Introduction
Cholangiocarcinoma (CCA) is a malignancy of biliary
epithelium, which is the second most common primary liver cancer
worldwide. The global incidence of CCA is <6/100,000 people but
it is 85/100,000 people in Northeastern Thailand (1), where the high prevalence of the
carcinogenic liver fluke, Opisthorchis viverrini, infection
is reported (2). Nonetheless,
mortality rates of intrahepatic CCA have also increased in other
continents (3). CCA is a
devastating cancer in that patients are often diagnosed at a late
stage when complete resection is not a curative option (4). CCA is well known for the low
response to currently available chemotherapy (5). The effectiveness of available
pharmacological treatments is limited by several mechanisms of
chemoresistance (5,6).
The resistance to chemotherapy is a major challenge
in CCA treatment. The common mechanisms of CCA chemoresistance are
i) reduced intracellular drug concentration, ii) altered drug
metabolism, iii) changed drug targets, iv) enhanced DNA repair, v)
changes in cancer microenvironment and vi) altered balance between
survival and apoptotic signals (5). The present authors have previously
reported increased expression of the specific adenosine
triphosphate binding cassette (ABC) transporter superfamily member;
ABCC1, in CCA and its contribution to worse prognosis (7), but the clinical benefits of an ABCC1
inhibitor are yet to be proven. The disturbance of pro- and
anti-apoptotic signals and the potential in CCA treatment were
focused on in the current study.
The increased anti-apoptotic proteins, such as
B-cell lymphoma 2 (Bcl-2), B-cell lymphoma-extra-large (Bcl-xL),
cellular inhibitor of apoptosis (cIAP) (8), FADD-like IL-1β-converting
enzyme-inhibitory protein (FLIP) (9), myeloid cell leukemia-1 (Mcl-1),
X-linked inhibitor of apoptosis protein (XIAP) (10,11) and survivin (12) in CCA cells have been previously
reported and these contribute to apoptotic evasion. On the
contrary, suppression of selected anti-apoptotic proteins induce
apoptosis and suppress CCA growth in vivo (8-10,12). The increased expression of
multiple anti-apoptotic proteins usually occurred simultaneously.
Therefore, this study aimed to find an FDA-approved agent that
could downregulate anti-apoptotic proteins.
A number of anti-apoptotic genes (e.g. Mcl-1,
survivin and XIAP) are under the regulation of specificity protein
1 (Sp1) transcription factor (13). Sp1 binds to GC-rich sequences on
their promoters and promotes transcription (13). Even though Sp1 is highly
controlled during development, over-expression of Sp1 is often
reported in cancers and contributes to poor prognosis (14,15). Thus, suppression of Sp1 or
interference of Sp1 binding to a target promoter is considered a
novel strategy for cancer treatment (13,16). According to current understanding,
among FDA-approved anti-cancer agents, mithramycin A (MTA) is a
selective Sp1 inhibitor, which suppresses Sp1-related
anti-apoptotic gene expression and induces caspase-dependent
apoptosis (17,18). There are reports, however, that
chromomycin A3 (CMA3), an MTA analog, possesses higher DNA binding
capacity and has demonstrated similar effects on neurons (19,20). Therefore, CMA3 might exhibit
potent Sp1-related gene suppression. CMA3 was selected for the
current studies.
CMA3 is an anthraquinone glycosidic antibiotic,
produced by Streptomyces griseus (21). The anti-cancer effects of CMA3
have been proposed since the 1960s (22,23). Side effects of CMA3 in advanced
breast cancers have interrupted that use (24) but it is widely used for DNA
staining (25). DNA binding
ability of CMA3 has been elucidated; CMA3 binds to a GC-rich
sequence at a minor groove and inhibits DNA replication and
transcription (26). It has been
reported that CMA3 induced cervical cancer cell apoptosis but the
underlying mechanism is still obscure (27). Therefore, the anti-CCA potentials
of CMA3 and its effects on Sp1-related anti-apoptotic proteins were
focused on.
Materials and methods
Cell lines and cell culture
A total of three CCA cell lines, KKU-055, KKU-100
and KKU-213, were established as previously described (28). Cells were obtained from the
Japanese Collection of Research Bioresources Cell Bank. Cells were
maintained in Dulbecco's modified Eagle's medium (Wako Pure
Chemical Industries, Ltd.) containing 10% fetal bovine serum
(HyClone; GE Healthcare), 100 U/ml penicillin and 100 µg/ml
streptomycin. CCA cells were cultured in a humidified incubator at
37°C with 5% CO2.
Reagents and antibodies
CMA3 was purchased from Abcam, MTA was from
Sigma-Aldrich; Merck KGaA, MTT and propidium iodide (PI) were from
Sigma-Aldrich; Merck KGaA, RNase A was from Takara Bio, Inc., and
Pacific Blue™ Annexin V was from BioLegend, Inc.
Sources of antibodies were as follows: Rat
anti-Hsc70 (1B5 clone; cat. no. ADI-SPA-815B) monoclonal antibody
(mAb) was from EnZo Life Sciences, Inc.; rabbit anti-Bim (C34C5
clone; cat. no. 2933) mAb, rabbit anti-cleaved caspase-3 (Asp175;
5A1E clone; cat. no. 9664) mAb, rabbit anti-cleaved caspase-8
(Asp391; 18C8 clone; cat. no. 9496) mAb and rabbit anti-cleaved
caspase-9 (Asp315; D8I9E clone; cat. no. 20750) mAb, horseradish
peroxidase (HRP)-linked anti-rabbit IgG (cat. no. 7071), and
HRP-linked anti-mouse IgG (cat. no. 7076) were from Cell Signaling
Technology, Inc.; mouse anti-FLIP short and long forms
(FLIPS/L; G-11 clone; cat. no. sc-5276) mAb, mouse
anti-Mcl-1 (22 clone; cat. no. sc-12756) mAb, mouse anti-Noxa
(114C307 clone; cat. no. sc-56169) mAb, mouse anti-survivin (D8
clone; cat. no. sc-17779) mAb, rabbit anti-cIAP2 (H-85 clone; cat.
no. sc-7944) polyclonal antibody (pAb), rabbit anti-Sp1 (PEP 2
clone; cat. no. sc-59) pAb, and rabbit anti-XIAP (H-202 clone; cat.
no. sc-11426) pAb, were from Santa Cruz Biotechnology, Inc.; mouse
anti-BAX (2D2 clone; cat. no. 633601) mAb was from Biolegend, Inc.;
mouse anti-α-smooth muscle actin (α-SMA; 1A4 clone; cat. no. M0851)
mAb, and rabbit anti-rat (cat. no. P0450) pAb were from Dako;
Agilent Technologies, Inc.; mouse anti-cytokeratin (AE1&AE3
clone; cat. no. 313M) cocktail was from Cell Marque™
(Sigma-Aldrich; Merck KGaA); rabbit anti- cytokeratin 19 (CK19;
cat. no. HPA002465) was from Sigma-Aldrich (Merck KGaA); rabbit
anti-BAX (E63 clone; cat. no. ab32503) pAb, and rabbit
anti-caspase-9 (cat. no. ab2014) pAb were from Abcam and the rabbit
anti-Ki-67 (30-9 clone; cat. no. 790-4286) mAb was from Ventana
Medical Systems, Inc.
Tetrazolium dye MTT assay
The anti-proliferative effects of CMA3 on KKU-055,
KKU-100 and KKU-213 CCA cells were determined by MTT assay. CCA
cells were seeded at 4×103 cells per well into a 96-well
plate. At 24 h after seeding, CMA3 or MTA were added at different
concentrations (0, 2.5, 5, 10, 20, 40, 80 and 160 nM for CMA3 and
0, 12.5, 25, 50, 100 and 200 nM for MTA) and cells were incubated
for 24 and 48 h. MTT solution was added to obtain a final
concentration of 0.5 mg/ml. Then formazan crystals were dissolved
by acidified isopropanol (0.04 N HCl in isopropanol). The
absorbance was measured at 570 nm using a microplate reader (iMark;
Bio-Rad Laboratories, Inc.). The data were analyzed using the
GraphPad Prism 8 (GraphPad Software, Inc.). The optical
density570 of the control was set to 100%.
Cell cycle analysis
Cells were treated at indicated concentrations of
CMA3 for 24 h. After that cells were harvested and fixed with cold
70% ethanol overnight at 4°C. Before analysis, cells were washed
with PBS and incubated with 0.1 mg/ml of RNase A at 37°C for 1 h,
followed by PI staining (50 µg/ml) at 4°C for 30 min in the
dark. Samples were analyzed by LSR II flow cytometer (BD
Biosciences; Becton, Dickinson and Company) and data were analyzed
by FlowJo software version 10.4 (Tree Star, Inc.).
