Introduction
Extracellular acidosis is a common abnormality of
the cellular microenvironment, and can be caused by various
pathological states, including hypoxia, ischemia reperfusion,
severe infection and renal dysfunction (1). Extracellular acidosis may reduce
tissue pH to 6.5 or even 6.0 (2).
Cardiomyocytes are sensitive to extracellular acidosis, since they
become prone to apoptosis, which leads to cardiac dysfunction
(3-5). Previous studies have indicated that
mitochondrial damage caused by extracellular acidosis plays an
important role in cell death (6).
Tumor necrosis factor receptor-associated protein 1
(TRAP1) belongs to the heat shock protein 90 family and is located
in the mitochondria. It was first identified for its role in
anti-oxidative stress and maintaining mitochondrial integrity
(7,8). TRAP1 is widely distributed in
various types of cells, including cardiomyocytes. Previous studies
have demonstrated that the expression of TRAP1 is increased by
multiple pathological stimuli, such as hypoxia, ischemia, glucose
deprivation and oxidative stress, in order to prevent cell death in
vital organs (9-11). However, whether the expression of
TRAP1 is protectively increased in cardiomyocytes under
extracellular acidosis remains unknown (7,12,13).
The present study thus aimed to investigate whether
TRAP1 protects cardiomyocytes in acidic medium, and whether
mitochondrial permeability transition pore (MPTP) opening is
involved in this process.
Materials and methods
Cell culture and acidosis treatment
Cell culture methods were performed as previously
described (14). The rat heart
cell line, H9C2, was purchased from Cell Bank of the Chinese
Academy of Sciences (cat. no. GNR 5). The cells were cultured in
Dulbecco's modified Eagle's medium (DMEM; cat. no. 11965175; Gibco;
Thermo Fisher Scientific, Inc.) with high glucose supplemented with
10% fetal bovine serum (BSA; cat. no. 10099141C Gibco; Thermo
Fisher Scientific, Inc.) and 1% penicillin/streptomycin (cat. no.
516106; Sigma-Aldrich; Merck KGaA). After two passages and at
60-70% confluency, cells were transferred to 96- or 6-well plates
or other dishes according to different assays. The pH of the cell
culture medium was adjusted to the desired values (7.4, 7.0, 6.5 or
6.0) using 2-(N-morpholino)ethanesulfonic acid (MES) buffer (cat.
no. M8010; Beijing Solarbio Science & Technology Co.,
Ltd.).
Western blot analysis
Western blot analysis was conducted as previously
described (15). Cell lysates
were prepared with radioimmunoprecipitation assay lysis buffer
(cat. no. P0013B; Beyotime Institute of Biotechnology) in the
presence of a protease inhibitor cocktail (cat. no. CW2200S;
Cwbio). Protein concentrations of cell lysates were quantified
using a Pierce BCA Protein assay kit (cat. no. 23227; Thermo Fisher
Scientific, Inc.). A total of 30 µg total proteins were
loaded onto each lane and separated via 10% sodium dodecyl sulfate
polyacrylamide gel electrophoresis (SDS-PAGE). After the proteins
were transferred to a PVDF membrane (cat. no. IPVH00010; Milipore;
Merck KGaA), the membrane were blocked in 5% non-fat milk (cat. no.
P0216; Beyotime Institute of Biotechnology) in TBST (150 mM
Tris-HCl, pH 7.4, 1.5 M NaCl, 0.5% Tween-20) for 1 h at room
temperature, incubated with primary antibodies in blocking solution
at 4°C overnight, washed with TBST (five times, 5 min each),
incubated with secondary antibodies for 1 h at room temperature and
washed again with TBST (5 times, 5 min each). Antibodies against
TRAP1 (1:1,000) and Bcl-2 (1:1,000) were purchased from Abcam (cat.
nos. ab64182 and ab196495, respectively). Antibodies against Bax
(1:1,000), cleaved caspase-3 (1:1,000) and caspase-3 (1:1,000) were
purchased from Cell Signaling Technology, Inc. (cat. nos. 5023,
9661 and 9662, respectively). Antibody against GAPDH (1:1,000) was
purchased from ProteinTech Group, Inc. (cat. no. 10494-1-AP).
HRP-conjugated secondary anti-body (1:5,000) was obtained from
Cwbio (cat. no. CW0103; Cwbio). Bands were visualized by
chemiluminescence with Immobilon Western Chemiluminescent HRP
Substrate (cat. no. WBKLS; Millipore; Merck KGaA) and ChemiDoc
Imaging System (Bio-Rad Laboratories, Inc.). Blots were
semi-quantified using ImageJ (v2.1.4.8; National Institute of
Health).
Mitochondrial membrane potential (MMP)
assay
MMP was detected with tetramethylrhodamine, methyl
ester (TMRM; cat. no. I34361; Thermo Fisher Scientific, Inc.)
according to the manufacturer's instructions. Briefly, cells were
cultured with 50 nmol/l TMRM in serum-free medium at 37°C for 30
min, washed with PBS 3 times and then observed under a laser
scanning confocal microscope (Leica TCS SP8; Leica Microsystems,
Inc.) or analyzed by flow cytometry (FACSCanto II; BD
Biosciences).
Cell viability detection
Cell viability was quantified using a Cell Counting
kit-8 (CCK-8; cat. no. CK04; Dojindo Molecular Technologies, Inc.)
as previously described (16).
Cells were seeded into 96-well plates at a density of 2,000 cells
per well in complete culture medium. Three replicates were set up
for each group. After 24 h, the culture media were changed to DMEM
with different pH values (7.4 and 6.5), and the cells were cultured
for the required time according to the corresponding experimental
group settings. Subsequently, 10 µl CCK-8 was added to each
well and incubated for an additional 1.5 h. Optical density values
were measured at a 450-nm wavelength on a microplate reader
(Multiskan™ FC; Thermo Fisher Scientific, Inc.).
Cell apoptosis detection
7-Aminoactinomycin D (7-AAD) and Annexin
V-allophycocyanin (APC) flow cytometry assay (cat. no.
70-AP105-100; MultiSciences) were used to detect cell apoptosis
according to the manufacturer's instructions. Briefly, cells were
incubated in media at different pH values (7.4, 7.0, 6.5 and 6.0)
for a certain periods of time (0, 2, 4, 8, 12 and 24 h) and
harvested in PBS. The cell pellet was then resuspended in binding
buffer and stained with APC-conjugated Annexin V and 7-AAD for 10
min. The cells were then analyzed using a flow cytometer (FACSCanto
II; BD Biosciences) and the apoptotic rate of the cells was
analyzed using FLOWJO (v10; BD Biosciences).
Recombinant lentivirus vector for TRAP1
overexpression and silencing
The recombinant lentivirus vector for the
overexpression and silencing of TRAP1 was purchased from
GeneCopoeia, Inc. The targeting sequence of the small interfering
RNA (siRNA) against rat TRAP1 was 5′-AGACCAAGGCTACGGATAT-3′. A
green fluorescent protein (GFP)-expressing sequence and an
anti-puromycin sequence were also constructed into all vectors. The
multiplicity of infection in H9C2 myocardial cells was 10. As
1×106 cells were seeded for transfection, 107
vg lentivirus and 5 µg/ml polybrene (GeneCopoeia, Inc.) were
added to the culture medium without fetal bovine serum. At 72 h
post-infection, the cells were selected by puromycin to obtain H9C2
cells stably overexpressing TRAP1 or cells in which TRAP1 was
silenced.
Immunofluorescence
Immunofluorescence assay was performed as previously
described (17). Cells were
seeded and proliferated for 24 h until 50% confluence. The cells
were then treated with media at different pH values (7.4 and 6.5)
for the required periods of time (12 h) and fixed with 4%
paraformaldehyde (cat. no. P0099; Beyotime Institute of
Biotechnology) for 15 min. Subsequently, 0.1% Triton X-100 (cat.
no. T9284; Sigma-Aldrich; Merck KGaA) was used for cell
permeabilization. The cells were then blocked with 3% BSA (cat. no.
