Introduction
The epigenetic regulation of cancer-associated
microRNAs (miRNAs) was reported to be indispensable during tumor
initiation and cancer progression (1,2).
In terms of tumor-suppressive miRNAs, aberrant methylation in the
CpG islands of their promoters, was demonstrated to be one of the
most common epigenetic mechanisms that modulated their expression
and biological function (2). A
number of miRNAs, including miR-132, miR-148a, miR-107, miR-181 and
miR-34, have been identified to be epigenetically silenced by the
hyper-methylation of CpG islands in/near the promoter region
(3-5). However, miRNA methylation status in
endometrial carcinoma (EC) has not been comprehensively
investigated.
miR-638, a newly characterized cancer-associated
miRNA, is differentially expressed in various types of human cancer
and participates in a number of biological processes during
tumorigenesis (6-9). However, the data concerning its
exact biological roles in different cancers are conflicting and
poorly understood. miR-638 is upregulated and acts as a tumor
promoting miRNA in prostate cancer (7), esophageal carcinoma (9), breast cancer (9) and melanoma (10). Nevertheless, miR-638 was
demonstrated to be downregulated and to function as a tumor
suppressive miRNA in gastric (8,11),
cervical (12) and breast cancer
(13), and hepatocellular
carcinoma (14). The methylation
status, expression level and biological roles of miR-638 have not
been investigated in EC.
The present study identified potential antitumor
miRNAs that were significantly downregulated by promoter
methylation using a microarray, among which miR-638 was selected to
be further investigated. The methylation status, expression level,
biological function, and molecular targets of miR-638 were
examined. The aim of the present study was to comprehensively
identify potential promoter methylation-silenced cancer-associated
miRNAs in EC and to specifically study the regulatory mechanisms of
miR-638 in EC in depth.
Materials and methods
Cell culture and demethylation
treatment
The human EC KLE and Ishikawa (ISH) cell lines,
which have already been authenticated, were purchased from the
American Type Culture Collection, which had already been
authenticated. Both cell lines were maintained in DMEM medium. For
DNA demethylation treatment, EC cell lines were treated with 3
µM 5-aza-2-deoxycytidine for 72 h at 37°C.
Clinical samples
A total of 68 consecutive paired EC tissues with
cancerous and non-cancerous tissues were obtained from patients who
underwent primary resection without neoadjuvant therapy at the
Fudan University Shanghai Cancer Center (FUSCC) between January
2014 and April 2014 and who were pathologically diagnosed with
endometrioid adenocarcinoma or serous carcinoma of the endometrium.
All specimens were frozen in liquid nitrogen immediately following
surgical removal and were then stored at −80°C. Subsequently,
tissue samples from 42 patients were randomly selected for further
research. Informed consent was obtained from the patients and the
study was approved by the Committee for the Ethical Review of
Research at FUSCC. The characteristics of the patients are
summarized in Table SI.
RNA isolation, miRNA microarray analysis
and semi-quantitative polymerase chain reaction (PCR)
Total RNA was extracted from freshly-frozen tissue
samples and cell lines using TRIzol® reagent (Thermo
Fisher Scientific, Inc.). miRNAs were isolated using an mirVana™
miRNA isolation kit (Thermo Fisher Scientific, Inc.). For the
microarray analysis, purified total RNAs from EC cells lines
treated with 5-aza-2-deoxycytidine or saline, and then hybridized
with a microchip using an miRNA Complete Labeling and Hyb kit
(Agilent Technologies, Inc.). The signals were then detected using
the Agilent Microarray Scanner and the images were analyzed using
the Agilent Feature Extraction Software version 10.5.1.1 (Agilent
Technologies, Inc.). Significantly upregulated miRNAs following
5-aza-2-deoxycytidine treatment were identified. TaqMan miRNA
assays (Applied Biosystems; Thermo Fisher Scientific, Inc.) were
used to quantify the relative expression of miR-638.
Semi-quantitative PCR with SYBR green I was used to compare the
relative expression of specific mRNAs using a SYBR®
Premix Ex Taq™ kit (Takara Biotechnology Co., Ltd.). The
thermocycling conditions for miR-638 were as follows: An initial
step at 95°C for 15 sec followed by 40 cycles of 95°C for 5 sec and
60°C for 30 sec. Amplified fragments were then detected on 4%
agarose gel electrophoresis containing ethidium bromide using a
ChemiDoc XRS system and Quantity One 1.0 software (Bio-Rad
Laboratories, Inc.). The primers used are listed in Table SII.
DNA methylation analysis
Genomic DNA was extracted from patient tissues and
cell lines using the QIAamp DNA formalin-fixed paraffin-embedded
Tissue kit (Qiagen, Inc.). Sodium bisulfite modification of DNA was
performed using the EpiTect Bisulfite kit (Qiagen, Inc.). PCR
products were recovered using Qiagen gel DNA kits (Qiagen, Inc.)
and then sequenced at GeneTech (Shanghai) Co., Ltd. The primer
sequences are listed in Table
SII.
Vector construction and cell
transfection
miR-638 mimic (5′-AGG GAT CGC GGG CG GGT GGC GGC
CT-3′), miR-638 mimic negative control (5′-AGG TAC GAA ACG CTA AGA
AT-3′), short hairpin RNA targeting MEF2C (shMEF2C; 5′-GGA CAA GGA
ATG GGA GGA TAT-3′) and shMEF2C negative control (5′-UCU CCG AUG
CAG GCU CAAC-3′) were synthesized by Shanghai GenePharma Co., Ltd.
For transfection, 100 ng miR-638 mimic, miR-638 mimic negative
control, shMEF2C or shMEF2C negative control was transfected using
Lipofectamine™ 2000 Transfection Reagent (Invitrogen; Thermo Fisher
Scientific, Inc.). In addition, a specific vector containing the
entire coding sequence of MEF2C but without its 3′-untranslated
region (UTR), as well as its negative control, were designed by
Shanghai GenePharma Co., Ltd. The lentiviral packing kit used to
transfect the various vectors were purchased from the Open
Biosystems, Inc. and transfection was performed at a final
concentration of 50 nM. 293T cells were purchased from the Cell
Bank of Chinese Academy of Sciences. Generally, EC cell lines with
5×104 cells were infected with 1×108
lentivirus-transducing units. Following transfection for 24 h, the
cells were cultured with 1 µg/ml puromycin for 2 weeks, and
then stably transduced EC cell lines were selected.
Cell viability, migration and invasion
assays and apoptosis analysis
Cell viability was investigated using Cell Counting
Kit-8 and colony formation assays. Cell migration and invasion
assays were performed using Transwell cell migration assay kits and
BioCoat™ Matrigel® Invasion Chambers (Corning
Incorporated), respectively. For the migration assay,
5×104 cells were seeded into the upper chamber and
incubated for 24 h at 37°C in 5% CO2. For the invasion
assay, 1×105 cells were plated on chambers pre-coated
with 1.6 mg Matrigel (Corning, Inc.) and incubated for 36 h at 37°C
in 5% CO2. Subsequently, cells were fixed, stained with
0.5% crystal violet solution at room temperature for 10 min,
photographed and counted under a light microscope (magnification,
×200).
Cell apoptosis was analyzed with the Annexin
V-fluorescein isothiocyanate/propidium iodide Apoptosis Detection
kit. Briefly, 1×105 cells were re-suspended in 500
µl binding buffer and stained with 5 µl Annexin V and
5 µl PI in the dark at room temperature for 20 min. The
samples were then analyzed using a FACScan flow cytometer and the
CellQuest™ Pro 1.0 software (BD Biosciences), according to the
manufacturer's instructions.
Western blot analysis
Cells were harvested and lysed in ice-cold
radioimmunoprecipitation assay lysis buffer. Protein concentrations
were determined by the BCA Protein assay reagent and measured using
a NanoDrop2000 Spectrophotometer (Thermo Fisher Scientific, Inc.).
Subsequently, 10 µg protein/lane was separated by 10%
SDS-PAGE gels and then transferred to PVDF membranes. The membranes
were blocked with 5% non-fat milk for 2 h at room temperature and
then incubated with primary antibodies against MEF2C (cat. no.
