Introduction
Age-related macular degeneration (AMD) is the
leading cause of blindness in the elderly, which manifests by
progressive loss of central vision through degeneration at the
ocular interface between the retina and the underlying choroid
(1). A typical characteristic
non-neovascular (dry) form of AMD is the formation of drusen and
geographic atrophy, whereas the exudative (wet) form involves blood
vessel invasion. Vascular endothelial growth factor (VEGF) is known
to play a pivotal role in the growth of abnormal blood vessels that
characterize the wet form of AMD (2).
The retinal pigment epithelium (RPE) is firmly
attached to the underlying choroid and is a monolayer of cells that
plays critical roles in retinal homeostasis, eye development and
vision. Dysfunction of RPE cells induces photoreceptor dystrophy,
retinal diseases and blindness. It was previously demonstrated that
the blue light-induced pathological changes of RPE cells resemble
AMD (3). Since RPE cells are
sensitive to blue light, which may trigger cellular apoptosis, blue
light exposure is considered as a suitable model for AMD study.
Exacerbated oxidative stress and inflammation in the
RPE contribute to the pathogenesis of AMD, eventually leading to
RPE degeneration (4). The
migration of macrophages and lymphocytes to the posterior
compartment of the eye and the secretion of proinflammatory
cytokines are characteristic during ocular inflammation (5). RPE cells are a component of the
outer blood-retinal barrier and respond to the macrophage-secreted
proinflammatory cytokine tumor necrosis factor (TNF)α (6). Leukocyte recruitment and adhesion to
the retina are mediated by intercellular adhesion molecule (ICAM)-1
(7). The inflammatory nuclear
factor (NF)-κB pathway plays a key role in TNFα-activated ICAM-1
expression (8).
The RPE is sensitive to blue light, which causes
oxidative stress and RPE cell damage at certain doses (9). Nuclear factor erythroid 2-related
factor 2 (Nrf2)-derived heme oxygenase (HO)-1 and the synthesis of
glutathione (GSH) have been found to act as critical antioxidants
in vivo and in vitro (10-12). GSH maintains a reduced cellular
environment and is part of a protective mechanism against numerous
cellular stressors (13).
Therefore, protecting RPE cells from blue light or oxidative stress
through engendering a Nrf2-regulated cell redox state may provide a
potential target for AMD treatment.
Carbon monoxide-releasing molecules (CORMs) have
been demonstrated to act pharmacologically by mimicking the
bioactive effects of HO-1 and CO gas (14-16). Low concentrations of CO have been
found to increase resistance to cell damage and apoptosis in
various model systems (17).
Since CO has exhibited the ability to mediate a number of
biological functions, including anti-inflammation, cell cycle
arrest and vasodilation, it has shown potential for use in various
therapeutic applications (17,18). However, the cytoprotective
mechanism of CO in RPE cells remains unclear. Thus, the present
study was designed to determine the molecular mechanisms underlying
the cytoprotective properties of CORMs in RPE cells. There are two
widely used CORMs: The lipid-soluble CORM2
{[Ru(CO)3Cl2]2} and the
water-soluble CORM3 [Ru(CO)3Cl2
(H2NCH2CO)2] (19). It was herein investigated whether
these CORMs possess protective properties that may contribute to
the CO-regulated cytoprotective effects.
Materials and methods
Materials
NF-κB/Luc vectors were constructed as described
previously (20). ICAM-1/CD54
antibody (cat. no. 4915S; 1:1,000) was purchased from Cell
Signaling Technology, Inc. NF-κB/p65 antibody (cat. no. KAS-TF110;
1:1,000) was purchased from Stressgen Biotechnologies. Antibodies
against IκBa (cat. no. sc-847; 1:1,000), poly(ADP-ribose)
polymerase 1 (PARP-1) (cat. no. sc-136208; 1:200) and lamin (cat.
no. sc-6217; 1:1,000) were purchased from Santa Cruz Biotechnology,
Inc. Tubulin antibody (cat. no. T568; 1:1,000) was obtained from
Sigma-Aldrich; Merck KGaA. Peroxidase-conjugated anti-rabbit (cat.
no. G-21040; 1:1,000) and anti-mouse (cat. no. 31460; 1:2,500)
antibodies were obtained from Invitrogen (Thermo Fisher Scientific,
Inc.) and nitrocellulose was obtained from Schleicher and Schuell.
The luciferase assay kit (cat. no. E1500) was purchased from
Promega Corporation. All other reagents, including TNF-α and VEGF-A
proteins, were purchased from Sigma-Aldrich; Merck KGaA.
RPE cell culture and blue light
exposure
The human RPE cell line ARPE-19 was obtained from
ATCC and cultured in DMEM-Ham's F12 (1:1; Invitrogen; Thermo Fisher
Scientific, Inc.) containing 10% FBS (Invitrogen; Thermo Fisher
Scientific, Inc.). The cells were grown for 3 days until reaching
90-100% confluence. The medium was replaced with fresh serum-free
DMEM-Ham's F12, and the cells were grown for an additional 12 h
prior to experimental treatment. ARPE-19 cells were cultured in the
dark or irradiated with blue light (400 nm) at an intensity of
2,000±500 lux for 24 h to establish the light-induced injury
model.
Endothelial cell and THP-1 cell
cultures
The human umbilical vein cell line EA.hy926 (ATCC
CRL-2922) was cultured in DMEM (Gibco-BRL; Thermo Fisher
Scientific, Inc.) supplemented with 10% FBS at 37°C under 5%
CO2. The THP-1 cells (ATCC® TIB202™) were
cultured in RPMI-1640 medium containing 10% FBS at 37°C under 5%
CO2.
Cell viability assay
Cell viability was assayed using Alamar Blue
(Serotec) according to the manufacturer's instructions. This assay
identifies live cell metabolic activity by detecting redox activity
in cells. The excitation/emission wavelength settings were adjusted
to 530/590 nm.
Morphological analysis after DAPI
staining
Cells were fixed with 4% paraformaldehyde for 15 min
at room temperature and then stained with DAPI for 5 min in the
dark at room temperature. After washing with PBS for 5 min, the
cells were analyzed under a fluorescence microscope (Axiovert S100;
Carl Zeiss AG) at a magnification of ×200. The normal cell nucleus
is round, has clear borders and is uniformly stained, whereas
apoptotic cells exhibit nuclear pyknosis, irregular edges and
strong staining.
Analysis of intracellular reactive oxygen
species (ROS)
To detect intracellular ROS production, RPE cells
were seeded into 96-well plates (2×104 cells/well) in
DMEM–F12 for 24 h and incubated in serum-free DMEM-F12 for 12 h
prior to experimental treatment. Next, the cells were incubated
with 20 µM peroxide-sensitive fluorescent probe
carboxy-H2DCFDA (Molecular Probes, LLC) for 30 min.
After washing with PBS twice (5-10 min per wash), the cells were
solubilized with 1% SDS and 5 mM Tris-HCl (pH 7.4). Fluorescence
was measured with a spectrofluorophotometer (model Rf-5301PC;
Shimadzu Corporation) with excitation and emission wavelengths of
450 and 520 nm, respectively. The cells were observed and images
were captured with a fluorescence microscope (Axiovert S100; Carl
Zeiss AG) at a magnification of ×200.
Determination of GSH and oxidized
glutathione (GSSG) levels
The intracellular level of reduced GSH was
determined as previously described (21). Briefly, cells were incubated with
the fluorescent probe monochlorobimane (40 mM; cat. no. HY-101899;
Sigma-Aldrich; Merck KGaA) for 20 min at room temperature in the
dark. After washing with PBS twice (5-10 min per wash), the cells
were solubilized with 5 mM Tris-HCl (pH 7.4) and 1% SDS.
Fluorescence was measured using a spectrofluorophotometer
(Rf-5301PC; Shimadzu Corporation) at an excitation wavelength of
390 nm and an emission wavelength of 520 nm. For GSSG analysis, the
GSH scavenger 1-methyl-2-vinylpyridinium trifluoromethane sulfonate
was added, and the GSSG levels were then determined
spectrophotometrically using the GSH reductase-linked
5,5′-dithiobis(2-nitrobenzoic acid).
Western blotting
Total protein was extracted from ARPE-19 cells using
RIPA buffer (1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS) and a
protease inhibitor mixture and separated by 10% SDS-PAGE. The
nitrocellulose membrane (EMD Millipore) needed for the transfer was
hydrated in cold Tris-glycine buffer (pH 8.3) at 10 V for 1.5 h.
