Introduction
As a common type of inflammatory arthritis, gout is
typically characterized by an elevated concentration of serum uric
acid (hyperuricemia) and a subsequent deposition of monosodium
urate (MSU) crystals in and around joints (1-3).
The prevalence of gout in developed countries has been reported to
be 1-3.9% (1-3). In addition, due to changes in
dietary habits and an increasing aging population, the prevalence
of gout has increased over past decades in China (4,5).
Patients with gout often present with metabolic diseases, including
diabetes mellitus, hypertriglyceridemia and obesity, in addition to
hypertension (6). In total, 16%
of patients with gout exhibit ischemic heart disease and metabolic
syndrome (7). Insulin resistance
serves a key function in the pathogenesis of metabolic syndrome.
Hyperuricemia in patients with metabolic syndrome is likely caused
by insulin resistance, since insulin promotes uric acid and sodium
reabsorption in the proximal tubule (8,9).
Gout is etiologically heterogeneous as environmental
and genetic components are involved (1-3).
Although an unhealthy diet and lifestyle are risk factors for
hyperuricemia, genetic factors were revealed to serve a greater
function in the development of gout compared with environmental
factors (10-12). Various previous genome-wide
association studies (GWASs) have improved the understanding of the
genes that regulate serum uric acid levels and increase
susceptibility to gout (13-15). Among the loci revealed to be
associated with gout, solute carrier family 2 member 9 and
ATP-binding cassette super-family G member 2 (ABCG2) are the
two most important genes identified to serve a function in gout
development (10-12). Two important pathways, including
renal and gut excretion of uric acid, have been revealed to
regulate hyperuricemia levels, and each of these pathways are
associated with glycolysis (16).
However, previous studies based on GWAS research in independent
individuals were only able to explain ~7% of the variance in serum
urate concentrations, and only a portion of those loci were
confirmed to be associated with the risk of gout (17,18).
Gout is heritable and has tendency to cluster in
families (12,19,20). In total, ~20% of patients with
gout were reported to have a family history of this disease. The
risk of gout is significantly higher in patients who have
first-degree relatives affected by this disease compared with the
normal population (19). A
previous segregation and linkage analysis of familial gout revealed
an autosomal-arbitrary major gene model, which indicated a genetic
basis for familial gout (20). A
previous population-based study was performed in a Taiwanese
population to estimate the degree of familial aggregation of gout,
and it was revealed that genetic factors serve a substantial
function in the development of gout (19).
The present study aimed to identify the potential
pathogenic genetic causes of 3 pedigrees with a familial
aggregation of gout using whole-exome sequencing (WES) technology
and diverse bioinformatics analysis, including genetic interaction
networks, disease ontology (DO) and gene ontology analysis.
Materials and methods
Patient recruitment
Patients (n=10) who were attending a clinic for
arthritis were recruited between July 2016 and June 2017.
Additionally, three families were recruited in the present study
as: i) Presenting with autosomal dominant inheritance and ii)
healthy family members agreed to participation in the present
study. Patient information is summarised in Table I. The present study was conducted
according to the principles outlined in the Declaration of
Helsinki. The study protocol was approved by the Ethics Committee
of The First Affiliated Hospital, Wenzhou Medical University
(Wenzhou, China; approval no. 2018-020). Written informed consent
was obtained from all participants or their guardians. Patients
with gout were clinically evaluated by physicians based on the 2015
gout classification criteria by the American College of
Rheumatology/European League Against Rheumatism Collaborative
Initiative (21). Patients with
inherited metabolic disorders, including Lesch-Nyhan syndrome, were
excluded from the present study. Autoantibodies and human leukocyte
antigen B27 (HLA-B27) tests were negative in all patients and X-ray
analyses of the affected joints were performed to exclude other
potential diseases. The classical symptoms of patients with gout
are characterized by the rapid development of monoarticular
arthritis, which is accompanied by swelling and redness of the
first metatarsophalangeal joint (MTP1).
 | Table IClinical data of patients with gout
in three large pedigrees. |
Table I
Clinical data of patients with gout
in three large pedigrees.
| Individual ID | Sex | Age (years) | Age at onset
(years) | Uric acid
(mg/l) | Hyperuricemia
(+/−) | Arthritis | Tophi | Comorbidities
|
|---|
| Hypertension |
Hyperlipidaemia | Hyperglycaemia | Obesity (BMI) |
Hyperbilirubinaemia |
|---|
| F1_I:1 | M | 48 | 30 | 579 | + | + | + | + | + | + | + | − |
| F1_II:1 | M | 20 | 15 | 801 | + | + | + | + | + | − | + | − |
| F2_II:1 | M | 67 | 55 | 617 | + | + | + | + | − | − | + | + |
| F2_II:7 | M | 51 | 38 | 580 | + | + | + | + | − | − | + | + |
| F2_II:11 | F | 68 | − | 364 | − | − | − | − | − | − | + | − |
| F2_III:1 | M | 40 | 35 | 499 | + | + | + | + | − | − | − | + |
| F2_III:9 | M | 34 | 28 | 610 | + | + | − | − | − | − | + | + |
| F3_II:2 | F | 78 | 70 | 647 | + | + | + | + | + | − | − | + |
| F3_III:2 | F | 52 | 52 | 474 | + | + | − | + | + | − | + | + |
| F3_III:3 | F | 62 | − | 463 | + | − | − | + | + | − | + | + |
WES and variant calling
The human genome hg19 was used as the reference
genome. Genomic DNA was extracted from peripheral blood leukocytes.
In total, 2 µg genomic DNA from each sample was sheared to
fragment with a length of 150 base pairs (bp) using the Covaris
S220. Subsequently, ligation of small fragments of DNA with A-tail
and adapters was conducted. A genomic library was constructed
subsequent to the amplification of adapter-ligated DNA using an
Agilent SureSelect Library Prep kit (Agilent Technologies, Inc.)
according to the manufacturer's protocol, and the samples from each
individual were marked with a unique index. Whole-exome capture was
performed using the Agilent SureSelect Human All Exon v5 kit
(Agilent Technologies, Inc.) according to the manufacturer's
protocol. High throughput sequencing was performed using an
Illumina HiSeq 4000 sequencer (Illumina, Inc.).
All raw sequencing data obtained from these three
families were processed in a similar manner, according to a
customized bioinformatics pipeline (22). To remove sequence adapters and
low-quality reads, the raw reads were filtered using the FastQC
software program, version 1.11.4 (http://www.bioin-formatics.babraham.ac.uk/projects/fastqc/).
The filtration criteria excluded Phred-scaled quality scores <30
and read lengths <80 bp. Subsequent to removing the low-quality
reads, the remaining reads were used for further analysis. FastQ
reads were aligned to the human reference genome (GRCh37/hg19)
using the Burrows-Wheeler alignment tool (23) and further visualized using
SplicingViewer software (24).
Then, Genome Analysis ToolKit (GATK; version 4.0.10.0; https://gatk.broadinstitute.org/hc/en-us) was used to
remove duplicated reads and reads mapped to multiple genome
locations. In addition, local realignment and map quality score
recalibration were performed. Candidate variants were then
identified using the GATK Unified Genotype (version 4.0.10.0;
https://gatk.broadinstitute.org/hc/en-us).
Variant annotation and
prioritization
mirTrios with an integrated ANNOVAR tool were used
to annotate all the detected mutations according to an in-house
pipeline (25). The minor allele
frequency (MAF) was obtained for each variant from publicly
available databases (25),
including ExAC, UK10K, dbSNP147, 1000 Genomes and ESP6500, and from
in-house exome data. The detected variants with a MAF >0.01%
present in any of the aforementioned databases were removed
(26). Subsequently, the effects
of the detected variants were predicted using four tools: i)
Polymorphism Phenotyping v2 (PolyPhen2) (27); ii) Likelihood Ratio Test (LRT)
(https://evomics.org/resources/likelihood-ratio-test/);
iii) MutationTaster (28); and
iv) Functional Analysis through Hidden Markov Models (FATHMM)
(29). A missense variant was
considered deleterious if the variant was predicted to be
deleterious or damaging by ≥3 of the four genetic prediction tools.
The remaining variants were considered to be high-confidence
causative variants.
Protein structural modeling
The amino acid sequence of human protein kinase
CGMP-dependent 2 (PRKG2) was retrieved from Uniprot (http://www.uniprot.org/uniprot/Q13237).