5030; Cell Signaling Technology, Inc.), matrix metalloproteinase
(MMP)2 (cat. no. 40994; Cell Signaling Technology, Inc.), MMP9
(cat. no. 13667; Cell Signaling Technology, Inc.), vascular
endothelial growth factor (VEGF; cat. no. ab69479; Abcam), cyclin
D1 (cat. no. 2922; Cell Signaling Technology, Inc.),
cyclin-dependent kinase (CDK)2 (cat. no. 2546; Cell Signaling
Technology, Inc.), CDK4 (cat. no. 12790; Cell Signaling Technology,
Inc.) and tubulin (cat. no. 5568; Cell Signaling Technology, Inc.)
overnight at 4°C. All aforementioned antibodies were used at
1:1,000. Then, the membranes were incubated with the corresponding
secondary antibodies overnight at 4°C. The secondary antibodies
used in the present study included: Horseradish
peroxidase-conjugated secondary antibodies against rabbit (cat. no.
SA00001-2; 1:10,000) and mouse (cat. no. SA00001-1; 1:10,000),
purchased from Proteintech Group, Inc. Finally, protein bands were
visualized using a chemiluminescence detection system (Bio-Rad
Laboratories, Inc.). Protein expression was quantified using
Image-Pro® Plus software (version 6.0; Media
Cybernetics, Inc.).
In vivo tumorigenicity
A total of 100 4-week-old female nude mice were
purchased from the Shanghai Institute of Materia Medica, Chinese
Academy of Sciences and placed in laminar flow cabinets under
specific pathogen-free conditions for 1 week. Then, 5-week-old mice
were randomly grouped (6 mice/treatment group) and 1×106
cells were injected subcutaneously into each mouse. The tumor size
was recorded weekly. A total of 50 days following cancer cell
injection, the mice were euthanized and the tumor weight was
determined. All of the mouse experiments were performed under the
guidelines of the National Institutes of Health for the Care and
Use of Laboratory Animals. The study protocol was approved by the
Committee on the Use of Live Animals in Teaching and Research of
Fudan University.
Target prediction and luciferase
assay
DIANA-microT-CDS (http://diana.imis.athena-innovation.gr/DianaTools/index.php),
TargetScan (http://www.targetscan.org/) (15) and miRNA (www.miRNA.org) were used for the prediction of miR-638
target genes. Luciferase constructs were generated by ligating
oligonucleotides containing the wild-type or a 4 bp mutation in the
putative target site of the MEF2C 3′-UTR into the Psi-CHECK2 vector
(Promega Corporation) downstream of a luciferase gene. The
wild-type/mutant type vectors were co-transfected with miR-638
mimics/negative control into 293T cells (Cell Bank of Chinese
Academy of Sciences), using Lipofectamine (Thermo Fisher
Scientific, Inc.). After 48 h, the cell lines were harvested and
the luciferase activity of each group was detected using the
dual-luciferase reporter assay system (Promega Corporation), with
Renilla luciferase selected as the internal control.
Statistical analysis
All results are presented as the mean ± standard
deviation. All statistical analyses were performed using GraphPad
Prism software 7.0 (GraphPad Software, Inc.) and SPSS v.22.0 (IBM
Corp.). For comparisons, one-way analysis of variance,
t-test, and Pearson chi-square test were performed as
indicated. Tukey's post hoc test was used for multiple comparisons
test. Disease free survival (DFS), defined as the time interval
between surgery to disease recurrence or mortality due to any
cause, was estimated using the Kaplan-Meier method and differences
among subgroups were investigated by the log-rank test. The
correlations between relative miR-638 expression and DNA
methylation level, as well as between relative miR-638 expression
and relative MEF2C mRNA expression, were determined using the
Spearman correlation coefficient. P<0.05 was considered to
indicate a statistically significant difference.
Results
miR-638 gene is hyper-methylated in
EC
To identify potential cancer-associated miRNAs whose
promoters were highly methylated in EC, the EC ISH and KLE cell
lines were treated with demethylation agents for 72 h, and the
expression levels of 1,347 common human miRNAs were measured using
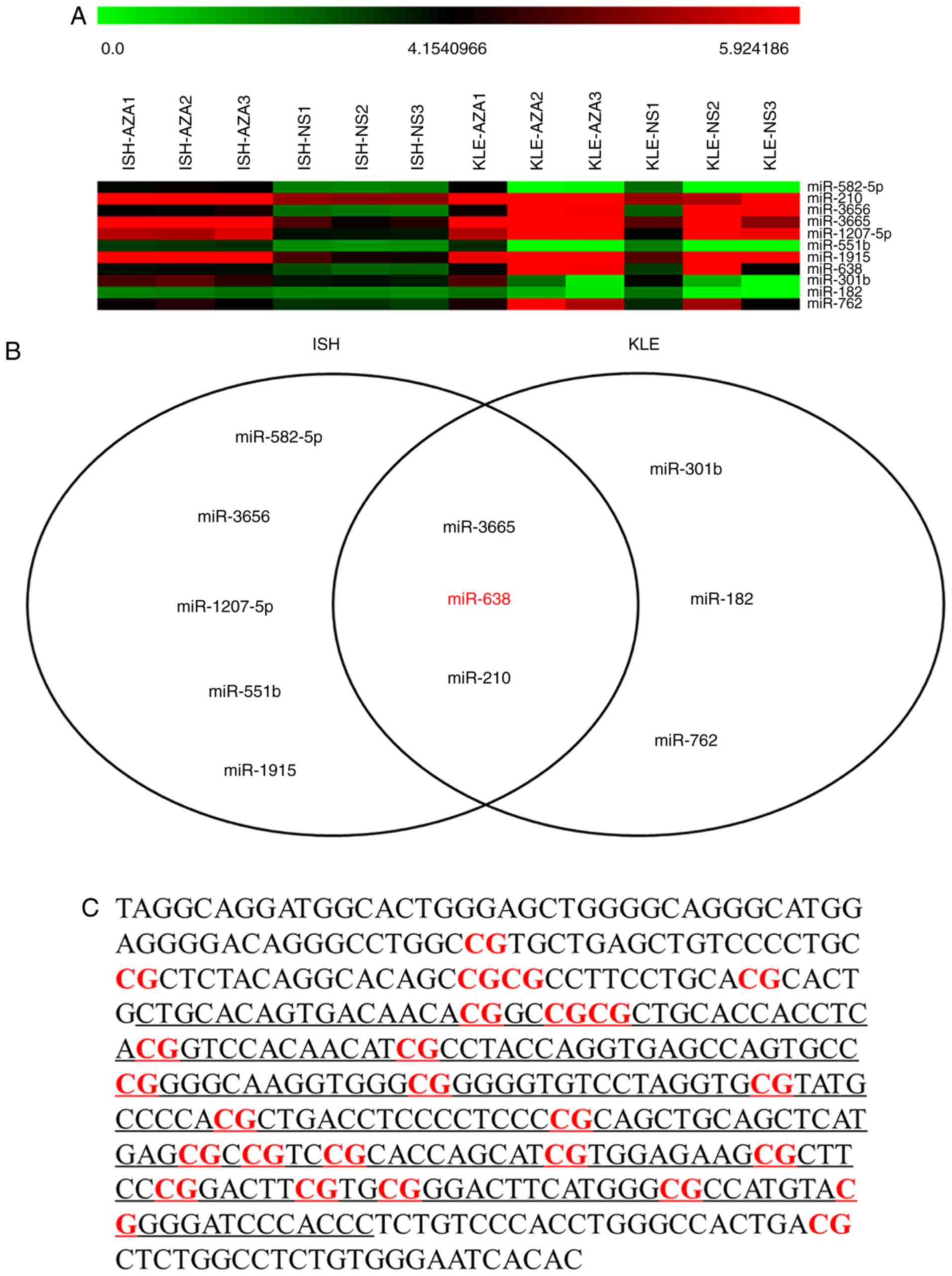
micro-array analysis (Fig. 1A).
With the cut-off criteria of P<0.05 and fold change >2,
miR-638, miR-3665 and miR-210 were revealed to be significantly
upregulated in both cell lines (Fig.