The membranes were blocked with TBS containing 5% non-fat milk and
incubated for 2 h at room temperature, followed by incubation with
primary antibodies against ICAM-1 (cat. no. 4915S; 1:1,000; Cell
Signaling Technology, Inc.), NF-κB/p65 (cat. no. KAS-TF110;
1:1,000; Stressgen Biotechnologies), IκBa (cat. no. sc-847;
1:1,000; Santa Cruz Biotechnology, Inc.), PARP-1 (cat. no.
sc-136208; 1:200; Santa Cruz Biotechnology, Inc.), lamin (cat. no.
sc-6217; 1:1,000; Santa Cruz Biotechnology, Inc.) and tubulin (cat.
no. T568; 1:1,000; Sigma-Aldrich; Merck KGaA) overnight at 4°C with
gentle shaking. After incubation with the primary antibodies, the
membranes were washed with TBS with 5% non-fat milk and incubated
with horseradish peroxidase-conjugated anti-rabbit (cat. no.
G-21040; 1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.) and
anti-mouse (cat. no. 31460; 1:2,500; Invitrogen; Thermo Fisher
Scientific, Inc.) antibodies for 2 h at 4°C. Immunoreactive bands
were visualized with enhanced chemiluminescence solution (EMD
Millipore).
Mitochondrial membrane potential
assay
ARPE-19 cells (1.5×104 cells/well) were
cultured in a 12-well plate and exposed to blue light for 24 h. The
measurement of mitochondrial membrane potential was performed using
JC-1 dye (Mitochondrial Membrane Potential Probe; Invitrogen;
Thermo Fisher Scientific, Inc.). The cells were washed with PBS for
5 min and incubated with 10 µg/ml JC-1 at 37°C for 15 min in
the dark. Images were captured using a fluorescence microscope
(Axiovert S100; Carl Zeiss AG; magnification, ×400), which detects
healthy cells with JC-1 J-aggregates (excitation/emission, 540/605
nm) and unhealthy cells with mostly JC-1 monomers
(excitation/emission, 480/510 nm). The quantification of the images
was performed with ImageJ software, version 1.8.0 (National
Institutes of Health).
Monocyte adhesion assay
Cells grown to 90-100% confluence in a 96-well plate
were pretreated with CORMs and/or TNFα for 6 h to allow for the
expression of ICAM-1. The cells were co-cultured with
5×105 calcein-labeled THP-1 cells for 30 min. After
washing twice with RPMI-1640 medium, adherent cells were examined
using an ELISA plate reader (FLx800, Bio-Tek Instruments, Inc.) at
485 nm excitation and 538 nm emission wavelengths (22).
Immunofluorescence staining
ARPE-19 cells were pretreated with the test
compounds for 2 h at 37°C prior to exposure to TNFα. The cells were
fixed in 80% ethanol at room temperature for 10 min and treated
with 0.1% (v/v) Triton X-100 in PBS. After blocking with 3% BSA
(Sigma-Aldrich; Merck KGaA) for 1 h at room temperature, the cells
were incubated with mouse monoclonal antibody (anti-NF-κB/p65
antibody diluted 1:100 in PBS) overnight at 4°C. Subsequently, the
cells were incubated with Alexa Fluor 488 goat anti-mouse IgG (H+L;
A28175; 1:1,000; Invitrogen; Thermo Fisher Scientific, Inc.) for 1
h. Cell nuclei were stained with propidium iodide (10 ng/ml in PBS)
at room temperature for 30 min in the dark and images were captured
using a by Zeiss Axiovert S100 fluorescence microscope (Carl Zeiss
AG).
Nuclear and cytoplasmic protein
extraction
ARPE-19 cells were collected by scraping in cold PBS
and pelleted by centrifugation at 1,000 × g for 5 min at 4°C. The
cell pellet was resuspended in the cell lysis buffer (including 10
mM HEPES, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM
dithiothreitol, 0.5 mM phenylmethanesulfonyl fluoride and 0.3%
Nonidet P-40) and then centrifuged at 12,000 × g for 5 min at 4°C.
The collected supernatant was designated as the cytoplasmic
fraction. Nuclear proteins were then extracted using a buffer
containing 20 mM HEPES, 25% glycerol, 1.5 mM MgCl2, 0.6
M KCl and 0.2 mM EDTA.
RNA isolation and reverse
transcription-PCR (RT-PCR) analysis
Total RNA was isolated using the TRIzol reagent
according to the manufacturer's instructions (Invitrogen; Thermo
Fisher Scientific, Inc.). Equal quantities (5 µg) of RNA
from the cells receiving various treatments were then
reverse-transcribed using 50 units of Superscript II (Invitrogen;
Thermo Fisher Scientific, Inc.) for 50 min at 42°C. PCR was
performed in a 25-µl reaction mixture containing 10 mM
Tris-HCl, 50 mM KCl, 5 mM MgCl2, 0.1% Triton X-100 (pH
9.0) and 0.6 U Taq DNA polymerase (Promega Corporation). The
primers (30 pmol) used for the amplification of glutamate-cysteine
ligase (GCL) modifier subunit (GCLM), GCL catalytic subunit (GCLC),
ICAM-1 and GAPDH were as follows: GCLM forward, 5′-CAG CGA GGA GCT
TCA TGA TTG-3′ and reverse, 5′-TGA TCA CAG AAT CCA GCT GTG C-3′;
GCLC forward, 5′-GTT CTT GAA ACT CTG CAA GAG AAG-3′ and reverse,
5′-ATG GAG ATG GTG TAT TCT TGT CC-3′; ICAM-1 forward, 5′-AGC AAT
GTG CAA GAA GAT AGC CAA-3′ and reverse, 5′-GGT CCC CTG CGT GTT CCA
CC-3′; GAPDH forward, 5′-TAT CGT GGA AGG ACT CAT GAC C-3′ and
reverse, 5′-TAC ATG GCA ACTG TGA GGG G-3′. The thermocycling
conditions were as follows: 1 cycle of 5 min at 95°C, followed by
30 cycles of 40 sec at 95°C, 30 sec at 59°C (62°C for GCLC), and 50
sec at 72°C, with a final extension at 72°C for 5 min. Reaction
products were separated electrophoretically on a 2.5% agarose gel
and stained with ethidium bromide.
Plasmids, transfections, and measurement
of luciferase activity
The ARPE-19 cells were transfected with 1 µg
of NF-κB/Luc or p3xARE/Luc using Lipofectamine 3000 (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. Briefly, the reporter DNA (2 µg) and
β-galactosidase DNA (0.5 µg) were mixed with 5 µl
Lipofectamine 3000 for 10 min at room temperature. The mixture was
added to ARPE-19 cells and, 4 h later, 10% FBS DMEM/F-12 was added
for 24 h. For the luciferase assays, the cell lysate was mixed with
luciferase substrate solution (Promega Corporation), and then the
resultant luciferase activity was measured using the FB12 Tube
Luminometer (Titertek-Berthold). The luciferase activities were
standardized to β-galactosidase activity.
Scratch assay for endothelial cell (EC)
migration
For the wound healing assay, 1.5×105 ECs
per well were cultured for 24 h in a 12-well plate with serum-free
DMEM. When the cells had grown to 90-100% confluence, the cell
monolayer was scratched with a 200-µl pipette tip, washed
twice with DMEM, and incubated in a serum-free medium with 10 ng/ml
VEGF-A alone, or 10 ng/ml VEGF-A with CORMs. Cell migration and
wound closure was monitored, and images were captured at 0, 12 and
24 h using a phase-contrast Axiovert S100 microscope (Carl Zeiss
AG). Photographs were taken at 12 and 24 h after wounding. The
distance migrated by the ECs to close the wounded area was
measured. The quantification of the area was analyzed with ImageJ
software, version 1.8.0 (National Institutes of Health). Results
are expressed as a migration index calculated from the slope of the
distance-time curve at 0, 6, 12, 18 and 24 h relative to the slope
of the VEGF-treated cells.
Transwell migration assay
Cell migration assays were performed using Transwell
plates with 8-µm pore filters (Corning, Inc.), following the
manufacturer's protocol. Briefly, for the cell migration assay,
cells (2×105) were suspended in 200 µl of
serum-free medium and seeded into the upper chambers of the
Transwell plates; serum-free medium supplemented with 10 ng/ml
VEGF-A was applied to the lower chamber as a chemoattractant to
induce migration. After incubation for 24 h at 37°C, cells were
fixed with 4% cold paraformaldehyde for 15 min at room temperature
and stained with 0.1% crystal violet solution for 5 min at room
temperature. Non-migrating cells remaining on the upper surface of
the filter membrane were scraped off gently with a cotton swab. For
quantification, the stained cells that had migrated to the other
side of the membrane were extracted with 33% acetic acid. The
absorbance of the eluted stain was determined at 570 nm.
Statistical analysis
Values are expressed as the mean ± standard error of
at least three experiments. Statistical significance was assessed
through one-way analysis of variance, followed by Tukey's post hoc
test using SigmaPlot version 12 (Systat Software, Inc.). P<0.05
was considered to indicate statistically significant
differences.
