Introduction
Acute lung injury (ALI) or acute respiratory
distress syndrome (ARDS) is a clinical condition that imposes a
substantial health burden (1-6). A
prospective epidemiologic study estimated an annual incidence of
ALI/ARDS of 190,000 adult patients in the United States with an
associated 74,500 deaths per year (3-6).
Despite progress in the understanding of the molecular and cellular
mechanisms of ALI/ARDS, and significant advances in supporting
therapies, ALI/ARDS-related mortality rate remains high (1-6).
ALI/ARDS is characterized by inflammation, disruption of
endothelial and epithelial barrier integrity, and tissue injury in
the lungs, resulting in lung interstitial edema and leakage of
proteins and immune cells into the alveolar space (1-3).
Accelerating lung repair, and promoting the resolution of lung
inflammation and edema is a potential therapeutic strategy for
ALI/ARDS (2,3). It is, therefore, essential to
understand the mechanisms and signaling path-ways that regulate
these processes. However, the mechanisms and pathways underlying
the regulation of lung repair and inflammation resolution are
poorly understood (2,3,6).
The Hippo-YAP pathway is an evolutionarily conserved
cell survival signaling complex that controls tissue growth and
organ size (7-11). Key components of this signaling
pathway include mammalian Ste20-like kinases 1/2 (Mst1/2), large
tumor suppressor kinases 1/2 (LATS1/2) and downstream effectors,
including Yes-associated protein (YAP) and transcriptional
coactivator with PDZ-binding motif (TAZ) (7-11).
YAP and TAZ are transcriptional coactivators that regulate gene
expression by interacting with TEA/ATTS domain transcription factor
1-4 (TEAD1-4) (12). YAP/TAZ
activity is regulated by their phosphorylation and subcellular
translocation (7-11). When the Hippo pathway is
activated, Mst1/2 phosphorylate and activate LATS1/2. LATS1/2
kinases, in turn, phosphorylate YAP and TAZ and cause them to be
transported out of the nucleus, leading to the inhibition of their
activity and thus, to the inhibition of expression of their target
genes (7-11). When the Hippo pathway is
inactivated, Mst1/2 and LATS1/2 activities are inhibited, and
YAP/TAZ are dephosphorylated and translocated into the nucleus
where they interact with the TEADs to induce the expression of
their target genes (7-12).
The Hippo-YAP pathway plays a regulatory role in
organ development and lung regeneration after pneumonectomy.
Hippo-YAP integrates with upstream growth signals (13,14) to stimulate cell proliferation,
differentiation and migration (15-17), promote stem and progenitor cell
self-renewal, lineage commitment and expansion (18-21), and stimulate angiogenesis
(22), leading to organogenesis,
tissue growth and regeneration. Moreover, Hippo-YAP senses physical
and geographic cues in growing tissue and thus mediates
tissue-derived feedback signals that counteract the upstream growth
signals (15,16,23-26). The reciprocal regulation of
upstream growth signals and tissue-derived feedback signals
mediated by the Hippo-YAP pathway ensures a balance between tissue
growth and maturation (27) and
prevents organ overgrowth (8,9).
Although the Hippo-YAP pathway is known to regulate
multiple biological processes that play important roles in organ
development and tissue growth (7-22),
there have been few studies examining the roles of Hippo-YAP
activity in organ repair and organ function recovery after injury.
YAP activity has been reported to mediate peripheral nerve repair
after injury (28), and to
regulate hepatic repair following ischemia reperfusion liver injury
(29). The roles of the Hippo-YAP
pathway in lung injury and repair are just beginning to be studied.
Compared to cells with normal level of LATS2 expression,
mesenchymal stem cells with downregulated LATS2 expression are more
effective in protecting against lipopolysaccharide (LPS)-induced
ALI (30). Naphthalene-mediated
lung injury causes nuclear translocation of YAP, which then
stimulates expansion of basal stem/progenitor cells to promote
regeneration (31). LPS- or
Streptococcus pneumoniae-induced lung injury is associated
with increased YAP/TAZ nuclear translocation (32,33). Selective knockdown of YAP or TAZ
in alveolar type II epithelial cells (AECII) impairs epithelial
regeneration in mouse models of pneumonia (32). However, new epithelial cell
generation is only one step in the process of epithelial barrier
repair. The newly generated epithelial cells must be incorporated
into the epithelial layer and form cell-cell junctions with their
neighboring cells on the epithelial layer to restore epithelial
barrier function (34-36). It is unclear whether the Hippo-YAP
pathway regulates interepithelial junction formation. Moreover,
lung repair involves multiple components, including repair of the
endothelial barrier, restoration of lung structure, and resolution
of lung inflammation and edema (3,34,35,37,38). To the best of our knowledge,
whether and how the Hippo-YAP pathway may regulate these biological
processes is not known. It is also unclear whether the Hippo-YAP
pathway modulates lung injury. Thus, the roles of this pathway in
inflammatory lung injury and repair remain to be elucidated.
In the current study, YAP activity was systemically
inhibited or stimulated to examine the effects of the Hippo-YAP
pathway on lung injury and repair in a mouse model of LPS-induced
ALI/ARDS. The results of the present study demonstrated that the
Hippo-YAP activity protected against lung injury by activating
multiple mechanisms.
Materials and methods
Animal studies
Adult male ICR mice (age, 8-10 weeks) were supplied
by Shanghai SLAC Laboratory Animal Co., Ltd. Mice were housed in a
temperature-controlled room (22±2°C) with 12-h light/dark cycle and
relative humidity of 40-60%. Mice had free access to food and
water. A total of 320 mice (25-30 g) were used for the studies. All
animal experiments were approved by the Institutional Animal Care
and Use Committee of Wenzhou Medical University (Wenzhou, China)
and were performed in accordance with the Guide for the Care and
Use of Laboratory Animals of the National Institute of Health
(39). For a dose-response study,
mice (n=30/group) were injected with equal volume of saline or 5,
7.5 or 10 mg/kg Escherichia coli LPS [cat. no. 0111:B4;
intraperitoneal injection (i.p.); Sigma-Aldrich; Merck KGaA]. The
survival rate was monitored for 7 days. Mice were closely observed
to identify animals that have reached humane endpoints and required
euthanasia. Those endpoints included slow, rapid, labored or agonal
breathing, cardiac failure, impaired ambulation, inability to
obtain food/water, ruffled fur, severe lethargy and signs of severe
distress. Those mice were euthanized and counted as non-surviving
animals in the survival rate analysis.
For time course studies, mice were injected with an
equal volume of saline or Escherichia coli LPS (7.5 mg/kg;
i.p.), and lung inflammation and injury, cell proliferation and YAP
activity were assessed at 0, 6, 12, 24, 48, 72, 120 and 168 h
post-LPS injection (n=5/time-point). Mice were anesthetized with
pentobarbital sodium (50 mg/kg; i.p.; cat. no. 4579/50; R&D
Systems, Inc.). At the end of each experiment, mice were euthanized
by CO2 asphyxia. Mice were put into a euthanasia
chamber. CO2 was allowed to flow into the chamber at a
displacement rate of 40% chamber volume/minute until mice were no
longer breathing. Death was confirmed by loss of vital signs,
including heartbeat and breathing. Euthanasia was performed when
mice have reached humane endpoints for survival study, and at the
end of each experiment for other studies.