1B). miR-638 was selected for further study as it was a newly
identified cancer-associated miRNA, and data concerning its
specific biological roles in different types of human cancer are
conflicting (9). A total of 2
CpG-rich regions at/near the promoter of miR-638 gene were
identified using CpG Island Searcher (15). The first CpG island overlapped
with the open reading frame sequence of the miR-638 gene and the
second CpG island was located about 5kb upstream of the miR-638
gene (Fig. 1C). The methylation
statuses of consecutive CpG sites in the second CpG island were
highly concordant (Fig. 1D) and
the mean value was used as representative of the methylation level
of the miR-638 gene. A significantly higher degree of DNA
methylation was identified in the cancerous tissues compared with
the non-cancerous tissues among clinical samples from 42 patients
with EC (Fig. 1E). The
associations between the DNA methylation level of miR-638 in
cancerous tissues with various clinicopathological parameters,
including age, primary tumor size, Federation of Gynecology and
Obstetrics (FIGO) stage (2014 version) (16), tumor differentiation and tumor
histology were also analyzed. A significant positive correlation
between miR-638 promoter methylation level and FIGO stage was
identified (Fig. 1F). Of note, no
statistical significance was identified when comparing the
methylation (P=0.679) or the relative expression (P=0.523) levels
of miR-638 in the cancerous tissue from the 36 patients with
endometrial adenocarcinoma with that from the 6 patients with
serous carcinoma.
miR-638 expression is downregulated in
EC
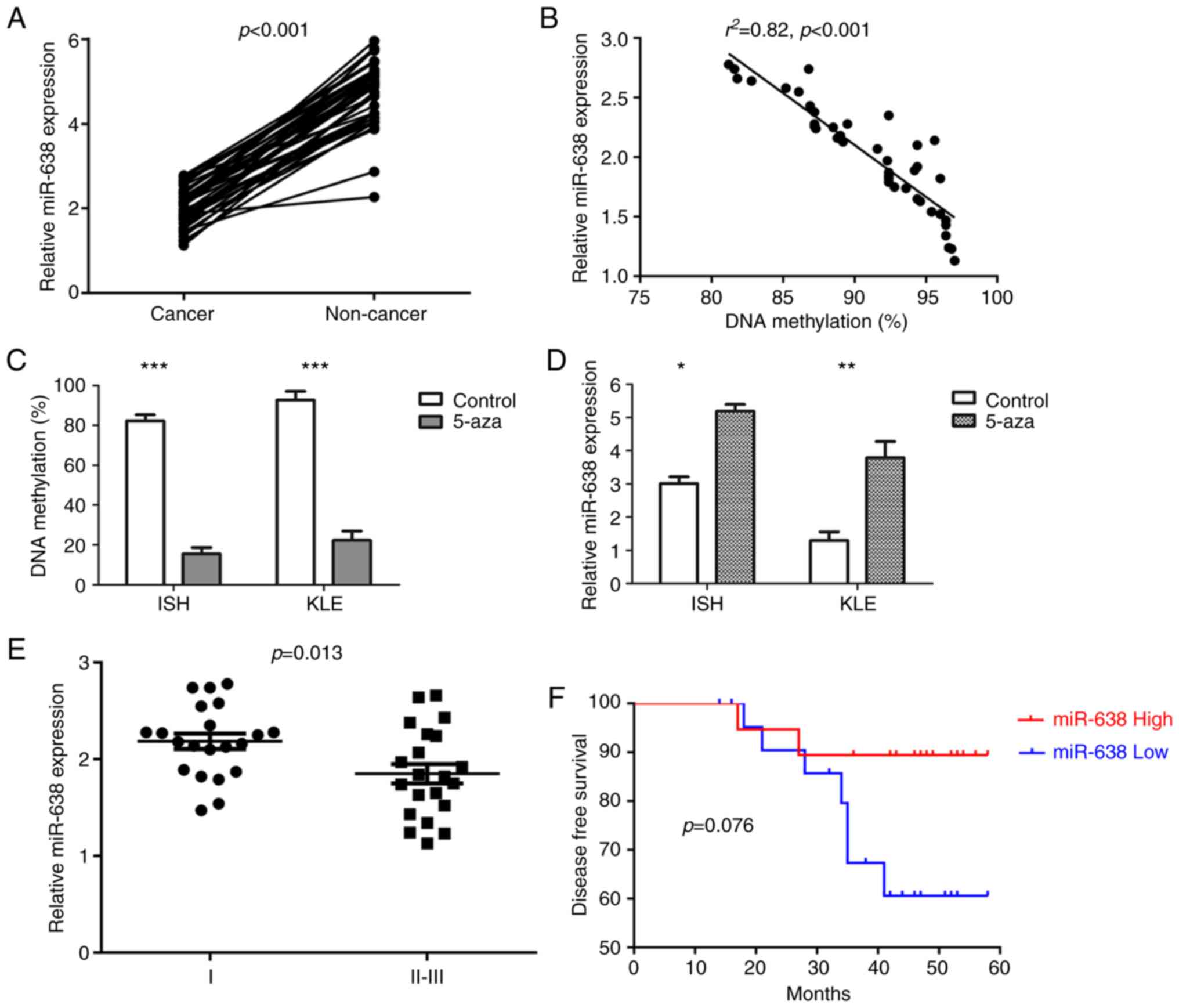
To examine the effect of miR-638 promoter region
methylation on miR-638 expression, the relative miR-638 expression
levels were measured in 42 paired EC samples. A significant
downregulation of miR-638 expression in cancerous tissues was
detected (Fig. 2A). Furthermore,
a significant inverse correlation between miR-638 DNA methylation
and miR-638 expression was demonstrated (Fig. 2B). Additionally, DNA methylation
and miR-638 expression levels were investigated in ISH and KLE cell
lines treated with 5-aza-2′-deoxycitidine. Both cell lines
exhibited hypermethylation of the miR-638 promoter region prior to
treatment and the DNA methylation levels decreased significantly
(Fig. 2C), whereas miR-638
expression was increased significantly following treatment
(Fig. 2D).
When examining the association between miR-638
expression with the aforementioned common clinical variables, a
significant negative correlation between miR-638 expression and
FIGO stage was identified (Fig.
2E). Furthermore, with a median follow-up of 48.0 months
(range, 14-58), 9 patients exhibited disease recurrence, and those
with increased expression levels of miR-638 exhibited a prolonged
DFS (Fig. 2F).
miR-638 functions as a tumor suppressive
miRNA in EC
EC cell lines transfected with miR-638 mimics
(miR-638) or its corresponding negative control (miR-638-NC), were
constructed (Fig. 3A).
Overexpression of miR-638 repressed cell proliferation (Fig. 3B and C), migration (Fig. 3D) and invasion (Fig. 3E), and promoted cell apoptosis
(Fig. 3F). Furthermore, miR-638
overexpression led to the downregulation of MMP2, MMP9, VEGF,
cyclin D1, CDK2 and CDK4 (Fig.
3G).