ST023; Beyotime Institute of Biotechnology) for 1 h. After washing
with PBS 3 times, a primary anti-TRAP1 rabbit antibody (1:300; cat.
no. ab64182; Abcam) was incu-bated with the cells at 4°C overnight,
and then a secondary antibody conjugated to Cy3 (1:200; cat. no.
93-6903-250; MultiSciences) was incubated with the cells at room
temperature for 1 h. The fluorescence intensity was observed using
a laser scanning confocal microscope (Leica TCS SP8; Leica
Microsystems, Inc.).
Reactive oxygen species (ROS) assay
Cell ROS levels were detected using CellROS
Oxidative Stress Reagents (cat. no. C10443; Thermo Fisher
Scientific, Inc.) as previously described (18). The cells were cultured in media
with different pH values (7.4 and 6.5) for the corresponding time
periods (12 and 24 h). The cells were then incubated with
serum-free medium containing 5 µm CellROX reagent for 30 min
at 37°C and washed with PBS 3 times. Subsequently, the cells were
analyzed using a flow cytometer (FACSCanto II; BD Biosciences) and
FLOWJO (v10; BD Biosciences).
ATP assay
Cell ATP levels were detected using an Enhanced ATP
Assay kit (cat. no. S0027; Beyotime Institute of Biotechnology)
according to the manufacturer's instructions. Briefly, the cells
were cultured in media with different pH values (7.4 and 6.5) for
the corresponding time periods (12 h). Subsequently, every
106 cells in different groups were added to 100
µl lysis buffer provided with the kit and the cell
supernatant was centrifuged at 12,000 × g for 5 min at 4°C to
remove cell debris. A total of 10 µl of supernant was then
mixed with 100 µl of ATP detection solution diluted 4 times
with dilution buffer and the luminescence was measured using a
luminometer (Varioskan Flash; Thermo Fisher Scientific, Inc.). The
luminescence values of ATP standards were determined in a similar
manner. The ATP concentration was calculated according to an
ATP-standard curve and normalized to protein concentration of the
supernatant.
Transmission electron microscopy
The mitochondrial ultrastructure were observed using
a transmission electron microscope (Transmission Electron
Microscope HT7700; Hitachi). H9C2 cells were collected and fixed in
4% glutaraldehyde for 1 h at room temperature and left at 4°C
overnight. The samples were dehydrated through a graded ethanol
series, then incubated in 100% ethanol and propylene oxide as well
as 2 exchanges of pure propylene oxide. Samples were embedded in
epoxy resin and polymerized at 60°C for 48 h. Specimens were cut
into 70-80-nm ultra-thin sections, then mounted on 300-mesh copper
grids. Sections were stained with uranyl acetate and leas citrate,
then subjected to observation.
Detection of MPTP opening
The MPTP opening of the H9C2 cells was detected
using a MitoProbe Transition Pore assay kit (cat. no. M34153;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Briefly, cells were seeded in a 6-well plate (cat.
no. 354773; Corning Inc.) and cultured in media with different pH
values (7.4 and 6.5) for the required time period (12 h) after
reaching 50-60% confluence. The cells were then collected and
resuspended in Hanks' Balanced Salt Solution (HBSS)/Ca buffer.
Subsequently, 5 µl Calcein AM working buffer (cat. no.
M34153; Thermo Fisher Scientific, Inc.) and CoCl2 were
added to the cell suspension solution, which was subsequently
incubated for 30 min at 37°C. After staining, the samples were
analyzed using a flow cytometer (FACSCanto II; BD Biosciences) with
488-nm excitation and 517-nm emission wavelengths and FLOWJO (v10;
BD Biosciences).
Chemical reagents
Mitochondrial permeabolity transition pore (MPTP)
opening inhibitor, cyclosporin A (cat. no. HY-B0579;
MedChemExpress), was used to inhibit MPTP opening (19). And MPTP opening promoter,
atractyloside (cat. no. HY-N0237; MedChemExpress), was used to
promote MPTP opening (20).
Briefly, 5 mg cyclosporin A powder were dissolved in 0.4158 ml
dimethyl sulfoxide (DMSO) to prepare stock solutions (10 mM). A
total of 5 mg atractyloside power was dissolved in 1.1148 ml DMSO
to prepare a stock solution (10 mM). Subsequently, 1 µl
stock solution (cyclosporin A or atractyloside respectively) was
added to 1 ml culture medium (pH 6.5) as a working solution (10
µM), and the cells were then cultured for 12 h,
respectively.
Statistical analysis
Data represent the means ± SD from 3 independent
experiments and were analyzed using SPSS v25.0 (IBM Corp.). The
Student's t-test was used to analyze the results of CCK-8 assay at
each time point between the 2 groups in Fig. 1C. One-way analysis of variance
followed by Tukey's post hoc test was carried out to measure
differences between groups. P<0.05 was considered to indicate a
statistically significant difference.
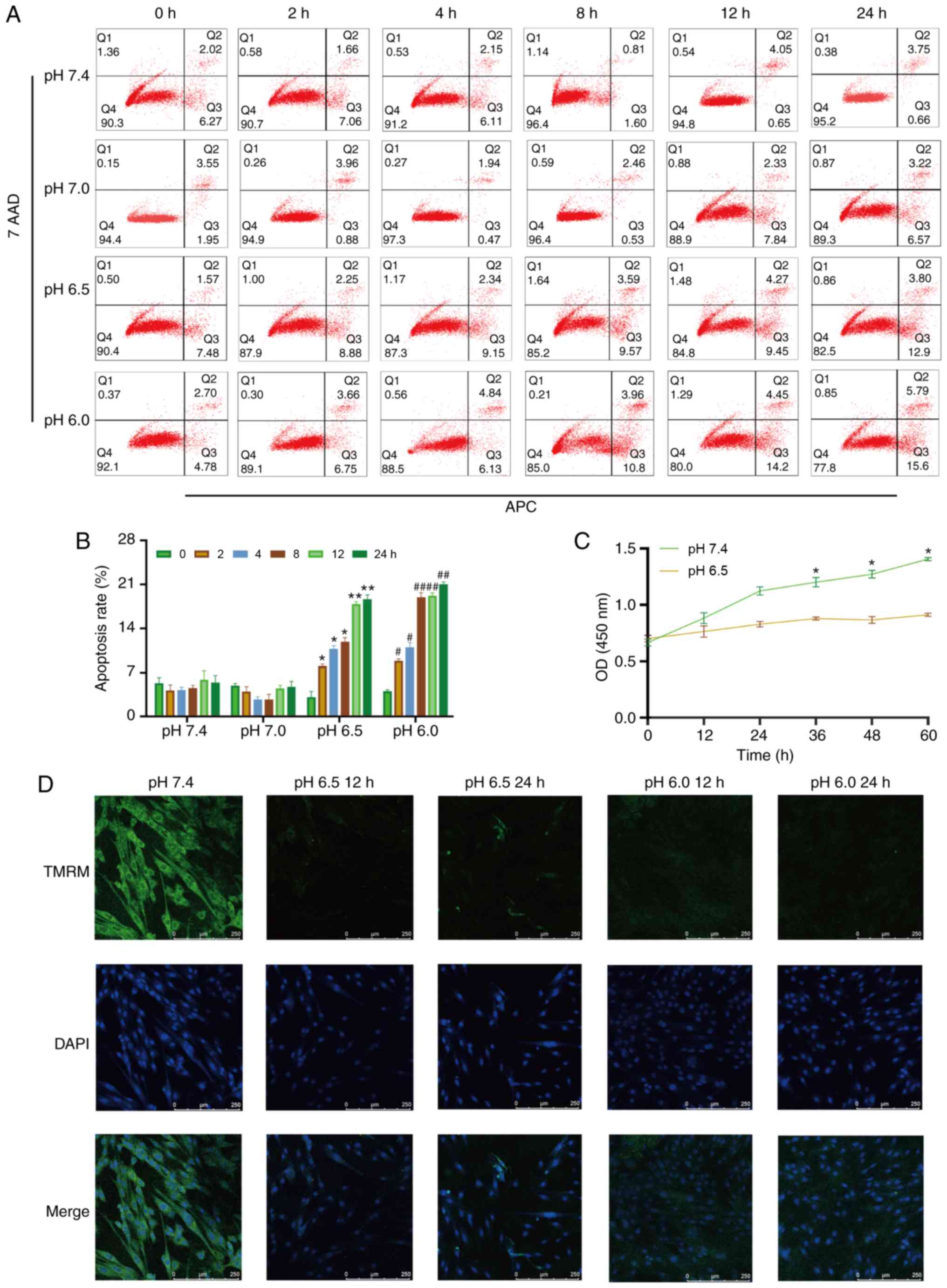 | Figure 1Extracellular acidification induces
H9C2 cell apoptosis and decreases MMP. (A) H9C2 cell apoptosis
induced by extracellular acidification. H9C2 cells were cultured in
different pH media (7.4, 7.0, 6.5 and 6.0) for 0, 2, 4, 8, 128 or
24 h. (B) Quantitative analysis of the results shown in (A). (C)
CCK-8 assay of H9C2 cells in different pH media. (D) MMP of H9C2
cells in different pH media. TMRM (green) is a type of
mitochondrial dye, and the fluorescence intensity is positively
associated with the cell MMP. Original magnification, ×100.