Annexin V binding assay
Cells were treated with various concentrations of
CMA3 for 24 h. Then cells were collected and incubated with Pacific
Blue™ Annexin V at room temperature for 30 min in the dark. PI was
added at the final concentration of 1 µg/ml before flow
cytometry analysis. Annexin V and PI-stained cells were determined
by flow cytometry. Annexin V-bound and PI-stained cells were
analyzed by FlowJo software.
Western blot analysis
KKU-213 cells were treated with CMA3 at indicated
concentrations and times. Protein lysate was prepared as described
elsewhere (29). Protein
concentrations were determined by the bicinchoninic acid protein
assay (Thermo Fisher Scientific, Inc.). A total of 20 µg of
protein was separated by SDS-PAGE (10-15%) and transferred to a
PVDF membrane (GE Healthcare Japan). The membrane was blocked with
5% skim milk in Tris-buffered saline containing 0.1% Tween-20
(TBST) at room temperature for 30 min and was incubated with
specific primary antibodies (1:1,000) at 4°C overnight. Membranes
were washed with TBST 3 times prior to the incubation with the
corresponding HRP-conjugated secondary antibody (1:2,000) for 1 h
at room temperature. Signals were detected using Chemi-Lumi One
Super reagents (Nacalai Tesque, Inc.) and visualized by ImageQuant
LAS 4000 system (GE Healthcare Japan). The quantitative analyses of
blots were quantified by Image J (30). Hsc70 was used as an internal
control.
Reverse-transcriptase-polymerase chain
reaction (RT-PCR)
KKU-213 cells were treated with 40 nM CMA3 or 200 nM
MTA for 0, 6, 12, 18 and 24 h. RNA preparation and RT-PCR were
performed as previously described (31). Gene expression was presented as
relative expression (fold, control at 0 h=1). The oligonucleotide
primers used in this study were previously reported elsewhere;
Mcl-1 (32), XIAP
(33), β-actin (34).
CMA3 toxicity testing
To demonstrate the toxic effects of CMA3 in the
mouse model, a total of eight 6-8 week-old male Balb/c Rag-2/Jak3
double deficient mice (35) were
randomly separated into 4 groups (n=2/group; body weight ~22-25
g/mouse); group 1 was intravenously injected with a vehicle,
dimethyl sulfoxide (DMSO) once a week, group 2 was injected with
0.1 mg/kg CMA3 once a week, group 3 was injected with 0.1 mg/kg
CMA3 twice a week and group 4 was injected with 0.5 mg/kg CMA3 once
a week. The vehicle or CMA3 was given for 3 weeks. The toxicity was
monitored by observation of the general appearance and
determination of the body weight.
Xenograft mouse model
KKU-213 cells (1×105 cells/site) were
subcutaneously injected into both flanks of 6-8 week-old male
Balb/c Rag-2/Jak3 double deficient mice. At 3 days after CCA
injection, 14 mice were randomly divided into 2 groups (n=7/group;
body weight ~22-25 g/mouse). CMA3 was administered to the treatment
group at 0.5 mg/kg intravenously, once a week for 3 weeks. DMSO was
given to the control group. Body weight and tumor volume were
monitored every 3 days. On day 22, mice were sacrificed and tumors
were collected for the immunohistochemistry staining.
Mice were housed and monitored in the animal
research facility according to the institutional guidelines. All
protocols were approved by the Institutional Animal Care and Use
Committee of Kumamoto University. All of the animal experiments
were conducted according to the Fundamental Guidelines for Proper
Conduct of Animal Experiment and Related Activities in Academic
Research Institutions under the jurisdiction of the Ministry of
Education, Culture, Sports, Science and Technology, Japan. In
brief, mice were housed in a strictly control environment (22±2°C,
50±10% humidity and 12-h light/dark cycle). Food and water were
provided ad libitum. Health and behavior of those mice were
monitored every 3 days to evaluate humane endpoints. If mice showed
loss of appetite, severe weight loss (>20% of the original
weight) or signs of pain associated behavior, mice would be
euthanized using isoflurane. Death was verified as follows: Lack of
respiration, heartbeat and corneal reflex. A total of 5% inhaled
isoflurane was used as an anesthetic.
Immunohistochemistry
Paraffin embeded xenograft tumor tissues were
prepared according to a standard protocol (36) and sectioned at 3-µm
thickness. Expression of cytokeratin, Ki-67 and α-SMA were detected
using BenchMark XT automated staining system (Ventana Medical
Systems, Inc.). EZ prep, Cell conditioning (CC1) and Inhibitor CM
(Ventana Medical Systems, Inc.) were used according to
manufacturer's protocol. Images were taken by an Eclipse Ni-E light
microscope (Nikon Corporation) using NIS Elements D software
version 4.5 (Nikon Corporation). Ki-67 positive nuclei per
cytokeratin areas were quantified by ImageJ (n=4/group, 3
images/tumor).
Caspase-9, CK19 and Bax expression was determined by
immunohistochemistry as previously described (37). The caspase-9 or Bax-positive areas
were determined by ImageJ (n=5/group, 5 images/tumor). The
expression of caspase-9 or Bax was presented as the marker-positive
area/CK19-positive nuclei/cancer area.
Statistical analysis
The data are presented as the mean ± standard
deviation from three independent experiments unless otherwise
specified. The statistical differences between the control and
experimental groups were determined by Student's t-test. For CMA3
toxicity testing, mean percentage of body weights among the groups
were analyzed using one-way analysis of variance with Tukey's post
hoc test. P<0.05 was considered to indicate a statistically
significant difference. Statistical analyses were performed using
the GraphPad Prism software version 8 (GraphPad Software, Inc.) and
SPSS software version 19.0 (IBM, Corp.).
Results
CMA3 and MTA inhibit CCA cell
proliferation
Effects of CMA3 and the prototype drug, MTA, on cell
proliferations of 3 CCA cell lines, KKU-213, KKU-055 and KKU-100,
were determined by MTT assay. Cells were treated with increasing
concentrations of CMA3 or MTA for 24 and 48 h. The dose-response
curves revealed CMA3 and MTA suppressed CCA cell proliferation in
dose- and time-dependent manners (Fig. 1). The half maximal inhibitory
concentrations (IC50s) of CMA3 on KKU-213 at 24 and 48 h
were 22.48±4.08 and 9.79±1.15 nM; IC50s of KKU-055 were
21.14±2.24 and 13.34±1.28 nM; and IC50s of KKU-100 were
30.52±2.91 and 14.74±1.34 nM. The IC50s of MTA were
higher than those of CMA3; IC50s of KKU-213 at 24 and 48
h were >200 and 46.08±1.63 nM; IC50s of KKU-055 were
>200 and 76.44±11.70 nM; and IC50s of KKU-100 were
>200 and 104.77±4.22 nM (Table
I).
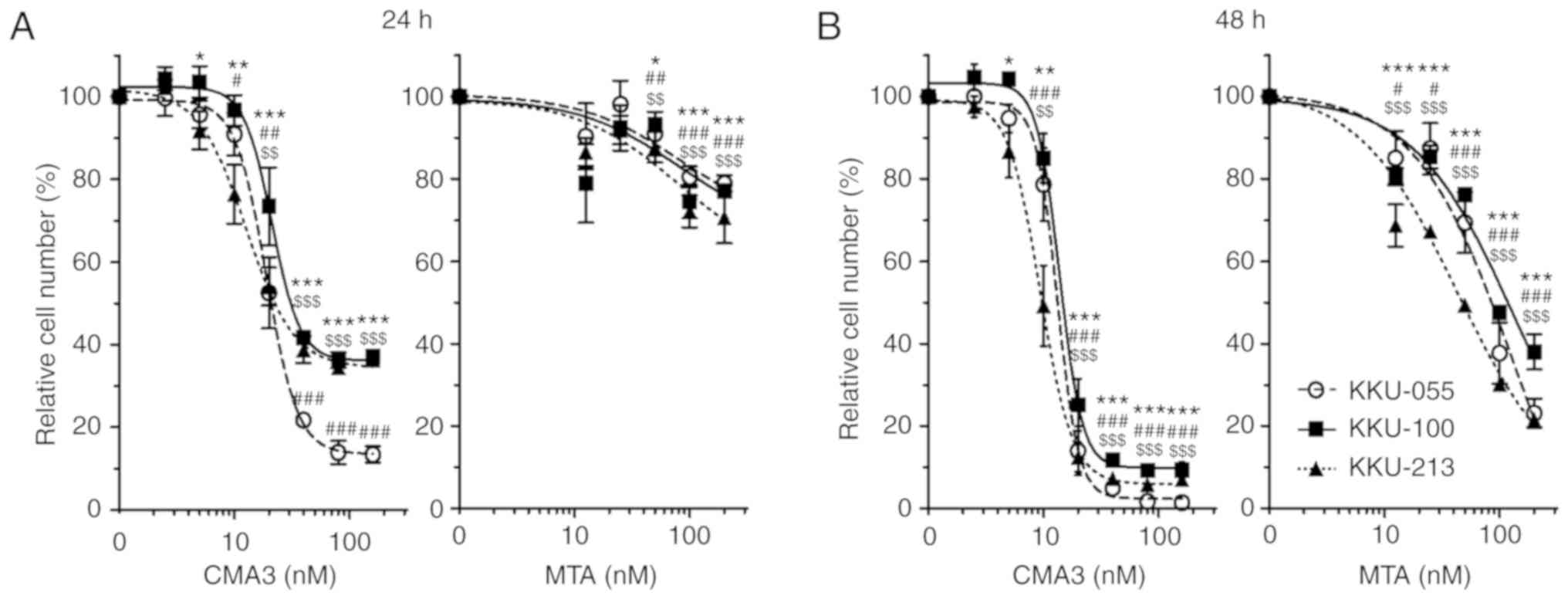 | Figure 1CMA3 and MTA inhibit CCA cell
proliferation in dose- and time-dependent manners.