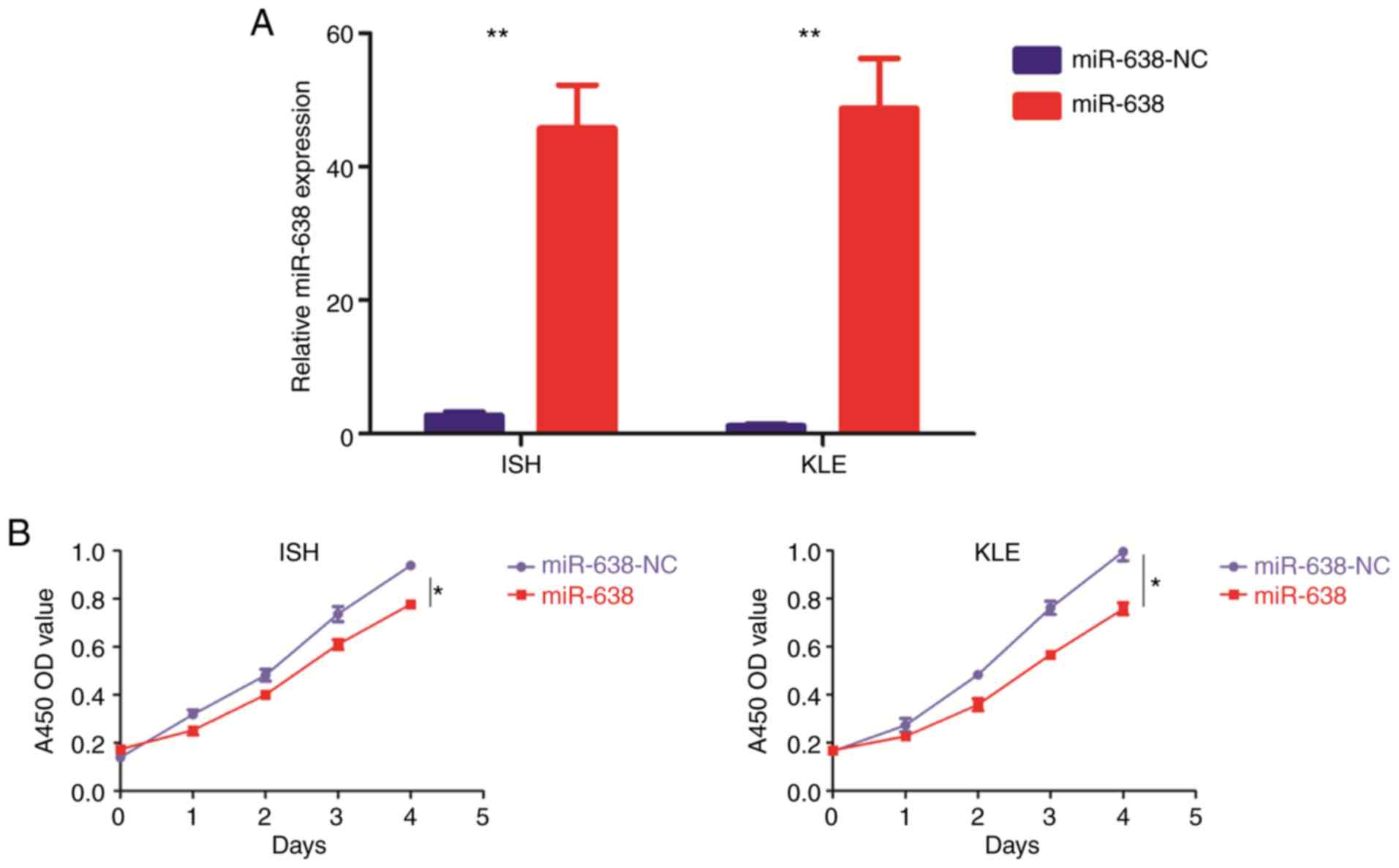 | Figure 3miR-638 functions as a
tumor-suppressive miRNA in endometrial carcinoma. The 2 endometrial
carcinoma ISH and KLE cell lines were stably transfected with an
miR-638 lentiviral vector (miR-638) or its negative control
(miR-638-NC), respectively. (A) Relative miR-638 expression was
examined by TaqMan quantitative polymerase chain reaction. (B and
C) Cell proliferation was examined using (B) Cell Counting Kit-8
and (C) colony formation assays. miR-638 functions as a
tumor-suppressive miRNA in endometrial carcinoma. The 2 endometrial
carcinoma ISH and KLE cell lines were stably transfected with an
miR-638 lentiviral vector (miR-638) or its negative control
(miR-638-NC), respectively. (D and E) Cell migration and invasion
were investigated by (D) migration and (E) invasion assays,
respectively. The number of cells that invaded through the membrane
was counted in 10 fields using a ×20 objective lens. Original
magnification, ×200. (F) At 48 h post-transfection, apoptosis was
measured by the flow cytometric analysis of cells stained with
Annexin V-FITC and PI. miR-638 functions as a tumor-suppressive
miRNA in endometrial carcinoma. The 2 endometrial carcinoma ISH and
KLE cell lines were stably transfected with an miR-638 lentiviral
vector (miR-638) or its negative control (miR-638-NC),
respectively. (G) Proteins that are frequently altered in various
types of human cancer were subjected to western blot analysis using
the indicated antibodies. (H) In vivo tumorigenicity was
investigated in a nude mouse xenograft model. Tumor size and weight
were measured on day 50 after cancer cell injection. All results
are presented as the mean ± standard deviation of values obtained
from 3 independent experiments. Statistical significance was
calculated using the Student's t-test. *P<0.05,
**P<0.01 and ***P<0.001. miR, microRNA;
FITC, fluorescein isothiocyanate; PI, propidium iodide; NC,
negative control. |
EC cell lines stably transfected with miR-638 mimics
or its negative control were subcutaneously implanted into nude
mice. It was identified that the overexpression of miR-638
significantly decreased tumor growth compared with controls
(Fig. 3H).
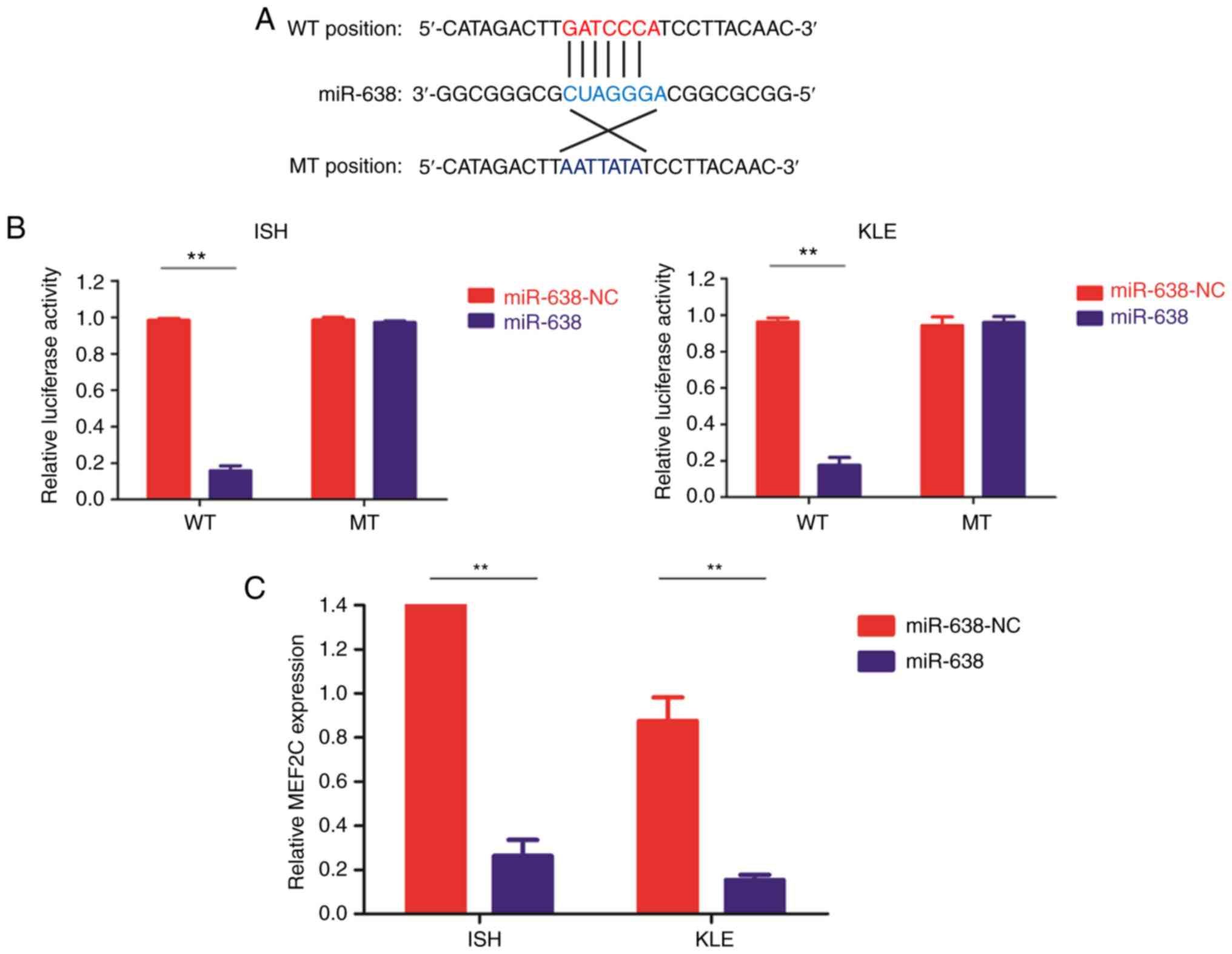
miR-638 directly targets MEF2C
MEF2C was identified to be one of the targets of
miR-638 using several commonly used microRNA database. To validate
this result, a luciferase reporter assay was employed. The entire
wild-type 3′-UTR of MEF2C or the mutant 3′-UTR with a 4 bp mutation
in the seed region was cloned downstream of the luciferase gene
open reading frame (Fig. 4A).
Luciferase activity was significantly decreased upon transfection
of miR-638 mimics in the wild type reporter. Conversely, luciferase
activity of mutant reporter was unaffected following transfection
with miR-638 mimics or the control vector (Fig. 4B). In addition, the MEF2C mRNA
(Fig. 4C) and protein (Fig. 4D) expression levels were
substantially decreased following miR-638 overexpression in both EC
cell lines. Furthermore, in the 42 paired clinical samples, MEF2C
mRNA levels were significantly increased in the cancerous tissues
(Fig. 4E) and a significant
inverse correlation between MEF2C mRNA level and miR-638 expression
was demonstrated (Fig. 4F). Of
note, no statistical significance was revealed when comparing the
MEF2C mRNA levels in the cancerous tissue from the 36 patients with
endometrial adenocarcinoma with that from the 6 patients with
serous carcinoma (P=0.472).