The effects of inhibiting YAP activity were studies
at 24, 48, 72 and 168 h post-LPS injection. There were four study
groups for each timepoint, control, verteporfin (VP), LPS and LPS +
VP groups (3 mice per group for biochemical analyses; 5 mice per
group for functional studies). Mice in control and LPS groups were
injected with 0.1% DMSO (solvent for VP), and mice in VP and LPS +
VP groups were injected with VP (100 mg/kg, i.p.; AdooQ
Bioscience). Mice in 24 h groups were injected with a single dose
of VP (100 mg/kg) 2 h before LPS injection. Mice in 48, 72 and 168
h groups were initially injected with VP (100 mg//kg) at 20 h after
LPS injection, followed by an additional 100 mg/kg injection every
24 h. Mice in control and VP groups were injected with equal volume
of saline, and mice in LPS and LPS + VP groups were injected with
LPS (7.5 mg/kg; i.p.). At 24, 48, 72 and 168 h after LPS or saline
injection, bronchoalveolar lavage (BAL) was performed, and lung
inflammation and injury, cell proliferation and YAP activity were
assessed.
The effects of stimulating YAP activity were studies
at 24, 48, 72 and 168 h post-LPS injection. There were four study
groups for each timepoint, control, XMU-MP-1, LPS and LPS +
XMU-MP-1 groups (3 mice per group for biochemical analyses; 5 mice
per group for functional studies). Mice in control and LPS groups
were injected with 0.1% citric acid containing 20% Kolliphor HS
(solvent for XMU-MP-1; cat. no. 42966; Sigma-Aldrich; Merck KGaA),
and mice in XMU-MP-1 and LPS + XMU-MP-1 groups were injected with
XMU-MP-1 (1 mg/kg; i.p.). Mice in 24 h groups were injected with a
single dose of XMU-MP-1 (1 mg/kg) 2 h before LPS injection. Mice in
48, 72 and 168 h groups were initially injected with XMU-MP-1 (1
mg/kg) at 20 h after LPS injection, followed by an additional dose
(1 mg/kg) injection every 24 h. Mice in control and XMU-MP-1 groups
were injected with saline, and mice in LPS and LPS+ XMU-MP-1 groups
were injected with LPS (7.5 mg/kg, i.p.). At 24, 48, 72 or 168 h
after saline or LPS injection, BAL was performed, and lung
inflammation and injury, cell proliferation and YAP activity were
assessed. XMU-MP-1 was synthesized as previously described
(40).
Histological examination
Lungs were perfused with PBS under deep anaesthesia,
fixed with 4% paraformaldehyde at 4°C for 24 h, processed by
passing through increasing concentrations of ethanol and paraffin
embedded, or embedded in optimal cutting temperature compound (cat.
no. 4583; Beijing Solarbio Science & Technology Co., Ltd.) and
snap frozen at −80°C for immunofluorescence staining, as described
below. For histological analysis, paraffin-embedded sections
(6-µm thick) were stained with hematoxylin and eosin at room
temperature for 5-10 min. Stained sections were examined under a
light microscope (Nikon Eclipse 80i; Nikon Corporation) at a
magnification of ×200.
BAL and analysis
BAL was performed by delivering 0.8 ml of pre-warmed
EDTA-saline into trachea followed by gentle chest massage and
suction. Following removal of debris, BAL fluid (BALF) from three
BALs was pooled and centrifuged at 4°C at 1,650 × g for 3 min. The
pellet and supernatant were collected.
BALF concentration of TNF-α (cat. no. JL10484-96T)
and IL-6 (cat. no. JL20268-96T) was quantified using ELISA kits
(J&L Biological, Inc.). BALF protein concentration, as an
indicator of epithelial permeability, was determined using a BCA
protein assay kit (Thermo Fisher Scientific, Inc.). To measure
myeloperoxidase (MPO) activity, as an indicator of neutrophil
infiltration, the cell pellets were homogenized in 5 mM phosphate
buffer and then centrifuged at 15,000 × g at 4°C for 20 min. The
pellets were resuspended in phosphate buffer containing 0.5%
hexadecyltrimethylammonium bromide, subjected to a cycle of
freezing at -80°C for 1 h and thawing at 4°C for 1 h, and assayed
for MPO activity (MPO assay kit; cat. no. A0441-1; Nanjing
Jiancheng Bio-Engineering Institute). MPO activity was measured by
continuously recording absorbance at 460 nm for 3 min. MPO activity
was presented as activity unit/l (U/l).
Reverse transcription quantitative-PCR
(RT-qPCR)
RNA was isolated from lungs using RNAiso Plus kit
(Takara Bio, Inc.) and reverse transcribed into cDNA using a
two-step cDNA synthesis kit (Takara Bio, Inc.) according to the
manufacturer's protocols. qPCR was performed using SYBR-Green PCR
master mix (Takara Bio, Inc.) and an ABI Prism 7900 sequence
detection system (Applied Biosystems; Thermo Fisher Scientific,
Inc). The following thermocycling conditions were used: Initial
denaturation at 95°C for 15 min; 40 cycles at 95°C for 10 sec, 60°C
for 20 sec and 72°C for 32 sec. The primer sequences are included
in Table I. Results were analyzed
using the 2−ΔΔCq method (41). The mRNA level of each target gene
was normalized to peptidylprolyl isomerase B (PPIB) level.
 | Table IPrimer sequences used for
quantitative PCR. |
Table I
Primer sequences used for
quantitative PCR.
| Target | Primer sequence
(5′-3′)
|
|---|
| Forward | Reverse |
|---|
| Sp-c |
ATGGACATGAGTAGCAAAGAGGT C |
ACGATGAGAAGGCGTTTGAG |
| CTGF |
CTCCACCCGAGTTACCAATG |
TGGCGATTTTAGGTGTCCG |
| CYR61 |
GGAGGTGGAGTTAACGAGAAAC |
GTGGTCTGAACGATGCATTTC |
| PDPN |
ACCGTGCCAGTGTTGTTCTG |
AGCACCTGTGGTTGTTATTTTGT |
| Yap |
ACGACTTCCTCAACAGTGTG |
TCATTGCATCTCCTTCCAGTG |
| E-cad |
CAGGTCTCCTCATGGCTTTGC |
CTTCCGAAAAGAAGGCTGTCC |
| IL-1β |
GCAACTGTTCCTGAACTCAACT |
ATCTTTTGGGGTCCGTCAACT |
| IL-6α |
TAGTCCTTCCTACCCCAATTTCC |
TTGGTCCTTAGCCACTCCTTC |
| TNF-α |
CCCTCACACTCAGATCATCTTCT |
GCTACGACGTGGGCTACAG |
| IL-10 |
GCTCTTACTGACTGGCATGAG |
CGCAGCTCTAGGAGCATGTG |
| Cyclin A2 |
GCCTTCACCATTCATGTGGAT |
TTGCTGCGGGTAAAGAGACAG |
| Ki-67 |
ATCATTGACCGCTCCTTTAGGT |
GCTCGCCTTGATGGTTCCT |
| PPIB |
GGCTCCGTCGTCTTCCTTTT |
ACTCGTCCTACAGATTCATCTCC |
Western blotting
Total, cytoplasmic or nuclear protein fractions were
extracted from lungs, as previously described (42,43). Equal amounts of proteins (30
µg/lane) were separated using SDS-PAGE (10% gel) and
transferred to polyvinylidene fluoride membranes (EMD Millipore).