Crystal structures of the wild-type PRKG2 protein were obtained
from the protein data bank (PDB; http://www.rcsb.org/structure/5BV6) and visualized
using PyMol software (version 1.8) (https://pymol.org/2/). Homology modelling of mutated
PRKG2 protein structures was performed using SWISS-MODEL
(https://swissmodelexpasy.org/.)
(30). In total, 120 residues
were modelled in the PRKG2 structure, including 102 residues in the
cGMP-binding region.
Collection of candidate genes for
hyperuricemia and gout
Various candidate genes for hyperuricemia and gout
were reported in previous studies. The literature search was
performed in June 2017 by searching 'gout' AND 'genes' in PubMed
(https://www.ncbi.nlm.nih.gov/pubmed).
Then all genes in all literature obtained were included. Following
the literature search, the candidate genes associated with
hyperuricemia and gout were collected and are listed in Table SI.
Construction of gene co-expression and
protein-protein interaction (PPI) networks
Temporally rich transcriptome data extracted from
the Genotype-Tissue Expression (GTEx) project (https://gtexportal.org/home/, accessed January 2018)
were used to build the co-expression network. The Pearson
correlation coefficients (r) for the gene co-expression levels were
calculated for each pairwise combination between different genes.
To investigate similarities among the genes forming the PPI network
and analyse their functions, significantly enriched DO terms were
identified using the R package DOSE (version 2.0) and a
hypergeometric test (31). To
assess function similarities between previously reported genes
(Table SI) associated with gout
and the core risk genes identified in the present study, a PPI
network was built. PPI data downloaded from STRING V10 (https://string-db.org) (32) were used for PPI network analysis.
To further investigate the gene functions in the PPI network,
biological processes (BP) analysis was conducted using ClueGO
v2.3.3 (33), a plug-in of
Cytoscape.
Results
Characteristics of the patient
cohort
The patient cohort for the present study was
composed of three families (Figs.
1A, 2A and 3). All patients experienced acute
monoarticular arthritis affecting the MTP1 and/or knee. The
affected joint made walking difficult, was painful to the touch and
was occasionally accompanied by fever. Tophus was observed in some
of the patients (Fig. 1B), and
metabolic disorders, including hypertension, diabetes and
hyperlipemia, were identified. The clinical data are presented in
Table I. The serum urate level
was >480 µmol/l in most cases, and in numerous cases it
was >600 µmol/l. Synovial fluid from a number of the
patients presented MSU crystals and erosions based on conventional
radiography of the affected joint. Autoantibodies and HLA-B27 tests
were negative in all patients (Table
SII). All affected patients were diagnosed with gout.
Detection of candidate deleterious
mutations
To determine the genetic etiology of these families,
WES was performed in the three probands. In total, ~13.53 Gb
high-quality data was obtained on mean subsequent to removing
sequencing adapters and low-quality sequences (Table SIII). For each sample, ≥98.68% of
the high-quality data was matched with the human reference genome
Hg19. Effective sequence on target was >4.91 Gb, with a minimum
of 39.10% fraction of effective bases on target following the
removal of polymerase chain reaction duplications. The mean
sequencing depth for each sample was 110.16-fold, with >99.00%
of target regions being covered at a 4-fold sequencing depth,
98.50% at 10-fold depth and 97.20% at 20-fold depth. Collectively,
the quality control data demonstrated a high reliability, which was
fundamental for the subsequent analyses.
Subsequent to removing low-quality reads, adapters
and duplicated reads from the raw sequencing data, a total of
541,954 single nucleotide variations (SNVs) and 84,415 indels were
identified using the GATK tool, which included 67,707 SNVs and
2,200 indels in exonic and splicing regions. Subsequent to applying
variant filtration against multiple databases, the number of rare
SNVs and indels causing protein change with MAF <0.001 was
reduced to 228 and 29, respectively. As a result, following effect
prediction by Polyphen2, LRT, Mutation Taster and FATHMM, the
variants predicted to be deleterious by >2 prediction tools were
validated by Sanger sequencing. Finally, three SNVs in the coding
regions of three gout-associated genes were retained and confirmed
following Sanger sequencing validation (Table II).
| Table IISummary of mutations detected by
whole-exome sequencing of patients with gout. |
Table II
Summary of mutations detected by
whole-exome sequencing of patients with gout.
| Individual ID | Chromosome | Gene | Position
(hg19) | Type | Protein change | ExAC | Polyphen2 | LRT | MutationTaster | FATHMM |
|---|
| F1:II:1 | Chr4 | ABCG2 | 89052275 | Missense | p.K157E |
8.24×10−6 | Likely
damaging | Deleterious | Disease
causing | Damaging |
| F2:II:1 | Chr4 | PRKG2 | 82090913 | Missense | p.N251S | - | Benign | Deleterious | Disease
causing | Damaging |
| F3:II:1 | Chr8 | ADRB3 | 37823976 | Missense | p.W4C |
6.49×10−5 | Likely
damaging | Neutral | Disease
causing | Tolerable |
Sanger sequencing validation and
co-segregation testing
Analysis of WES data indicated a missense mutation
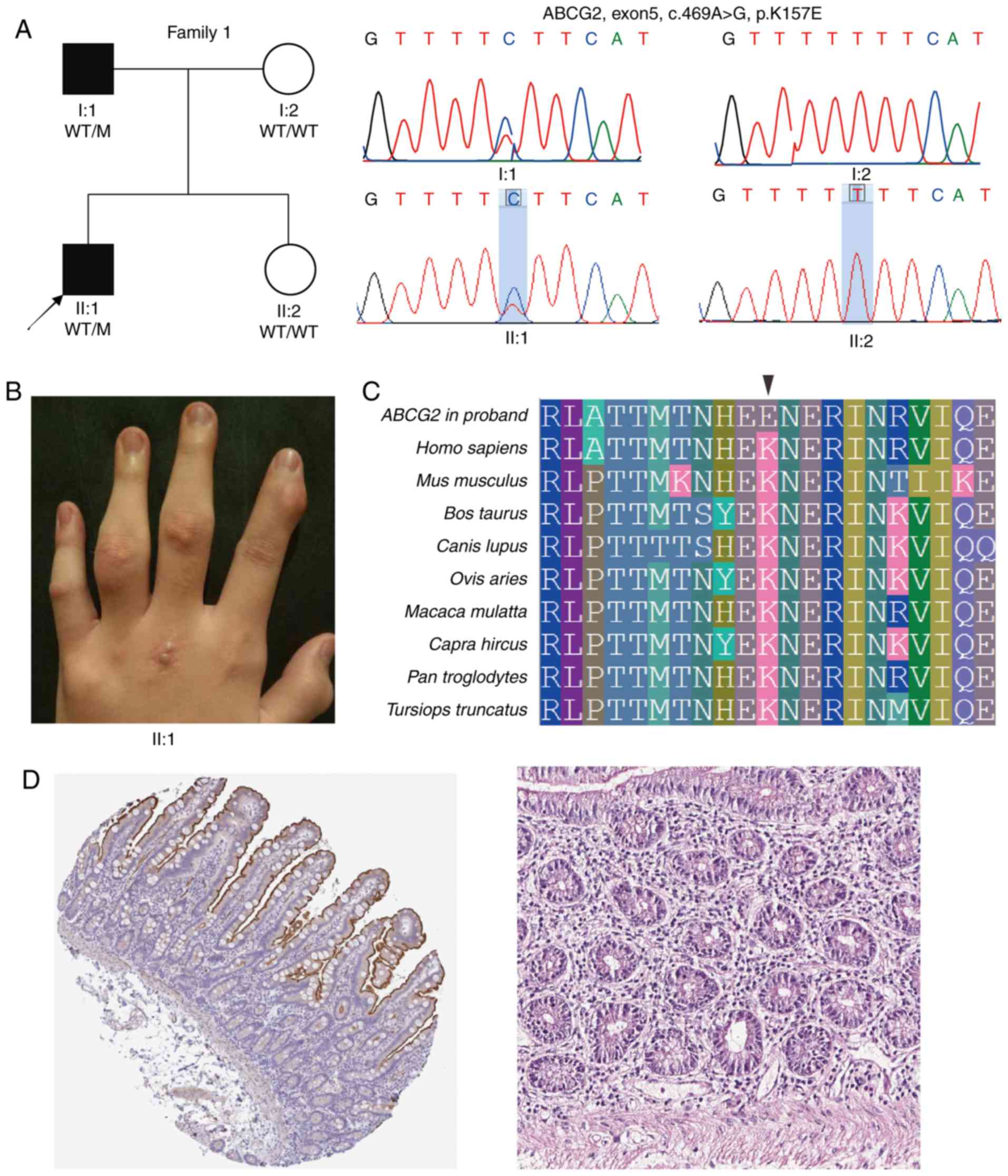
(p.Lys157Glu) in the ABCG2 gene in the proband F1:II:1 of
family 1 presenting typical symptoms of gout (Fig. 1 and Table I). The ABCG2 protein is involved
in the excretion of urate from the intestine and kidney, and its
dysfunction causes extrarenal urate underexcretion type (34) and/or renal urate underexcretion
type gout (35). The heterozygous
missense mutation (c.469A>G) causing a lysine (Lys) to glutamic
acid (Glu) substitution was located at the amino acid 157.