Results
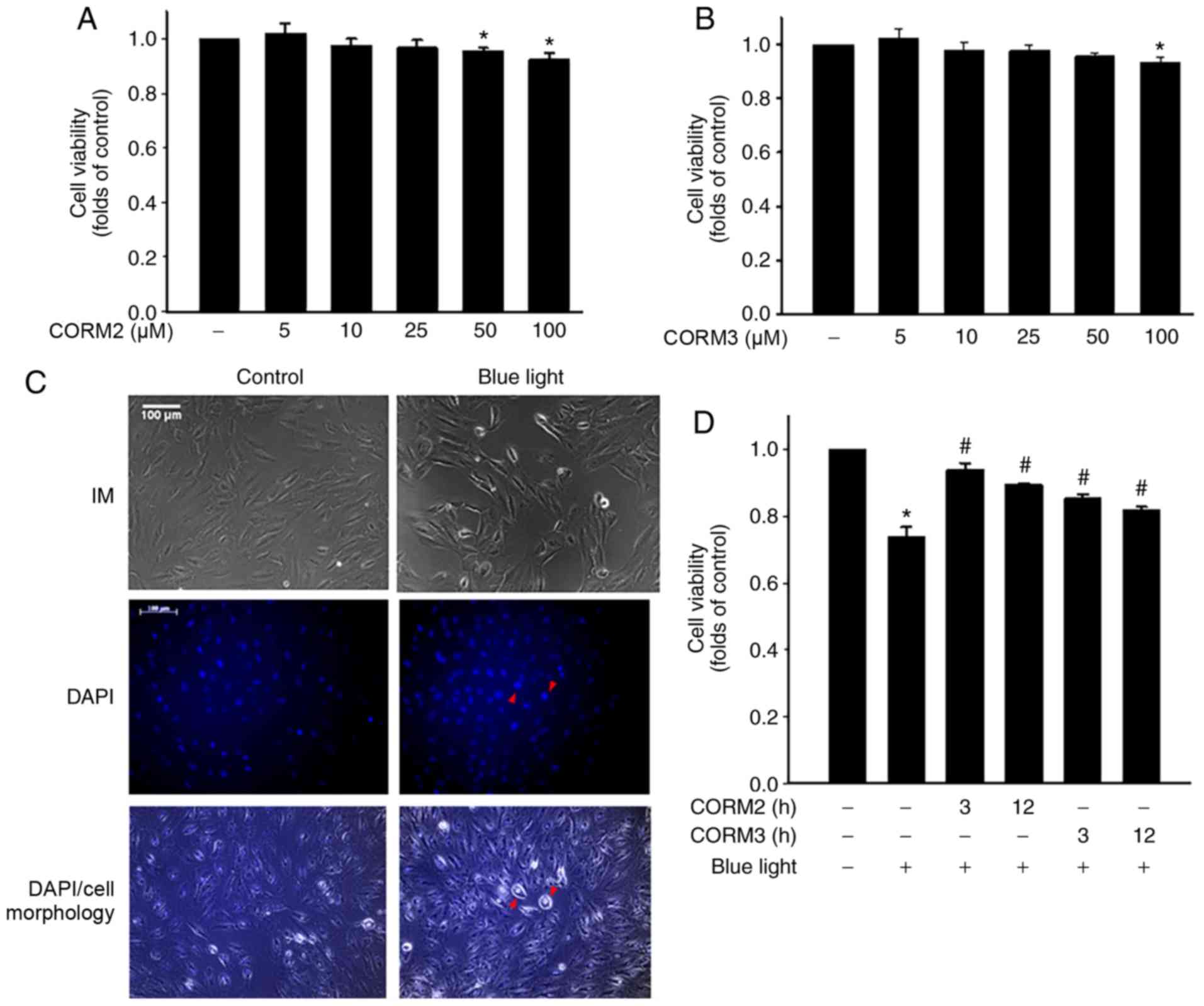
CORMs reduce blue light-induced
cytotoxicity in RPE cells
To determine whether CO could protect RPE cells from
stress, the viability of ARPE-19 cells treated with CORM2 and CORM3
was first determined. As shown in Fig. 1A and B, CORM2 and CORM3 at 25 mM
did not exert a cytotoxic effect on ARPE-19 cells. Subsequently,
the protective effects of CORMs against blue light-induced
cytotoxicity were examined in RPE cells. First, after exposure to
blue light for 24 h, some RPE cells were shrunk and their nuclei
were condensed, as detected by DAPI staining (Fig. 1C). Pretreating cells with 25 mM
CORM2 or CORM3 for 3 or 12 h significantly prevented this blue
light-induced cytotoxicity (Fig.
1D), with the protective effect of CORM2 being superior to that
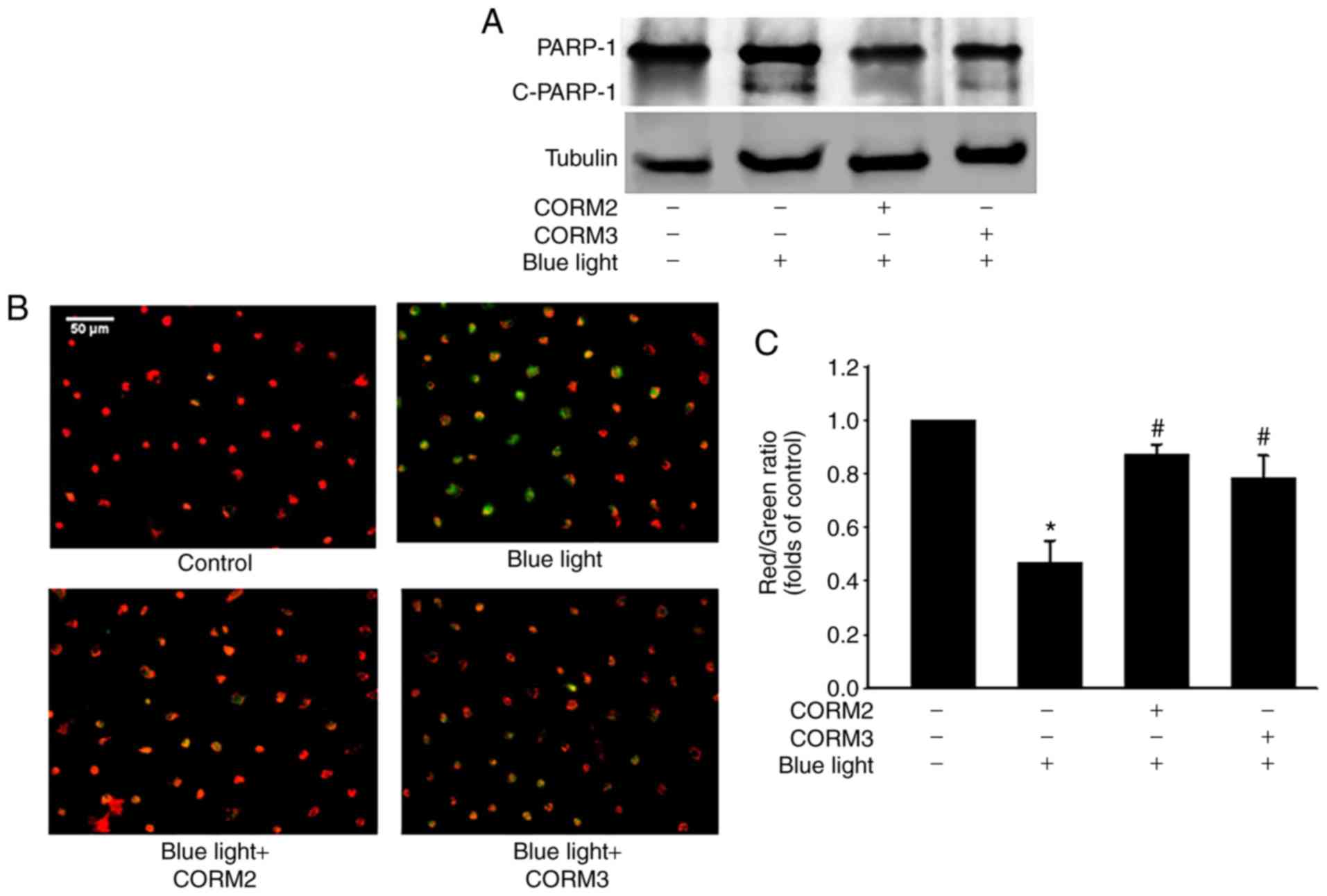
of CORM3. To elucidate whether CORM2 or CORM3 could attenuate blue
light-induced cytotoxicity, the expression of cleaved PARP-1, which
is one of the downstream effectors of caspase-3, was detected by
immunoblotting. As shown in Fig.