Membranes were blocked with 5% non-fat dry milk in TBST (10 mM/l
Tris-HCl, pH 7.5; 150 mM/l NaCl; 0.1% Tween-20) at room temperature
for 1 h, and incubated overnight at 4°C with antibodies against YAP
(1:1,000; cat. no. 4912S; Cell Signaling Technology, Inc.),
phosphorylated (p)-YAP (1:1,000; cat. no. 4912S; Cell Signaling
Technology, Inc.), connective tissue growth factor (CTGF; 1:500;
cat. no. sc-101586; Santa Cruz Biotechnology, Inc.), proliferating
cell nuclear antigen (PCNA; 1:500; cat. no. ab29; Abcam), pulmonary
surfactant apoprotein C (Sp-c; 1:1,000; cat. no. ab90716; Abcam),
aquaporin 5 (AQP-5; 1:1,000; cat. no. ab78486; Abcam), E-cadherin
(1:4,000, cat. no. 20874-1-AP; Proteintech Group, Inc.), GADPH
(1:5,000, cat. no. AB2302; EMD Millipore), β-actin (1:1,000; cat.
no. WH100959; ABclonal Biotech Co., Ltd.) and type B1 nuclear lamin
(lamin B1; 1:5,000; cat. no. ab16048; Abcam). The membranes were
washed, incubated at room temperature for 1.5 h with horseradish
peroxidase-conjugated secondary antibody (anti-mouse/rabbit IgG;
1:3,000; cat. nos. A2304 and A0545; Sigma-Aldrich; Merck KGaA), and
washed again. Immunoreactive bands were visualized using an
enhanced chemiluminescent reagent (Bio-Rad laboratories, Inc.). The
blot band intensities were semi-quantified using ImageJ version
1.46 (National Institutes of Health).
Immunofluorescence (IF) staining
Cryosections (6-µm thick) were fixed with 4%
paraformaldehyde at 4°C for 15 min, permeabilized with 0.1% Triton
X-100 (cat. no. T8200; Beijing Solarbio Science & Technology
Co., Ltd., https://solarbio.en.alibaba.com) at 4°C for 10 min,
blocked with 5% BSA (cat. no. A3858; Sigma-Aldrich; Merck KGaA) at
4°C for 15 min and stained with anti-AQP-5 (1;200, cat. no.
ab78486; Abcam), anti-podoplanin (PDPN, 1;200, cat. no. ab11936;
Abcam), anti-Sp-c (1:100, cat. no. sc-7706; Santa Cruz,
Biotechnology, Inc.) antibodies at 4°C for 4 h, followed by washing
and staining with Alexa Fluor 594-conjugated goat anti-rabbit
(1:500, cat. no. 33112ES60; Shanghai Yeasen Biotechnology Co.,
Ltd.), donkey anti-rabbit (1:500, cat. no. 34212ES60; Shanghai
Yeasen Biotechnology Co., Ltd.) or goat anti-hamster IgG (1:500,
cat. no. A-21113; Invitrogen; Thermo Fisher Scientific, Inc.) at
room temperature for 1 h. These sections were washed, blocked again
with 5% BSA at 4°C for 15 min and stained with anti-Ki-67 (1:25;
cat. no. AF7649; R&D Systems, Inc.) or anti-YAP (1:50, cat. no.
4912S; Cell Signaling Technology, Inc.) antibodies at 4°C
overnight, followed by washing and staining with Alexa Fluor
488-conjugated rabbit anti-goat (1:500; cat. no. 33706ES60), donkey
anti-rabbit (1:500; cat. no. 34206ES60) or FITC-conjugated rabbit
anti-sheep IgG (1:500; cat. no. 33807ES60) (all Shanghai Yeasen
Biotechnology Co., Ltd.) at room temperature for 1 h. The slides
were washed, nuclei counterstained with DAPI (Sigma-Aldrich; Merck
KGaA) at room temperature for 15 min, and mounted with mounting
medium (Beyotime Institute of Biotechnology). Slides were viewed
under a Zeiss confocal microscope (ZeissLSM800; Carl Zeiss AG), and
images were captured and analyzed using Zeiss imagine analyzing
system (ZEISS ZEN lite 2011; Carl Zeiss AG).
Assessment of endothelial
permeability
Microvascular endothelial permeability was assessed
using a dual albumin tracer method. A total of 2 h prior to
sacrifice, mice in control, 24, 48, 72, 120 and 168 h groups (total
18 mice, 3 mice per group) were anesthetized and injected with 100
µg mouse FITC-labeled BSA (FITC-BSA; cat. no. R-H-10026;
Xi'an Qiyue Biotechnology, Co.) and then injected with 100
µg mouse TRITC-labeled BSA (TRITC-BSA, cat. no. R-H-10085;
Xi'an Qiyue Biotechnology, Co.) 2 h later via the jugular vein. Two
minutes after TRITC-BSA injection, blood (0.2 ml) was withdrawn,
and lungs were excised and weighed. Lungs were homogenized,
centrifuged at 500 × g at 4°C for 5 min and supernatant was
prepared. FITC-BSA and TRITC-BSA fluorescence intensity in each
supernatant and plasma sample was read using a Varioskan Flash
multimode reader (Thermo Fisher Scientific, Inc.). Endothelial
permeability was assessed based on extravascular leakage of
FITC-BSA, expressed as FITC to TRITC fluorescence ratio.
Statistical analysis
Data are presented as the mean ± SEM. GraphPad Prism
5.0 (GraphPad Software, Inc.) was used to perform statistical
analyses. Comparisons between multiple groups with one and two
factors were made using one-way and two-way ANOVA. Post hoc
analyses were performed using Bonferroni post hoc test. Comparison
between two groups was made using two-sided unpaired Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Time course profile of lung injury and
repair in LPS-induced ALI
The repair process after lung injury can last days,
depending on the cause of the injury (37,38). One major challenge when studying
lung repair mechanisms is to develop an animal model that produces
significant lung injury, and yet has sufficient survival rate so
that the process and mechanisms of lung repair can be studied in
detail (37,38). For this purpose, a dose-response
study was initially performed (Fig.
1B). Injecting mice with 7.5 mg/kg LPS produced marked lung
injury (Fig. 1A) and resulted in
significant inflammation compared with the control group (Fig. 1C-F) but resulted in a 1-week
survival rate of ~65% (Fig. 1B).
This LPS dose was chosen for subsequent experiments.