Subsequently, Sanger sequencing confirmed the presence of this
mutation in the patient's affected father while his unaffected
mother and sister did not present the A to G change at cDNA
nucleotide 469 (Fig. 1A). The MAF
of p.Lys157Glu was 8.24×10−6 and was predicted to be
deleterious by all four effect prediction tools (Table II). Additionally, the residue
157Lys is highly conserved among different vertebrate species
(Fig. 1C). Subsequently, the
tissue specificity of the expression of the human ABCG2 was
examined in all major tissues and organs using the Human Protein
Atlas database (https://www.proteinatlas.org). ABCG2 exhibited
the most abundant protein expression in the small intestine
(Fig. 1D). The results reflected
abundant mRNA expression in the luminal membrane of the intestine
with 136.8 transcripts per million (TPM).
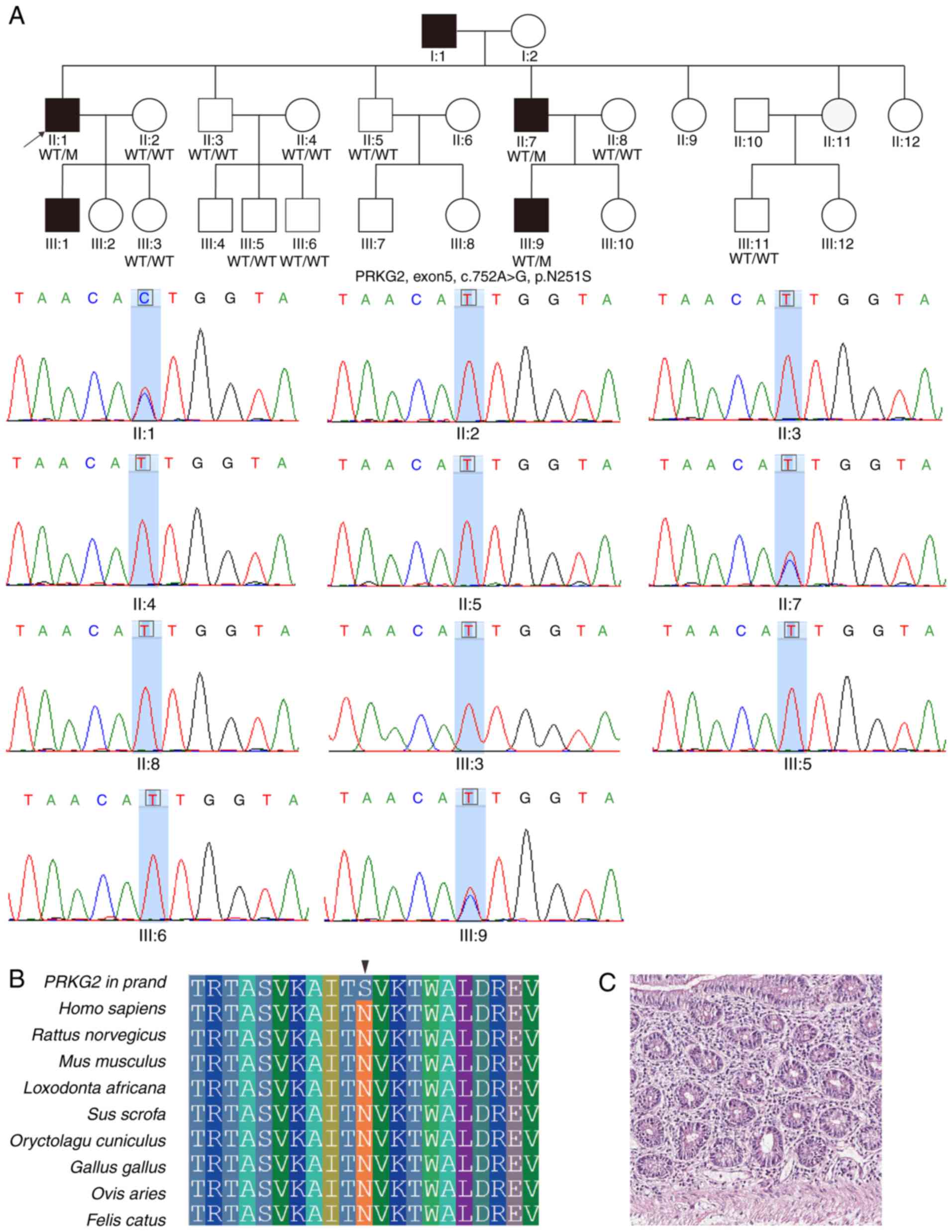
Another heterozygous missense mutation (c.752A>G;
p.Asn251Ser) located in exon 5 of PRKG2 gene was detected in
proband F2:II:1 and his affected brother F2:II:9, as well as in the
patient III9 (Fig. 2A).
cGMP-dependent protein kinase 2 (cGKII)/PRKG2 is involved in
the regulation of aldosterone and renin secretion (36). In total, nine unaffected family
members presented wild-type alleles (Fig. 2A). Importantly, this mutation was
not previously observed in any public database. The residue 251Asn
was revealed to be evolutionarily conserved (Fig. 2B) and PRKG2 expression was
identified to be enriched in the small intestine (18.36 TPM),
mainly in glandular cells (Fig.
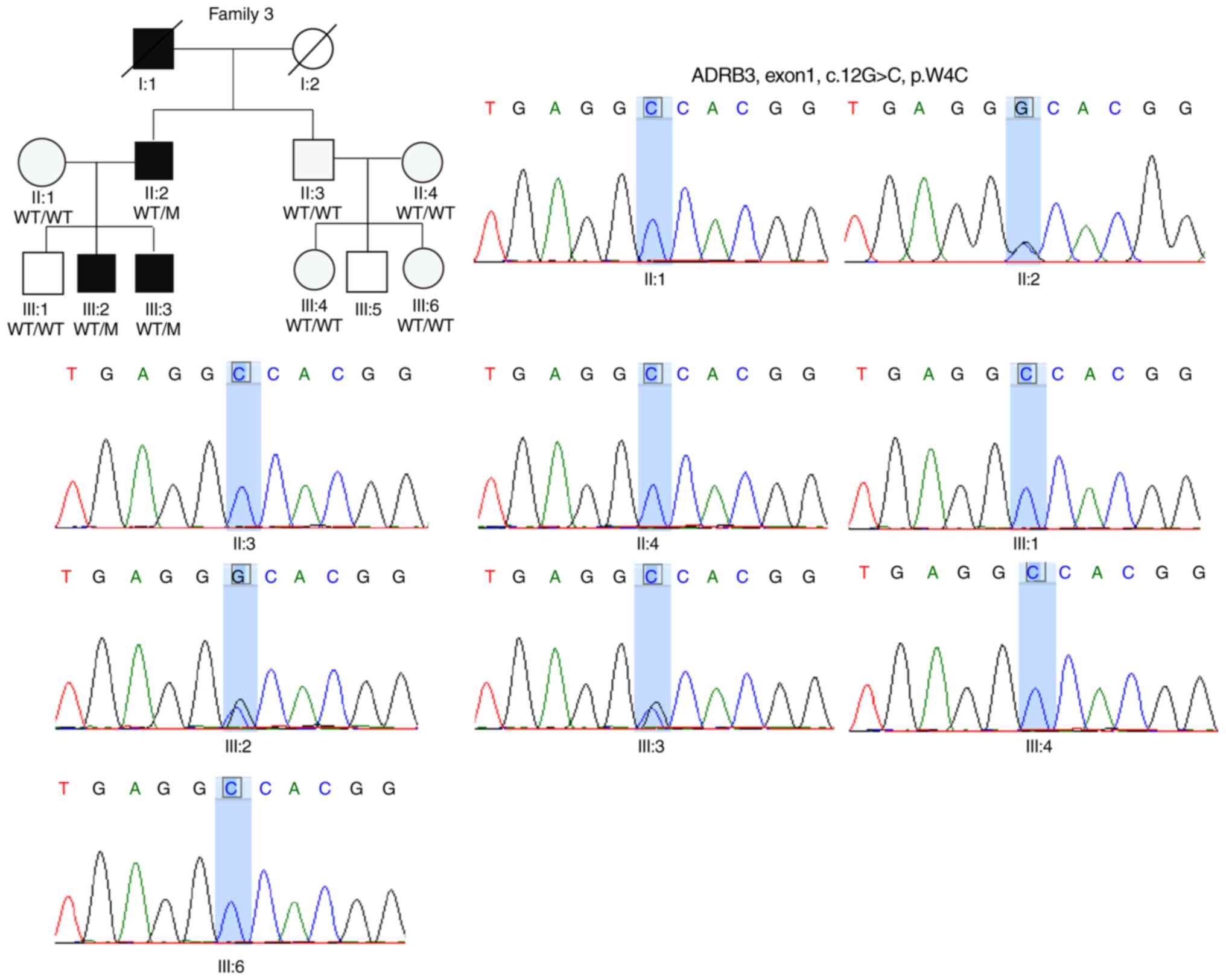
2C). In the F3, the proband F3:II:1 and his two affected sons
shared the same missense mutation consisting of a G to C
substitution (c.12G>C) in ADRB3 (Fig. 3). ADRB3 is part of the
adrenergic system, which is involved in the regulation of lipid
metabolism and glucose homeostasis (37). This single-nucleotide change
resulted in a non-synonymous substitution (p.Trp4Cys). PolyPhen2
predicted that this variant was likely damaging (Table II). Sanger sequencing validated
that six healthy family members presented homozygous wild-type
alleles for ADRB3 (Fig.