2A, the blue light-induced cleaved PARP-1 expression was
markedly suppressed by pre-incubation with CORM2 or CORM3.
Subsequently, the protective mechanisms of CORM2 and CORM3 were
compared to investigate the effect of blue light on mitochondrial
activity using JC-1 fluorescence and the results are shown in
Fig. 2B. As seen in the
representative images, the red fluorescence intensity was increased
by 25 mM CORM2 or CORM3 pretreatment compared to that of the blue
light-treated cells (Fig. 2C).
These results suggest that CORM2 and CORM3 effectively inhibited
blue light-induced apoptosis by maintaining the mitochondrial
membrane potential.
The antioxidant activity of CORM2 is
mediated through an increase in the GSH level
A Previous study demonstrated that inflammatory
cytokines increase the production of ROS through mitochondria and
NADPH oxidases in cultured RPE cells (23). To determine the antioxidant
properties of CORMs, the protective effects of CORM2 and CORM3
against oxidative stress were first examined by subjecting the
cells to 1 mM H2O2 for 1 h. Pretreating cells
with 25 mM CORM2 for 12 h significantly prevented oxidative
stress-induced cell death (Fig.
3A). As shown in Fig. 3B,
CORM2 treatment for 3 h increased the HO-1 level. GSH is the most
abundant antioxidant n maintaining cellular redox status (12). The enzyme involved in the de
novo synthesis of GSH is GCL, which comprises GCLC and GCLM
subunits. Treatment with CORM2 increased the GCLC and GCLM
expression levels over the course of the incubation period
(Fig. 3C). As indicated in
Fig. 3D, the reduced GSH levels
were increased after 3 h of CORM2 treatment. However, the GSSG
levels were found to have increased slightly at 3 h of CORM2
treatment (Fig. 3E). CORM2, but
not CORM3, caused an increase in Nrf2 transcriptional activity with
ARE-luciferase reporter construct in RPE cells (Fig. 3F). These findings suggested that
CORM2 pretreatment exerted a stronger antioxidant effect compared
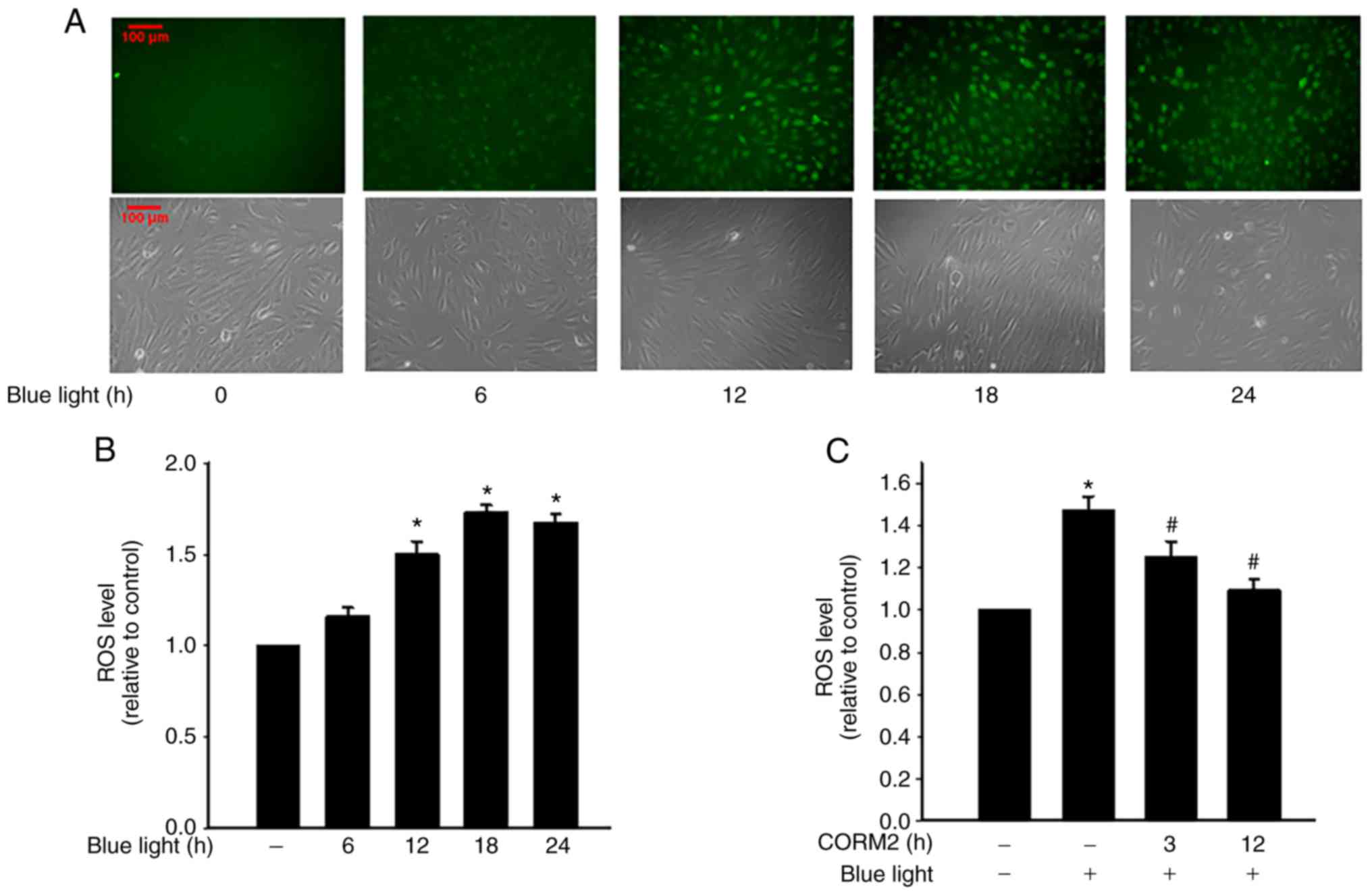
with CORM3. As shown in Fig. 4A and
B, blue light exposure induced ROS generation in RPE cells and
increased intracellular ROS levels in a time-dependent manner. In
addition, CORM2 inhibited blue light-induced oxidative stress
(Fig. 4C). Taken together, these
results demonstrated that CORM2 exerted an antioxidant effect and
abolished blue light-increased oxidative stress in RPE cells.
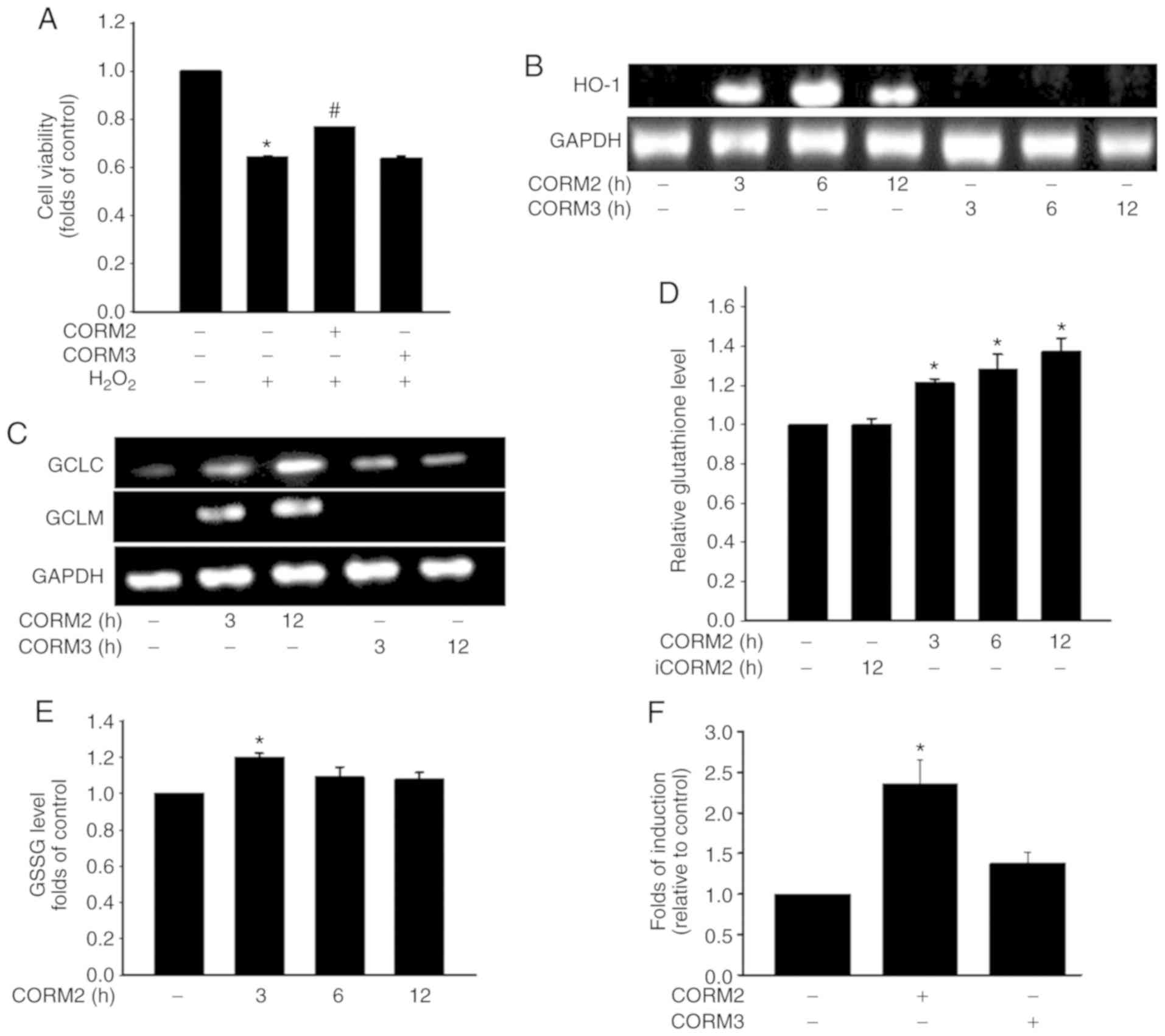 | Figure 3The antioxidant activity of CORM2 was
mediated through an increase of the GSH level. (A) RPE cells were
pretreated with 25 µM CORM2 or CORM3 for 12 h, then treated
with 1 mM of H2O2 for 1 h, and cell viability
was measured. The results shown are the mean ± standard error.