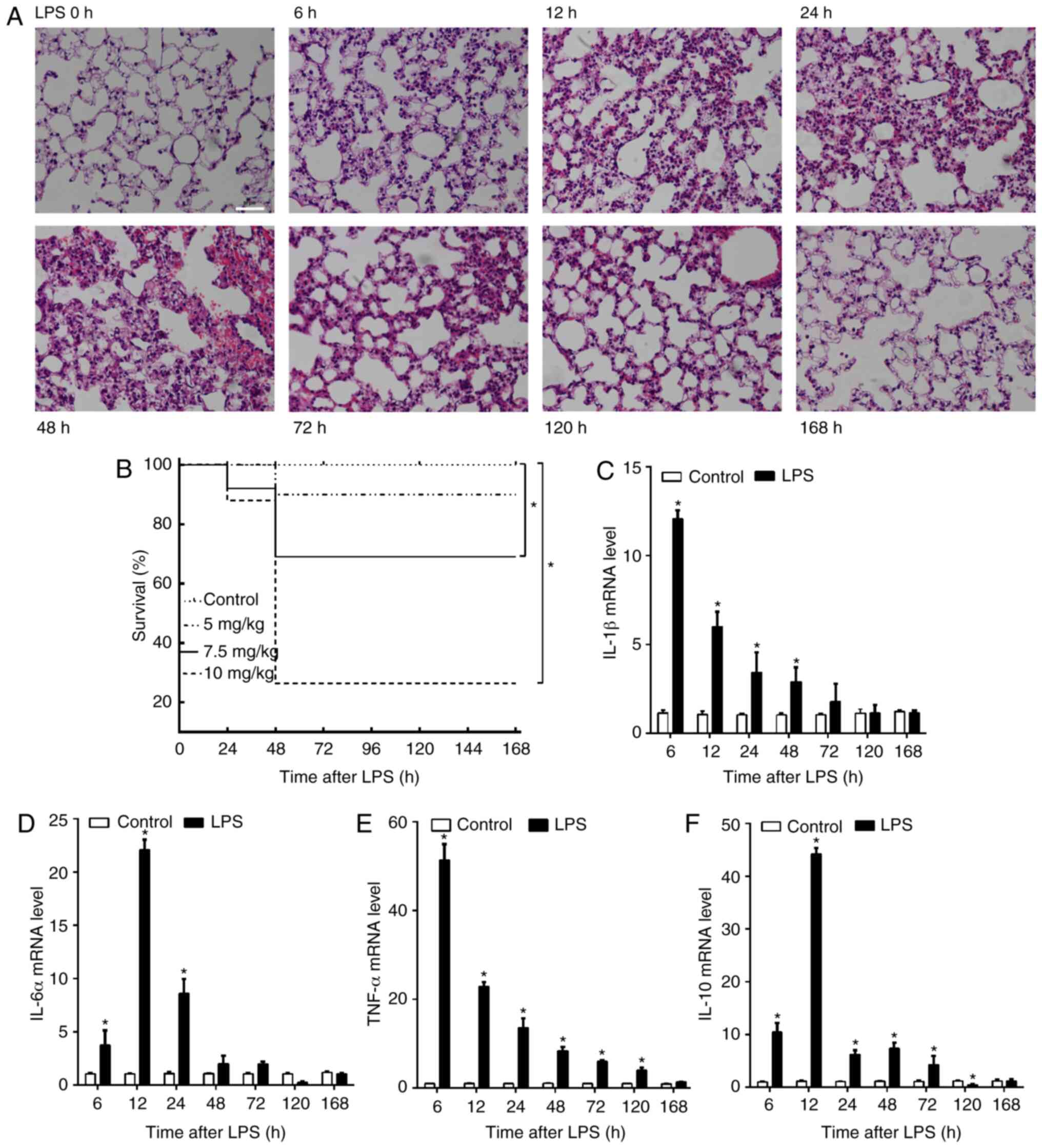 | Figure 1Time course of lung injury and
recovery in LPS-induced acute lung injury. (A) Histological
examination of lung injury in mice at 0, 6, 12, 24, 48, 72, 120 and
168 h post-LPS injection. Similar result were obtained in all 5
mice at each respective time-point. Magnification, ×200. (B)
Dose-response studies showed 1-week survival rate after injections
with different doses of LPS. *P<0.05 compared with
the control group (n=30 mice/group). Reverse
transcription-quantitative PCR analysis of lung tissue mRNA
expression levels of (C) IL-1β, (D) IL-6α, (E) TNF-α and (F) IL-10.
Data are presented as the mean ± SEM (n=3 mice/group; 2
experimental repeats). *P<0.05 vs. the corresponding
control group. LPS, lipopolysaccharide. |
The entire course of lung injury and recovery after
the LPS challenge was followed. The major pathological features of
ALI/ARDS, inflammation, tissue injury, and endothelial and
epithelial permeability changes were followed. Histological
examination of lungs from control, 6, 12, 24, 48, 72, 120 and 168 h
groups of mice showed that LPS caused marked lung tissue injury, as
evidenced by inflammatory cell infiltration, hemorrhage, increased
alveolar wall thickness and alveolar distortion. Lung injury
deteriorated progressively between 6 to 48 h, peaked at 48 h, and
then recovered gradually between 48 to 168 h to reach full recovery
at 168 h post-LPS injection (Fig.
1A). Lung inflammation, as assessed by tissue mRNA expression
levels of inflammatory and anti-inflammatory cytokines, peaked at
6-12 h and decreased rapidly after that to reach a low level within
48 h post-LPS injection (Fig.
1C-F). The low level of lung inflammation persisted until 120
h, and was not observed at 168 h post-LPS injection (Fig. 1C-F). Changes in lung epithelial
permeability followed a similar time course to lung tissue injury
and recovery, with damage peaking at 48 h, followed by gradual
recovery between 48 and 168 h post-LPS injection (Figs. 1A and 2B). An increase in lung endothelial
permeability followed a slightly different time course, peaking at
24 h, remained high till 48 h, and then decreased rapidly between
72 and 120 h, to finally reach the control level at 168 h post-LPS
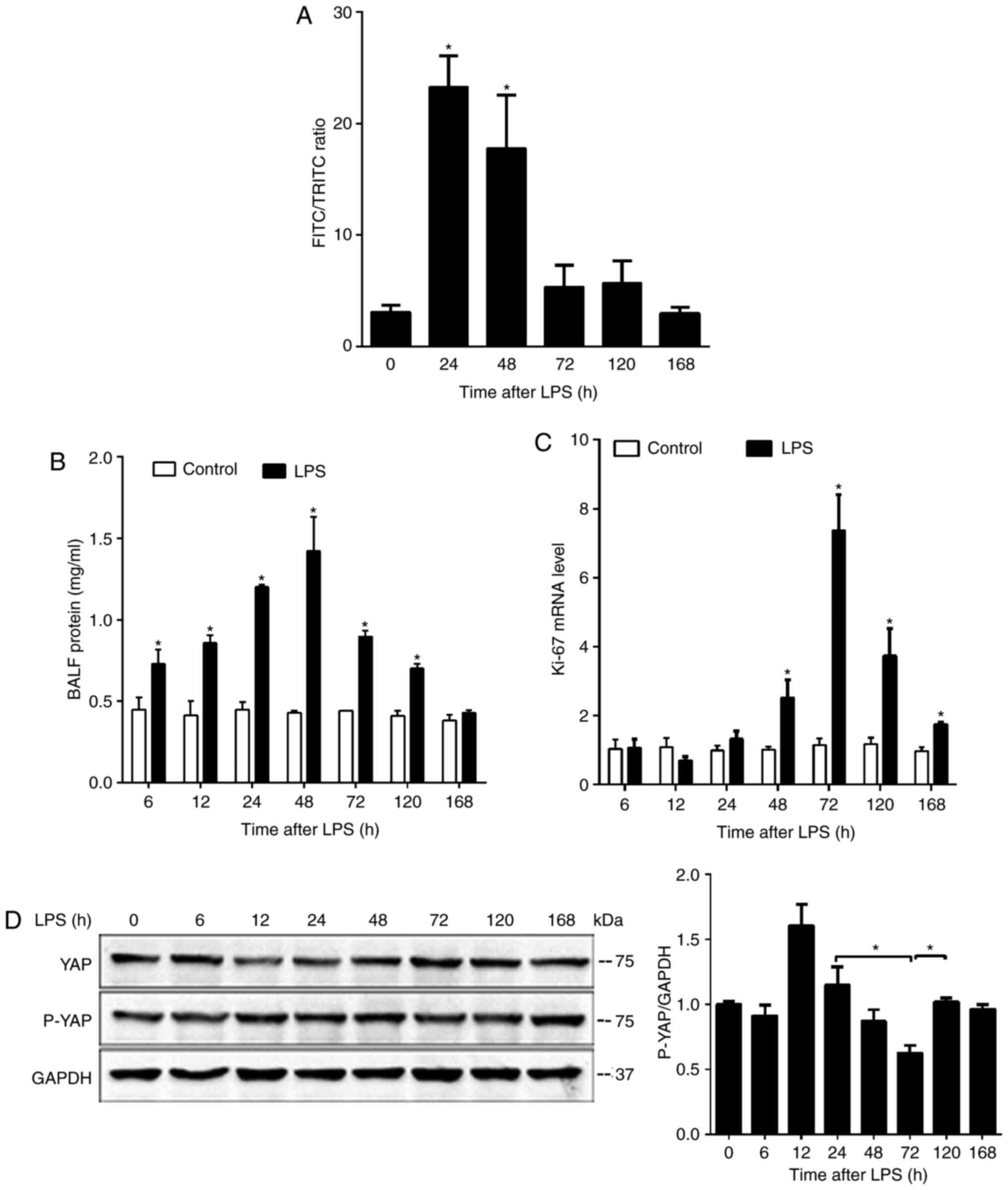
injection (Figs. 1A and 2A).