3). Similarly to the aforementioned two residues, this position
was predicted to be evolutionary conserved by the GERP++ tool
(https://omictools.com/gerp-tool).
Protein structural modeling
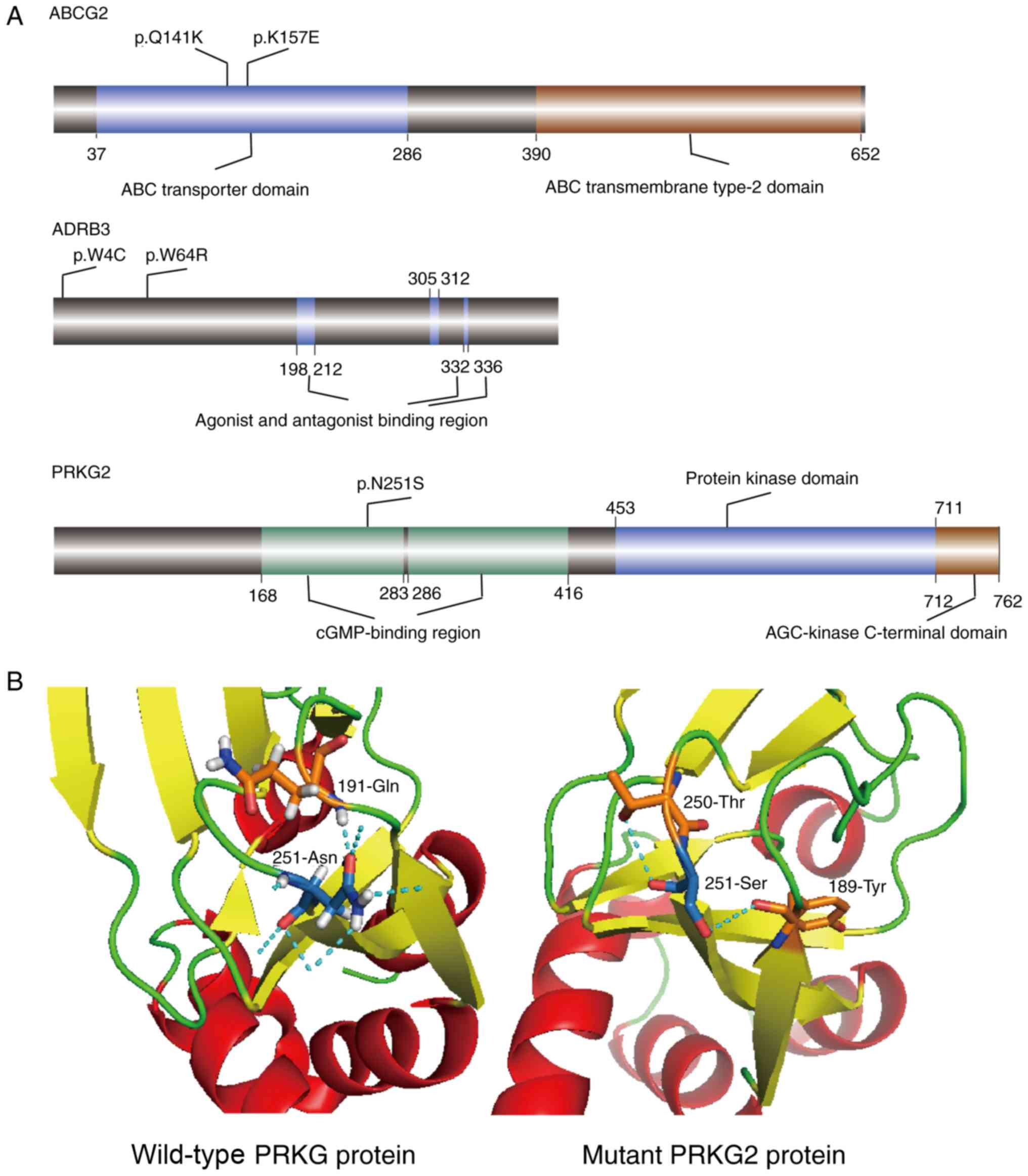
Previous GWAS studies identified single nucleotide
polymorphisms (SNPs) in these three genes that were associated with
gout (38-40). All SNPs and three candidate
mutations identified in the present study were mapped to schematic
representations of ABCG2, PRKG2 and ADRB2
genes (Fig. 4A). Except for the
two mutations in PRKG2, all the other mutations were located
in the protein functional domains. Furthermore, p.Lys157Glu
mutation in ABCG2 was located in the same highly conserved
ABC transporter domain as the pathogenic missense variant
Gln141Lys, which has been revealed to be associated with an
increase in serum uric acid levels (41). The Asn251Ser mutation was in the
cGMP-binding region of PRKG2; however, rs768867227 and
rs10033237 SNPs, which were previously reported to cause gout, were
present in non-coding regions (42).
In order to further study the functional defects
caused by mutations in the protein structure, the potential
structural differences between the wild-type and mutant proteins
were investigated. Therefore, numerous differences were identified
between the wild-type and mutant PRKG2 proteins. The
wild-type structure (5BV6) was downloaded from the PDB. In the
wide-type protein, there were seven hydrogen bonds at residue 251
(Asn); one bond was revealed between Asn251 and Glu191, and the
other six connected Asn251 to water molecules (Fig. 4B). The mutated structural
modelling revealed the formation of two different hydrogen bonds,
one between the mutated Ser251 and Thr250, and another between the
mutated Ser251 and Tyr189 (Fig.
4B).