*P<0.05 compared with untreated cells,
#P<0.05 compared with H2O2
alone (mean ± standard error). (B) RPE cells were treated with 25
µM CORM2 and CORM3 for the indicated time periods and then
subjected to RT-PCR analysis. (C) RPE cells were treated with 25
µM CORM2 and CORM3 for the indicated time periods and
subjected to RT-PCR analysis. (D and E) RPE cells were exposed to
25 µM CORM2 or iCORM2 for the indicated time periods and the
intracellular GSH and GSSG levels were then measured. The results
shown are the mean ± standard error. *P<0.05 compared
with untreated cells. (F) Cells were transfected with an
ARE-luciferase construct. After 12 h, the cells were treated with
25 µM CORM2 and CORM3. All values are presented as mean ±
standard error. *P<0.05. RPE, retinal pigment
epithelium; CORM, carbon monoxide-releasing molecule; iCORM2,
inactive CORM2; GSH, reduced glutathione; GSSG, oxidized GSH; HO-1,
heme oxygenase 1; GCL, glutamate-cysteine ligase; GCLC, GCL
catalytic subunit; GCLM, GCL modifier subunit; RT-PCR, reverse
transcription-PCR. |
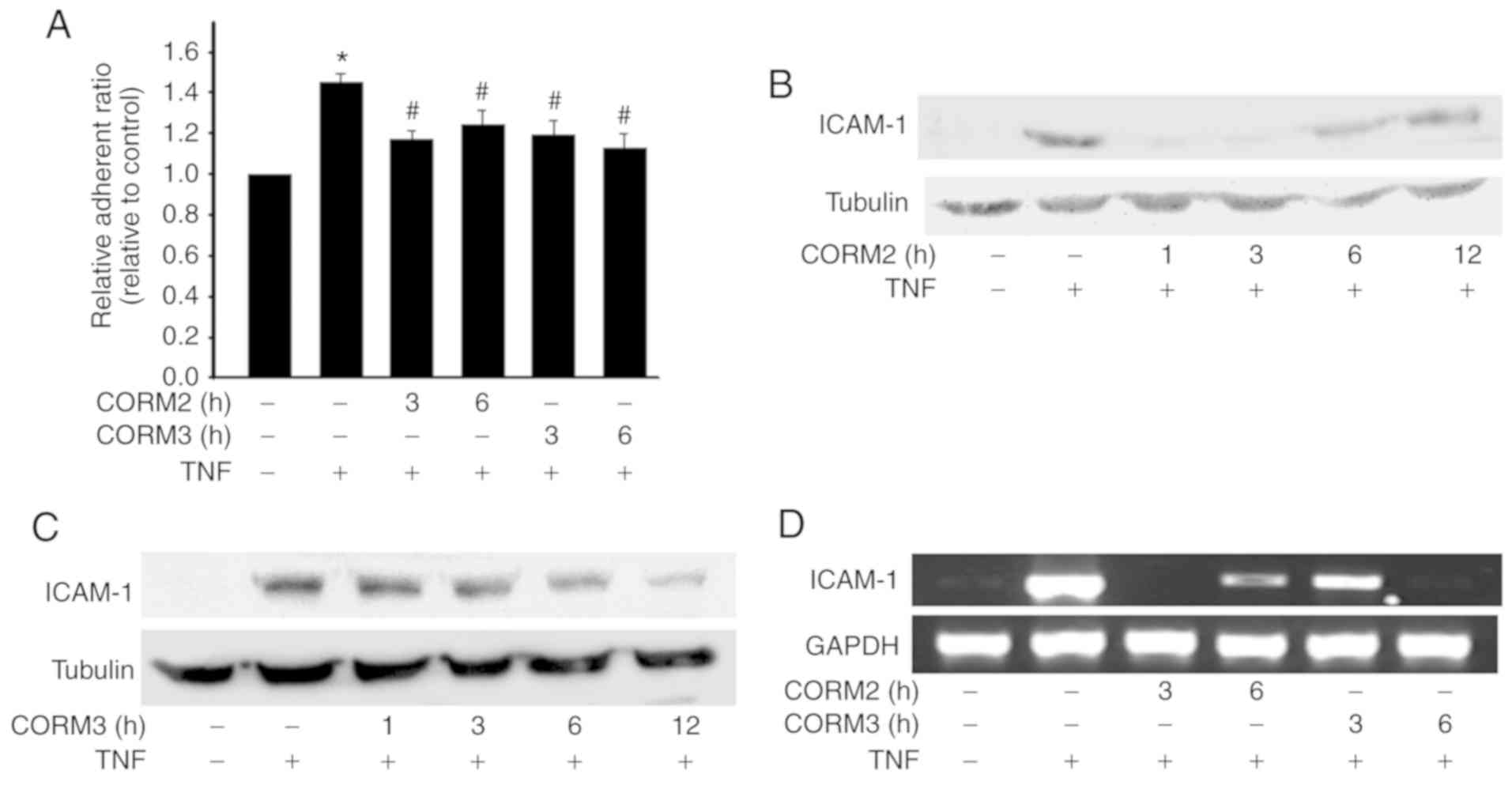
CORM2 and CORM3 inhibit monocyte adhesion
and ICAM-1 expression
To detect the anti-inflammatory properties of CORMs
in RPE cells, the effects of CORMs on monocyte adhesion to ARPE-19
were examined. We discovered that TNFα significantly increased
monocyte adhesion, which was inhibited by pretreatment with 25 mM
CORM2 and CORM3 for 3-6 h (Fig.
5A). ICAM-1 is a major component in leukocyte adhesion
(5). Therefore, we next examined
the effects of CORM2 and CORM3 on the expression of adhesion
molecules in ARPE-19 cells. It was observed that pretreatment of
ARPE-19 cells with CORM2 and CORM3 for 12 h significantly inhibited
TNFα-induced ICAM-1 expression. Pretreatment of ARPE-19 cells with
CORM2 was found to have inhibited TNFα-induced ICAM-1 expression
after 1 h, but its effect became weaker over time (Fig. 5B). By contrast, the inhibitory
effect of CORM3 did not begin until after 3 h of pretreatment, but
its effect lasted for up to 12 h (Fig. 5C). In addition, the ICAM-1 mRNA
levels were analyzed using RT-PCR. Pretreatments with CORM2 and
CORM3 were conducted at 3 and 6 h, and it was observed that these
treatments decreased the ICAM-1 mRNA levels consistently with the
protein levels (Fig. 5D).
