med Univ. 14:1–7. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Huang YP, Ye DY, Wu P, Huang YF, Ying HG
and Ma TY: The therapeutic effectiveness of inhaled DHAP in the
treatment of intrauterine growth retardation. Acta Med Univ Sci Et
Technol Huazhong. 31:192–194. 2002.In Chinese.
|
|
13
|
Wang Z, Gao HQ, An Y, Zhu GQ and Yang ZM:
Effect of 3,4-dihydroxyacetophenone on rabbit platelet function.
Zhongguo Yao Li Xue Bao. 5:187–192. 1984.In Chinese. PubMed/NCBI
|
|
14
|
Wang Z, Gao HQ, Zhu GQ, An Y and Huang RS:
Effect of 3,4-dihydroxyacetophenone on the generation of PGI2-like
substances in the rat aorta. Zhongguo Yao Li Xue Bao. 7:37–40.
1986.In Chinese. PubMed/NCBI
|
|
15
|
Xu H, Wang Z and An Y: Effect of
3,4-dihydroxyacetophenone on platelet phosphodiesterase in rabbits.
Acta Pharmacol Sin. 8:159–162. 1986.In Chinese.
|
|
16
|
Wang Z, An Y, Liu Z, Zhu GQ and Huang RS:
Effect of 3,4-dihy-droxyacetophenone on TXA2 release from rabbit
platelets. Yao Xue Xue Bao. 22:330–334. 1987.In Chinese. PubMed/NCBI
|
|
17
|
Li S, Li Y, Xiong Z and Wu X: Influence of
Qingxingtong on platelet function of PIH-patients. J Tongji Med
Univ. 23:305–307. 1994.In Chinese.
|
|
18
|
Wang Z, Huang RS, Gao YH, An Y and Qiang
ZG: 3,4-Dihy-droxyacetophenone-a cyclooxygenase inhibitor (brief).
Acta Pharmacol Sin. 1:351988.In Chinese.
|
|
19
|
Shi L, Qin ZH and Gao SX: Effect of
3,4-dihydroxyacetophe-none on the membrane fluidity of platelets by
fluorescence polarization analysis. Zhongguo Yao Li Xue Bao.
7:149–151. 1986.In Chinese. PubMed/NCBI
|
|
20
|
Shan Z, Chang C, Feng L and Wu X: The
regulatory effect of 3,4-dihydroxyacetophenone on tPA and PAI
activity of the vascular endothelial calls. Pharmacol Clin Chin
Materia Med. 6:33–35. 1995.In Chinese.
|
|
21
|
Beijing Institute of Pharmaceutical
Industry, The People's Liberation Army 157 Hospital:
Pharmacological study of bald holly leaves on coronary heart
disease. Chin Pharm J. 15:421980.In Chinese.
|
|
22
|
Wu P, Huang YP and Ye DJ: Advances in
research on the mechanism of qingxintong promoting blood
circulation and removing blood stasis. Chin Trad Herbal Drugs.
23:277–279. 2001.In Chinese.
|
|
23
|
Ullah F, Wang DX, Ming Z and Yu SB:
Effects of 3,4-dihydroxy-acetophenone (3,4-DHAP) on hypoxic
pulmonary and systemic vascular response in dogs. J Tongji Med
Univ. 15:26–30. 1995. View Article : Google Scholar
|
|
24
|
Hong ZG, Wang DX, Jin S, Zhang J, Wang XL
and Ye DY: Effect of 3,4-dihydroxyacetophenone on delayed rectifier
potassium current of the intrapulmonary artery smooth muscle cells
in rats. Chin Pharmacol Bull. 18:386–389. 2002.In Chinese.
|
|
25
|
Yang DS, Xi-rui W and Ting-yuan M: Effects
of 3,4-dihy-droxyacetophenone on the biosynthesis of TXA2 and PGI2
in human placental villus and umbilical artery segments in vitro.