MEF2C mediates the tumor suppressive
effect of miR-638 in EC
EC cell lines were transfected with lentivirus
encoding shMEF2C, which mediated the stable knockdown of endogenous
MEF2C. siRNA-mediated knockdown of MEF2C inhibited cell
proliferation, migration and invasion, but promoted cell apoptosis.
It also downregulated the expression levels of MMP2, MMP9, VEGF,
cyclin D1, CDK2 and CDK4 and repressed in vivo tumor growth
in mouse models (Fig. 5).
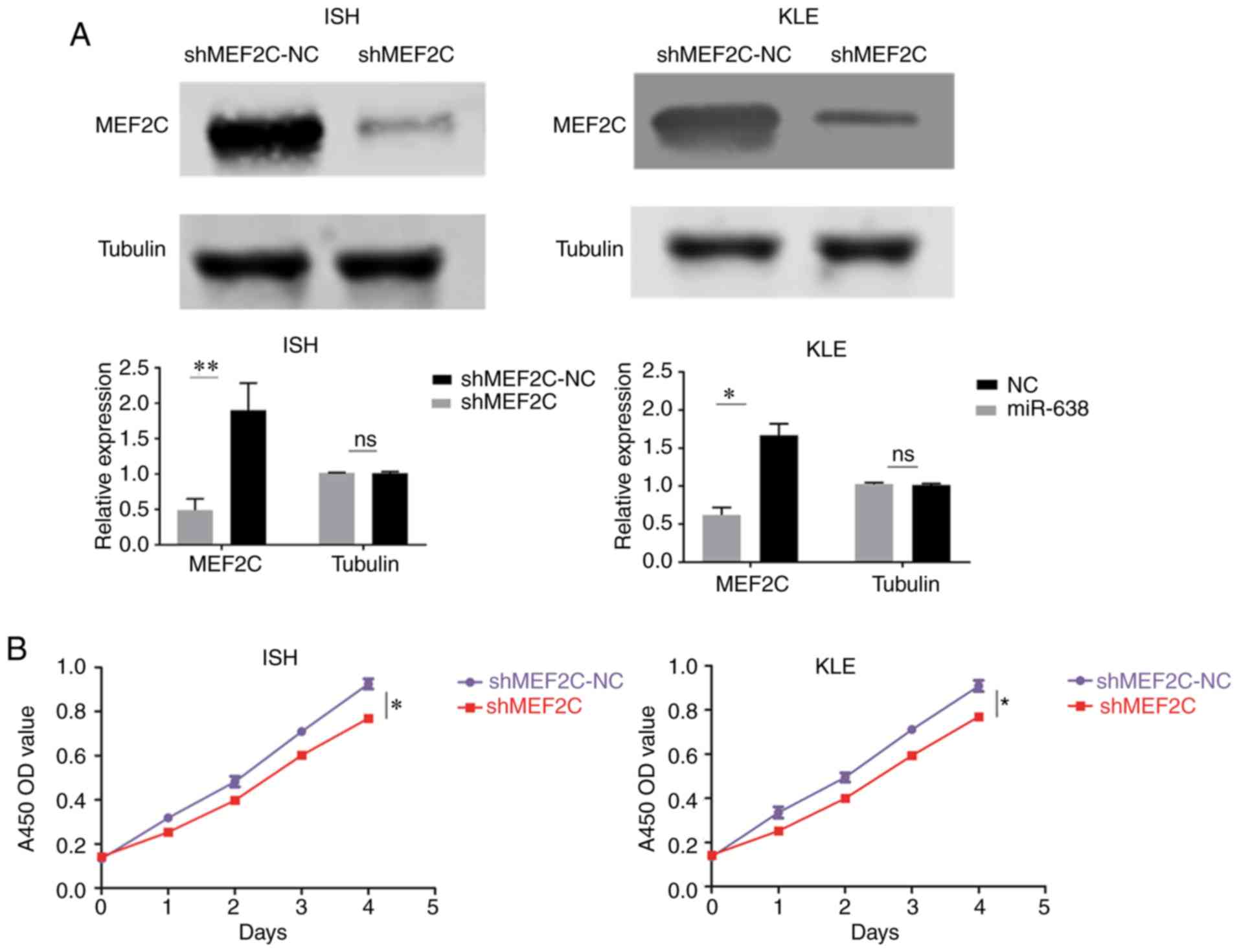 | Figure 5MEF2C functions as a onco-protein in
EC. The 2 EC ISH and KLE cell lines were stably transfected with
shMEF2C lentiviral vector (shMEF2C) or its negative control
(shMEF2C-NC), respectively. (A) MEF2C expression was examined by
western blot analysis using the indicated antibodies. (B and C)
Cell proliferation was examined by (B) Cell Counting Kit-8 and (C)
colony formation assays. (D) Cell migration and invasion were
investigated by migration and invasion assays, respectively. The
number of cells that invaded through the membrane was counted in 10
fields using a ×20 objective lens. Original magnification, ×200.
MEF2C functions as a oncoprotein in EC. (E) Cell migration and
invasion were investigated by migration and invasion assays,
respectively. The number of cells that invaded through the membrane
was counted in 10 fields using a ×20 objective lens. Original
magnification, ×200. (F) At 48 h post-transfection, apoptosis was
measured by the flow cytometric analysis of cells stained with
Annexin V-FITC and PI. Statistical significance was calculated
using the Student's t-test. (G) MMP2, MMP9, VEGF, cyclin D1, CDK2
and CDK4 expression levels were examined using western blot
analysis with the indicated antibodies. Tubulin expression was
selected as a control. MEF2C functions as a oncoprotein in EC. (H)
In vivo tumorigenicity was investigated in a nude mouse
xenograft model. Tumor size and weight were measured on day 50
after cancer cell injection. All results are presented as the mean
± standard deviation of values obtained from 3 independent
experiments. *P<0.05, **P<0.01 and
***P<0.001. MEF2C, myocyte enhancer factor 2C; EC,
endometrial carcinoma; sh, short hairpin; miR, microRNA; FITC,
fluorescein isothiocyanate; PI, propidium iodide; NC, negative
control; MMP, matrix metalloproteinase; VEGF, vascular endothelial
growth factor; CDK, cyclin-dependent kinase. |
Next, a rescue experiment was performed to examine
the biological and molecular associations between MEF2C and miR-638
in EC. A specific lentiviral vector (MEF2C analog) that encoded the
entire coding sequence of MEF2C but lacked its 3′-UTR was
constructed, in order to ectopically express MEF2C without
affecting the expression of miR-638 and its numerous other targets.
EC cell lines stably transfected with miR-683 mimics and/or MEF2C
analog were selected, and MEF2C expression levels in these cells is
presented in Fig. S1.
Overexpressing miR-638 led to significant downregulation of MEF2C,
while co-transfection of miR-638 mimics and the MEF2C analog
(miR-638 + MEF2C) rescued the expression of MEF2C. Using the 3
specifically designed EC cell lines (miR-638-NC, miR-638 and
miR-638 + MEF2C), it was demonstrated that the ectopic expression
of MEF2C significantly abrogated the tumor-suppressive effect
induced by miR-638 (Fig. 6).