Functional analysis of the co-expression
and PPI networks
Previous studies have identified that
gout-associated genes are closely associated with urate excretion
(10-12). To further investigate the
expression pattern of the three candidate genes (ABCG2,
PRKG2 and ADRB2) identified in the present study and
to examine whether their expression patterns were similar to other
gout-associated genes identified in previous studies, the
transcriptomic data in the small intestinal tissue from the GTEx
project were analysed. Based on the co-expression network, these
three genes demonstrated similar expression patterns with numerous
gout-associated genes, with a Pearson correlation coefficient
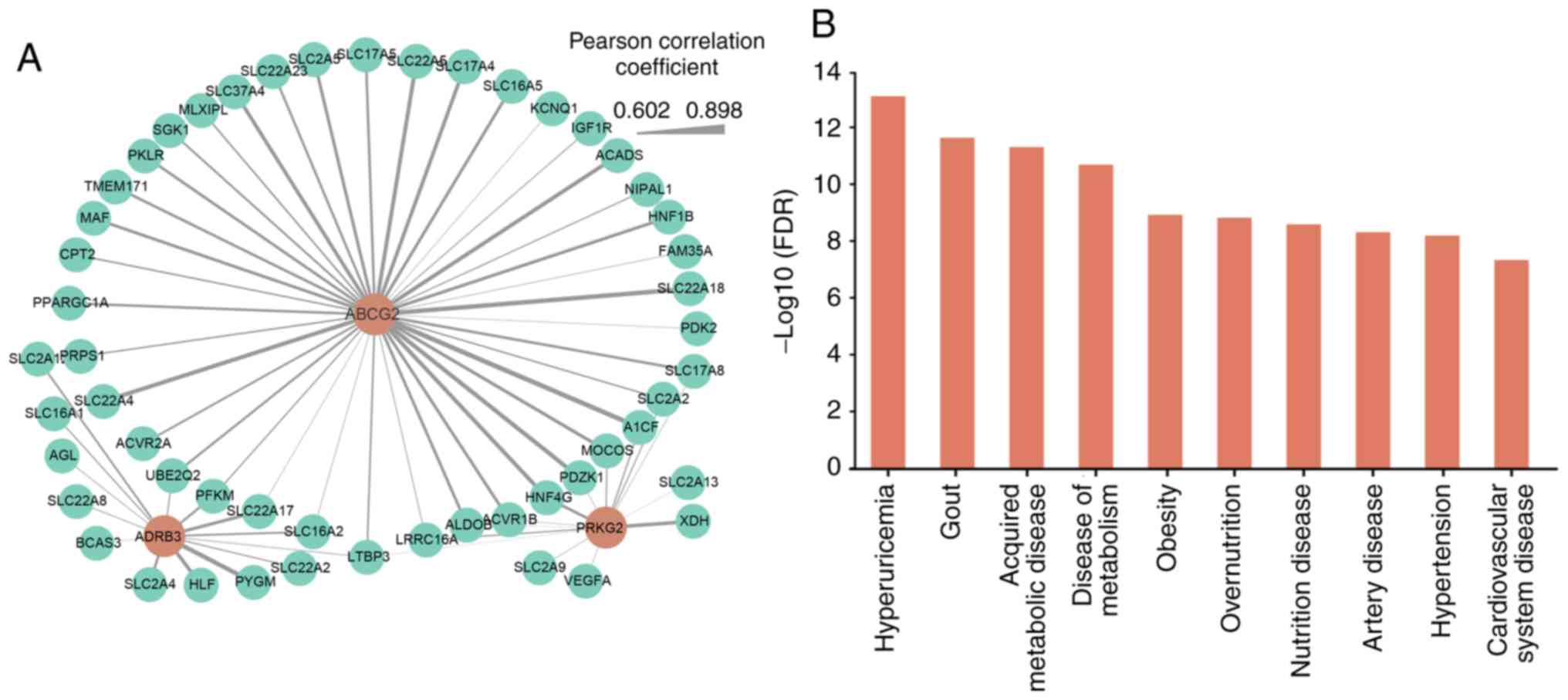
ranging between 0.602 and 0.898 (Fig.
5A). Among these three genes, the number of candidate genes
co-expressed with the ABCG2 gene was the largest (n≤39).
Furthermore, to identify functional similarities among these
co-expressed genes that may have a function in the development of
gout, significantly enriched DO terms were identified using the R
package DOSE. Among the top ten most statistically significant DO
terms with Bonferroni corrected P<0.05 (Fig. 5B; Table SIV), the majority of the terms
were associated with cardiovascular and metabolic diseases,
supporting the idea that gout is often accompanied by metabolic
syndrome. The two directly associated terms, hyperuricemia [false
discovery rate (FDR)=8.02×10−14, hypergeometric test]
and gout (FDR=2.32×10−12, hypergeometric test) were the
most statistically significant, indicating the significant
enrichment of the co-expressed genes in hyperuricemia and gout.
To investigate the association between the three
candidate genes and other gout candidate genes, data from the
STRING v10 database were analysed. The STRING database contains
PPIs, including physical and functional associations. As a result,
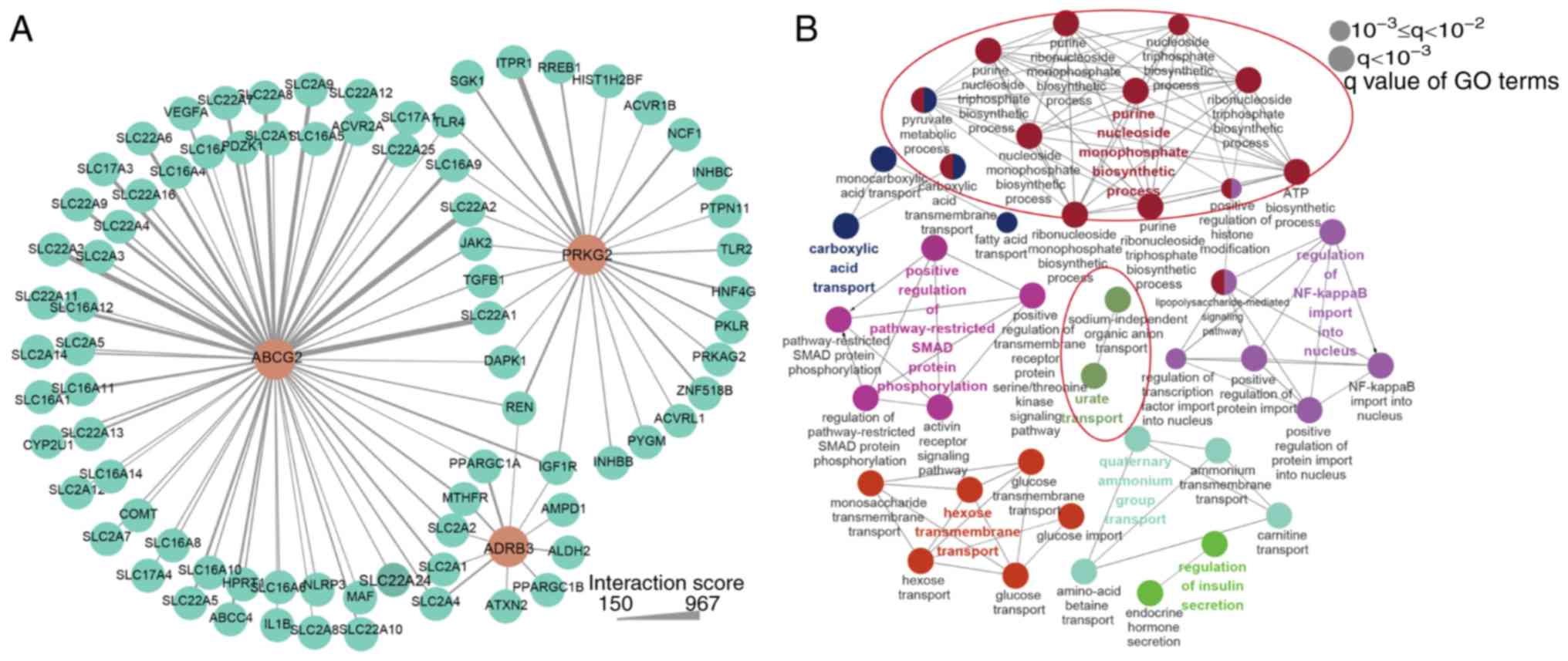
a total of 79 candidate genes were included in the PPI network
(Fig. 6A) with the interaction
score ranging between 150 and 967. Solute carrier family 22 member
1 exhibited the strongest interaction with ABCG2, which is
expressed in the kidney and mediates the transport of xenobiotics,
endogenous organic anions and urate (43). PRKG2 exhibited the
strongest interaction with inositol 1,4,5-trisphosphate receptor
type 1, which was previously reported to be associated with high
serum uric acid concentrations (44). In the next step, all 79 candidate
genes in the PPI network were used in the BP with ClueGO plugin. A
total of 40 enriched BP terms were divided into eight groups
(Fig. 6B) and terms in the same
group had similar biological functions. In total, two groups served
direct functions in the metabolism of uric acid, and the terms in
the two groups with the most significant statistical significance
were 'purine nucleoside monophosphate biosynthetic process' and
'urate transport'. 'Glycometabolism' and 'anion transport' groups
were also identified following data enrichment analysis. The
detailed results of the BP analysis are presented in Table SV.
Discussion
Gout is a complex disorder characterised by clinical
and genetic heterogeneity, and the genetic mechanism underlying
gout remains unclear, mainly because i) previous studies focused on
sporadic cases; and ii) genotyping chips were unable to identify
rare or novel variants. To address these two critical issues, the
present study aimed to reveal candidate rare/novel mutations in
large pedigrees with gout aggregation. A previous complex
segregation and linkage analysis of familial gout revealed an
autosomal-arbitrary major gene model (20). WES is able to provide insights
into the pathogeny of hereditary diseases and extend molecular
diagnosis (45-48). Therefore, WES was performed in
three families with gout. Subsequent to employing previously
established filtering strategies, three candidate variants were
identified in these three families.