#P<0.05 and ##P<0.01 vs. 0 h group.
*P<0.05 and **P<0.01 vs. pH 7.4 group.
MMP, mitochondrial membrane potential; TMRM, tetramethylrhodamine,
methyl ester. |
Results
Extracellular acidification induces H9C2
cell apoptosis, decreases cell viability and causes mitochondrial
dysfunction
To investigate whether metabolic acidosis induces
cell apoptosis and decreases cell viability, CCK-8 assay and
Annexin V-APC/7-AAD apoptosis kit were used. The results revealed
that H9C2 cell apoptosis was increased in lower pH culture medium
(pH 6.5 or 6.0) or longer acidic culture time (Fig. 1A and B), and cell viability
decreased in acidic medium (Fig.
1C). The MMP of the H9C2 cells was also decreased in acidic
medium, which indicated that mitochondrial function was damaged
(Fig. 1D).
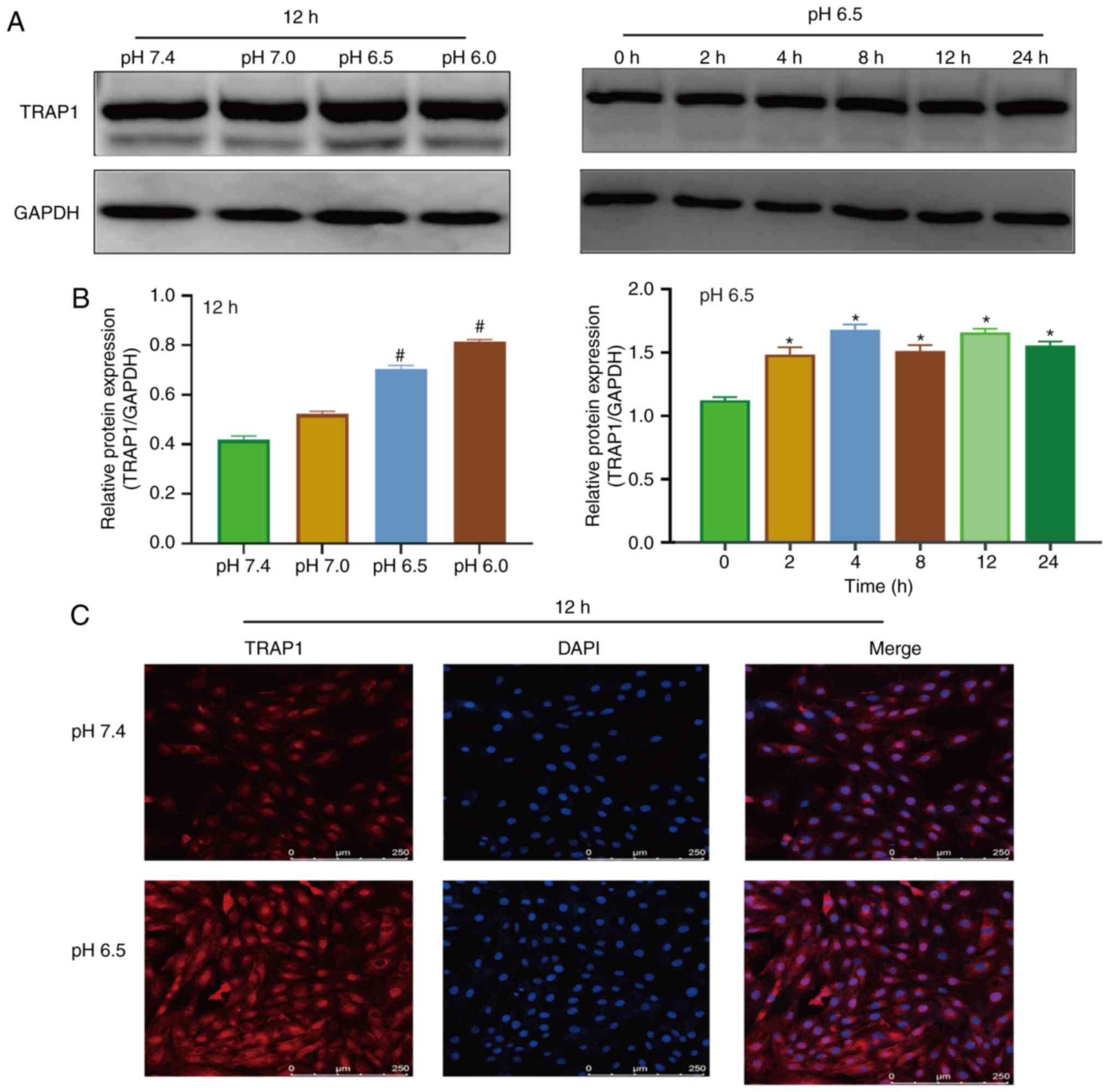
Extracellular acidification increases
TRAP1 expression in H9C2 cells
Western blot analysis and immunofluorescence were
used to detect the expression of TRAP1 in acidic medium. The
results of western blot analysis revealed that the expression of
TRAP1 increased after 4 h in acidic medium (Fig. 2A and B). Immunofluorescence assay
also demonstrated that the acidic environment increased the
expression of TRAP1 (Fig.
2C).