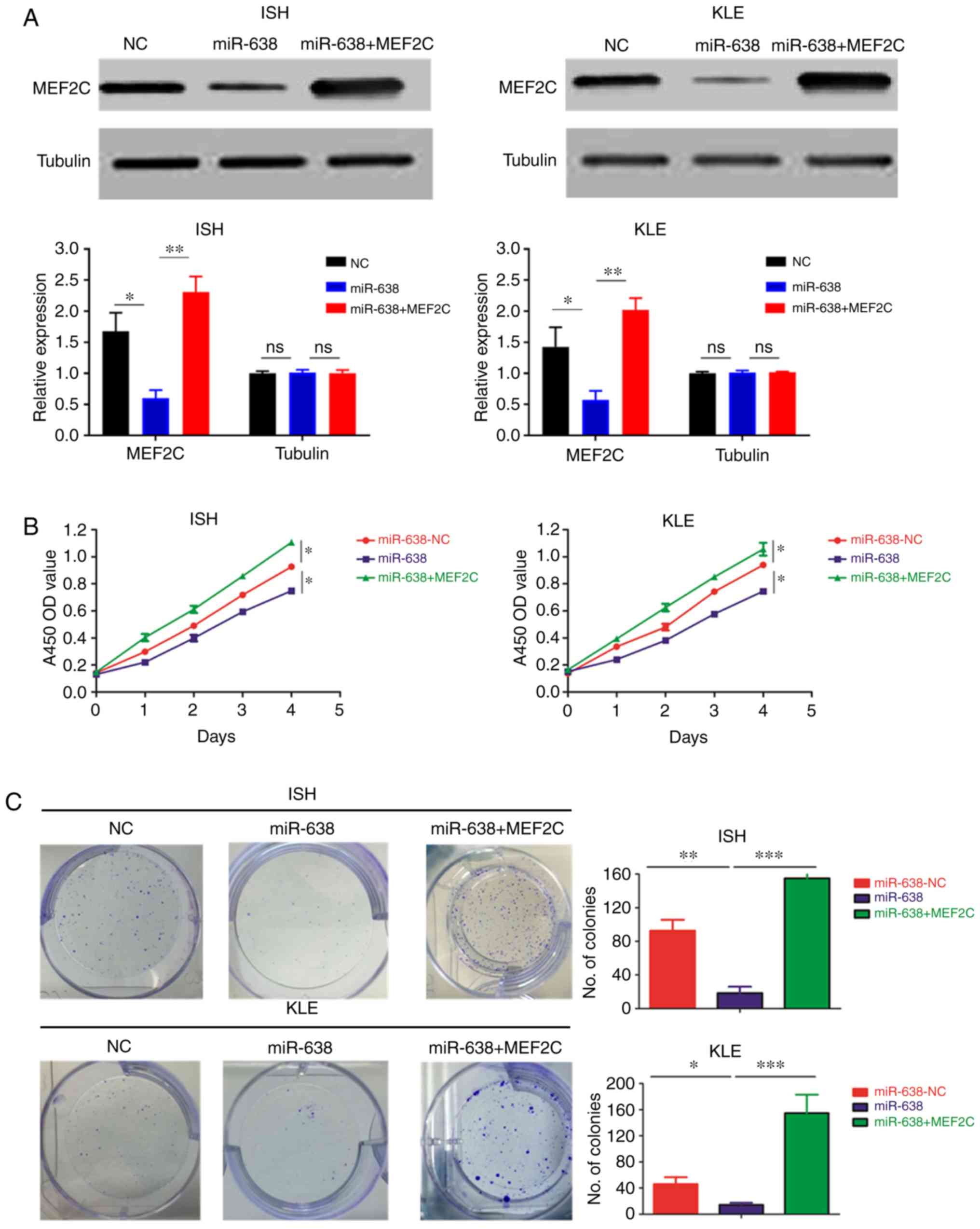 | Figure 6MEF2C mediates the tumor-suppressive
function of miR-638. A total of 3 cell lines were used in these
experiments: EC cell lines transfected with negative control
(miR-638-NC), miR-638 mimics (miR-638) and miR-638 mimics and a
MEF2C-expressing vector that encoded the entire coding sequence of
MEF2C but lacked the 3′-UTR (miR-638 + MEF2C). (A) MEF2C expression
was examined by western blot analysis using the indicated
antibodies. (B and C) Cell proliferation was examined by (B) Cell
Counting Kit-8 and (C) colony formation assays. MEF2C mediates the
tumor-suppressive function of miR-638. (D and E) Cell migration and
invasion were investigated using (D) migration and (E) invasion
assays, respectively. The number of cells that invaded through the
membrane was counted in 10 fields using a ×20 objective lens.
Original magnification, ×200. (F) At 48 h post-transfection,
apoptosis was measured by the flow cytometric analysis of cells
stained with Annexin V-FITC and PI. MEF2C mediates the
tumor-suppressive function of miR-638. (G) MMP2, MMP9, VEGF,
cyclin-D1, CDK2 and CDK4 expression levels were measured using
western blot analysis with the indicated antibodies. Tubulin
expression was selected as the control. (H) In vivo
tumorigenicity was investigated in a nude mouse xenograft model.
Tumor size and weight were measured on day 50 after cancer cell
injection. All results are presented as the mean ± standard
deviation of values obtained from 3 independent experiments.
Statistical significance was calculated using the Tukey's post hoc
test. *P<0.05, **P<0.01 and
***P<0.001. MEF2C, myocyte enhancer factor 2C; sh,
short hairpin; miR, microRNA; FITC, fluorescein isothiocyanate; PI,
propidium iodide; NC, negative control; MMP, matrix
metalloproteinase; VEGF, vascular endothelial growth factor; CDK,
cyclin-dependent kinase. |
Discussion
Epigenetic modifications of cancer-associated miRNAs
have been demonstrated to have important roles during tumorigenesis
(1,4). In the present study, miR-638 was
verified to be downregulated by promoter methylation in EC. The
promoter region of the miR-638 gene was hyper-methylated in samples
from patients with EC and EC cell lines, which is consistent with a
previous study of miR-638 in colorectal carcinoma (17). In addition, the promoter
methylation level of miR-638 and miR-638 expression were
significantly associated with FIGO stage. Furthermore, an increased
expression level of miR-638 was observed to be associated with
improved DFS. Notably, all of the patients in the present study
were grouped into FIGO stages I and II-III, and the majority of
patients were diagnosed FIGO stage I clinically, which was similar
to previous studies (18,19). Although a trend towards an
increased level of methylation and decreased expression of miR-638
gene were observed among patients with FIGO stage III disease when
compared with that of patients with FIGO stage II disease, no
statistical significance was identified, most likely due to the
small sample size. In order to further investigate the prognostic
significance of methylation status and expression level of miR-638
among patients with FIGO stage II and stage III separately, future
studies with larger sample size are required.
As is known, there are two main clinic-pathological
subtypes of EC; the estrogen-associated type I EC, primarily
endometrioid adenocarcinoma, and the non-estrogen-associated type
II EC, including uterine papillary serous carcinoma, clear cell
carcinoma and others. In the present study, down-regulation of
miR-638 by its promoter region methylation and upregulation of
MEF2C were observed in both endometrioid adenocarcinoma and serous
carcinoma, despite considerable differences in genetic backgrounds,
molecular aberrations and drug sensitivities being reported between
these 2 pathological subtypes of EC (20-22). Estrogen-associated type I EC
frequently exhibits mutations in PTEN, catenin beta 1, PIK3
catalytic subunit alpha, AT-rich interaction domain (ARID)1A,
KRAS and SWItch/Sucrose non-fermentable chromatin remodeling
complex genes such as ARID5B. Conversely, as one of the most common
type of non-estrogen-associated endometrial carcinoma, serous
carcinoma of the endometrium normally has extensive copy number
alterations, and often shares genomic features with ovarian serous
and basal-like breast carcinomas (20,21,23). Nevertheless, there are certain
similarities between these two different histologic subtypes. A
pooled analysis of individual-level data from 10 cohort and 14
case-control studies identified that patients with these 2
endometrial cancer types shared a number of common etiologic
factors, including parity, oral contraceptive use, cigarette
smoking, age at menarche and presence of diabetes (24). Furthermore, shared molecular
regulatory mechanisms among these 2 pathological subtypes of EC
have been previously reported, such as solute carrier family 1
member 5 regulating glutamine uptake and cell growth in EC
(25). In the present study, the
differences in methylation status and expression levels of miR-638,
as well as expression levels of MEF2C, were compared among
cancerous tissues and non-cancerous tissues in clinical samples
from patients with endometrioid adenocarcinoma and serous
carcinoma. Additionally, no statistical significance was identified
when comparing the methylation or expression levels of miR-638 gene
in the cancerous tissues from the patients with endometrioid
adenocarcinoma with that from the patients with serous carcinoma.
Furthermore, 2 different cell lines were selected for analysis in
the present study. ISH was isolated from a patient with estrogen
receptor-positive, well-differentiated endometrioid adenocarcinoma,
and KLE was isolated from a patient with less differentiated and
highly aggressive, type II endometrium carcinoma, and the results
were confirmed in both cell lines. Taken together, silencing
miR-638 through promoter methylation may be a shared epigenetic
regulatory mechanism among these 2 different histologic subtypes of
EC, which warrants future validation.
Additionally, miR-3665 and miR-210 were also
identified as potential miRNAs that may be epigenetically regulated
by promoter hypermethylation. miR-3665 is associated with influenza
virus (26) and tuberculosis
infection (27), but its
associations with human cancer has rarely been reported. However,
the downregulation of miR-210 expression by the hypermethylation of
its promoter region has been reported (28,29).