Anti-proliferative effects of CMA3 and MTA on three CCA cell lines
were determined by MTT assay. KKU-213, KKU-055 and KKU-100 were
treated with increasing concentrations of CMA3 (0, 2.5, 5, 10, 20,
40, 80 and 160 nM) or MTA (0, 12.5, 25, 50, 100 and 200 nM) for (A)
24 and (B) 48 h. Data are represented as the mean ± standard
deviation from three independent experiments. The statistical
significances of each cell line compared with the control are shown
as follows: *P<0.05, **P<0.01 and
***P<0.001 vs. 0 nM for KKU-213 cells;
#P<0.05, ##P<0.01 and
###P<0.001 vs. 0 nM for KKU-005 cells;
$$P<0.01 and $$$P<0.001 vs. 0 nM for
KKU-100 cells. CMA3, chromomycin A3; MTA, mithramycin A; CCA,
cholangiocarcinoma. |
 | Table IIC50s of CMA3 and MTA for
three CCA cell lines at 24 and 48 h. |
Table I
IC50s of CMA3 and MTA for
three CCA cell lines at 24 and 48 h.
| Cell lines | CMA3
| MTA
|
|---|
| 24 h | 48 h | 24 h | 48 h |
|---|
| KKU-213 | 22.48±4.08 | 9.79±1.15 | >200 | 46.08±1.63 |
| KKU-055 | 21.14±2.24 | 13.34±1.28 | >200 | 76.44±11.70 |
| KKU-100 | 30.52±2.91 | 14.74±1.34 | >200 | 104.77±4.22 |
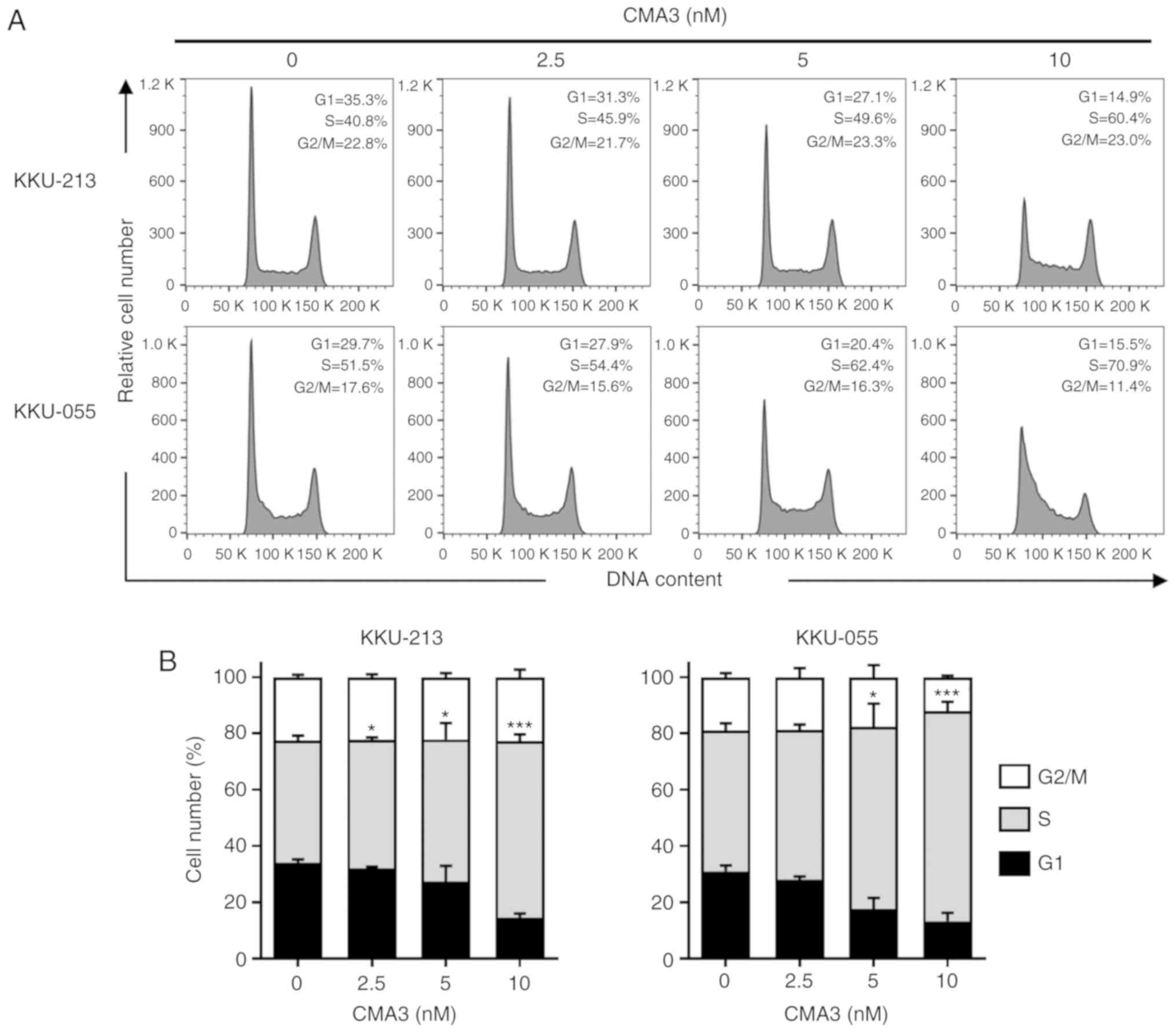
CMA3 promotes S phase arrest in KKU-213
and KKU-055
CMA3 is a DNA-binding agent that binds to DNA at a
minor groove and inhibits DNA replication and transcription
(26). To verify the effects of
CMA3 on cell cycle distribution, KKU-213 and KKU-055 were treated
with 0, 2.5, 5, and 10 nM CMA3 for 24 h. Cells were stained with PI
and the cell cycle distributions were analyzed by flow cytometry.
The results showed increased concentrations of CMA3 significantly
increased cells in the S phase. Fig.
2A shows the representative histograms. CMA3 caused the
accumulation of cells in S phase, while the reduction of cells in
G1 phase was observed (Fig. 2A).
A total of 42.5±1.7, 45.8±0.9, 50.4±5.9 and 62.8±2.4% of KKU-213
and 50.0±2.6, 53.2±1.9, 64.9±8.1 and 74.6±3.3% of KKU-055 were in S
phase of the cell cycle when cells were treated with 0, 2.5, 5, and
10 nM CMA3 (P<0.05 and P<0.001; Fig. 2B).