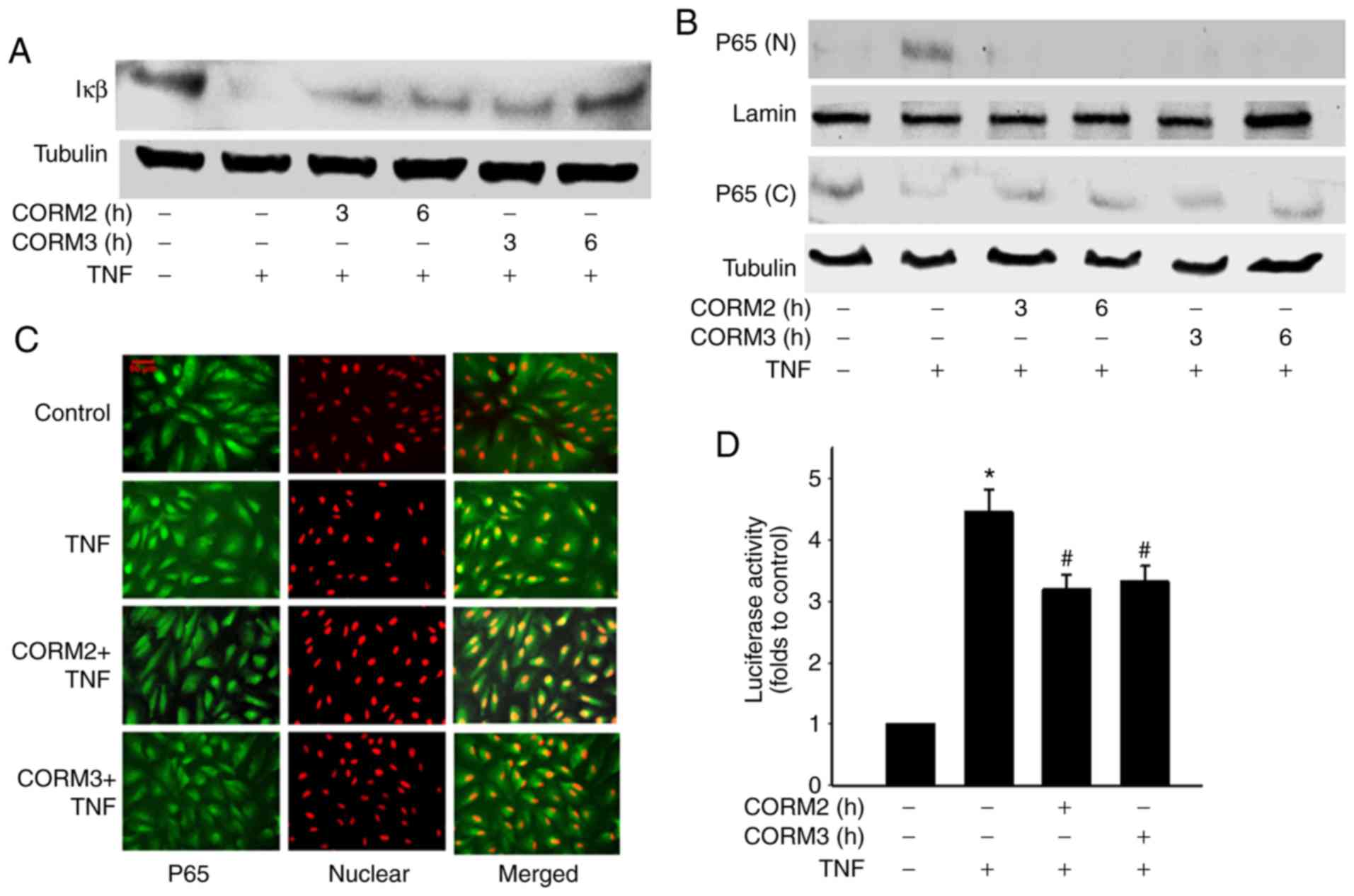
Effect of CORMs on TNFα-induced IκBα
degradation and NF-κB nuclear translocation
A previous study demonstrated that TNFa induces the
upregulation of ICAM-1 through an NF-κB activation pathway
(7). In the present study, it was
examined whether CORM2 and CORM3 regulate TNFα-induced NF-κB
activation. Degradation of the inhibitory protein IκBα was blocked
by CORM2 and CORM3 treatment (Fig.
6A). From 3 to 6 h after CORM2 and CORM3 pretreatment,
TNFα-induced NF-κB/p65 nuclear translocation was decreased
(Fig. 6B). The effect of CORM2
and CORM3 on nuclear translocation were further tested by
immunostaining with a p65 antibody to determine NF-κB
translocation, and an inhibitory effect was found to be associated
with the 6-h pretreatment (Fig.
6C). In addition, we tested whether CORM2 and CORM3 inhibit
TNFα-induced p65 activation at the transcriptional level by using
an NF-κB reporter assay. After pretreatment and a subsequent delay
of 12 h, a luciferase assay was employed (Fig. 6D) to confirm whether TNFα-induced
NF-κB activation was inhibited. Collectively, our experiments
demonstrated that CORM2 and CORM3 inhibited NF-κB nuclear
translocation and activation.
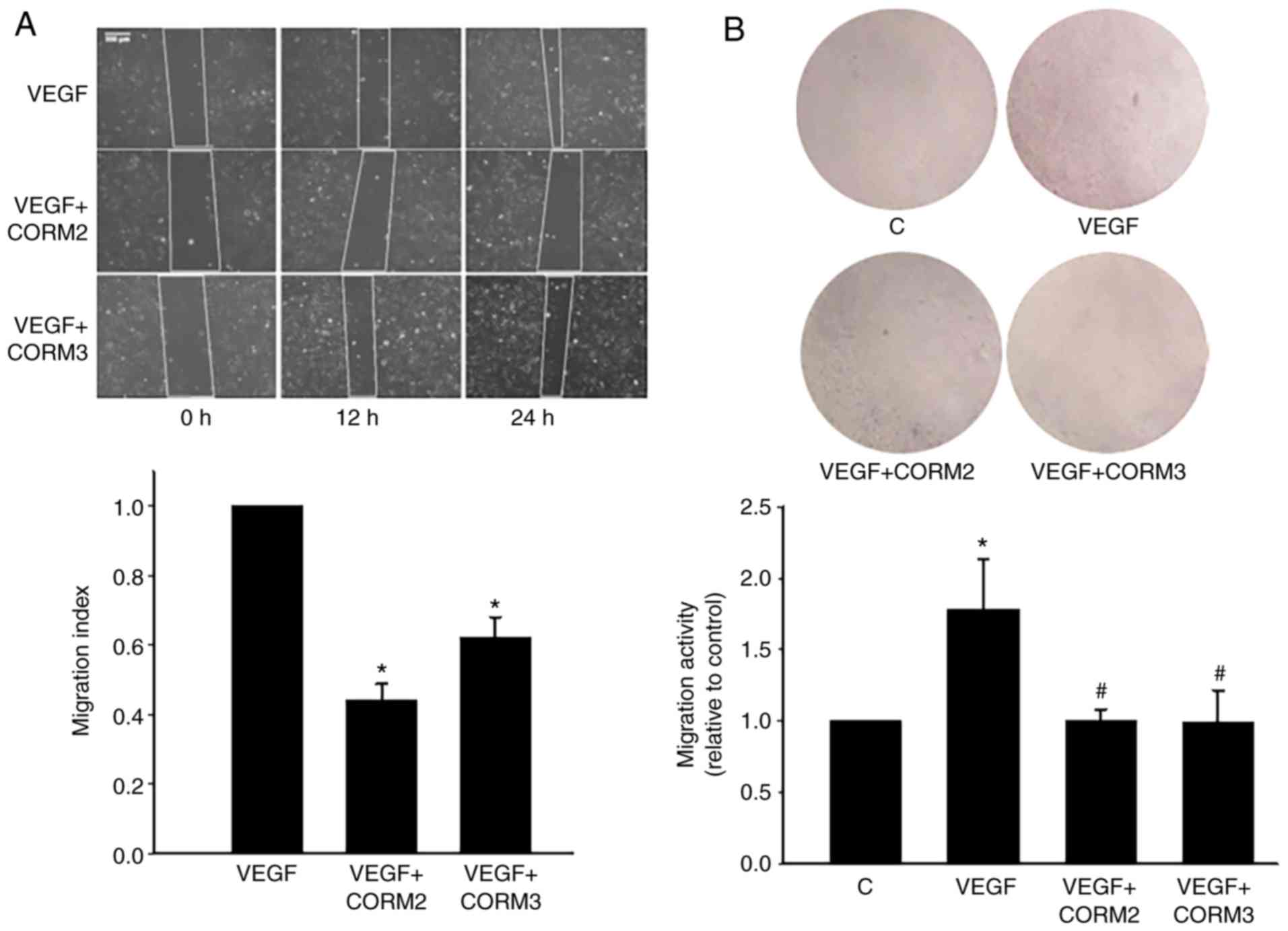
CORMs prevented the migration of ECs
VEGF-induced angiogenesis plays a pivotal role in
the choroidal neovascularization that characterizes the wet form of
AMD (2). To determine whether CO
could inhibit EC migration, a scratch assay was used, and it
demonstrated that VEGF induced the migratory capacity of ECs.