Lung injury and repair are associated
with decreased and increased YAP activity, respectively
Based on the aforementioned time course profile,
0-48 h post-LPS injection was considered as the injury phase and
48-168 h as the repair phase. Within these phases, 6-24 h was
termed the progressive injury phase, and 72-120 h was termed the
active repair phase. This definition is consistent with the time
course of cell proliferation (Fig.
2C), a major mechanism of lung repair. The tissue mRNA
expression level of the proliferation marker Ki-67 remained stable
at the 6-24 h progressive injury phase, increased at the 48 h
post-LPS injection transition phase, markedly increased at the
72-120 h active repair phase and decreased to near the control
level at 168 h fully recovery (Fig.
2C). Whether lung injury and recovery were associated with
alterations in YAP activity was also determined. YAP activity is
controlled by its phosphorylation which reduces YAP nuclear
translocation and transactivation activity (5-9).
The p-YAP/YAP ratio in the lung tissue was significantly increased
at 12 h (progressive injury phase) and significantly decreased at
72 h (active repair phase) (Fig.
2D), suggesting that lung injury and repair are associated with
a decreased or increased YAP activity, respectively (Fig. 2).
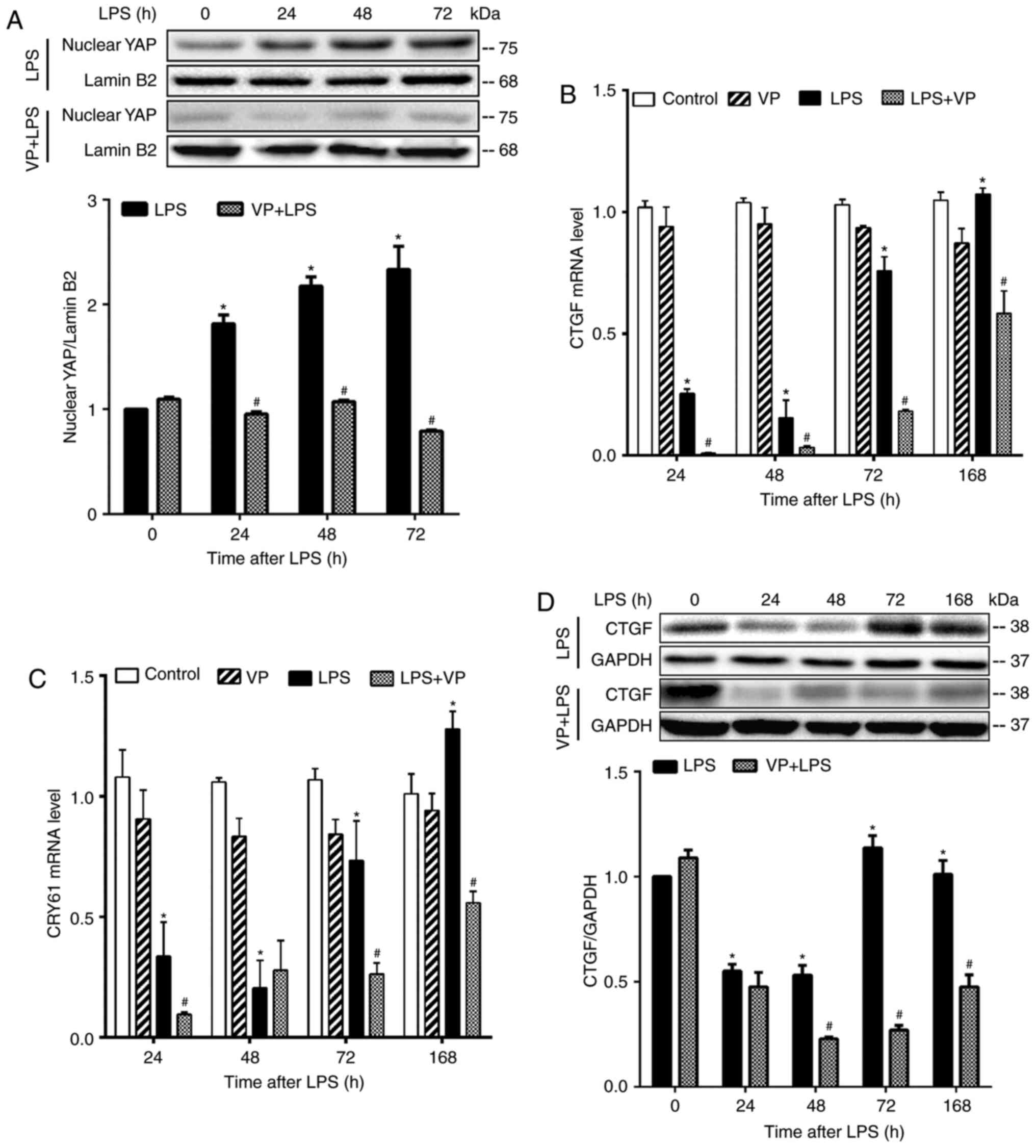
Inhibition of YAP activity exacerbates
lung injury and impedes lung recovery
To understand the roles of YAP activity in lung
injury and repair, LPS-challenged mice were treated with VP, a
small molecule that suppresses YAP activity by binding to it, and
thereby preventing the formation of the TEAD-YAP complex in the
nucleus (44). Treatment of mice
with VP markedly reduced nuclear YAP protein content in LPS-treated
lungs, but not in control lungs (Fig.