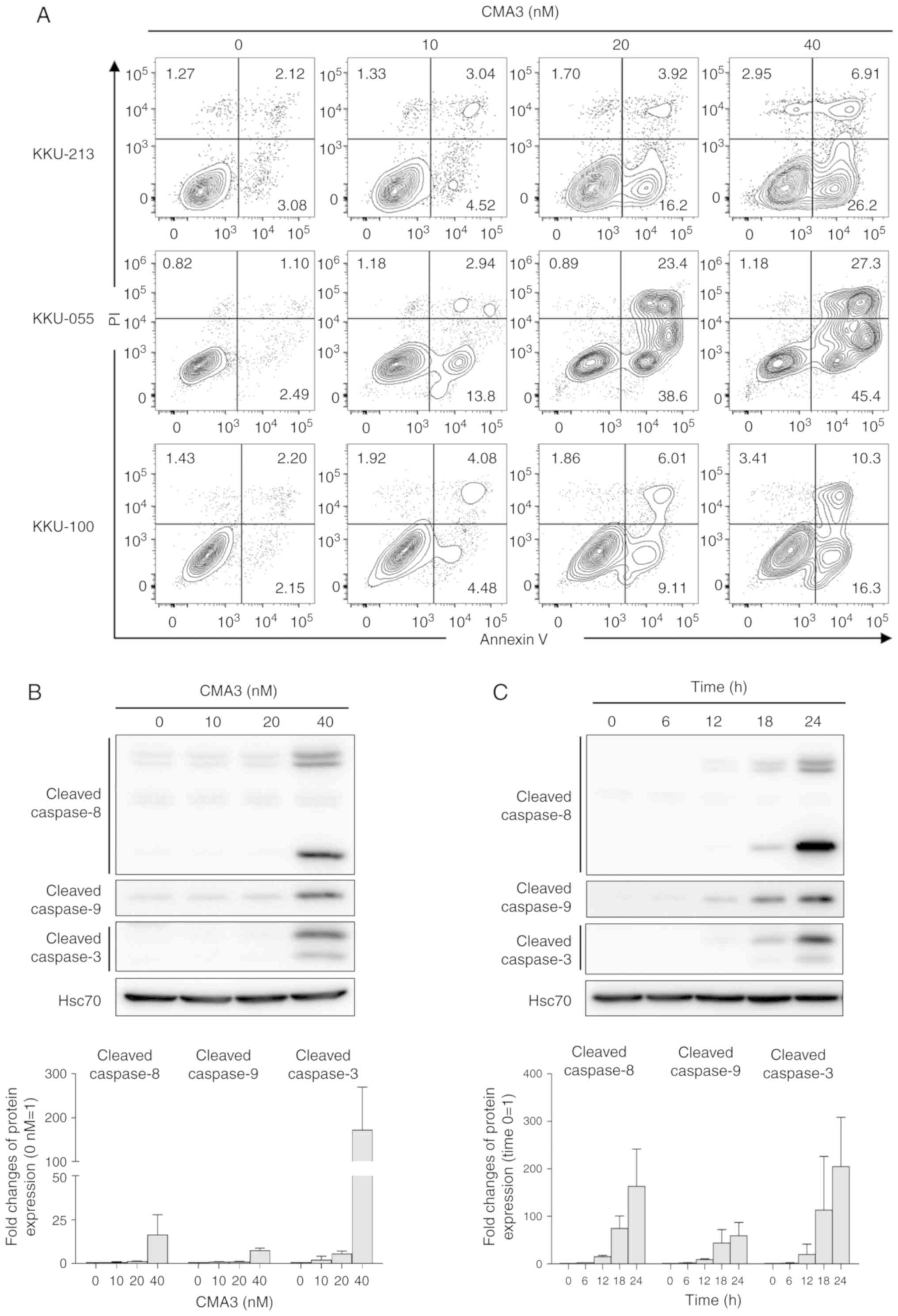
CMA3 induces caspase-dependent apoptosis
in CCA cells
To investigate the effect of CMA3 on apoptotic
induction, Annexin V/PI staining was performed in KKU-213, KKU-055
and KKU-100 cells after treatment with 0, 10, 20 and 40 nM of CMA3
for 24 h. Annexin V and PI stained cells were evaluated by flow
cytometry. Annexin V-positive and PI-negative cells were early
apoptotic cells, whereas Annexin V and PI double-positive cells
were late apoptotic. The percentages of early and late apoptotic
cells were dramatically increased when cells were treated with
higher concentrations of CMA3 (Fig.
3A). The percentages of Annexin V-positive cells were
quantified by combining of percentages of early and late apoptotic
cells and are presented in Fig. S1A
and B (P<0.05). The results showed increased percentages of
apoptotic cells in all three CCA cell lines in a dose-dependent
manner. To elucidate the mechanism of CMA3-induced apoptosis,
caspase activations were investigated. Caspase-8, -9 and -3 were
selected to be the representatives of the extrinsic, intrinsic and
common apoptotic pathways (38).
The results demonstrated CMA3 induced caspase-dependent apoptosis
in KKU-213 through both extrinsic (cleaved caspase-8), intrinsic
(cleaved caspase-9) and eventually common (cleaved caspase-3)
pathways in dose- and time-dependent manners (Fig. 3B and 3C). Similar results of CMA3-induced
caspase-dependent apoptosis were observed in KKU-055 (Fig. S2A and B). Increased cleaved
caspase-8, -9 and -3 levels were observed when cells were treated
with 10-20 nM CMA3, with the highest levels observed following 12 h
of 20 nM CMA3 treatment.
CMA3 downregulates Sp1-related
anti-apoptotic proteins in KKU-213 and KKU-055
CMA3 is a structural analog of MTA. GC-rich,
Sp1/Sp3-specific binding of MTA and CMA3 are established in neurons
(20). Moreover, the roles of MTA
on inhibition of Sp1 binding to the XIAP promoter and
caspase-dependent apoptosis induction have been demonstrated in
various cancer cells (17).
Functions of CMA3 on Sp1-related anti-apoptotic protein expression
were of interest. Sp1-related anti-apoptotic proteins including
FLIP, cIAP2, Mcl-1, survivin and XIAP were selected (13,39,40). The results showed
FLIPs, cIAP2, Mcl-1, survivin and XIAP were reduced in
KKU-213 cells treated with 40 nM CMA3 (Fig. 4A). CMA3 treatment had no effects
on either Sp1 or Sp1-related Bcl-2 family pro-apoptotic proteins,
e.g., Bcl-2-associated X-protein, Bax, Bcl-2-like protein 11, Bim
and Noxa (Fig. 4B and 4C). The quantitative analysis of Sp1,
Sp1-related proteins was reported in Fig. 4D. Similar effects of
CMA3-suppressed Sp1-related anti-apoptotic proteins were observed
in KKU-055 when treated with 20 nM CMA3 (Fig. S2C). The inhibitory effects of
CMA3 and MTA on Mcl-1 and XIAP expression were
confirmed by RT-PCR. KKU-213 cells were treated with 40 nM CMA3 or
200 nM MTA for the indicated times and then the expression levels
of Mcl-1 and XIAP were measured. The results showed
CMA3 and MTA efficiently inhibited Mcl-1 and XIAP
expression and the reductions of expression were observed at 6 h
onward. Moreover, CMA3 possessed a stronger effect on Mcl-1
expression (Fig. S3).