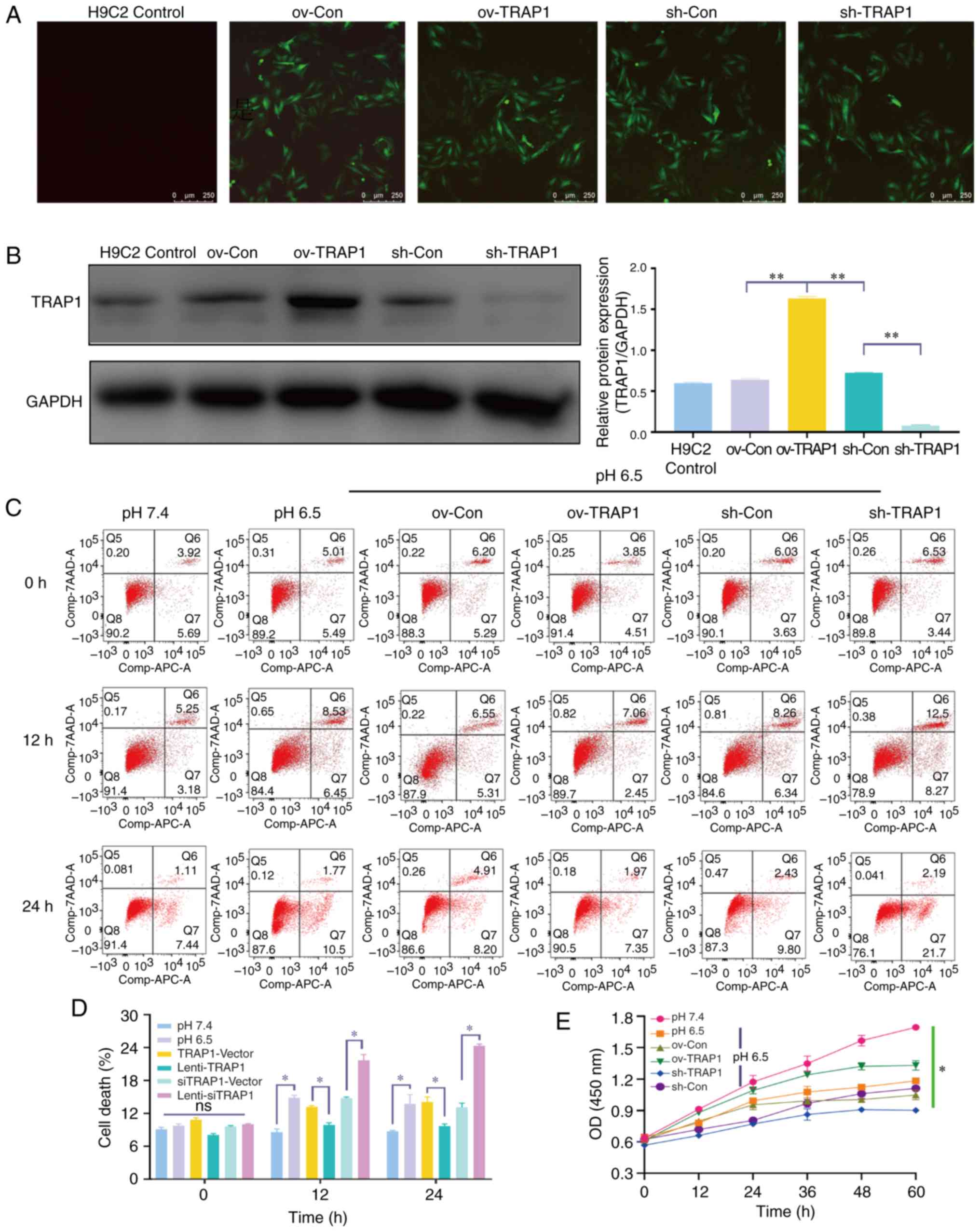
TRAP1 inhibits acid-induced H9C2 cell
apoptosis and increases cell viability
Considering the increase observed in TRAP1
expression in acidic medium, the present study aimed to confirm
that TRAP1 is a protective factor for H9C2 cells in acidic medium.
TRAP1-GFP lentiviral vector was constructed for TRAP1
overexpression (ov-TRAP1) or TRAP1 silencing (sh-TRAP1). At 5 days
post-transfection, H9C2 cells expressing a high level of green
fluorescence were observed (Fig.
3A). The results of western blot analysis further confirmed
that the expression of TRAP1 was significantly increased by
Lenti-TRAP1 and decreased by Lenti-TRAP1 siRNA (Fig. 3B). Following culture in acidic
medium for 12 or 24 h, TRAP1 overexpression inhibited H9C2 cell
apoptosis (Fig. 3C and D) and
increased cell viability (Fig.
3E) in acidic medium, while TRAP1 silencing aggravated the
damage to the cells induced by acidic medium (Fig. 3C and E).
TRAP1 reverses the acid-induced
mitochondrial dysfunction of H9C2 cells
H9C2 cell mitochondrial function was detected by ATP
assay and MMP assay. The results revealed that the H9C2 cell MMP
(Fig. 4A and B) and ATP levels
(Fig. 4D) in acidic medium were
significantly lower than those in the cells in neutral pH medium.
TRAP1 overexpression reverse the decrease in MMP and the ATP level
induced by acidic medium (Fig. 4A, B
and D). However, the levels of ROS were not markedly altered
(Fig. 4E).
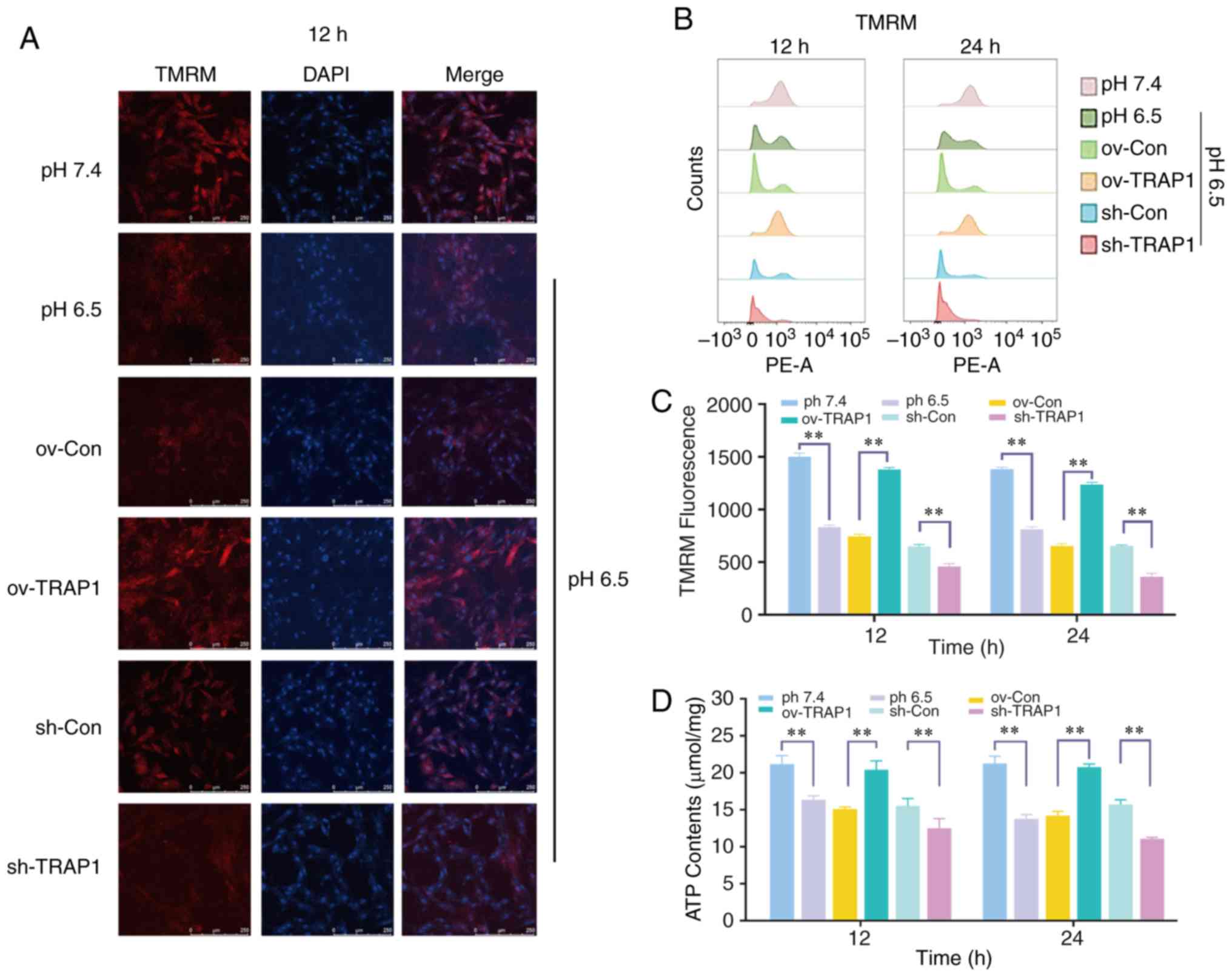 | Figure 4Effect of the overexpression or
silencing of TRAP1 on the mitochondrial function. (A) MMP of H9C2
cells in pH 6.5 at 12 h following transfection. TMRM (red) is a
type of mitochondrial dye, and the fluorescence intensity is
positively associated with the cell MMP. Original magnification,
×100. (B) MMP of H9C2 cells detected using a flow cytometer. After
staining cells for 45 min with TMRM, the fluorescence intensity was
detected by flow cytometry. (C) Quantitative analysis of the
results shown in (B). (D) ATP levels of H9C2 cells in pH 6.5 at 12
h post-transfection. (E) ROS levels of H9C2 cells in pH 6.5 at 12 h
following transfection. (F) Western blot analysis of Bax, Bcl-2,
caspase-3 and cleaved caspase-3, and semiquantitative analysis of
the protein levels. (G) Mitochondrial ultrastructure of H9C2 cells
was severely damaged under extracellular acidification, including
the reduction of mitochondrial cristae and mitochondrial
vacuolization. TRAP1 overexpression significantly reversed the
damage to the normal ultrastructure of the mitochondria.