3A), and inhibited the mRNA expression of CTGF and cellular
communication network factor 1 (CYR61), two YAP target genes, in
LPS-treated lungs (Fig. 3B and
C). Compared to LPS alone group, VP also inhibited CTGF protein
expression (Fig. 3D). These
results confirmed that VP suppressed LPS-stimulated YAP activity in
the lungs.
The effects of YAP activity inhibition on lung
inflammation and lung injury and recovery were subsequently
examined at different pathological stages. Compared with the LPS
alone group, mice in the LPS + VP group displayed more severe
tissue injury (Fig. 4A),
significantly higher tissue expression levels of IL-1β, IL-6α,
TNF-α and IL-10 (Fig. 4B-E),
significantly elevated BALF levels of IL-6α and TNF-α (Fig. 4F-G), significantly increased BALF
MPO activity (Fig. 4H), and
significantly increased BALF protein content indicating higher
epithelial permeability (Fig. 4I)
at all time-points examined. Exacerbated lung inflammation and
injury caused by VP treatment led to a worse outcome. Compared with
mice in LPS alone group, mice in the LPS + VP group showed a
delayed return to the control level in inflammatory cytokine
expression (Fig. 4B-E) and
delayed recovery from the injury (Fig. 4A and F-I). The 1-week survival
rate has decreased from 76% in the LPS group to 48% in the LPS + VP
group (Fig. 4J).
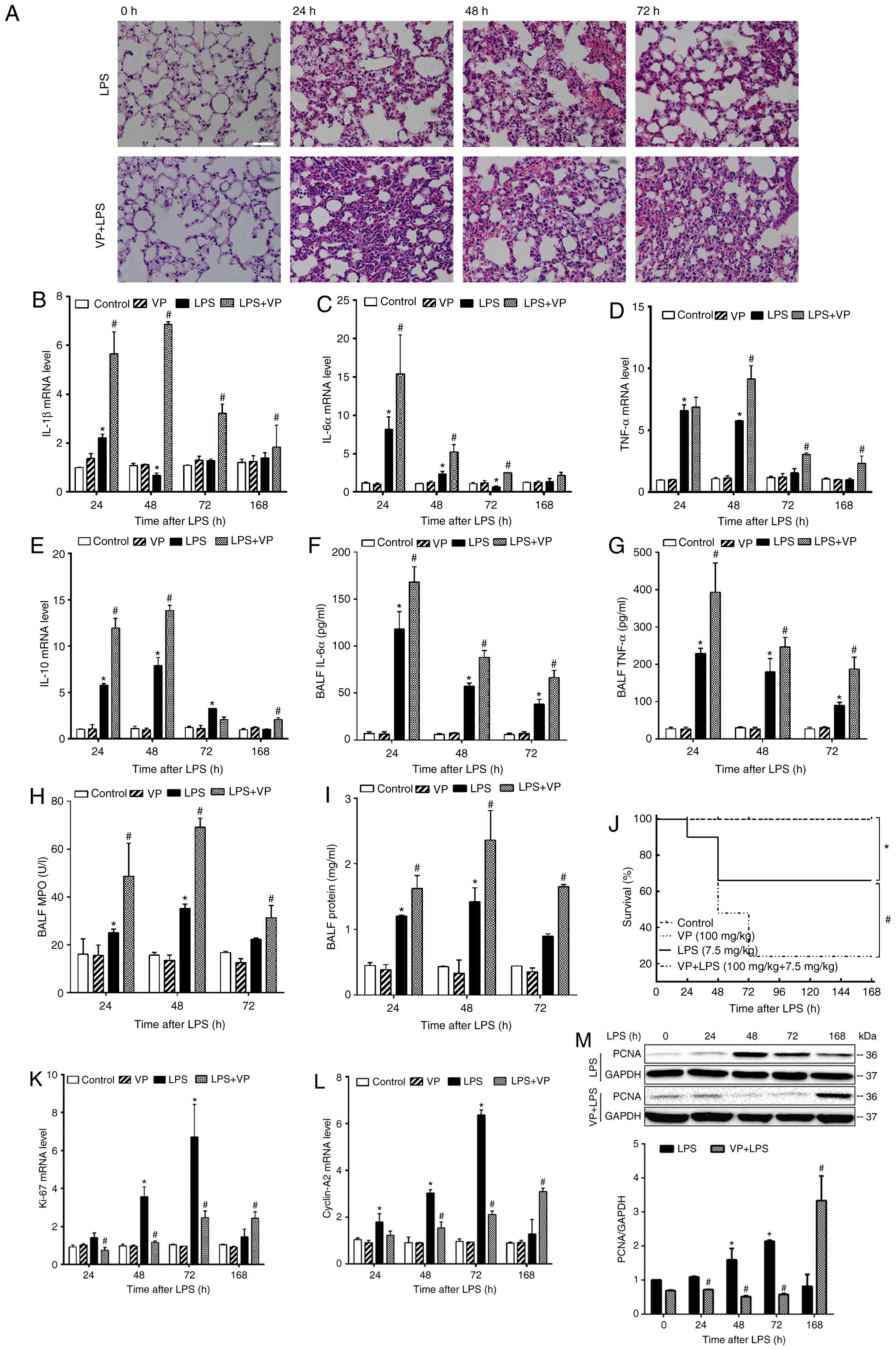 | Figure 4Continued. Inhibition of YAP activity
exacerbates lung injury and impedes lung recovery. Mice were
intraperitoneally injected with DMSO (solvent for VP), VP (100
mg/kg), LPS or LPS + VP. The effects of inhibiting YAP activity on
lung injury and recovery, and on lung inflammation were evaluated
at injury and repair phases. (A) Histological examination of lung
injury. Similar result were obtained in all 5 mice at each
respective time-point. Magnification, ×200. Reverse
transcription-quantitative PCR analysis of tissue expression levels
of (B) IL-1β, (C) IL-6α, (D) TNF-α and (E) IL-10. Data are
presented as the mean ± SEM (n=3 mice per group; 2 experimental
repeats/animal). Analyses of BALF levels of cytokines, including
(F) IL-6α and (G) TNF-α, (H) MPO activity, and (I) total protein
concentration. Data are presented as the mean ± SEM (n=5
mice/group). (J) Inhibition of YAP activity significantly reduced
the survival rate (n=30 mice/group). Analyses of tissue expression
levels of (K) Ki-67 and (L) cyclin-A2 mRNA, and (M) PCNA protein.
Data are presented as the mean ± SEM (n=3 mice/group; 2
experimental repeats/animal). *P<0.05 vs. the control
group and #P<0.05 vs. the LPS alone group. LPS,
lipopolysaccharide; YAP, Yes-associated protein; BALF,
bronchoalveolar lavage fluid; MPO, myeloperoxidase; PCNA,
proliferating cell nuclear antigen; VP, verteporfin. |
Inhibition of YAP activity in VP-treated mice
significantly decreased tissue expression levels of cell
proliferation markers, including Ki-67, cyclin-A2 and PCNA
(Fig. 4K-M). YAP inhibition by VP
appeared to have different effects on the expression of cell
proliferation markers at different timepoints. Compared with mice
in the LPS alone group, mice treated with LPS + VP had
significantly reduced expression levels of Ki-67 and PCNA at 24, 48
and 72 h, and a reduced level of cyclin-A2 at 48 and 72 h post LPS
injection (Fig. 4K-M). By
contrast, mice in the LPS + VP group had significantly increased
levels of Ki-67, PCNA and cyclin-A2 expression at 168 h post LPS
injection (Fig. 4K-M).