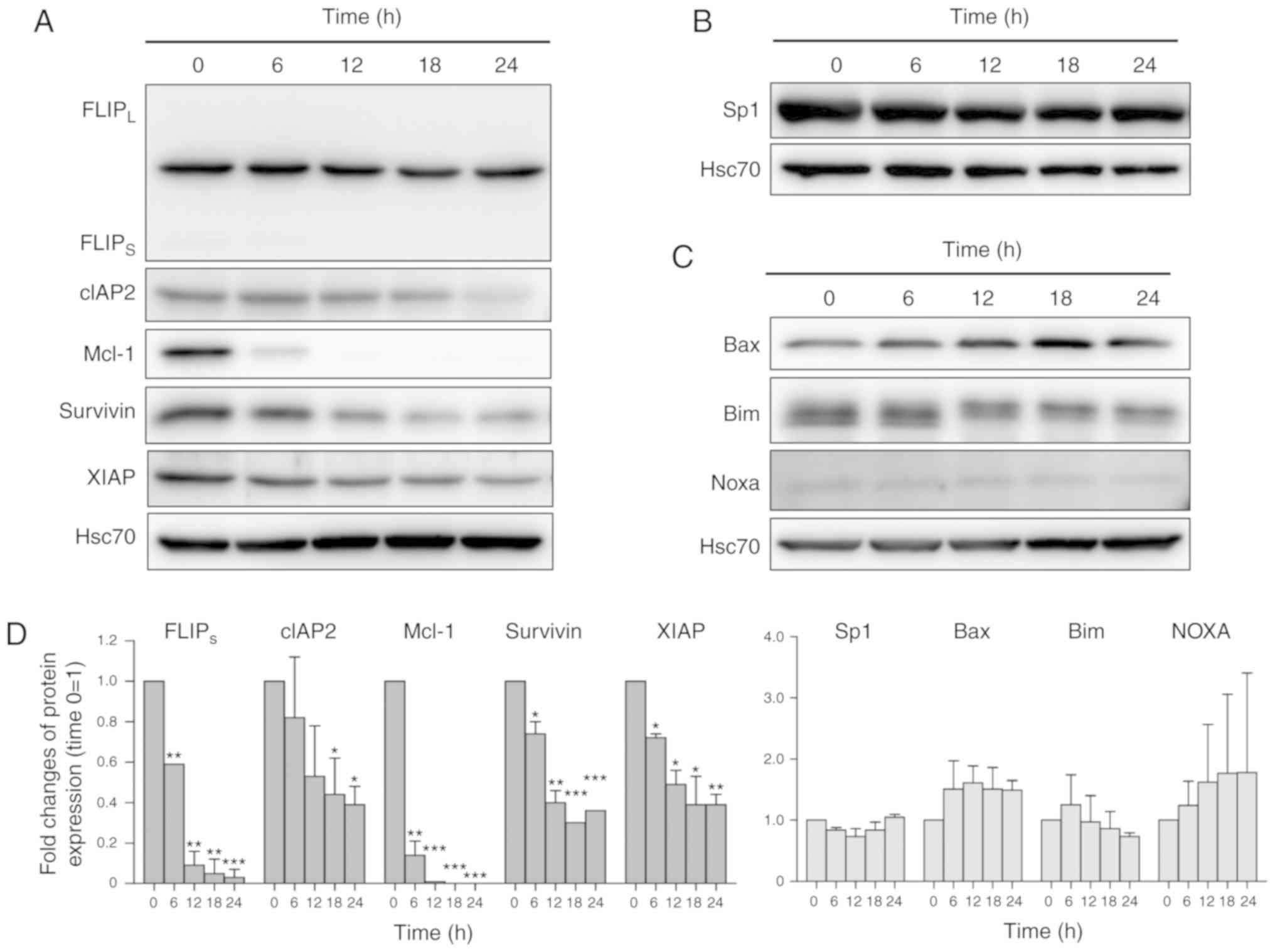 | Figure 4CMA3 induces apoptosis in CCA cells
via downregulation of Sp1-related anti-apoptotic proteins. KKU-213
was treated with 40 nM CMA3 for 0-24 h. (A) The expression of
Sp1-related anti-apoptotic proteins: FLIPs, cIAP2, Mcl-1, survivin
and XIAP was demonstrated by western blotting. (B) The expression
of Sp1 transcription factor and (C) pro-apoptotic proteins; Bax,
Bim and Noxa, is revealed. Intensities of protein bands were
normalized with Hsc70 and compared relative to those without CMA3
(0 h=1). (D) Normalized intensities of Sp1 and Sp1-related protein
expression from three independent experiments were shown. For
FLIPS/L, only the FLIPS isoform was
quantitated. CMA3, Chromomycin A3; CCA, cholangiocarcinoma; Sp1,
specificity protein 1; FLIP, FADD-like IL-1β-converting
enzyme-inhibitory protein; cIAP2, cellular inhibitor of apoptosis;
XIAP, X-linked inhibitor of apoptosis protein; Mcl-1, myeloid cell
leukemia-1. *P<0.05, **P<0.01,
***P<0.001 vs. 0 h. |
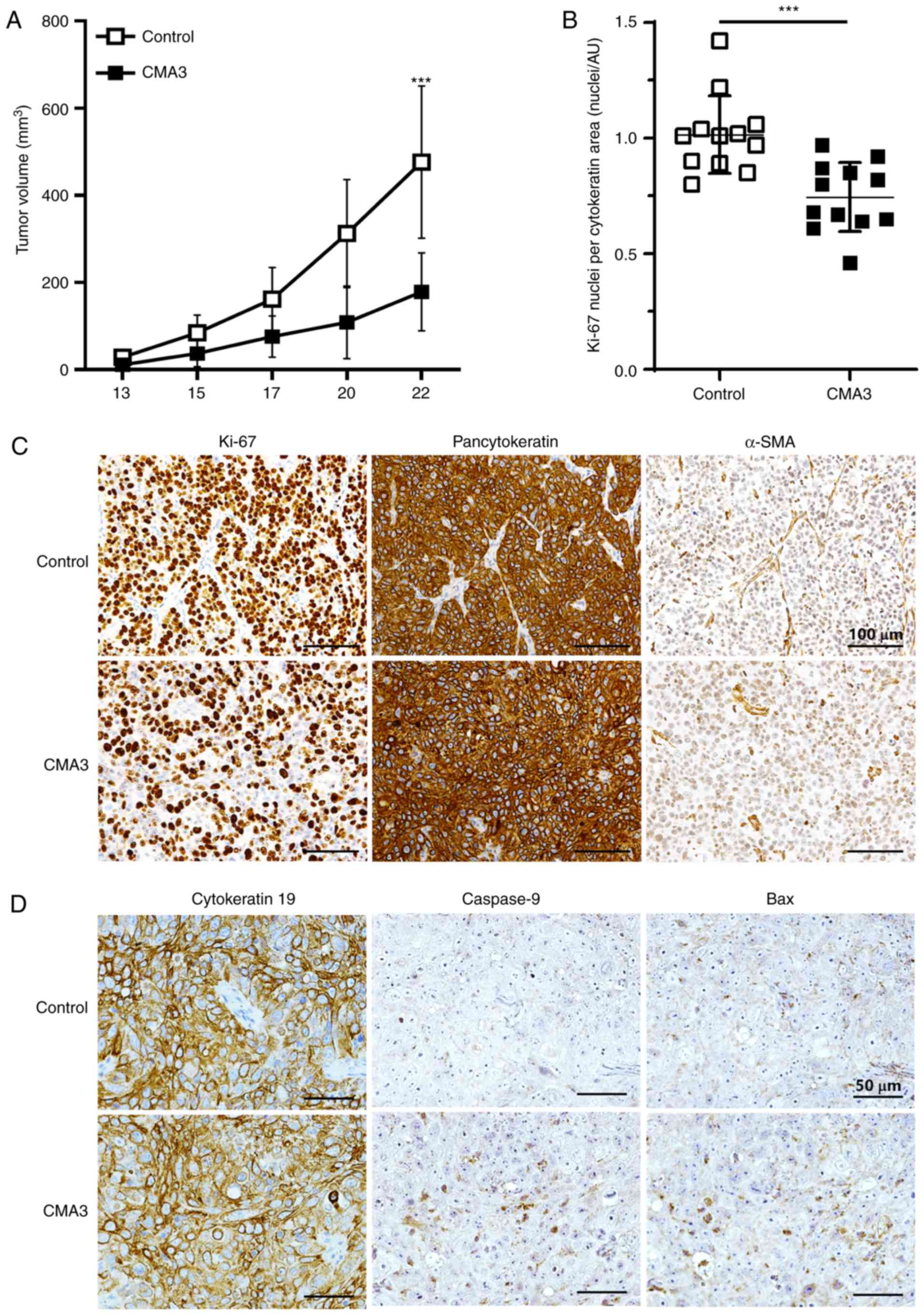
CMA3 suppresses CCA growth and induces
apoptosis in the xenograft mouse model
To determine the therapeutic potential of CMA3 on
CCA in vivo, KKU-213 cells were injected subcutaneously into
flanks of Balb/c RJ mice. Mice were randomly assigned to the
control and CMA3-treated groups and the treatments were given for 3
weeks. No noticeable toxicity of CMA3 was observed during the
experiment (Fig. S4C), which
corresponded with the results of the CMA3 toxicity test (Fig. S5). Tumor volumes were measured.
The results demonstrated that on day 22, the tumor volumes of the
treatment group were significantly smaller than those of the
control group (P<0.001; 178.3±89.4 vs. 476.0±174.7
mm3; Fig. 5A). All
mice were subsequently euthanized and xenograft tumors were
weighed. The average tumor weights of the CMA3-treated group were
significantly decreased compared with control (P<0.05; 0.34±0.22
vs. 0.56±0.20 g; Fig. S4A and
B). Moreover, when tumors were removed, they were then examined
for the densities of cancer and stromal cell compartments. Ki-67
was used as proliferative marker, while pancytokeratin and α-SMA
were used as epithelial (CCA) and active stromal markers. Ki-67
nuclei/cytokeratin-positive areas were assessed and compared
between groups. The results demonstrated that the
Ki-67/cytokeratin-positive areas were smaller in the CMA3-treated
group when compared with the control group [P<0.001; 1.02±0.17
vs. 0.75±0.15 nuclei/arbitrary units (AU); Fig. 5B]. Representative images of Ki-67,
pancytokeratin and α-SMA staining are shown in Fig. 5C.