**P<0.01. ov-Con, H9C2 cells transfected with vector;
ov-TRAP1, H9C2 cells overexpressing TRAP1; sh-Con, H9C2 cells
transfected with scrambled siRNA; sh-TRAP1, H9C2 cells transfected
with siRNA; C3, caspase-3; CC3, cleaved caspase-3; TRAP1, tumor
necrosis factor receptor-associated protein 1. |
TRAP1 inhibits the acid-induced
mitochondrial apoptotic pathway and maintained normal mitochondrial
ultrastructure of H9C2 cells
The mitochondrial apoptotic pathway is an important
pathway of cell apoptosis and is activated following mitochondrial
injury. The pro-apoptotic factor, Bax, and the anti-apoptotic
factor, Bcl-2, are currently recognized as important components of
the mitochondrial apoptotic pathway. They activate caspase-3
(forming cleaved caspase-3) to regulate mitochondrial apoptosis and
eventually lead to cell apoptosis. The present study demonstrated
that the expression levels of Bax and Bcl-2 increased under
extracellular acidification, and the levels of cleaved caspase-3
exhibited a similar trend (Fig.
4F). TRAP1 overexpression significantly inhibited the
expression of Bax and cleaved caspase-3, and maintained the
Bax/Bcl-2 ratio (Fig. 4F). The
mitochondrial ultrastructure of H9C2 cells was severely damaged
under extracellular acidification, including the reduction of
mitochondrial cristae and mitochondrial vacuolization. TRAP1
overexpression significantly reversed the damage to the normal
ultrastructure of the mitochondria (Fig. 4G).
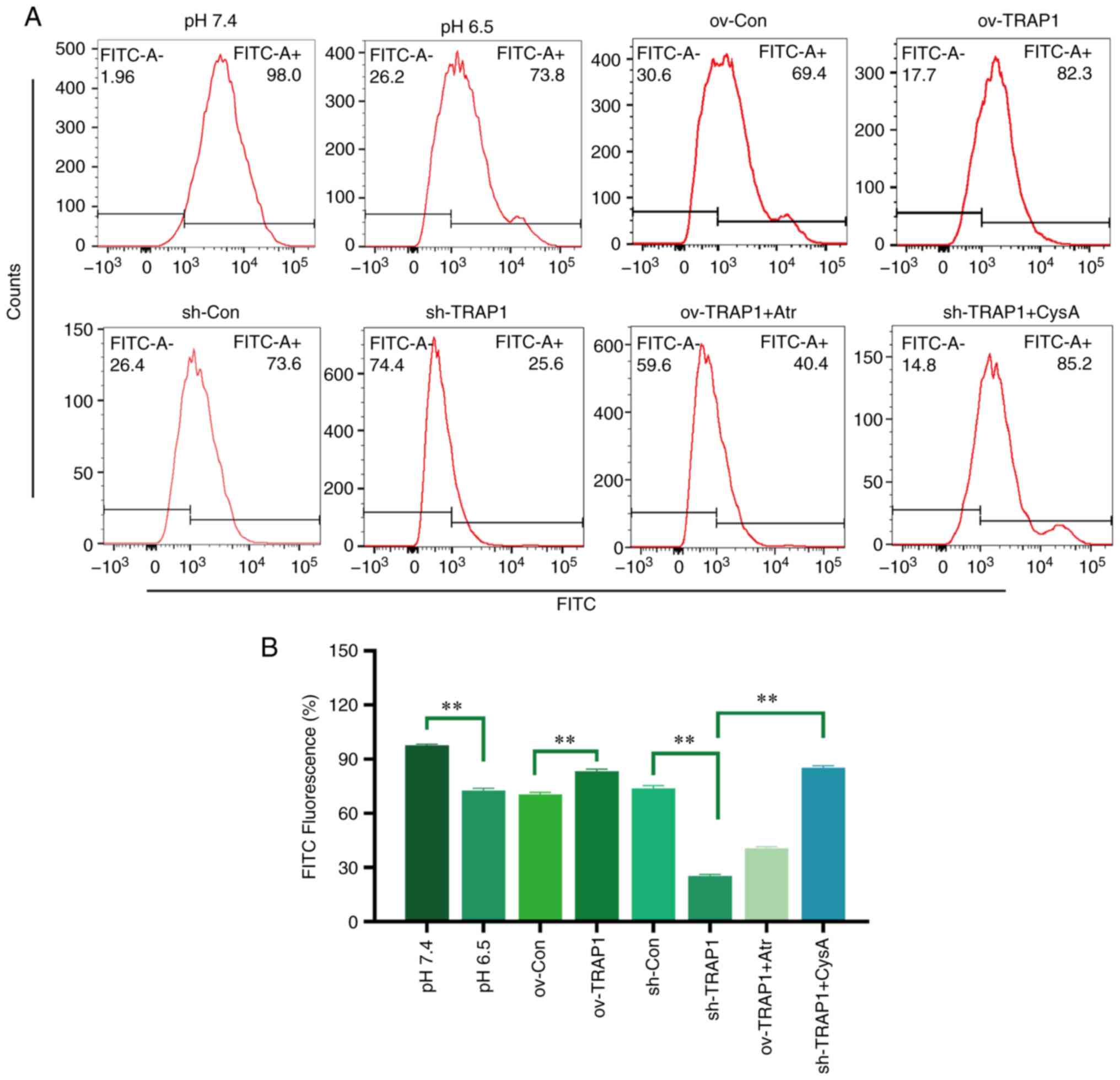
TRAP1 inhibits the MPTP opening of H9C2
cells in acidic medium
The MPTP is an important component of the
mitochondrial membrane and is an initiator of the mitochondrial
apoptotic pathway. Thus, the present study aimed to investigate the
effects of TRAP1 on MPTP opening. The results revealed that the
MPTP opening of H9C2 cells was increased in acidic medium compared
with normal pH medium (Fig. 5).
The overexpression of TRAP1 decreased MPTP opening, while the
silencing of TRAP1 further increased MPTP opening (Fig. 5).