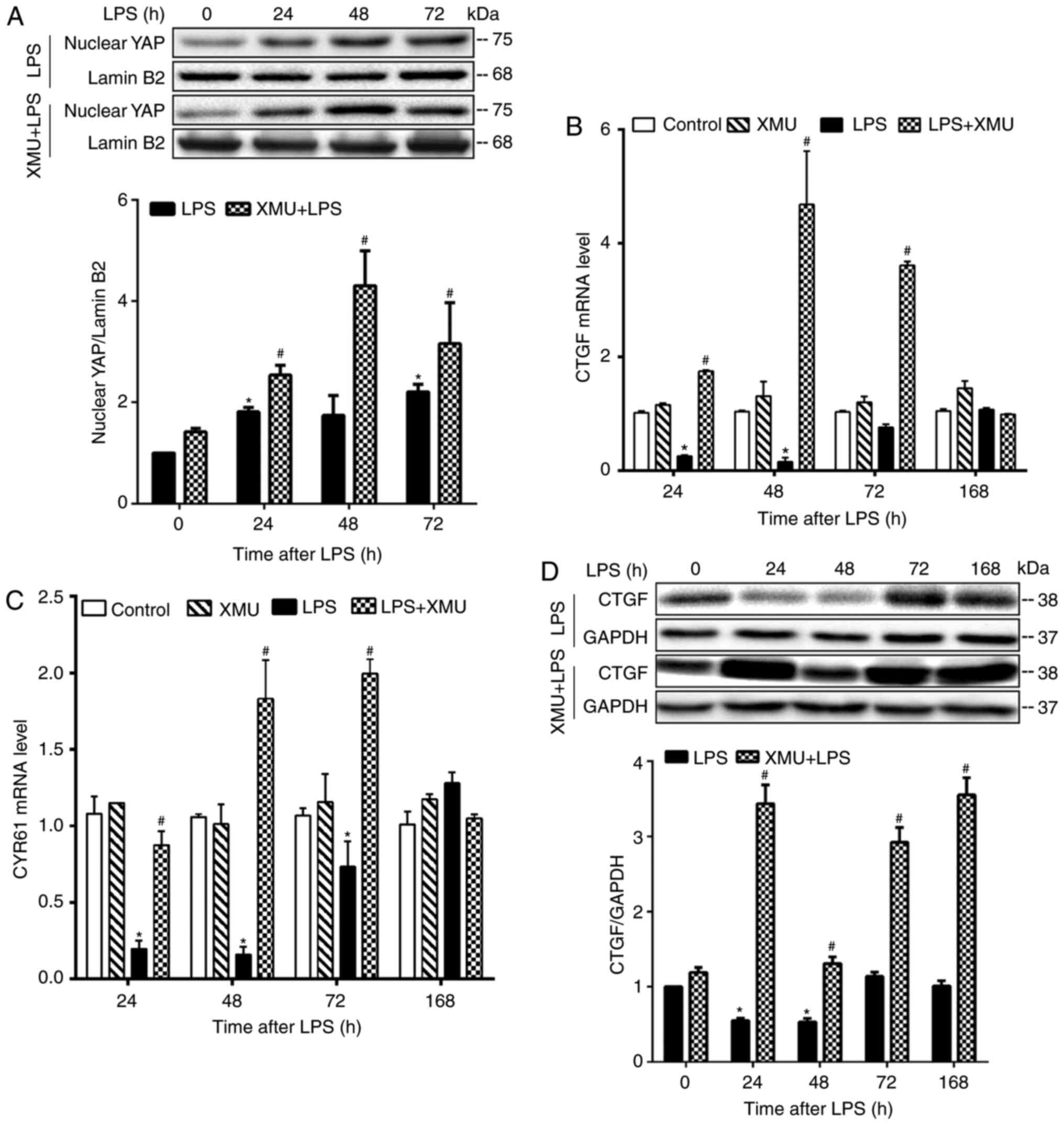
Stimulation of YAP activity attenuates
lung injury and promotes lung recovery
YAP activity was stimulated by treating control and
LPS-challenged mice with XMU-MP-1. The effects of this stimulation
on lung inflammation, injury and recovery were examined at
different pathological stages. XMU-MP-1 is a selective Mst1/2
inhibitor that prevents YAP phosphorylation and stimulates its
nuclear translocation and activity (40). Compared with mice in LPS alone
groups, mice in LPS + XMU-MP-1 groups showed a significantly
increased nuclear content of YAP (Fig. 5A), increased lung mRNA expression
of CTGF and CYR61 (Fig. 5B and
C), and increased lung CTGF protein expression (Fig. 5D) at 24, 48 and 72 h post LPS
injection. Thus, these results confirmed that XMU-MP-1
significantly stimulated YAP activity in the lungs.
The effects of stimulating YAP activity on lung
injury, recovery and inflammation were subsequently studied.
Compared with mice injected with LPS alone, mice treated with LPS +
XMU-MP-1 showed a markedly attenuated tissue injury (Fig. 6A), lower tissue expression levels
of TNF-α and Il-10 (Fig. 6B and
C), lower BALF levels of IL-6α and TNF-α (Fig. 6D and E), lower BALF MPO activity
(Fig. 6F), and lower BALF protein
content (Fig. 6G) as an indicator
for lower epithelial permeability (3), at 24, 48 and 72 h post-LPS
injection. Alleviation of lung inflammation and injury by XMU-MP-1
treatment led to an improved outcome. Compared with mice injected
with LPS alone, mice treated with LPS + XMU-MP-1 showed an earlier
return of tissue inflammatory cytokine expression to the control
level (Fig. 6B and C),
accelerated recovery from lung injury (Fig. 6A and D-G) and a higher survival
rate (Fig. 6H).