The effects of CMA3 on the apoptotic proteins,
caspase-9 and Bax, were determined in xenograft tumors. The results
revealed that CMA3 treatment potentiated caspase-9 and Bax
expression (Fig. 5D). The
expression of caspase-9 and Bax in the CMA3-treated group were
significantly increased compared with DMSO-treated control;
caspase-9 expression were 3.25±1.10 vs. 2.14±0.76 in control and
Bax were 3.60±0.95 vs. 2.86±1.16 in control (P<0.05 and
P<0.001; AU/cells/field; Fig.
S6).
Discussion
CCA is an aggressive cancer, in which patients are
often diagnosed at the advanced stages and curative surgery is not
an option (4). Several
chemotherapies have been tried to improve the clinical outcome
(41). Nevertheless, the
efficacies of the treatments are restricted by CCA chemoresistance
(5). Increased expression of
multiple anti-apoptotic proteins including Bcl-2, Bcl-xL, FLIP,
cIAP, Mcl-1, survivin and XIAP have been reported accompanied by
acquired chemoresistance (8-12,42). The potential role of
anti-apoptotic protein modulation on CCA treatment was demonstrated
in the present study using CMA3 as a candidate. CMA3 effectively
inhibited CCA cell proliferation in the nanomolar ranges and the
effects were stronger than the prototype drug, MTA. The DNA binding
property of CMA3 (19)
contributed to cell cycle arrest in the S phase in doses lower than
IC50. That CMA3 promoted CCA apoptosis was demonstrated.
This was partially proven through Sp1-related anti-apoptotic
protein suppression, e.g., FLIP, cIAP2 Mcl-1, survivin and XIAP,
and caspase activation. The benefit of CMA3 treatment on CCA was
established in a xenograft model. Suppression of CCA growth was
demonstrated histologically and grossly. To the best of the
authors' knowledge, this is the first study to demonstrate
Sp1-related anti-apoptotic protein suppression leading to apoptotic
induction and suppression of tumor growth by CMA3.
The anti-cancer effects of CMA3 were previously
reported in cancer (23,24,27). CMA3 has been tried in a phase 1
clinical trial in patients with breast cancer, Hodgkin's disease,
lung adenocarcinoma, melanoma and rhabdomyosarcoma but the results
were inconsistent in tolerable doses and side effects (23,24). The suspected toxicity might have
led to 2 patient deaths (24),
however, it has been put to an alternative use as DNA staining dye
(25). Currently, the repurposing
program in oncology has provided an opportunity for previously
tested agents such as CMA3 (43).
The anti-proliferation of CMA3 was previously reported in cervical
cancer cells, ME180 and HeLa (27). It is worth mentioning that the
stronger effects of CMA3 were observed in the current study. The
IC50s in cervical cancer cells were 4-10 times higher.
The differences could be affected by the different assessments; ATP
quantitation, as reported by Miller et al (27) and mitochondrial enzyme activity in
the present study. This requires further attention and was beyond
the scope of this study.
The roles of CMA3 in CCA inhibition have been
explored. CCA cells treated with CMA3 were accumulated at the S
phase of the cell cycle. The effects of CMA3 on the cell cycle have
never been reported to the best of our knowledge. There is a report
that chromomycin A2, a CMA3 analog, inhibits the melanoma cell
cycle at the G0/G1 phase (44).
This effect was related to the induction of cyclin D1 and D3 and
the suppression of cyclin A2 and B1. By comparison, no significant
alteration was observed in CMA3 treated melanoma. The discrepancy
of CMA analogs on the cell cycle might be due to the DNA binding
specificities (20). The
functions of CMA3 in cell cycle control in cancer require further
investigation. CMA3-induced CCA apoptosis was demonstrated in the
present study. CMA3 induced phosphatidylserine exposure and
caspase-8, -9, and -3 activations. CMA3-promoted caspase-3/7
activation was previously demonstrated in cervical cancer (27). CMA3 activated caspase-8, -9 and
-3-related apoptotic induction was observed in MTA-treated cervical
cancer (18). Activations of both
extrinsic and intrinsic apoptotic pathways were first demonstrated
in the current study, to the best of our knowledge.
Mechanisms in which CMA3-induced apoptosis were
proposed to be Sp1-related based on the mechanism of the
structurally related agent, MTA (17,18). Sp1-related anti-apoptotic
proteins; FLIP, cIAP2, Mcl-1, survivin and XIAP, were selected
candidates (13,39,40). The results showed CMA3 suppressed
certain Sp1-related anti-apoptotic proteins. These alterations were
not observed in Sp1 and Sp1-related pro-apoptotic proteins; Bax,
Bim, and Noxa. The inhibitory effects of CMA3 and MTA on
Sp1-related anti-apoptotic gene expression were confirmed by
determination of Mcl-1 and XIAP mRNA expression in
CMA3- or MTA-treated KKU-213. The results corresponded to the
previous report that CMA3 possesses stronger DNA binding activity
than MTA (19,20). The potent inhibition was observed
on Mcl-1 expression. It is worth mentioning that the
expression of XIAP in CMA3-treated KKU-213 rebounded to the
original state at 24 h. The differential effects of CMA3 on
Mcl-1 and XIAP promoter bindings and expression
require further investigation. The effects of MTA on XIAP
suppression were demonstrated in renal carcinoma (Caki), colon
cancer (HT29), breast cancer (MDA321), prostate cancer (PC3) and
astroglioma (U87) (17). The
similar effects of MTA and CMA3 on XIAP expression were proposed
through the inhibition of Sp1 binding on the promoter. The role of
MTA on Sp1 expression at mRNA and protein levels were previously
reported in cervical cancer cells, HEp-2 and KB, (18) but it was not observed in the
current study. This might be due to the higher concentrations
(IC70-IC90 vs. IC60 in this study)
and the longer treatments (48 vs. 24 h in this study) than in the
other studies. Longer treatments with higher drug concentrations
might possess a stronger effect on Sp1 suppression.
Anti-CCA effects of CMA3 were supported by the
xenograft model. Treatment with intravenous CMA3 to CCA-bearing
mice inhibited tumor growth by inhibition of cell proliferation.
Lower densities of Ki-67-positive CCA cells were observed in the
treatment group. It is worth mentioning that larger nuclei were
observed in the treatment group consistent with S phase arrest
observed in the cell cycle analysis. The growth inhibitory effects
of CMA3 in vivo were partly due to apoptosis induction.
Increased caspase-9 and Bax expression in CMA3-treated tumors was
demonstrated in the present study. Only 3 doses of CMA3 were
administered to mice and no observable toxicity was noticed.
Therefore, these results confirmed the potent anti-CCA effect of
CMA3. The toxicity of CMA3 is of concern. From in vivo
toxicity testing in the current study, no obvious effects were
observed in mouse general well-being and body weights. In
vitro toxicity testing using peripheral blood mononuclear cells
or other normal cell types and the comparative study of CMA3 and
MTA effects in vivo are required prior to a trial in
patients. It is not possible in the current study due to limited
resources.