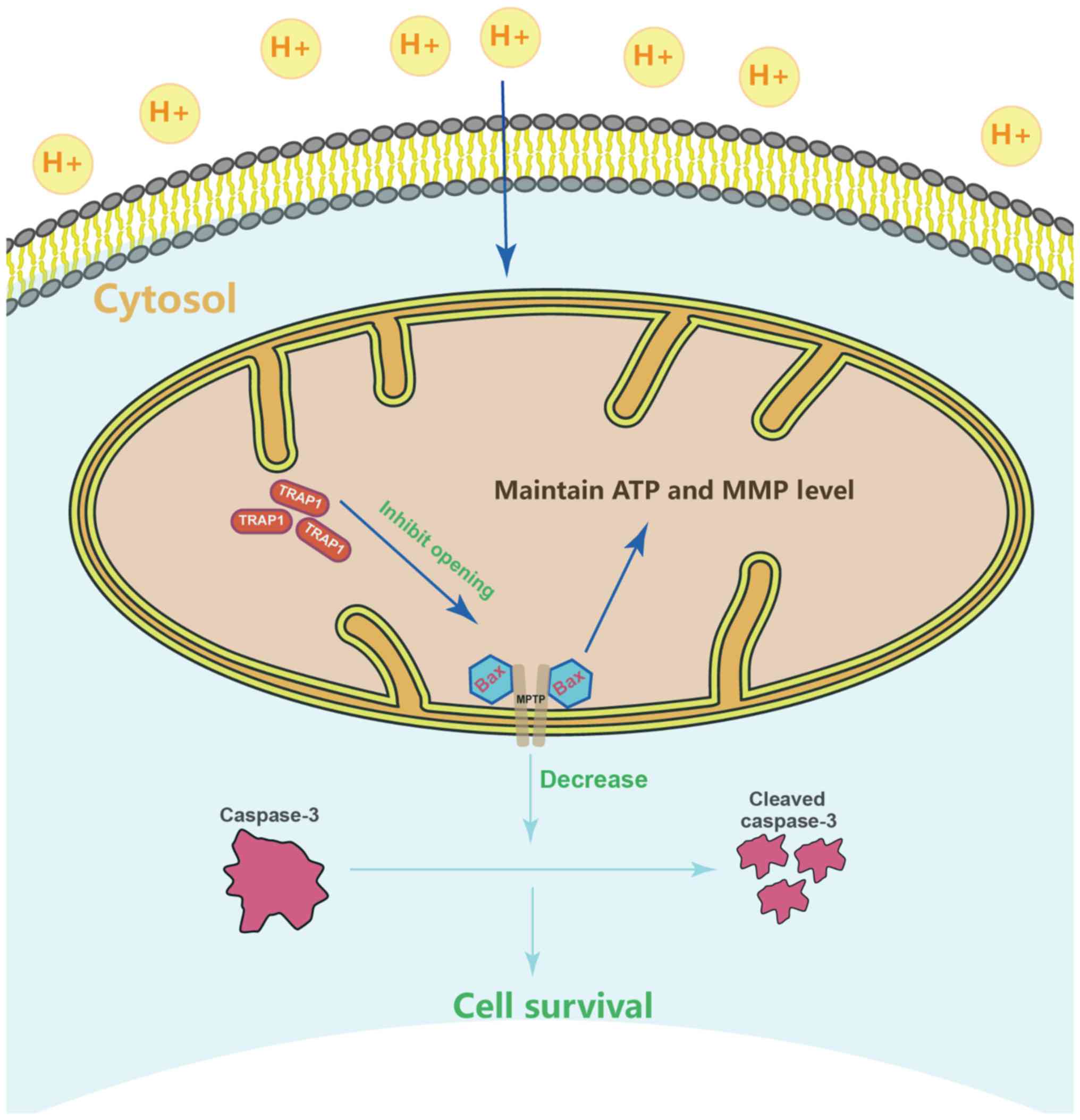
TRAP1 inhibits the mitochondrial
apoptotic pathway, stabilizes mitochondrial function, and thus
inhibits apoptosis, via the inhibition of MPTP opening
After identifying that TRAP1 decreased acid-induced
MPTP opening, atractyloside (Atr, 10 µM, a specific MPTP
opening promoter) and cyclosporin A (CysA, 10 µM, a specific
MPTP opening inhibitor) were used to further identify whether TRAP1
protects H9C2 cells in acidic medium by inhibiting MPTP opening. It
was observed that the MPTP opening promoter abolished the
protective effects of TRAP1 on H9C2 cells (Fig. 6A and C–F). On the other hand, the
MPTP opening inhibitor reversed the increased expression of Bax,
caspase-3 and cleaved caspase-3, further reversing mitochondrial
dysfunction and cell apoptosis induced by the silencing of TRAP1
(Fig. 6A and C–F). The normal
mitochondrial ultrastructure maintained by TRAP1 overexpression was
abolished following Atr treatment. Unexpectedly, it was not
observed that CysA significantly reversed the mitochondrial
ultrastructure damage induced by TRAP1 silencing, which may
indicate that TRAP1 maintained mitochondrial ultrastructure in a
mPTP-independent manner (Fig.
6G). On the whole, TRAP1 protected mitochondrial function and
attenuated cell apoptosis under extracellular acidification via the
inhibition of MPTP opening (Fig.
7).
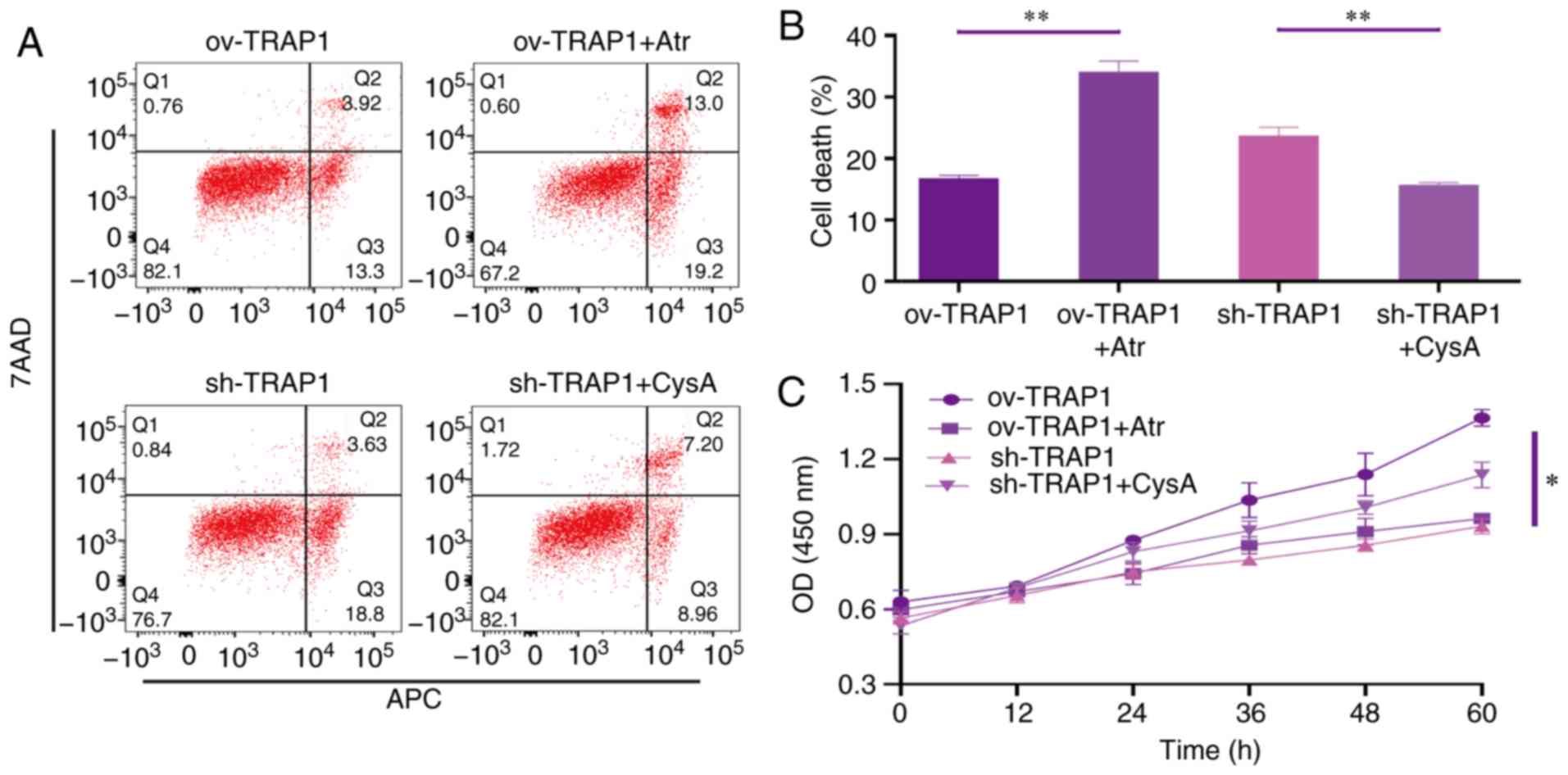 | Figure 6TRAP1 protects H9C2 cells in acidic
medium by inhibiting MPTP. (A) H9C2 cell apoptosis induced by
extracellular acidification after transfection and addition of
drugs. (B) MMP of H9C2 cells in pH 6.5 for 12 h after transfection.