In the proband F1:II:1, one novel missense mutation
was revealed in ABCG2 (c.469A>G, p.Lys157Glu), and it was
predicted to be deleterious by all four functional prediction
tools. ABCG2 has been reported to be an important factor
involved in the reduction of urate transport rates (40) and in a pathway regulating fructose
metabolism, which is associated with obesity (13,49). The patient in F1 presenting the
ABCG2 mutation exhibited early onset-gout and was
overweight, and these symptoms are in line with the pathological
features of gout. The two most commonly reported dysfunctional SNPs
are Gln126Ter (rs72552713) and Gln141LysK (rs2231142), which are
located in the ABC domain, which is considered to be critical for
the interactions between the intracellular loops of the
transmembrane portion of the protein (40). The presence of the Q141K
polymorphism in the ABC transporter domain was previously reported
to induce a 2-fold decrease in urate efflux (50). The mutation p.Lys157Glu identified
in the present study is located in the ABC transporter domain,
indicating that the pathogenic mechanism of this mutation is caused
by dysfunctions in this functional domain. Furthermore, a previous
study has identified three common and 19 rare non-synonymous
variants of ABCG2 in patients with gout and functional
assays were performed to determine the urate transport activity of
each ABCG2 variant (51).
Almost all rare variants identified in the present study exhibited
lower urate transport and almost completely inhibited ABCG2
activity compared with the wild-type protein, as assessed by
functional assays performed to determine the urate transport
activity of each ABCG2 variant.
The second mutation identified was present in
ADRB3, which encodes a β-3-adrenergic receptor and has been
revealed to serve an important function in the regulation of
lipolysis and glucose homeostasis (37,39). Hyperuricemia and gout are closely
associated with metabolic disorders, including obesity,
dyslipidaemia, glucose intolerance and hypertension (52,53). Wang et al (20) and Huang et al (54) reported that a p.Trp64Arg variant
was associated with hyperuricemia in Chinese male patients. A
similar study investigating Spanish patients identified that a
p.Trp64Arg polymorphism in ADRB3 gene may increase the risk
of hyperuricemia (55).
Therefore, the missense mutation c.12G>C in the ADRB3
gene revealed in the present study was spatially near the same
intracellular loop affected in the p.Trp64Arg mutation, and it is
thus expected to be involved in the development of basal metabolic
diseases including dyslipidaemia, which was confirmed by the fact
that patients with gout in Family 3 had hyperlipemia and
hyperbilirubinemia.
The third mutation identified in the present study
was located in the PRKG2 gene. Accumulating evidence has
demonstrated that the cGMP signalling pathway serves important
functions in urate cycles, and PRKG2 is a cGKII gene
(42). One case-control study
revealed that there was a correlation between the polymorphisms
rs768867227 and rs10033237 in PRKG2, and gout susceptibility
(56). Each of these
polymorphisms are located in non-coding regions. Although the
biological function of combined rs10033237 and rs7688672 in the
PRKG2 gene has not been elucidated, dysfunctions in
PRKG2 may result in hypertension by destroying the
renin-angiotensin-aldosterone system, and may result in
hyperuricemia (42). The
renin-angiotensin-aldosterone system in the patients in Family 3
carrying heterozygous missense mutations (c.752A>G and
Asn251Ser) in PRKG2 was potentially impaired. An impaired
renin-angiotensin-aldosterone system may cause hyperuricemia, which
was consistent with the presence of hypertension in the patients in
Family 3. Furthermore, a previous study (57) demonstrated that PRKG2
serves a key function in mediating M1 polarization and phagocytotic
activity by regulating the levels of monosodium urate and
lipopolysaccharides.
Excretion of uric acid requires specialized
transporters located in renal tubule cells and intestinal
epithelial cells (58).
Accordingly, direct intestinal secretion is considered as a
substantial contributor to the extra-renal elimination of uric acid
(59). It is estimated that ~30%
of uric acid is excreted by the intestine (60). Numerous gout-associated genes are
highly expressed in the small intestine, including ABCG2 and
PRKG2 detected in the present study (10-12). The co-expression network revealed
that gout-associated genes share similar expression patterns with
ABCG2, PRKG2 and ADRB3 in the small intestine.
ABCG2 is a well-characterized urate transporter in the
intestine, and the molecular function of ABCG2 was
previously identified (10-12). The functional interactions between
proteins are crucial for their biological function, and their
systematic characterization may increase current understanding of
molecular systems biology (61).
The STING network constructed using ABCG2, PRKG2 and
ADRB3 contained a number of gout-associated genes,
suggesting that the pathogenic mechanism of these genes may share
the same pathway.
Gout is a metabolic disorder caused by urate
overproduction and/or reduced urate excretion (62). According to previous studies, gout
is associated with five major metabolic syndromes and/or
consequences of metabolic syndrome: Hypertension, cardiovascular
disease, insulin resistance and diabetes, obesity and
hyperlipidaemia (63-65). In the present study, a DO analysis
was performed on the genes identified in the co-expression network,
and three DO terms associated with the cardiovascular system,
including cardiovascular system disease, hypertension and artery
disease, were in the top 10 significantly enriched terms. By
directly impairing the vascular endothelium and the renal system,
high serum urate levels may increase blood pressure (66). Other enriched terms were mainly
associated with obesity. Following a data analysis performed on
data from 517 participants in the Bogalusa Heart Study, Muntner
et al (67) suggested that
an elevated body mass index was associated with high levels of uric
acid. Furthermore, the BP analysis of the genes in the PPI network
contained terms including 'hexose transmembrane transport' and
'regulation of insulin secretion', and the two groups involved in
glycose metabolism have been reported to affect the transport of
uric acid (68-70). The majority of the enriched BP
terms were revealed to be involved in metabolite transports, which
are closely associated with urate transport.
Despite recent advancements in the understanding of
the mechanism underlying gout, the number of families involved in
the present study was small; therefore, the statistical power for
the autosomal dominant inheritance model in gout was limited,
although the model was previously proposed according to statistical
analyses based on large sample sizes (19,20). In addition, no functional genomics
studies were performed in the present study. Therefore, the sample
size of the study should be expanded to provide stronger evidence
for the Mendelian genetic inheritance of gout. Copy number
variations were not analysed in the present study (71,72). Furthermore, functional experiments
are necessary to further determine the function of the mutations
detected in gout pathogenesis.
Collectively, to the best of our knowledge, the
present study is the first to use WES to dissect rare or novel
genetic variants using pedigree and family aggregation analyses.
Among the three families, three deleterious mutations in three
different gout pathogenic genes were identified. The present
results increase the current knowledge of the genotypic
heterogeneity underlying the phenotypic heterogeneity of gout,
which assists not only clinical diagnoses but also potential
personalized therapy.
Supplementary Data
Funding
The present study was supported by the Zhejiang
Provincial Natural Science Foundation (grant no. LY20H100001) and
the Wenzhou Science and Technology Bureau (grant no.
Y20180129).
Availability of data and materials
All the datasets generated and/or analysed in the
present study are available from the corresponding author upon
reasonable request.
Authors' contributions
XX and JJ conceived and designed the study. ZC and
ZZ performed the experiments. HC and CZ collected and analysed the
experimental data. LZ wrote the article. LS helped revise the
article. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
This study was conducted according to the principles
outlined in the Declaration of Helsinki. The study protocol was
approved by the Ethics Committee of The First Affiliated Hospital,
Wenzhou Medical University (Wenzhou, China; approval no. 2018-020).
Written informed consent was obtained from all participants or
their guardians.
Patient consent for publication
The patients provided written informed consent
regarding the publication of the case details and any associated
images.
Competing interests
The authors declare that they have no competing
interests.
Acknowledgments
Not applicable.
References
|
1
|
Bardin T and Richette P: The epidemiology
and genetic of gout. Presse Med. 40:830–835. 2011.In French.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kuo CF, Grainge MJ, Zhang W and Doherty M:
Global epidemiology of gout: Prevalence, incidence and risk
factors. Nat Rev Rheumatol. 11:649–662. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhu Y, Pandya BJ and Choi HK: Prevalence
of gout and hyperuricemia in the US general population: The
national health and nutrition examination survey 2007-2008.