The biological roles of miR-638 appear to be cancer
type-dependent (6-9) and it functions as a tumor
suppressive miRNA in EC. Liu et al (30) reported that miR-638, along with 3
other miRNAs, predicted survival of patients with nasopharyngeal
carcinoma. miR-638 was upregulated in esophageal squamous cell
carcinoma and breast cancer cells, and it promoted starvation- and
rapamycin-induced autophagy by targeting DACT3/Wnt/β-catenin
signaling. Ma et al (6)
revealed that miR-638 was downregulated in colorectal cancer and
served as a tumor suppressive miRNA by targeting SRY-box
transcription factor 2. Similarly, Zhang et al (8) demonstrated that miR-638 mediated its
tumor suppressive function by targeting phospholipase D1 in gastric
cancer. All these data suggest that miR-638 is associated with
various processes during tumorigenesis, but mediates its versatile
effects by targeting distinct downstream genes in different types
of human cancer. To the best of our knowledge, the present study is
the first comprehensive investigation of the status and biological
role of miR-638 in EC. The results demonstrated that miR-638 was
downregulated by promoter hypermethylation and functioned as a
powerful tumor suppressive miRNA by targeting MEF2C in EC.
MEF2C, a myogenic differentiation transcription
factor, was recently characterized as a potent oncoprotein in
various types of human cancer, in particular leukemia (31,32). MEF2C was activated by NK2 homeobox
5 or BCR-ABL (33,34), participated in the regulation of
homing and invasiveness (35) and
was associated with chemotherapy resistance (36) or patient prognosis in various
types of leukemias (37).
Furthermore, several studies have investigated its expression and
biological function in solid tumors. Zhang et al (38) identified that Yin Yang-1
suppressed the invasion and metastasis of pancreatic ductal
adenocarcinoma by downregulating MMP10 via a mucin 4, cell surface
associated/Erb-B2 receptor tyrosine kinase 2/p38 mitogen-activated
protein kinase (MAPK)/MEF2C-dependent mechanism. Bai et al
(39) demonstrated that MEF2C
mediated VEGF-induced vasculogenic mimicry, angiogenesis and
migration/invasion, with involvement of the p38 MAPK and protein
kinase C signaling pathways, and that the nuclear translocation of
MEF2C inhibited cancer proliferation via blockade of Wnt/β-catenin
signaling in hepatocellular carcinoma. Ignatius et al
(40) revealed that the Notch
receptor 1/snail family transcriptional repressor 1/MEF2C pathway
regulated growth and self-renewal of tumor-propagating cells in
embryonal rhabdomyosarcoma. The present study demonstrated that the
downregulation of MEF2C inhibited cell proliferation, migration,
invasion and promoted cell apoptosis in EC, partly through the
upregulation of MMP2, MMP9, VEGF, cyclin D1, CDK2 and CDK4. It is
important to note that the expression of MEF2C was not successfully
significantly upregulated by transfecting a vector overexpressing
MEF2C. We hypothesized that there may be certain feedback
regulatory systems that affect the expression of MEF2C and it may
be difficult to further upregulate the expression of MEF2C in a
state of sustained high expression in EC cells. VEGF, MMP2 and MMP9
are well-recognized angiogenic factors in EC (41), whereas cyclin D1, CDK2 and CDK4
are common regulators of the cell cycle and apoptosis (42). Therefore, we hypothesized that
MEF2C promotes EC progression partly through the regulation of
angiogenesis, cell cycle and apoptosis, which warrant further
research. Additionally, CDK4/6 inhibitors such as palbociclib and
abemaciclib, were demonstrated to exhibit preliminary anti-tumor
potency in EC cell lines and animal models, and particularly in
patients with genomic aberrations that activate cyclin D1 (43,44). Further research focusing on the
detailed molecular networks of miR-638/MEF2C/cyclin D1 signaling
may lead to the improved identification of patients who could
benefit the most from CDK4/6 inhibitors.
Supplementary Data
Acknowledgements
Not applicable.
Funding
This study was supported by the Laboratory Animal
Project funded by the Shanghai Science and Technology Commission
(grant no. 14140901100) and by the National Natural Science
Foundation of China (grant no. 81902643).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HW and YR designed the study. JN and SL conducted
the experiments and wrote the manuscript. BS and WT performed the
experiments. JN, SL and YR analyzed the data. All authors read and
approved the final version of the manuscript.
Ethics committee approval and patient
consent
The study was approved by the Committee for the
Ethical Review of Research at Fudan University Shanghai Cancer
Center, and written informed consent was obtained from the
patients. The protocol for the animal study was approved by the
Committee on the Use of Live Animals in Teaching and Research,
Fudan University, Shanghai.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Holubekova V, Mendelova A, Jasek K,
Mersakova S, Zubor P and Lasabova Z: Epigenetic regulation by DNA
methylation and miRNA molecules in cancer. Future Oncol.
13:2217–2222. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ma J, Hong L, Chen Z, Nie Y and Fan D:
Epigenetic regulation of microRNAs in gastric cancer. Dig Dis Sci.
59:716–723. 2014. View Article : Google Scholar
|
|
3
|
Wang P, Chen L, Zhang J, Chen H, Fan J,
Wang K, Luo J, Chen Z, Meng Z and Liu L: Methylation-mediated
silencing of the miR-124 genes facilitates pancreatic cancer
progression and metastasis by targeting Rac1. Oncogene. 33:514–524.
2014. View Article : Google Scholar
|
|
4
|
Loginov VI, Rykov SV, Fridman MV and Braga
EA: Methylation of miRNA genes and oncogenesis. Biochemistry
(Mosc). 80:145–162. 2015. View Article : Google Scholar
|
|
5
|
Mei Q, Li X, Zhang K, Wu Z, Li X, Meng Y,
Guo M, Luo G, Fu X and Han W: Genetic and methylation-induced loss
of miR-181a2/181b2 within chr9q33.3 facilitates tumor growth of
cervical cancer through the PIK3R3/Akt/FoxO signaling pathway. Clin
Cancer Res. 23:575–586. 2017. View Article : Google Scholar
|
|
6
|
Ma K, Pan X, Fan P, He Y, Gu J, Wang W,
Zhang T, Li Z and Luo X: Loss of miR-638 in vitro promotes cell
invasion and a mesenchymal-like transition by influencing SOX2
expression in colorectal carcinoma cells. Mol Cancer. 13:1182014.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tay Y, Tan SM, Karreth FA, Lieberman J and
Pandolfi PP: Characterization of dual PTEN and p53-targeting
microRNAs identifies microRNA-638/Dnm2 as a two-hit oncogenic
locus. Cell Rep. 8:714–722. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang J, Bian Z, Zhou J, Song M, Liu Z,
Feng Y, Zhe L, Zhang B, Yin Y and Huang Z: MicroRNA-638 inhibits
cell proliferation by targeting phospholipase D1 in human gastric
carcinoma. Protein Cell. 6:680–688. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ren Y, Chen Y, Liang X, Lu Y, Pan W and
Yang M: MiRNA-638 promotes autophagy and malignant phenotypes of
cancer cells via directly suppressing DACT3. Cancer Lett.
390:126–136. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bhattacharya A, Schmitz U, Raatz Y,
Schönherr M, Kottek T, Schauer M, Franz S, Saalbach A, Anderegg U,
Wolkenhauer O, et al: miR-638 promotes melanoma metastasis and
protects melanoma cells from apoptosis and autophagy. Oncotarget.
6:2966–2980. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhao LY, Tong DD, Xue M, Ma HL, Liu SY,
Yang J, Liu YX, Guo B, Ni L, Liu LY, et al: MeCP2, a target of
miR-638, facilitates gastric cancer cell proliferation through
activation of the MEK1/2-ERK1/2 signaling pathway by upregulating
GIT1. Oncogenesis. 6:e3682017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wei H, Zhang JJ and Tang QL: MiR-638
inhibits cervical cancer metastasis through Wnt/β-catenin signaling
pathway and correlates with prognosis of cervical cancer patients.
Eur Rev Med Pharmacol Sci. 21:5587–5593. 2017.PubMed/NCBI
|
|
13
|
Li M, Wang J and Liu H: Downregulation of
miR-638 promotes progression of breast cancer and is associated
with prognosis of breast cancer patients. OncoTargets Ther.
11:6871–6877. 2018. View Article : Google Scholar
|
|
14
|
Zhang Y, Zhang D, Jiang J and Dong L: Loss
of miR-638 promotes invasion and epithelial-mesenchymal transition
by targeting SOX2 in hepatocellular carcinoma. Oncol Rep.
37:323–332. 2017. View Article : Google Scholar
|
|
15
|
Agarwal V, Bell GW, Nam JW and Bartel DP:
Predicting effective microRNA target sites in mammalian mRNAs.
Elife. 4:2015. View Article : Google Scholar
|
|
16
|
FIGO Committee on Gynecologic Oncology:
FIGO staging for carcinoma of the vulva, cervix, and corpus uteri.