TMRM (red) is a type of mitochondrial dye, and the fluorescence
intensity is positively associated with the cell MMP. (C) CCK-8
assay of H9C2 cells in acidic medium following transfection and
addition of drugs. (D) MMP of H9C2 cells was detected using a flow
cytometer. After staining the cells for 45 min with TMRM, the
fluorescence intensity was detected by flow cytometry. Original
magnification, ×100. (E) ATP levels of H9C2 cells in pH 6.5 and 12
h following transfection and the addition of drugs. (F) Western
blot analysis of Bax, Bcl-2, caspase-3 and cleaved caspase-3, and
semi-quantitative analysis of the protein levels. (G) Mitochondrial
ultrastructure of the cells following transfection with sh-TRAP1
and CysA. *P<0.05 and **P<0.01.
ov-TRAP1, H9C2 cells overexpressing TRAP1; sh-TRAP1, H9C2 cells
transfected with TRAP1-siRNA; ov-TRAP1 + Atr, H9C2 cells
overexpressing TRAP1 after adding Atr (10 µM) to the culture
medium; sh-TRAP1 + CysA, H9C2 cells transfected with TRAP1-siRNA
after adding CysA (10 µM) to the culture medium; C3,
caspase-3; CC3, cleaved caspase-3; TRAP1, tumor necrosis factor
receptor-associated protein 1. |
Discussion
The present study firstly reported that
extracellular acidosis increased the expression of TRAP1 in H9C2
cells, which is parallel to other pathological models such as
hypoxia or ischemia reperfusion injury (21-23). Subsequently, lentiviral vectors
were used to overexpress and silence TRAP1 in H9C2 cells. The
overexpression of TRAP1 inhibited cell apoptosis and increased cell
viability, whereas the silencing of TRAP1 deteriorated the cell
damage induced by extracellular acidosis. These results demonstrate
that TRAP1 protects H9C2 cells in extracellular acidosis, which
enhances the protective effects of TRAP1 against cell injury
(11,13,24).
TRAP1 has been reported to maintain MMP and cell ATP
production efficiency, and inhibit cell ROS, thus protecting cells
under pathological conditions (22,25). Zhang et al used siRNA to
silence TRAP1 expression in H9C2 cells, observing that
mitochondrial function was deteriorated in high glucose medium
(26). Consistent with the
findings of previous studies, the present study observed that the
overexpression of TRAP1 maintained MMP and the cellular ATP level,
while the silencing of TRAP1 decreased MMP and the cellular ATP
level. Severe mitochondrial damage activates the mitochondrial
apoptotic pathway and induces cell apoptosis. The findings of the
present study demonstrated that extracellular acidosis activated
the cell mitochondrial apoptotic pathway, while the overexpression
of TRAP1 inhibited this activation. These results strongly indicate
that TRAP1 protects the physiological function of the mitochondria
under conditions of extracellular acidosis, and inhibits the
activation of the mitochondrial apoptotic pathway, thus preventing
cell death. Furthermore, TRAP1 overexpression can maintain the
normal mitochondrial ultrastructure, while TRAP1 silencing leads to
further damage to the ultrastructure. This indicates that TRAP1
protects both mitochondrial function and morphology under
conditions of extracellular acidification. Moreover, the present
study did not detect any increase in cell ROS levels during
extracellular acidosis. The association between ROS and
extracellular acidosis remains controversial. Previous studies have
suggested that there is a positive association. Teixeira et
al reported that extracellular acidification increases cell ROS
level and induces protein carbonylation (27). However, few studies, such as the
one by Wang et al have reported that extracellular
acidification directly reconstructs acid-sensing ion channel 1a
(ASIC1a) conformation to induce cell injury, which was different
from the ROS-dependent cell injury pathological model, including
hypoxia or ischemia reperfusion injury (28). These results suggest that
extracellular acidosis may exert a ROS-independent effect on cell
damage. These results confirmed this hypothesis. Moreover,
different pathological processes may be involved in different
disease models may involve, which leads to different phenotypes.
Therefore, the association between extracellular acidification and
ROS warrants further investigation.
Furthermore, the present study identified the
mechanisms underlying the protective effects of TRAP1 on
mitochondrial function under conditions of extracellular acidosis.
MPTP is an important channel protein across the mitochondrial inner
and outer membranes (29-31). Previous studies have identified
that MPTP opening is a key step to induce cell mitochondrial damage
and activates the mitochondrial apoptotic pathway (32-34). The present study revealed that
extracellular acidosis increased MPTP opening, while the
overexpression of TRAP1 reversed this effect. The addition of the
specific MPTP opening promoter, Atr, to increase MPTP opening,
abolished the protective effects of TRAP1. The specific MPTP
opening inhibitor, CysA, alleviated cell injury and inhibited the
mitochondrial apoptotic pathway in acidic medium when TRAP1 was
silenced. The present study, to the best of our knowledge, is the
first to report the TRAP1-MPTP opening-mitochondrial apoptotic
pathway in cardiomyocytes, which may provide a novel approach for
extracellular acidosis treatment. Previous studies have shown that
inhibiting MPTP opening can regulate mitochondrial morphology
(35). Unexpectedly, specific
MPTP opening inhibitor CysA did not significantly reversed the
mitochondrial ultrastructure damage induced by TRAP1 silencing
under conditions of extracellular acidosis. This may indicate that
TRAP1 maintained mitochondrial ultrastructure in an
MPTP-independent manner; however, further studies are required to
elucidate the detailed mechanisms involved. In conclusion, the
present study demonstrates that TRAP1 protects cardiomyocytes
against extracellular acidification by regulating MPTP opening
(Fig. 7).
Acknowledgements
Not applicable.
Funding
The present study was supported by the Science and
Technology Planning Project of Guangzhou, China (grant no.
201604020119).
Availability of data and materials
The datasets used and/or analysed in the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
LZ contributed to the conception of the study,
performed the majority of the experiments and wrote the manuscript.
TZ, LL and XG designed the present study, interpreted the results,
revised the manuscript and approved the final version of the
manuscript. XZ collected the experimental data, performed the main
statistical analysis and assisted with the revision of the
manuscript. XL, JZ, YL and XG performed the CCK-8, apoptosis, ROS
and MMP assays, respectively and assisted with the revision of the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Abbreviations:
|
TRAP1
|
tumor necrosis factor
receptor-associated protein 1
|
|
MPTP
|
mitochondrial permeability transition
pore
|
|
MMP
|
mitochondrial membrane potential
|
|
TMRM
|
tetramethylrhodamine, methyl ester
|
|
CCK-8
|
Cell Counting kit-8
|
|
7-AAD
|
7-aminoactinomycin D
|
|
APC
|
allophycocyanin
|
|
ROS
|
reactive oxygen species
|
|
MES
|
4-morpholineethanesulfonic acid
|
|
BSA
|
bovine serum albumin
|
|
C3
|
caspase-3
|
|
CC3
|
cleaved caspase 3
|
|
TEM
|
transmission electron microscopy
|
References
|
1
|
Kraut JA and Madias NE: Metabolic
acidosis: Pathophysiology, diagnosis and management. Nat Rev
Nephrol. 6:274–285. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kraut JA and Madias NE: Treatment of acute
metabolic acidosis: A pathophysiologic approach. Nat Rev Nephrol.
8:589–601. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Teplinsky K, O'Toole M, Olman M, Walley KR
and Wood LD: Effect of lactic acidosis on canine hemodynamics and
left ventricular function. Am J Physiol. 258:H1193–H1199.
1990.PubMed/NCBI
|
|
4
|
Gunnerson KJ, Saul M, He S and Kellum JA:
Lactate versus non-lactate metabolic acidosis: A retrospective
outcome evaluation of critically ill patients. Crit Care.
10:R222006. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Mitchell JH, Wildenthal K and Johnson RJ
Jr: The effects of acid-base disturbances on cardiovascular and
pulmonary function. Kidney Int. 1:375–389. 1972. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wagner CA, Imenez Silva PH and Bourgeois
S: Molecular pathophysiology of acid-base disorders. Semin Nephrol.