Arthritis Rheum. 63:3136–3141. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu R, Han C, Wu D, Xia X, Gu J, Guan H,
Shan Z and Teng W: Prevalence of hyperuricemia and gout in mainland
China from 2000 to 2014: A systematic review and meta-analysis.
Biomed Res Int. 2015:7628202015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lu X, Li X, Zhao Y, Zheng Z, Guan S and
Chan P: Contemporary epidemiology of gout and hyperuricemia in
community elderly in Beijing. Int J Rheum Dis. 17:400–407. 2014.
View Article : Google Scholar
|
|
6
|
Yu TF, Dorph DJ and Smith H:
Hyperlipidemia in primary gout. Semin Arthritis Rheum. 7:233–244.
1978. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vázquez-Mellado J, García CG, Vázquez SG,
Medrano G, Ornelas M, Alcocer L, Marquez A and Burgos-Vargas R:
Metabolic syndrome and ischemic heart disease in gout. J Clin
Rheumatol. 10:105–109. 2004. View Article : Google Scholar
|
|
8
|
Facchini F, Chen YD, Hollenbeck CB and
Reaven GM: Relationship between resistance to insulin-mediated
glucose uptake, urinary uric acid clearance, and plasma uric acid
concentration. JAMA. 266:3008–3011. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kaplan NM: The deadly quartet. Upper-body
obesity, glucose intolerance, hypertriglyceridemia, and
hypertension. Arch Intern Med. 149:1514–1520. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Choi HK, Zhu Y and Mount DB: Genetics of
gout. Curr Opin Rheumatol. 22:144–151. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mikuls TR and Saag KG: New insights into
gout epidemiology. Curr Opin Rheumatol. 18:199–203. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Riches PL, Wright AF and Ralston SH:
Recent insights into the pathogenesis of hyperuricaemia and gout.
Hum Mol Genet. 18:R177–R184. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Dehghan A, Kottgen A, Yang Q, Hwang SJ,
Kao WL, Rivadeneira F, Boerwinkle E, Levy D, Hofman A, Astor BC, et
al: Association of three genetic loci with uric acid concentration
and risk of gout: A genome-wide association study. Lancet.
372:1953–1961. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kolz M, Johnson T, Sanna S, Teumer A,
Vitart V, Perola M, Mangino M, Albrecht E, Wallace C, Farrall M, et
al: Meta-analysis of 28,141 individuals identifies common variants
within five new loci that influence uric acid concentrations. PLoS
Genet. 5:e10005042009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Q, Kottgen A, Dehghan A, Smith AV,
Glazer NL, Chen MH, Chasman DI, Aspelund T, Eiriksdottir G, Harris
TB, et al: Multiple genetic loci influence serum urate levels and
their relationship with gout and cardiovascular disease risk
factors. Circ Cardiovasc Genet. 3:523–530. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Merriman TR: An update on the genetic
architecture of hyperuricemia and gout. Arthritis Res Ther.
17:982015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kottgen A, Albrecht E, Teumer A, Vitart V,
Krumsiek J, Hundertmark C, Pistis G, Ruggiero D, O'Seaghdha CM,
Haller T, et al: Genome-wide association analyses identify 18 new
loci associated with serum urate concentrations. Nat Genet.
45:145–154. 2013. View Article : Google Scholar :
|
|
18
|
Li C, Li Z, Liu S, Wang C, Han L, Cui L,
Zhou J, Zou H, Liu Z, Chen J, et al: Genome-wide association
analysis identifies three new risk loci for gout arthritis in Han
Chinese. Nat Commun. 6:70412015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kuo CF, Grainge MJ, See LC, Yu KH, Luo SF,
Valdes AM, Zhang W and Doherty M: Familial aggregation of gout and
relative genetic and environmental contributions: A nationwide
population study in Taiwan. Ann Rheum Dis. 74:369–374. 2015.
View Article : Google Scholar :
|
|
20
|
Wang WH, Chang SJ, Wang TN, Cheng LS, Feng
YP, Chen CJ, Huang CH and Ko YC: Complex segregation and linkage
analysis of familial gout in Taiwanese aborigines. Arthritis Rheum.
50:242–246. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Neogi T, Jansen TL, Dalbeth N, Fransen J,
Schumacher HR, Berendsen D, Brown M, Choi H, Edwards NL, Janssens
HJ, et al: 2015 Gout classification criteria: An American college
of rheumatology/European league against rheumatism collaborative
initiative. Arthritis Rheumatol. 67:2557–2568. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang T, Liu Q, Li X, Wang X, Li J, Zhu X,
Sun ZS and Wu J: RRBS-analyser: A comprehensive web server for
reduced representation bisulfite sequencing data analysis. Hum
Mutat. 34:1606–1610. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li H and Durbin R: Fast and accurate
long-read alignment with Burrows-Wheeler transform. Bioinformatics.
26:589–595. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Q, Chen C, Shen E, Zhao F, Sun Z and
Wu J: Detection, annotation and visualization of alternative
splicing from RNA-Seq data with SplicingViewer. Genomics.
99:178–182. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li J, Jiang Y, Wang T, Chen H, Xie Q, Shao
Q, Ran X, Xia K, Sun ZS and Wu J: mirTrios: An integrated pipeline
for detection of de novo and rare inherited mutations from
trios-based next-generation sequencing. J Med Genet. 52:275–281.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Huang XF, Huang F, Wu KC, Wu J, Chen J,
Pang CP, Lu F, Qu J and Jin ZB: Genotype-phenotype correlation and
mutation spectrum in a large cohort of patients with inherited
retinal dystrophy revealed by next-generation sequencing. Genet
Med. 17:271–278. 2015. View Article : Google Scholar
|
|
27
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Schwarz JM, Cooper DN, Schuelke M and
Seelow D: MutationTaster2: Mutation prediction for the
deep-sequencing age. Nat Methods. 11:361–362. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shihab HA, Gough J, Mort M, Cooper DN, Day
IN and Gaunt TR: Ranking non-synonymous single nucleotide
polymorphisms based on disease concepts. Hum Genomics. 8:112014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Waterhouse A, Bertoni M, Bienert S, Studer
G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C,
Bordoli L, et al: SWISS-MODEL: Homology modelling of protein
structures and complexes. Nucleic Acids Res. 46:W296–W303. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yu G, Wang LG, Yan GR and He QY: DOSE: An
R/Bioconductor package for disease ontology semantic and enrichment
analysis. Bioinformatics. 31:608–609. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Szklarczyk D, Gable AL, Lyon D, Junge A,
Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork
P, et al: STRING v11: Protein-protein association networks with
increased coverage, supporting functional discovery in genome-wide
experimental datasets. Nucleic Acids Res. 47:D607–D613. 2019.
View Article : Google Scholar
|
|
33
|
Bindea G, Mlecnik B, Hackl H, Charoentong
P, Tosolini M, Kirilovsky A, Fridman WH, Pagès F, Trajanoski Z and
Galon J: ClueGO: A Cytoscape plug-in to decipher functionally
grouped gene ontology and pathway annotation networks.
Bioinformatics. 25:1091–1093. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ichida K, Matsuo H, Takada T, Nakayama A,
Murakami K, Shimizu T, Yamanashi Y, Kasuga H, Nakashima H, Nakamura
T, et al: Decreased extra-renal urate excretion is a common cause
of hyperuricemia. Nat Commun. 3:7642012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Matsuo H, Nakayama A, Sakiyama M, Chiba T,
Shimizu S, Kawamura Y, Nakashima H, Nakamura T, Takada Y, Oikawa Y,
et al: ABCG2 dysfunction causes hyperuricemia due to both renal
urate underexcretion and renal urate overload. Sci Rep. 4:37552014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Vaandrager AB, Hogema BM and de Jonge HR:
Molecular properties and biological functions of cGMP-dependent
protein kinase II. Front Biosci. 10:2150–2164. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Krief S, Lönnqvist F, Raimbault S, Baude
B, Van Spronsen A, Arner P, Strosberg AD, Ricquier D and Emorine
LJ: Tissue distribution of beta 3-adrenergic receptor mRNA in man.