Int J Gynaecol Obstet. 125:97–98. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang J, Fei B, Wang Q, Song M, Yin Y,
Zhang B, Ni S, Guo W, Bian Z, Quan C, et al: MicroRNA-638 inhibits
cell proliferation, invasion and regulates cell cycle by targeting
tetraspanin 1 in human colorectal carcinoma. Oncotarget.
5:12083–12096. 2014.PubMed/NCBI
|
|
18
|
Pecorelli S: Revised FIGO staging for
carcinoma of the vulva, cervix, and endometrium. Int J Gynaecol
Obstet. 105:103–104. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ouldamer L, Bendifallah S, Body G, Touboul
C, Graesslin O, Raimond E, Collinet P, Coutant C, Lavoué V, Lévêque
J, et al: Predicting poor prognosis recurrence in women with
endometrial cancer: A nomogram developed by the FRANCOGYN study
group. Br J Cancer. 115:1296–1303. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Cancer Genome Atlas Research Network;
Kandoth C, Schultz N, Cherniack AD, Akbani R, Liu Y, Shen H,
Robertson AG, Pashtan I, Shen R, et al: Integrated genomic
characterization of endometrial carcinoma. Nature. 497:67–73. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hecht JL and Mutter GL: Molecular and
pathologic aspects of endometrial carcinogenesis. J Clin Oncol.
24:4783–4791. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
McConechy MK, Ding J, Cheang MC, Wiegand
K, Senz J, Tone A, Yang W, Prentice L, Tse K, Zeng T, et al: Use of
mutation profiles to refine the classification of endometrial
carcinomas. J Pathol. 228:20–30. 2012.PubMed/NCBI
|
|
23
|
Murali R, Soslow RA and Weigelt B:
Classification of endometrial carcinoma: More than two types.
Lancet Oncol. 15:e268–278. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Setiawan VW, Yang HP, Pike MC, McCann SE,
Yu H, Xiang YB, Wolk A, Wentzensen N, Weiss NS, Webb PM, et al:
Type I and II endometrial cancers: Have they different risk
factors? J Clin Oncol. 31:2607–2618. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Marshall AD, van Geldermalsen M, Otte NJ,
Lum T, Vellozzi M, Thoeng A, Pang A, Nagarajah R, Zhang B, Wang Q,
et al: ASCT2 regulates glutamine uptake and cell growth in
endometrial carci-noma. Oncogenesis. 6:e3672017. View Article : Google Scholar
|
|
26
|
Gao J, Gao L, Li R, Lai Z, Zhang Z and Fan
X: Integrated analysis of microRNA-mRNA expression in A549 cells
infected with influenza A viruses (IAVs) from different host
species. Virus Res. 263:34–46. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Das K, Saikolappan S and Dhandayuthapani
S: Differential expression of miRNAs by macrophages infected with
virulent and avirulent mycobacterium tuberculosis. Tuberculosis
(Edinb). 93(Suppl): S47–S50. 2013. View Article : Google Scholar
|
|
28
|
Kiga K, Mimuro H, Suzuki M,
Shinozaki-Ushiku A, Kobayashi T, Sanada T, Kim M, Ogawa M, Iwasaki
YW, Kayo H, et al: Epigenetic silencing of miR-210 increases the
proliferation of gastric epithelium during chronic Helicobacter
pylori infection. Nat Commun. 5:44972014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xiong L, Wang F, Huang X, Liu ZH, Zhao T,
Wu LY, Wu K, Ding X, Liu S, Wu Y, et al: DNA demethylation
regulates the expression of miR-210 in neural progenitor cells
subjected to hypoxia. FEBS J. 279:4318–4326. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu N, Cui RX, Sun Y, Guo R, Mao YP, Tang
LL, Jiang W, Liu X, Cheng YK, He QM, et al: A four-miRNA signature
identified from genome-wide serum miRNA profiling predicts survival
in patients with nasopharyngeal carcinoma. Int J Cancer.
134:1359–1368. 2014. View Article : Google Scholar
|
|
31
|
Cante-Barrett K, Pieters R and Meijerink
JP: Myocyte enhancer factor 2C in hematopoiesis and leukemia.
Oncogene. 33:403–410. 2014. View Article : Google Scholar
|
|
32
|
Homminga I, Pieters R, Langerak AW, de
Rooi JJ, Stubbs A, Verstegen M, Vuerhard M, Buijs-Gladdines J, Kooi
C, Klous P, et al: Integrated transcript and genome analyses reveal
NKX2-1 and MEF2C as potential oncogenes in T cell acute
lymphoblastic leukemia. Cancer Cell. 19:484–497. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Nagel S, Meyer C, Quentmeier H, Kaufmann
M, Drexler HG and MacLeod RA: MEF2C is activated by multiple
mechanisms in a subset of T-acute lymphoblastic leukemia cell
lines. Leukemia. 22:600–607. 2008. View Article : Google Scholar
|
|
34
|
Agatheeswaran S, Singh S, Biswas S, Biswas
G, Chandra Pattnayak N and Chakraborty S: BCR-ABL mediated
repression of miR-223 results in the activation of MEF2C and PTBP2
in chronic myeloid leukemia. Leukemia. 27:1578–1580. 2013.
View Article : Google Scholar
|
|
35
|
Schwieger M, Schuler A, Forster M,
Engelmann A, Arnold MA, Delwel R, Valk PJ, Löhler J, Slany RK,
Olson EN and Stocking C: Homing and invasiveness of MLL/ENL
leukemic cells is regulated by MEF2C. Blood. 114:2476–2488. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Brown FC, Still E, Koche RP, Yim CY, Takao
S, Cifani P, Reed C, Gunasekera S, Ficarro SB, Romanienko P, et al:
MEF2C phosphorylation is required for chemotherapy resistance in
acute myeloid leukemia. Cancer Discov. 8:478–497. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Laszlo GS, Alonzo TA, Gudgeon CJ,
Harrington KH, Kentsis A, Gerbing RB, Wang YC, Ries RE, Raimondi
SC, Hirsch BA, et al: High expression of myocyte enhancer factor 2C
(MEF2C) is associated with adverse-risk features and poor outcome
in pediatric acute myeloid leukemia: A report from the Children's
Oncology Group. J Hematol Oncol. 8:1152015. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Zhang JJ, Zhu Y, Xie KL, Peng YP, Tao JQ,
Tang J, Li Z, Xu ZK, Dai CC, Qian ZY, et al: Yin Yang-1 suppresses
invasion and metastasis of pancreatic ductal adenocarcinoma by
down-regulating MMP10 in a MUC4/ErbB2/p38/MEF2C-dependent
mechanism. Mol Cancer. 13:1302014. View Article : Google Scholar
|
|
39
|
Bai XL, Zhang Q, Ye LY, Liang F, Sun X,
Chen Y, Hu QD, Fu QH, Su W, Chen Z, et al: Myocyte enhancer factor
2C regulation of hepatocellular carcinoma via vascular endothelial
growth factor and Wnt/β-catenin signaling. Oncogene. 34:4089–4097.
2015. View Article : Google Scholar
|
|
40
|
Ignatius MS, Hayes MN, Lobbardi R, Chen
EY, McCarthy KM, Sreenivas P, Motala Z, Durbin AD, Molodtsov A,
Reeder S, et al: The NOTCH1/SNAIL1/MEF2C pathway regulates growth
and self-renewal in embryonal rhabdomyosarcoma. Cell Rep.
19:2304–2318. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mahecha AM and Wang H: The influence of
vascular endothelial growth factor-A and matrix metalloproteinase-2
and -9 in angiogenesis, metastasis, and prognosis of endometrial
cancer. OncoTargets Ther. 10:4617–4624. 2017. View Article : Google Scholar
|
|
42
|
Golsteyn RM: Cdk1 and Cdk2 complexes
(cyclin dependent kinases) in apoptosis: A role beyond the cell
cycle. Cancer Lett. 217:129–138. 2005. View Article : Google Scholar
|
|
43
|
Dosil MA, Mirantes C, Eritja N, Felip I,
Navaridas R, Gatius S, Santacana M, Colàs E, Moiola C,
Schoenenberger JA, et al: Palbociclib has antitumour effects on
Pten-deficient endometrial neoplasias. J Pathol. 242:152–164. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gong X, Litchfield LM, Webster Y, Chio LC,
Wong SS, Stewart TR, Dowless M, Dempsey J, Zeng Y, Torres R, et al:
Genomic aberrations that activate D-type cyclins are associated
with enhanced sensitivity to the CDK4 and CDK6 inhibitor
abemaciclib. Cancer Cell. 32:761–776. e7662017. View Article : Google Scholar : PubMed/NCBI
|