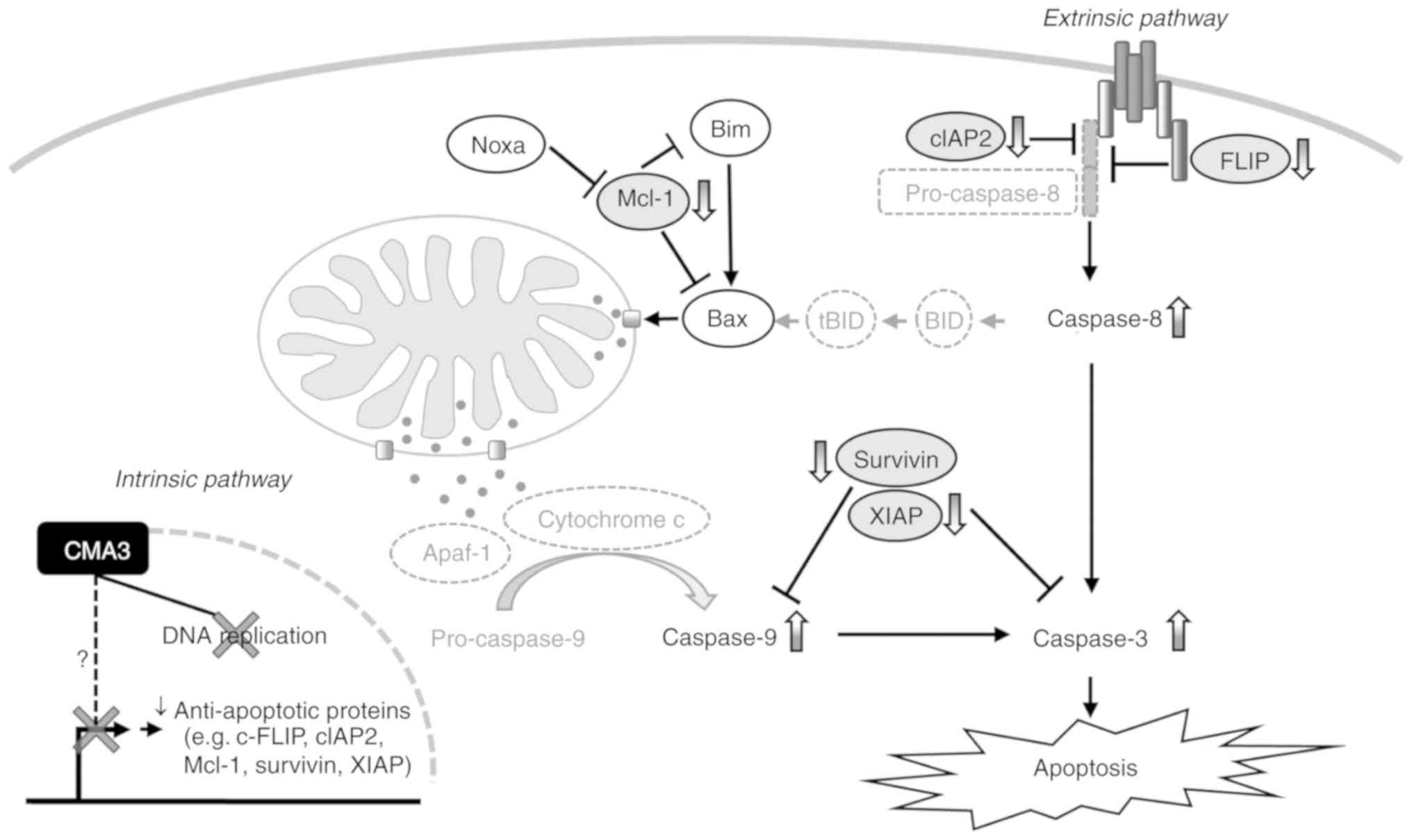
The anti-cancer effect of CMA3 on CCA is summarized
in Fig. 6. The proposed mechanism
of how CMA3 inhibited CCA was through selective DNA binding, which
resulted in 2 outcomes; i) inhibition of DNA replication, which
later causes S phase arrest; ii) reductions of Sp1-related
anti-apoptotic proteins (e.g., FLIP, cIAP2, Mcl-1, survivin and
XIAP), which cause an imbalance between pro-apoptotic and
anti-apoptotic signals, and subsequently promotes caspase
activation and apoptotic induction. No significant alterations of
Sp1 and Sp1-related pro-apoptotic proteins (e.g., Bax, Bim and
Noxa) were observed in the current study. Altogether, the
underlying mechanism in which CMA3-induced CCA apoptosis through
alleviation of Sp1-related anti-apoptotic proteins was demonstrated
for the first time in the current study. The mechanistic studies
using an Sp1 reporter assay or immunoprecipitation might directly
establish the effects of CMA3 and require further investigations.
This may attenuate the chemoresistance of CCA to currently
available chemotherapeutic drugs. The adjunct treatment with CMA3
or CMA3 derivatives might be a novel strategy for CCA
treatment.
Supplementary Data
Funding
The present study was supported in part by the
Faculty of Medicine Khon Kaen University, Thailand (grant no.
IN61304 to PS and KV), Khon Kaen University, Thailand (grant no.
6200020001 to KV) and Grant-in-Aid for Scientific Research from the
Ministry of Education, Culture, Sport Science and Technology (MEXT)
of Japan (grant no. 16K08742 to SO). PS was supported by a
scholarship from the Graduate School, Khon Kaen University (grant
no. 591H109) and by the student exchange support from the Japan
Student Services Organization, Japan and the Study and Research in
Abroad Scholarship of Khon Kaen University.
Availability of data and materials
All data generated or analyzed during this study are
included in this published article.
Authors' contributions
PS, SO and KV made substantial contributions to the
conception and design of the study, acquisition of data, and
analysis of data. RK, GS, PB and TB performed experiments and
interpreted data. KS, CW and SW contributed to the conception of
the study and experimental design. PS, SO and KV drafted the
article and critically reviewed it for the intellectual content.
All authors approved the final version of the article. All authors
agree to be held accountable for all aspects of the work in
ensuring that questions related to the accuracy or integrity of the
work are appropriately investigated and resolved.
Ethics approval and consent to
participate
The study protocol was reviewed and approved by the
Ethical Committee for the Human Research of Khon Kaen University
(policy no. HE611034), based on the Declaration of Helsinki of
1975. All animal protocols were approved by the Institutional
Animal Care and Use Committee of Kumamoto University (policy no. A
29-046), and conducted according to the Fundamental Guidelines for
Proper Conduct of Animal Experiment and Related Activities in
Academic Research Institutions under the jurisdiction of the
Ministry of Education, Culture, Sports, Science and Technology,
Japan.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
The authors would like to thank Ms Sawako Fujikawa
and and Ms Yoshie Kanagawa (Division of Hematopoiesis, Joint
Research Center for Human Retrovirus Infection, Kumamoto
University) for their technical assistance and secretarial
assistance, respectively.
References
|
1
|
Banales JM, Cardinale V, Carpino G,
Marzioni M, Andersen JB, Invernizzi P, Lind GE, Folseraas T, Forbes
SJ, Fouassier L, et al: Expert consensus document:
Cholangiocarcinoma: Current knowledge and future perspectives
consensus statement from the European network for the study of
cholangiocarcinoma (ENS-CCA). Nat Rev Gastroenterol Hepatol.
13:261–280. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sripa B, Bethony JM, Sithithaworn P,
Kaewkes S, Mairiang E, Loukas A, Mulvenna J, Laha T, Hotez PJ,
Brindley PJ, et al: Opisthorchiasis and opisthorchis-associated
cholangiocarcinoma in Thailand and Laos. Acta Trop. 120(Suppl 1):
S158–S168. 2011. View Article : Google Scholar
|
|
3
|
Khan SA, Taylor-Robinson SD, Toledano MB,
Beck A, Elliott P and Thomas HC: Changing international trends in
mortality rates for liver, biliary and pancreatic tumours. J
Hepatol. 37:806–813. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bridgewater J, Galle PR, Khan SA, Llovet
JM, Park JW, Patel T, Pawlik TM and Gores GJ: Guidelines for the
diagnosis and management of intrahepatic cholangiocarcinoma. J
Hepatol. 60:1268–1289. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Marin JJ, Romero MR and Briz O: Molecular
bases of liver cancer refractoriness to pharmacological treatment.
Curr Med Chem. 17:709–740. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Dhanasekaran R, Hemming AW, Zendejas I,
George T, Nelson DR, Soldevila-Pico C, Firpi RJ, Morelli G, Clark V
and Cabrera R: Treatment outcomes and prognostic factors of
intra-hepatic cholangiocarcinoma. Oncol Rep. 29:1259–1267. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Srimunta U, Sawanyawisuth K, Kraiklang R,
Pairojkul C, Puapairoj A, Titipungul T, Hahnvajanawong C,
Tassaneeyakul W, Wongkham C, Wongkham S and Vaeteewoottacharn K:
High expression of ABCC1 indicates poor prognosis in intrahepatic
chol-angiocarcinoma. Asian Pac J Cancer Prev. 13(Suppl): S125–S130.
2012.
|
|
8
|
Prakobwong S, Gupta SC, Kim JH, Sung B,
Pinlaor P, Hiraku Y, Wongkham S, Sripa B, Pinlaor S and Aggarwal
BB: Curcumin suppresses proliferation and induces apoptosis in
human biliary cancer cells through modulation of multiple cell
signaling pathways. Carcinogenesis. 32:1372–1380. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pawar P, Ma L, Byon CH, Liu H, Ahn EY,
Jhala N, Arnoletti JP, McDonald JM and Chen Y: Molecular mechanisms
of tamoxifen therapy for cholangiocarcinoma: Role of calmodulin.