J Clin Invest. 91:344–349. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Sakiyama M, Matsuo H, Chiba T, Nakayama A,
Nakamura T, Shimizu S, Morita E, Fukuda N, Nakashima H, Sakurai Y,
et al: Common variants of cGKII/PRKG2 are not associated with gout
susceptibility. J Rheumatol. 41:1395–1397. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Wang B, Meng D, Wang J, Jia Z, Zhoub S,
Liu S, Chu N, Han L, Zhang K, Ma X and Li C: Positive correlation
between Beta-3-Adrenergic Receptor (ADRB3) gene and gout in a
Chinese male population. J Rheumatol. 38:738–740. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Woodward OM, Köttgen A, Coresh J,
Boerwinkle E, Guggino WB and Köttgen M: Identification of a urate
transporter, ABCG2, with a common functional polymorphism causing
gout. Proc Natl Acad Sci USA. 106:10338–10342. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Woodward OM, Tukaye DN, Cui J, Greenwell
P, Constantoulakis LM, Parker BS, Rao A, Köttgen M, Maloney PC and
Guggino WB: Gout-causing Q141K mutation in ABCG2 leads to
instability of the nucleotide-binding domain and can be corrected
with small molecules. Proc Natl Acad Sci USA. 110:5223–5228. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guo M, Cheng Z, Li C, Li S, Li M, Wang M,
Xu J, Tang Y, Wang Y, Qiu W and Liu X: Polymorphism of rs7688672
and rs10033237 in cGKII/PRKG2 and gout susceptibility of Han
population in northern China. Gene. 562:50–54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Sakiyama M, Matsuo H, Shimizu S, Nakashima
H, Nakayama A, Chiba T, Naito M, Takada T, Suzuki H, Hamajima N, et
al: A common variant of organic anion transporter 4 (OAT4/SLC22A11)
gene is associated with renal underexcretion type gout. Drug Metab
Pharmacokinet. 29:208–210. 2014. View Article : Google Scholar
|
|
44
|
Chittoor G, Kent JW Jr, Almeida M, Puppala
S, Farook VS, Cole SA, Haack K, Göring HH, MacCluer JW, Curran JE,
et al: GWAS and transcriptional analysis prioritize ITPR1 and CNTN4
for a serum uric acid 3p26 QTL in Mexican Americans. BMC Genomics.
17:2762016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Bergmann C: Advances in renal genetic
diagnosis. Cell Tissue Res. 369:93–104. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wu J, Shen E, Shi D, Sun Z and Cai T:
Identification of a novel Cys146X mutation of SOD1 in familial
amyotrophic lateral sclerosis by whole-exome sequencing. Genet Med.
14:823–826. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Huang XF, Xiang L, Cheng W, Cheng FF, He
KW, Zhang BW, Zheng SS, Han RY, Zheng YH, Xu XT, et al: Mutation of
IPO13 causes recessive ocular coloboma, microphthalmia, and
cataract. Exp Mol Med. 50:532018. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Huang XF, Xiang L, Fang XL, Liu WQ, Zhuang
YY, Chen ZJ, Shen RJ, Cheng W, Han RY, Zheng SS, et al: Functional
characterization of CEP250 variant identified in nonsyndromic
retinitis pigmentosa. Hum Mutat. 40:1039–1045. 2019.PubMed/NCBI
|
|
49
|
Dalbeth N, House ME, Gamble GD, Pool B,
Horne A, Purvis L, Stewart A, Merriman M, Cadzow M, Phipps-Green A
and Merriman TR: Influence of the ABCG2 gout risk 141 K allele on
urate metabolism during a fructose challenge. Arthritis Res Ther.
16:R342014. View
Article : Google Scholar : PubMed/NCBI
|
|
50
|
Basseville A and Bates SE: Gout, genetics
and ABC transporters. F1000 Biol Rep. 3:232011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Higashino T, Takada T, Nakaoka H, Toyoda
Y, Stiburkova B, Miyata H, Ikebuchi Y, Nakashima H, Shimizu S,
Kawaguchi M, et al: Multiple common and rare variants of ABCG2
cause gout. RMD Open. 3:e0004642017. View Article : Google Scholar :
|
|
52
|
Choi HK and Ford ES: Prevalence of the
metabolic syndrome in individuals with hyperuricemia. Am J Med.
120:442–447. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Choi HK, Ford ES, Li C and Curhan G:
Prevalence of the metabolic syndrome in patients with gout: The
third national health and nutrition examination survey. Arthritis
Rheum. 57:109–115. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Huang Q, Zhang LF, Cheng Y, Zhao YC, Si L,
Gao Y and Wei W: Trp64Arg (rs4994) polymorphism of β3-adrenergic
receptor gene is associated with hyperuricemia in a Chinese male
population. Clin Chem Lab Med. 51:1755–1760. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Morcillo S, Rojo-Martinez G, Martin-Nunez
GM, Gómez- Zumaquero JM, García-Fuentes E, Ruiz de Adana M, de la
Cruz Almaraz M and Soriguer F: Trp64Arg polymorphism of the ADRB3
gene predicts hyperuricemia risk in a population from southern
Spain. J Rheumatol. 37:417–421. 2010. View Article : Google Scholar
|
|
56
|
Chang SJ, Tsai MH, Ko YC, Tsai PC, Chen CJ
and Lai HM: The cyclic GMP-dependent protein kinase II gene
associates with gout disease: Identified by genome-wide analysis
and case-control study. Ann Rheum Dis. 68:1213–1219. 2009.
View Article : Google Scholar
|
|
57
|
Liao WT, You HL, Li C, Chang JG, Chang SJ
and Chen CJ: Cyclic GMP-dependent protein kinase II is necessary
for macrophage M1 polarization and phagocytosis via toll-like
receptor 2. J Mol Med (Berl). 93:523–533. 2015. View Article : Google Scholar
|
|
58
|
Xu X, Li C, Zhou P and Jiang T: Uric acid
transporters hiding in the intestine. Pharm Biol. 54:3151–3155.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Hosomi A, Nakanishi T, Fujita T and Tamai
I: Extra-renal elimination of uric acid via intestinal efflux
transporter BCRP/ABCG2. PLoS One. 7:e304562012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
So A and Thorens B: Uric acid transport
and disease. J Clin Invest. 120:1791–1799. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Szklarczyk D, Franceschini A, Kuhn M,
Simonovic M, Roth A, Minguez P, Doerks T, Stark M, Muller J, Bork
P, et al: The STRING database in 2011: Functional interaction
networks of proteins, globally integrated and scored. Nucleic Acids
Res. 39:D561–D568. 2011. View Article : Google Scholar :
|
|
62
|
Yamanaka H: Gout and hyperuricemia in
young people. Curr Opin Rheumatol. 23:156–160. 2011. View Article : Google Scholar
|
|
63
|
Billiet L, Doaty S, Katz JD and Velasquez
MT: Review of hyperuricemia as new marker for metabolic syndrome.
ISRN Rheumatol. 2014:8529542014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Puig JG and Martinez MA: Hyperuricemia,
gout and the metabolic syndrome. Curr Opin Rheumatol. 20:187–191.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Thottam GE, Krasnokutsky S and Pillinger
MH: Gout and metabolic syndrome: A tangled web. Curr Rheumatol Rep.
19:602017. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kang DH, Han L, Ouyang X, Kahn AM,
Kanellis J, Li P, Feng L, Nakagawa T, Watanabe S, Hosoyamada M, et
al: Uric acid causes vascular smooth muscle cell proliferation by
entering cells via a functional urate transporter. Am J Nephrol.
25:425–433. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Muntner P, Srinivasan S, Menke A, Patel
DA, Chen W and Berenson G: Impact of childhood metabolic syndrome
components on the risk of elevated uric acid in adulthood: The
Bogalusa Heart Study. Am J Med Sci. 335:332–337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Fam AG: Gout, diet, and the insulin
resistance syndrome. J Rheumatol. 29:1350–1355. 2002.PubMed/NCBI
|
|
69
|
Gheita TA, El-Fishawy HS, Nasrallah MM and
Hussein H: Insulin resistance and metabolic syndrome in primary
gout: Relation to punched-out erosions. Int J Rheum Dis.
15:521–525. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Perez-Ruiz F, Aniel-Quiroga MA,
Herrero-Beites AM, Chinchilla SP, Erauskin GG and Merriman T: Renal
clearance of uric acid is linked to insulin resistance and lower
excretion of sodium in gout patients. Rheumatol Int. 35:1519–1524.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Huang XF, Mao JY, Huang ZQ, Rao FQ, Cheng
FF, Li FF, Wang QF and Jin ZB: Genome-wide detection of copy number
variations in unsolved inherited retinal disease. Invest Ophthalmol
vis Sci. 58:424–429. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Huang XF, Wu J, Lv JN, Zhang X and Jin ZB:
Identification of false-negative mutations missed by
next-generation sequencing in retinitis pigmentosa patients: A
complementary approach to clinical genetic diagnostic testing.
Genet Med. 17:307–311. 2015. View Article : Google Scholar : PubMed/NCBI
|