39:340–352. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Masgras I, Ciscato F, Brunati AM, Tibaldi
E, Indraccolo S, Curtarello M, Chiara F, Cannino G, Papaleo E,
Lambrughi M, et al: Absence of neurofibromin induces an oncogenic
metabolic switch via mitochondrial ERK-mediated phosphorylation of
the chaperone TRAP1. Cell Rep. 18:659–672. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lettini G, Sisinni L, Condelli V, Matassa
DS, Simeon V, Maddalena F, Gemei M, Lopes E, Vita G, Del Vecchio L,
et al: TRAP1 regulates stemness through Wnt/β-catenin pathway in
human colorectal carcinoma. Cell Death Differ. 23:1792–1803. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Xiang F, Huang YS, Shi XH and Zhang Q:
Mitochondrial chaperone tumour necrosis factor receptor-associated
protein 1 protects cardiomyocytes from hypoxic injury by regulating
mitochondrial permeability transition pore opening. FEBS J.
277:1929–1938. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yoshida S, Tsutsumi S, Muhlebach G,
Sourbier C, Lee MJ, Lee S, Vartholomaiou E, Tatokoro M, Beebe K,
Miyajima N, et al: Molecular chaperone TRAP1 regulates a metabolic
switch between mitochondrial respiration and aerobic glycolysis.
Proc Natl Acad Sci USA. 110:E1604–E1612. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen JF, Wu QS, Xie YX, Si BL, Yang PP,
Wang WY, Hua Q and He Q: TRAP1 ameliorates renal tubulointerstitial
fibrosis in mice with unilateral ureteral obstruction by protecting
renal tubular epithelial cell mitochondria. FASEB J. 31:4503–4514.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lau AT, He QY and Chiu JF: A proteome
analysis of the arsenite response in cultured lung cells: Evidence
for in vitro oxidative stress-induced apoptosis. Biochem J.
382:641–650. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Palladino G, Notarangelo T, Pannone G,
Piscazzi A, Lamacchia O, Sisinni L, Spagnoletti G, Toti P, Santoro
A, Storto G, et al: TRAP1 regulates cell cycle and apoptosis in
thyroid carcinoma cells. Endocr Relat Cancer. 23:699–709. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tomecka E, Wojasinski M, Jastrzebska E,
Chudy M, Ciach T and Brzozka Z: Poly(l-lactic Acid) and
polyurethane nanofibers fabri-cated by solution blow spinning as
potential substrates for cardiac cell culture. Mater Sci Eng C
Mater Biol Appl. 75:305–316. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bass JJ, Wilkinson DJ, Rankin D, Phillips
BE, Szewczyk NJ, Smith K and Atherton PJ: An overview of technical
considerations for Western blotting applications to physiological
research. Scand J Med Sci Spor. 27:4–25. 2017. View Article : Google Scholar
|
|
16
|
Wang L, Feng Y, Xie X, Wu H, Su XN, Qi J,
Xin W, Gao L, Zhang Y, Shah VH and Zhu Q: Neuropilin-1 aggravates
liver cirrhosis by promoting angiogenesis via VEGFR2-dependent
PI3K/Akt pathway in hepatic sinusoidal endothelial cells.
EbioMedicine. 43:525–536. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Odell ID and Cook D: Immunofluorescence
techniques. J Invest Dermatol. 133:e42013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kang T, Lu W, Xu W, Anderson L, Bacanamwo
M, Thompson W, Chen YE and Liu D: MicroRNA-27 (miR-27) targets
prohibitin and impairs adipocyte differentiation and mitochondrial
function in human adipose-derived stem cells. J Biol Chem.
288:34394–34402. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Basit F, van Oppen LM, Schöckel L,
Bossenbroek HM, van Emst-de Vries SE, Hermeling JC, Grefte S,
Kopitz C, Heroult M, Hgm Willems P and Koopman WJ: Mitochondrial
complex I inhibition triggers a mitophagy-dependent ROS increase
leading to necroptosis and ferroptosis in melanoma cells. Cell
Death Dis. 8:e27162017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu Z, Lv XA, Dai Q, Lu M and Jin Z:
Exogenous BDNF increases mitochondrial pCREB and alleviates
neuronal metabolic defects following mechanical injury in a
MPTP-dependent way. Mol Neurobiol. 55:3499–3512. 2018. View Article : Google Scholar
|
|
21
|
Hua G, Zhang Q and Fan Z: Heat shock
protein 75 (TRAP1) antagonizes reactive oxygen species generation
and protects cells from granzyme m-mediated apoptosis. J Biol Chem.
282:20553–20560. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xu L, Voloboueva LA, Ouyang Y, Emery JF
and Giffard RG: Overexpression of mitochondrial Hsp70/Hsp75 in rat
brain protects mitochondria, reduces oxidative stress, and protects
from focal ischemia. J Cereb Blood Flow Metab. 29:365–374. 2009.
View Article : Google Scholar
|
|
23
|
Zhang P, Lu Y, Yu D, Zhang D and Hu W:
TRAP1 provides protection against myocardial ischemia-reperfusion
injury by ameliorating mitochondrial dysfunction. Cell Physiol
Biochem. 36:2072–2082. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Costantino E, Maddalena F, Calise S,
Piscazzi A, Tirino V, Fersini A, Ambrosi A, Neri V, Esposito F and
Landriscina M: TRAP1, a novel mitochondrial chaperone responsible
for multi-drug resistance and protection from apoptotis in human
colorectal carcinoma cells. Cancer Lett. 279:39–46. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Tian X, Ma P, Sui CG, Meng FD, Li Y, Fu
LY, Jiang T, Wang Y and Jiang YH: Suppression of tumor necrosis
factor receptor-associated protein 1 expression induces inhibition
of cell proliferation and tumor growth in human esophageal cancer
cells. FEBS J. 281:2805–2819. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang X, Zhong Z and Li W: Downregulation
of TRAP1 aggravates injury of H9c2 cardiomyocytes in a
hyperglycemic state. Exp Ther Med. 18:2681–2686. 2019.PubMed/NCBI
|
|
27
|
Teixeira J, Basit F, Swarts HG, Forkink M,
Oliveira PJ, Willems PHGM and Koopman WJH: Extracellular
acidification induces ROS- and mPTP-mediated death in HEK293 cells.
Redox Biol. 15:394–404. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang YZ, Wang JJ, Huang Y, Liu F, Zeng WZ,
Li Y, Xiong ZG, Zhu MX and Xu TL: Tissue acidosis induces neuronal
necroptosis via ASIC1a channel independent of its ionic conduction.
Elife. 4:e056822015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kwong JQ and Molkentin JD: Physiological
and pathological roles of the mitochondrial permeability transition
pore in the heart. Cell Metab. 21:206–214. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bernardi P, Rasola A, Forte M and Lippe G:
The mitochondrial permeability transition pore: Channel formation
by F-ATP synthase, integration in signal transduction, and role in
pathophysiology. Physiol Rev. 95:1111–1155. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rottenberg H and Hoek JB: The path from
mitochondrial ROS to aging runs through the mitochondrial
permeability transition pore. Aging Cell. 16:943–955. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li X, Jia P, Huang Z, Liu S, Miao J, Guo
Y, Wu N and Jia D: Lycopene protects against myocardial
ischemia-reperfusion injury by inhibiting mitochondrial
permeability transition pore opening. Drug Des Devel Ther.
13:2331–2342. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Šileikytė J, Devereaux J, de Jong J,
Schiavone M, Jones K, Nilsen A, Bernardi P, Forte M and Cohen M:
Second-generation inhibitors of the mitochondrial permeability
transition pore with improved plasma stability. ChemMedChem.
14:1771–1782. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Panel M, Ruiz I, Brillet R, Lafdil F,
Teixeira-Clerc F, Nguyen CT, Calderaro J, Gelin M, Allemand F,
Guichou JF, et al: Small-molecule inhibitors of cyclophilins block
opening of the mitochondrial permeability transition pore and
protect mice from hepatic ischemia/reperfusion injury.
Gastroenterology. 157:1368–1382. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Hom JR, Quintanilla RA, Hoffman DL, de
Mesy Bentley KL, Molkentin JD, Sheu SS and Porter GA Jr: The
permeability transition pore controls cardiac mitochondrial
maturation and myocyte differentiation. Dev Cell. 21:469–478. 2011.
View Article : Google Scholar : PubMed/NCBI
|