Clin Cancer Res. 15:1288–1296. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Taniai M, Grambihler A, Higuchi H,
Werneburg N, Bronk SF, Farrugia DJ, Kaufmann SH and Gores GJ: Mcl-1
mediates tumor necrosis factor-related apoptosis-inducing ligand
resistance in human cholangiocarcinoma cells. Cancer Res.
64:3517–3524. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Yamagiwa Y, Marienfeld C, Meng F, Holcik M
and Patel T: Translational regulation of X-linked inhibitor of
apoptosis protein by interleukin-6: A novel mechanism of tumor cell
survival. Cancer Res. 64:1293–1298. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chang Q, Liu ZR, Wang DY, Kumar M, Chen YB
and Qin RY: Survivin expression induced by doxorubicin in
cholangiocar-cinoma. World J Gastroenterol. 10:415–418. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Beishline K and Azizkhan-Clifford J: Sp1
and the 'hallmarks of cancer'. FEBS J. 282:224–258. 2015.
View Article : Google Scholar
|
|
14
|
Wang L, Wei D, Huang S, Peng Z, Le X, Wu
TT, Yao J, Ajani J and Xie K: Transcription factor Sp1 expression
is a significant predictor of survival in human gastric cancer.
Clin Cancer Res. 9:6371–6380. 2003.PubMed/NCBI
|
|
15
|
Jiang NY, Woda BA, Banner BF, Whalen GF,
Dresser KA and Lu D: Sp1, a new biomarker that identifies a subset
of aggressive pancreatic ductal adenocarcinoma. Cancer Epidemiol
Biomarkers Prev. 17:1648–1652. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vizcaino C, Mansilla S and Portugal J: Sp1
transcription factor: A long-standing target in cancer
chemotherapy. Pharmacol Ther. 152:111–124. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee TJ, Jung EM, Lee JT, Kim S, Park JW,
Choi KS and Kwon TK: Mithramycin A sensitizes cancer cells to
TRAIL-mediated apoptosis by down-regulation of XIAP gene promoter
through Sp1 sites. Mol Cancer Ther. 5:2737–2746. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choi ES, Nam JS, Jung JY, Cho NP and Cho
SD: Modulation of specificity protein 1 by mithramycin A as a novel
therapeutic strategy for cervical cancer. Sci Rep. 4:71622014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Barcelo F, Ortiz-Lombardia M, Martorell M,
Oliver M, Méndez C, Salas JA and Portugal J: DNA binding
characteristics of mithramycin and chromomycin analogues obtained
by combinatorial biosynthesis. Biochemistry. 49:10543–10552. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chatterjee S, Zaman K, Ryu H, Conforto A
and Ratan RR: Sequence-selective DNA binding drugs mithramycin A
and chromomycin A3 are potent inhibitors of neuronal apoptosis
induced by oxidative stress and DNA damage in cortical neurons. Ann
Neurol. 49:345–354. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
21
|
Gause GF: Olivomycin, mithramycin,
chromomycin: Three related cancerostatic antibiotics. Adv
Chemother. 2:179–195. 1965. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Schmitz H, Heinemann B, Lein J and Hooper
IR: NSC A-649, an antitumor antibiotic. Antibiot Chemother
(Northfield). 10:740–746. 1960.
|
|
23
|
Reynolds RD, Fisher JI, Jensen PA, Pajak
TF and Bateman JR: Phase I alternate-day dose study of chromomycin
A3. Cancer Treat Rep. 60:1251–1255. 1976.PubMed/NCBI
|
|
24
|
Samal B, Jones S, Brownlee RW, Morrison F,
Hoogstraten B, Caoili E and Baker L: Chromomycin A3 for advanced
breast cancer: A Southwest oncology group study. Cancer Treat Rep.
62:19–22. 1978.PubMed/NCBI
|
|
25
|
Iranpour FG, Nasr-Esfahani MH, Valojerdi
MR and al-Taraihi TM: Chromomycin A3 staining as a useful tool for
evaluation of male fertility. J Assist Reprod Genet. 17:60–66.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boer DR, Canals A and Coll M: DNA-binding
drugs caught in action: The latest 3D pictures of drug-DNA
complexes. Dalton Trans. 399–414. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miller SC, Huang R, Sakamuru S, Shukla SJ,
Attene-Ramos MS, Shinn P, Van Leer D, Leister W, Austin CP and Xia
M: Identification of known drugs that act as inhibitors of
NF-kappaB signaling and their mechanism of action. Biochem
Pharmacol. 79:1272–1280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sripa B, Leungwattanawanit S, Nitta T,
Wongkham C, Bhudhisawasdi V, Puapairoj A, Sripa C and Miwa M:
Establishment and characterization of an opisthorchiasis-associated
cholan-giocarcinoma cell line (KKU-100). World J Gastroenterol.
11:3392–3397. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Saisomboon S, Kariya R, Vaeteewoottacharn
K, Wongkham S, Sawanyawisuth K and Okada S: Antitumor effects of
flavopiridol, a cyclin-dependent kinase inhibitor, on human
cholangiocar-cinoma in vitro and in an in vivo xenograft model.
Heliyon. 5:e016752019. View Article : Google Scholar
|
|
30
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Phoomak C, Silsirivanit A, Park D,
Sawanyawisuth K, Vaeteewoottacharn K, Wongkham C, Lam EW, Pairojkul
C, Lebrilla CB and Wongkham S: O-GlcNAcylation mediates metastasis
of cholangiocarcinoma through FOXO3 and MAN1A1. Oncogene.
37:5648–5665. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Cerella C, Muller F, Gaigneaux A, Radogna
F, Viry E, Chateauvieux S, Dicato M and Diederich M: Early
downregulation of Mcl-1 regulates apoptosis triggered by cardiac
glycoside UNBS1450. Cell Death Dis. 6:e17822015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cai J, Wang D, Bai ZG, Yin J, Zhang J and
Zhang ZT: The long noncoding RNA XIAP-AS1 promotes XIAP
transcription by XIAP-AS1 interacting with Sp1 in gastric cancer
cells. PLoS One. 12:e01824332017. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Obchoei S, Weakley SM, Wongkham S,
Wongkham C, Sawanyawisuth K, Yao Q and Chen C: Cyclophilin A
enhances cell proliferation and tumor growth of liver
fluke-associated cholangiocarcinoma. Mol Cancer. 10:1022011.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ono A, Hattori S, Kariya R, Iwanaga S,
Taura M, Harada H, Suzu S and Okada S: Comparative study of human
hematopoietic cell engraftment into BALB/c and C57BL/6 strain of
rag-2/jak3 double-deficient mice. J Biomed Biotechnol.
2011:5397482011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Harlow E and Lane D: Preparing paraffin
tissue sections for immunostaining. CSH Protoc.
2006:pdb.prot43292006.PubMed/NCBI
|
|
37
|
Vaeteewoottacharn K, Kariya R, Dana P,
Fujikawa S, Matsuda K, Ohkuma K, Kudo E, Kraiklang R, Wongkham C,
Wongkham S and Okada S: Inhibition of carbonic anhydrase
potentiates bevacizumab treatment in cholangiocarcinoma. Tumour
Biol. 37:9023–9035. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Parrish AB, Freel CD and Kornbluth S:
Cellular mechanisms controlling caspase activation and function.
Cold Spring Harb Perspect Biol. 5:a0086722013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bedolla RG, Gong J, Prihoda TJ, Yeh IT,
Thompson IM, Ghosh R and Kumar AP: Predictive value of Sp1/Sp3/FLIP
signature for prostate cancer recurrence. PLoS One. 7:e449172012.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lau R, Niu MY and Pratt MA: cIAP2
represses IKKα/β-mediated activation of MDM2 to prevent p53
degradation. Cell Cycle. 11:4009–4019. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Patel T: Cholangiocarcinoma-controversies
and challenges. Nat Rev Gastroenterol Hepatol. 8:189–200. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Kobayashi S, Werneburg NW, Bronk SF,
Kaufmann SH and Gores GJ: Interleukin-6 contributes to Mcl-1
up-regulation and TRAIL resistance via an Akt-signaling pathway in
cholangiocarcinoma cells. Gastroenterology. 128:2054–2065. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sleire L, Forde HE, Netland IA, Leiss L,
Skeie BS and Enger PO: Drug repurposing in cancer. Pharmacol Res.
124:74–91. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Guimaraes LA, Jimenez PC, Sousa Tda S,
Freitas HP, Rocha DD, Wilke DV, Martín J, Reyes F, Deusdênia Loiola
Pessoa O and Costa-Lotufo LV: Chromomycin A2 induces autophagy in
melanoma cells. Mar Drugs. 12:5839–5855. 2014. View Article : Google Scholar : PubMed/NCBI
|