VEGF-induced migration was decreased through treatment with 25 mM
CORM2 and CORM3 (Fig. 7A). The
results of Transwell assay revealed that CORMs considerably reduced
the number of migrating cells on the membrane filter compared with
the VEGF-containing medium (Fig.
7B). Collectively, these results suggest that CORM2 and CORM3
contribute to the inhibition of the migration capacity of ECs.
Discussion
RPE cells are a component of the outer blood-retinal
barrier located between the blood vessels of the choroid and the
outer segments of the photoreceptors. They are vital for protecting
the retina from excessive light exposure, oxidative stress and
immune responses (24,25). In several retinal diseases,
including AMD, the dysfunction of RPE cells is a crucial event in
the disease process (4). How
CORMs can cross the blood-retinal barrier is not fully understood,
but released CO gas is considered to cross biological membranes
(19). This suggests that CORMs
may act as a therapeutic target to initiate protective mechanisms
in the retina. The present study investigated the protective
effects of CORMs against blue light-induced cell injury,
TNF-α-induced inflammation and VEGF-induced cell migration.
RPE cells are sensitive to blue
light
The present study demonstrated that blue light
exposure induced cell injury and caused cell apoptosis (Fig. 1C). Our results also confirmed an
increase in ROS levels in blue light-exposed RPE cells (Fig. 4A). A previous study demonstrated
that blue light-induced mitochondrial ROS accumulation may lead to
cell death (26), and oxidative
stress may be responsible for the blue light-induced cell
apoptosis. Consistently, we found that CORMs effectively reduced
cleaved PARP-1 and maintained mitochondrial membrane potential in
blue light-exposed RPE cells (Fig.
2). However, the protective mechanisms of CORM2 and CORM3 were
compared with respect their effects of blue light-induced
cytotoxicity. It was observed that CORM2 exerted a stronger
protective effect compared with that of CORM3 in preventing blue
light-induced cytotoxicity (Fig.
1D). A possible explanation is that the lipid-soluble CORM2 was
more efficient than the water-soluble CORM3 in its antioxidant role
(Fig. 3A).
Inflammation is a defense mechanism that is
triggered by tissue damage due to various causes. Chronic
inflammation is a prolonged condition in which damaged tissues
attempt to self-repair, leading to tissue remodeling and possible
dysfunction. Chronic inflammation is the common pathological basis
for certain age-associated diseases, such as AMD. Local
inflammation and immune-mediated processes play a key role in AMD
pathogenesis (4,5). The results of the present study
demonstrated that treatment with CORMs conferred anti-inflammatory
protection on cytokine-treated RPE cells, and that CORM2 and CORM3
enhanced the inhibition of TNF-α-induced monocyte-RPE interaction
by downregulating the expression of ICAM-1. It was demonstrated
that CORM2 and CORM3 exerted an anti-inflammatory effect on RPE
cells, and this effect was characterized by inhibition of
TNF-α-induced expression of ICAM-1 with respect to the molecular
mechanisms of NF-κB translocation and IκBa degradation. A
particularly notable finding was that the lipid-soluble CORM2 and
water-soluble CORM3 displayed different patterns in their
anti-inflammatory effects by blocking ICAM-1 expression (Fig. 5) and degradation of the inhibitory
protein IκBα (Fig. 6A). A shown
in Fig. 5B and C, CORM2
pretreatment exert its inhibitory effect on ICAM-1 expression in 1
and 3 h, while the inhibitory effect of CORM3 is observed after 6 h
of pretreatment. A possible explanation is the lipid solubility of
CORM2, which enables it to exert its anti-inflammatory effects more
quickly compared with the water-soluble CORM3. The lipid-soluble
CORM2 may pass through cell membranes and more efficiently release
CO at the targets, whereas CORM3 may release CO at a certain
concentration outside the cells.
The effects of CORM2 are mediated through HO-1
induction in various systems (14,15,27). In the present analyses, a
significant increase in HO-1 protein level was observed following
treatment with CORM2, but not CORM3, for 3 h (Fig. 3B). This finding suggests the
involvement of HO-1 in the CORM2-mediated antioxidant effect during
long-term treatment. Although the half-life of CORM2 is short (~20
min), CORM2 triggers a positive feedback loop through increasing
the HO-1 expression to maintain the antioxidant effect of CO for 12
h (Fig. 3A).
GSH plays numerous roles in protecting cells from
oxidants and maintaining cellular thiol redox state. An imbalance
in the cellular thiol redox state has been implicated in the
progression of AMD (28). Our
earlier study reported that lycopene, a natural carotenoid, can
increase GSH, which further protects the RPE cells from oxidative
stress-induced damage (29).
Another study demonstrated that curcumin protects retinal cells
from light-induced and oxidative stress-induced damage through
Nrf2-dependent upregulation of antioxidative enzymes (30). Our previous studies demonstrated
that the GSH-dependent redox state is a major modulator of the
anti-inflammatory effects of cinnamaldehyde and CO in ECs (31,32). CO has been reported to generate
mild oxidative stress by inhibition of the function of cytochrome c
oxidase in the mitochondria; however, this mild stress may induce
the Nrf2 pathway and, thus, increase GSH levels (32). The present study determined that
CORM2 increased the gene expression of both GCLC and GCLM, which
are the key rate-limiting enzyme in GSH synthesis (Fig. 3C). Additional experiments
demonstrated that CORM2 increased the intracellular GSH level and
Nrf-2 activity in RPE cells (Fig. 3D
and E). There is a possibility that the lipid-soluble CORM2 can
more efficiently reach an intracellular concentration to increase
antioxidant activity. In the present study, it was demonstrated
that CORM2 could abolish blue light-induced oxidative stress
(Fig. 4C). Thus, the increased
GSH level is a key contributor to the cytoprotective effect of
CORM2.
AMD is a major cause of vision loss among elderly
people. The neovascular form (wet AMD) is characterized by the
growth of blood vessels from the choroid toward the retina
(1). VEGF plays a critical role
in the pathophysiological process of neovascular AMD (33). Anti-VEGF therapy is currently the
standard of care for wet AMD (34). A previous study demonstrated that
CO inhibits sprouting angiogenesis and VEGF receptor-2
phosphorylation (35). In
addition, our previous study demonstrated that CORM2 induced nitric
oxide (NO) production in ECs (36). NO acts as a potent vasodilator and
plays a key role in the physiological regulation of ocular blood
flow. The present study demonstrated that treatment with 25 mM
CORM2 and CORM3 enhanced the suppression of VEGF-induced migration
(Fig. 7A). In Transwell invasion
assays, CORMs reduced cell migration toward the VEGF-containing
medium (Fig. 7B). Collectively,
these results suggest that CORM2 and CORM3 contribute to the
inhibition of the migration and invasion capacity of ECs.
In conclusion, the present research demonstrated
that CORM2 and CORM3 exhibited different patterns of
anti-inflammatory effects and played pivotal roles in the
anti-inflammatory process through the suppression of NF-κB
activation in ARPE-19 cells. Compared with CORM3, CORM2 was more
efficient in increasing antioxidant enzyme expression, resulting in
more prominent cytoprotective effects. A better understanding of
the functional mechanisms of CORMs may contribute to the
development of an effective CO-releasing model for therapeutic
application in pathological eye conditions.
Acknowledgments
The authors would like to thank Kai-Qin Yang,
Yu-Zhang Chen, Jing-Yao Huang and Zi-Qiao Lan for their assistance
with selected experiments.
Funding
The present study was supported by grants from the
National Science Council of Taiwan (no. 105-2320-B-415-007) and the
Chiayi Christian Hospital (grant no. R107-20).
Availability of data and materials
The datasets used and/or analyzed during the present
study are available from the corresponding author on reasonable
request.
Authors' contributions
PMY, KCC and BSW conceived and designed the
experiments. PMY, KCC and SHY performed all experiments. PMY and
BSW analyzed the data. BSW wrote and revised the manuscript. All
authors have read and approved the final manuscript.
Ethics approval and consent to
participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
All the authors declare that they have no competing
interests.
References
|
1
|
Kokotas H, Grigoriadou M and Petersen MB:
Age-related macular degeneration: Genetic and clinical findings.
Clin Chem Lab Med. 49:601–616. 2011. View Article : Google Scholar
|
|
2
|
Kent D and Sheridan C: Choroidal
neovascularization: A wound healing perspective. Mol Vis.
9:747–755. 2003.
|
|
3
|
Vicente-Tejedor J, Marchena M, Ramírez L,
García-Ayuso D, Gómez-Vicente V, Sánchez-Ramos C, de la Villa P and
Germain F: Removal of the blue component of light significantly
decreases retinal damage after high intensity exposure. PLoS One.