Prostaglandins. 38:497–504. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hang YP, Ye DJ and Ma TJ: Effect of
Qingxintong on nitric oxide synthase and plasma endothelin in
placental vascular wall of patients with pregnancy induced
hypertension. Chin J Obstet Gynecol. 31:667–669. 1996.In
Chinese.
|
|
27
|
Li G, Wu P, Zhang DJ, Ye DY and Li F:
Mechanism of inhibition of 3, 4-dihydroxyacetophenone on
LPS-induced apoptosis of RAW264.7 macrophages in mice. Chin Trad
Herbal Drugs. 36:1835–1838. 2005.In Chinese.
|
|
28
|
Wu P, Zhang L, Zhou X, Li Y, Zhang D, Wan
J and Ye D: Inflammation pro-resolving potential of
3,4-dihydroxyaceto-phenone through 15-deoxy-Δ12,14-prostaglandin J2
in murine macrophages. Int Immunopharmacol. 7:1450–1459. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhang D, Liu T, Liu J, Cui X, Guo J, Wang
J and Ye D: Relationship between therapeutic and preventive effect
of dihydroxyaceto-phenone in treating atherosclerosis and the
Toll-like-receptor 4 pathway. Trad Chin Drug Res Clin Pharmacol.
20:404–407. 2009.In Chinese.
|
|
30
|
Zhang D, Liu T, Cui X, Liu J, Guo J, Wang
J and Ye D: Preventive effect of 3,4-dihydroxyacetophenone on
atherosclerosis and role of visfatin expression. Chin J
Pathophysiol. 26:1700–1703. 2010.In Chinese.
|
|
31
|
Zhang D, Gong Y, Liu J, Wang J, Wang L and
Liu T: Effects of 3,4-dihydroxyacetophenone on 5-lipoxygenase in
macrophages of atherosclerosis plaque. Trad Chin Drug Res Clin
Pharmacol. 23:243–246. 2012.In Chinese.
|
|
32
|
Zhang DJ, Liu JY, Wang L, Wang J, Li W,
Zhuang B, Hou J and Liu T: Effects of 3,4-dihydroxyacetophenone on
the hypercho-lesterolemia-induced atherosclerotic rabbits. Biol
Pharm Bull. 36:733–740. 2013. View Article : Google Scholar
|
|
33
|
Li SX, Li Y, Shan ZZ, Xiong ZM, Wu XR and
Yin XG: Pharmacokinetics of Qingxintong in rabbits. Acta
Universitatis Medictnae Tangji. 22:91–93. 1993.In Chinese.
|
|
34
|
Pang X, Fu G and Zuo M: Comparison of
bioavailability of Qingxintong by three administration routes. Trad
Chin Drug Res Clin Pharmacol. 13:31–32. 2001.In Chinese.
|
|
35
|
Eun JK, Jong WA, Hye SL and Wolfram C:
Determination of peripheral catecholO-methyltransferase (COMT)
activity in vivo using
(2-14C)-3′,4′-dihyroxyacetophenone. Arch Pharm Res.
14:290–294. 1991. View Article : Google Scholar
|
|
36
|
Montenegro L, Carbone C, Maniscalco C,
Lambusta D, Nicolosi G, Ventura CA and Puglisi G: In vitro
evaluation of quercetin-3-O acyl esters as topical prodrugs. Int J
Pharm. 336:257–262. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Jin YY, Zhang JS, Zhang Y and Zhang YH:
Studies on the intestinal adsorption of crocin in rats and
determination of the partition coefficient. J Chin Pharm Univ.
35:247–257. 2004.In Chinese.
|
|
38
|
Fang L: Pharmaceutics. 8th edition.
People's Medical Publishing House; Beijing: pp. 137–139. 2016, In
Chinese.
|
|
39
|
Zhou X, Zhang X, Han S, Dou Y, Liu M,
Zhang L, Guo J, Shi Q, Gong G, Wang R, et al: Yeast
microcapsule-mediated targeted delivery of diverse nanoparticles
for imaging and therapy via the oral route. Nano Lett.
17:1056–1064. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
You QD: Medicinal Chemistry. 8th edition.
People's Medical Publishing House; Beijing: pp. 462016, In
Chinese.
|
|
41
|
Liu CX: Introduction to Pharmacokinetics.
Liu D: China Academic Press Publication; Beijing: pp. 215–217.
1984, In Chinese.
|