13:e01942182018. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Strauss O: The retinal pigment epithelium
in visual function. Physiol Rev. 85:845–81. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Cousins SW, Espinosa-Heidmann DG and Csaky
KG: Monocyte activation in patients with age-related macular
degeneration: A biomarker of risk for choroidal neovascularization?
Arch Ophthalmol. 122:1013–1018. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang L, Froio RM, Sciuto TE, Dvorak AM,
Alon R and Luscinskas FW: ICAM-1 regulates neutrophil adhesion and
transcellular migration of TNF-alpha-activated vascular endothelium
under flow. Blood. 106:584–592. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ledebur HC and Parks TP: Transcriptional
regulation of the inter-cellular adhesion molecule-1 gene by
inflammatory cytokines in human endothelial cells. Essential roles
of a variant NF-kappaB site and p65 homodimers. J Biol Chem.
270:933–943. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tak PP and Firestein GS: NF-kappaB: A key
role in inflammatory diseases. J Clin Invest. 107:7–11. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Nakanishi-Ueda T, Majima HJ, Watanabe K,
Ueda T, Indo HP, Suenaga S, Hisamitsu T, Ozawa T, Yasuhara H and
Koide R: Blue LED light exposure develops intracellular reactive
oxygen species, lipid peroxidation, and subsequent cellular
injuries in cultured bovine retinal pigment epithelial cells. Free
Radic Res. 47:774–780. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Itoh K, Chiba T, Takahashi S, Ishii T,
Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, et
al: An Nrf2/small Maf heterodimer mediates the induction of phase
II detoxifying enzyme genes through antioxidant response elements.
Biochem Biophys Res Commun. 236:313–322. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Alam J, Stewart D, Touchard C, Boinapally
S, Choi AM and Cook JL: Nrf2, a Cap'n'Collar transcription factor,
regulates induction of the heme oxygenase-1 gene. J Biol Chem.
274:26071–26078. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McMahon M, Itoh K, Yamamoto M, Chanas SA,
Henderson CJ, McLellan LI, Wolf CR, Cavin C and Hayes JD: The
Cap'n'Collar basic leucine zipper transcription factor Nrf2 (NF-E2
p45-related factor 2) controls both constitutive and inducible
expression of intestinal detoxification and glutathione
biosynthetic enzymes. Cancer Res. 61:3299–3307. 2001.PubMed/NCBI
|
|
13
|
Townsend DM, Tew KD and Tapiero H: The
importance of glutathione in human disease. Biomed Pharmacother.
57:145–155. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kim KM, Pae HO, Zheng M, Park R, Kim YM
and Chung HT: Carbon monoxide induces heme oxygenase-1 via
activation of protein kinase R-like endoplasmic reticulum kinase
and inhibits endothelial cell apoptosis triggered by endoplasmic
reticulum stress. Circ Res. 101:919–927. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schwer CI, Mutschler M, Stoll P, Goebel U,
Humar M, Hoetzel A and Schmidt R: Carbon monoxide releasing
molecule-2 inhibits pancreatic stellate cell proliferation by
activating p38 mitogen-activated protein kinase/heme oxygenase-1
signaling. Mol Pharmacol. 77:660–669. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shin DY, Chung J, Joe Y, Pae HO, Chang KC,
Cho GJ, Wolf CR, Cavin C and Hayes JD: Pretreatment with
CO-releasing molecules suppresses hepcidin expression during
inflammation and endoplasmic reticulum stress through inhibition of
the STAT3 and CREBH pathways. Blood. 119:2523–2532. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ryter SW, Alam J and Choi AM: Heme
oxygenase-1/carbon monoxide: From basic science to therapeutic
applications. Physiol Rev. 86:583–650. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bannenberg GL and Vieira HL: Therapeutic
applications of the gaseous mediators carbon monoxide and hydrogen
sulfide. Expert Opin Ther Pat. 19:663–682. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Queiroga CS, Vercelli A and Vieira HL:
Carbon monoxide and the CNS: Challenges and achievements. Br J
Pharmacol. 172:1533–1545. 2015. View Article : Google Scholar :
|
|
20
|
Lian KC, Chuang JJ, Hsieh CW, Wung BS,
Huang GD, Jian TY and Sun YW: Dual mechanisms of NF-kappaB
inhibition in carnosol-treated endothelial cells. Toxicol Appl
Pharmacol. 245:21–35. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kamencic H, Lyon A, Paterson P and
Juurlink BH: Monochlorobimane fluorometric method to measure tissue
glutathione. Anal Biochem. 286:35–37. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Braut-Boucher F, Pichon J, Rat P, Adolphe
M, Aubery M and Font J: A non-isotopic, highly sensitive,
fluorimetric, cell-cell adhesion microplate assay using calcein
AM-labeled lymphocytes. J Immunol Methods. 178:41–51. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang D, Elner SG, Bian ZM, Till GO, Petty
HR and Elner VM: Pro-inflammatory cytokines increase reactive
oxygen species through mitochondria and NADPH oxidase in cultured
RPE cells. Exp Eye Res. 85:462–472. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sparrow JR, Hicks D and Hamel CP: The
retinal pigment epithelium in health and disease. Curr Mol Med.
10:802–823. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Pavan B and Dalpiaz A: Retinal pigment
epithelial cells as a therapeutic tool and target against
retinopathies. Drug Discov Today. 23:1672–1679. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
King A, Gottlieb E, Brooks DG, Murphy MP
and Dunaief JL: Mitochondria-derived reactive oxygen species
mediate blue light-induced death of retinal pigment epithelial
cells. Photochem Photobiol. 79:470–475. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Choi YK, Kim CK, Lee H, Jeoung D, Ha KS,
Kwon YG, Kim KW and Kim YM: Carbon monoxide promotes VEGF
expression by increasing HIF-1alpha protein level via two distinct
mechanisms, translational activation and stabilization of
HIF-1alpha protein. J Biol Chem. 285:32116–32125. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Samiec PS, Drews-Botsch C, Flagg EW, Kurtz
JC, Sternberg P Jr, Reed RL and Jones DP: Glutathione in human
plasma: Decline in association with aging, age-related macular
degeneration, and diabetes. Free Radic Biol Med. 24:699–704. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yang PM, Wu ZZ, Zhang YQ and Wung BS:
Lycopene inhibits ICAM-1 expression and NF-κB activation by
Nrf2-regulated cell redox state in human retinal pigment epithelial
cells. Life Sci. 155:94–101. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Mandal MN, Patlolla JM, Zheng L, Agbaga
MP, Tran JT, Wicker L, Kasus-Jacobi A, Elliott MH, Rao CV and
Anderson RE: Curcumin protects retinal cells from light-and oxidant
stress-induced cell death. Free Radic Biol Med. 46:672–679. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liao BC, Hsieh CW, Liu YC, Tzeng TT, Sun
YW and Wung BS: Cinnamaldehyde inhibits the tumor necrosis
factor-alpha-induced expression of cell adhesion molecules in
endothelial cells by suppressing NF-kappaB activation. Effects upon
IkappaB and Nrf2. Toxicol Appl Pharmacol. 229:161–171. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yeh PY, Li CY, Hsieh CW, Yang YC, Yang PM
and Wung BS: CO-releasing molecules and increased heme oxygenase-1
induce protein S-glutathionylation to modulate NF-kappaB activity
in endothelial cells. Free Radic Biol Med. 70:1–13. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hernández-Zimbrón LF, Zamora-Alvarado R,
Ochoa-De la Paz L, Velez-Montoya R, Zenteno E, Gulias-Cañizo R,
Quiroz-Mercado H and Gonzalez-Salinas R: Age-related macular
degeneration: New paradigms for treatment and management of AMD.
Oxid Med Cell Longev. 2018:83746472018. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Martin DF, Maguire MG, Ying GS, Grunwald
JE, Fine SL and Jaffe GJ: Ranibizumab and bevacizumab for
neovascular age-related macular degeneration. N Engl J Med.
364:1897–1908. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ahmad S, Hewett PW, Fujisawa T, Sissaoui
S, Cai M, Gueron G, Al-Ani B, Cudmore M, Ahmed SF, Wong MK, et al:
Carbon monoxide inhibits sprouting angiogenesis and vascular
endo-thelial growth factor receptor-2 phosphorylation. Thromb
Haemost. 113:329–337. 2015. View Article : Google Scholar
|
|
36
|
Yang PM, Huang YT, Zhang YQ, Hsieh CW and
Wung BS: Carbon monoxide releasing molecule induces endothelial
nitric oxide synthase activation through a calcium and
phosphati-dylinositol 3-kinase/Akt mechanism. Vascul Pharmacol.
87:209–218. 2016. View Article : Google Scholar : PubMed/NCBI
|