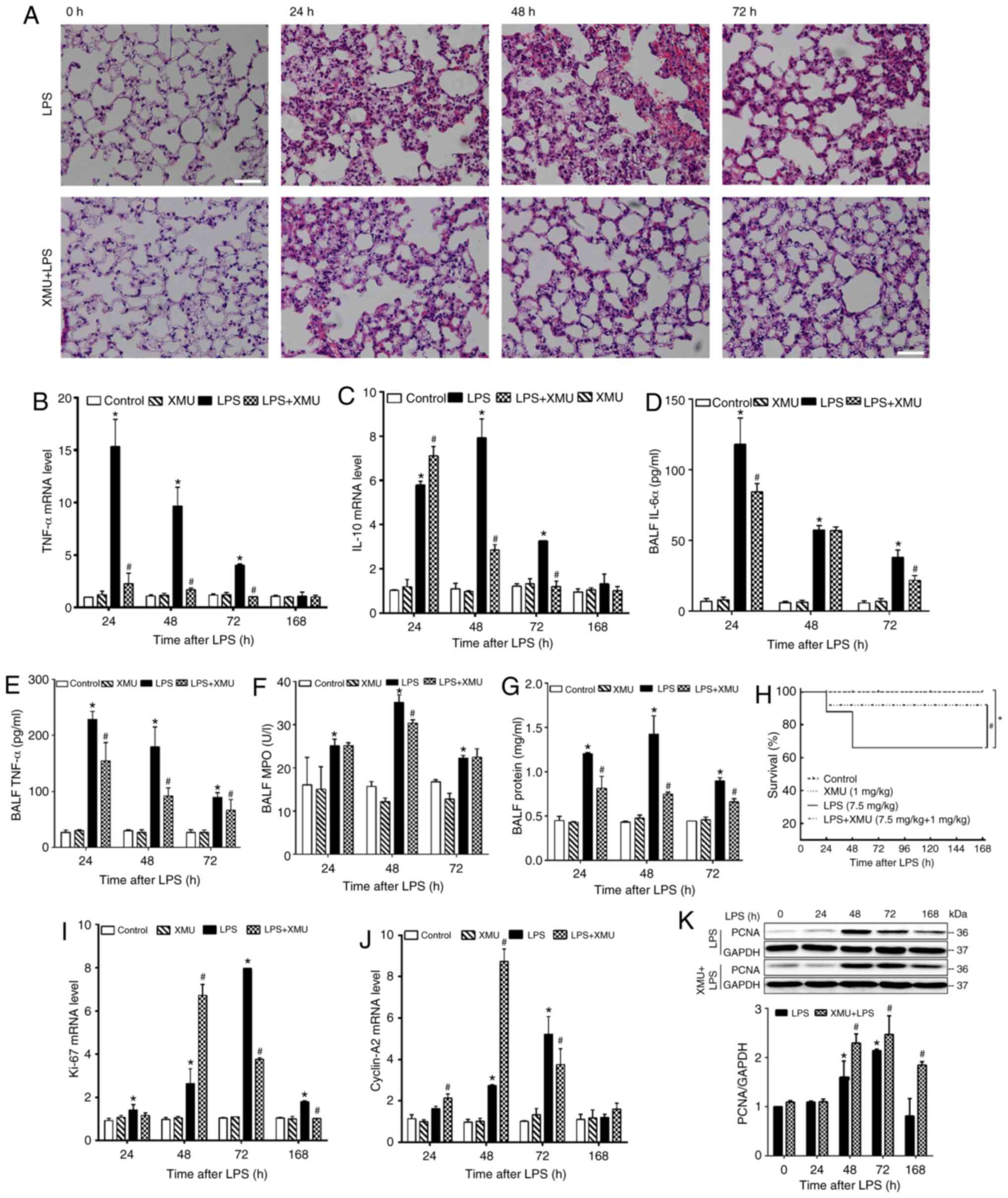 | Figure 6Stimulation of YAP activity
attenuates lung injury and promotes lung recovery. Mice were
intraperitoneally injected with 0.1% citric acid containing 20%
Kolliphor HS (solvent for XMU-MP-1), 1 mg/kg XMU-MP-1, LPS or LPS +
XMU-MP-1. The effects of stimulating YAP activity on lung injury
and recovery, and on lung inflammation were evaluated at injury and
repair phases. (A) Histological examination of lung injury. Similar
result were obtained in all 5 mice at each respective time-point.
Magnification, ×200. Reverse transcription-quantitative PCR
analysis of tissue mRNA expression levels of (B) TNF-α and (C)
IL-10. Data are presented as the mean ± SEM (n=3 mice/group; 2
experimental repeats/animal). Analysis of BALF levels of (D) IL-6α
and (E) TNF-α, (F) MPO activity and (G) total protein
concentration. Data are presented as the mean ± SEM (n=5
mice/group). (H) Stimulation of YAP activity improved the survival
rate. Analyses of tissue expression levels of (I) Ki-67 and (J)
cyclin-A2 mRNA, and (K) PCNA protein. Data are presented as the
mean ± SEM (n=3 mice/group; 2 experimental repeats/animal).
*P<0.05 vs. the control group; and
#P<0.05 vs. the LPS alone group. LPS,
lipopolysaccharide; XMU, XMU-MP-1; BALF, bronchoalveolar lavage
fluid; PCNA, proliferating cell nuclear antigen. |
Stimulation of YAP activity with XMU-MP-1 had no
effect on tissue expression levels of the cell proliferation
markers, including Ki-67, cyclin-A2 and PCNA (Fig. 6I-K) at 24 h (progressive injury
phase), but the expression levels of these markers were
significantly increased at 48 h post LPS injection (transition
phase) (Fig. 6I-K). XMU-MP-1 had
a varied effect on tissue expression levels of these cell
proliferation markers at 72 h post LPS injection (active repair
phase), augmenting the expression of PCNA (Fig. 6K), but inhibiting that of Ki-67
and cyclin-A2 (Fig. 6I and
J).
Inhibition of YAP activity suppresses
epithelial cell regeneration
Epithelial regeneration is an essential component of
lung repair (2,3,34,35). Using tissue protein expression
levels of alveolar type I epithelial cell (AECI)-specific markers,
PDPN and AQP-5 (45), and AECII
specific marker, Sp-c (46), as
indicators, the present study estimated the effects of LPS on the
abundance of AECI and AECII cells in the lungs and assessed the
effects of inhibiting the YAP activity on LPS-induced changes in
AECI and AECII cell abundance. Lung tissue mRNA expression levels
of PDPN and Sp-c in the LPS group decreased significantly at 24 and
48 h (injury phase) compared with that in the control group, but
partially returned to control levels at 72 and 168 h post LPS
injection (repair phase) (Fig. 7A and
B). Lung tissue protein expression levels of Sp-c and AQP-5 in
the LPS group followed a similar pattern to the aforementioned mRNA
expression levels (Fig. 7C and
D). Inhibition of YAP activity with VP inhibited LPS-induced
changes in PDPN mRNA expression at 24, 48, 72 and 168 h, and in
Sp-c mRNA expression at 24, 72 and 168 h post-LPS injection
(Fig. 7A and B). Inhibition of
YAP activity with VP had no effects on LPS-induced downregulation
of Sp-c and AQP-5 protein expression at 24 and 48 h (Fig. 7C and D), but significantly
inhibited LPS-induced upregulation of Sp-c protein expression at 72
and 168 h, and AQP-5 protein expression at 72 h post-LPS injection
(Fig. 7C and D). This result is
consistent with the hypothesis that lung injury is associated with
loss of AECI and AECII cells, and lung repair is associated with an
increased number of these cells in the lungs (3,34,35). Inhibition of YAP activity impedes
the restoration of AECI and AECII cell numbers in the lungs.
Suppression of Hippo-YAP activity appeared to impede AECI and AECII
proliferation. Compared with the numbers observed in the LPS group,
lung sections from mice treated with LPS + VP had markedly lower
numbers of AQP-5/Ki-67 or PDPN/Ki-67 double-positive proliferating
AECI cells, and Sp-c/Ki-67 positive proliferating AECII cells
associated with a reduced number of Sp-c/YAP double-positive cells
at 48 and 72 h post-LPS injection (Fig. 7E).
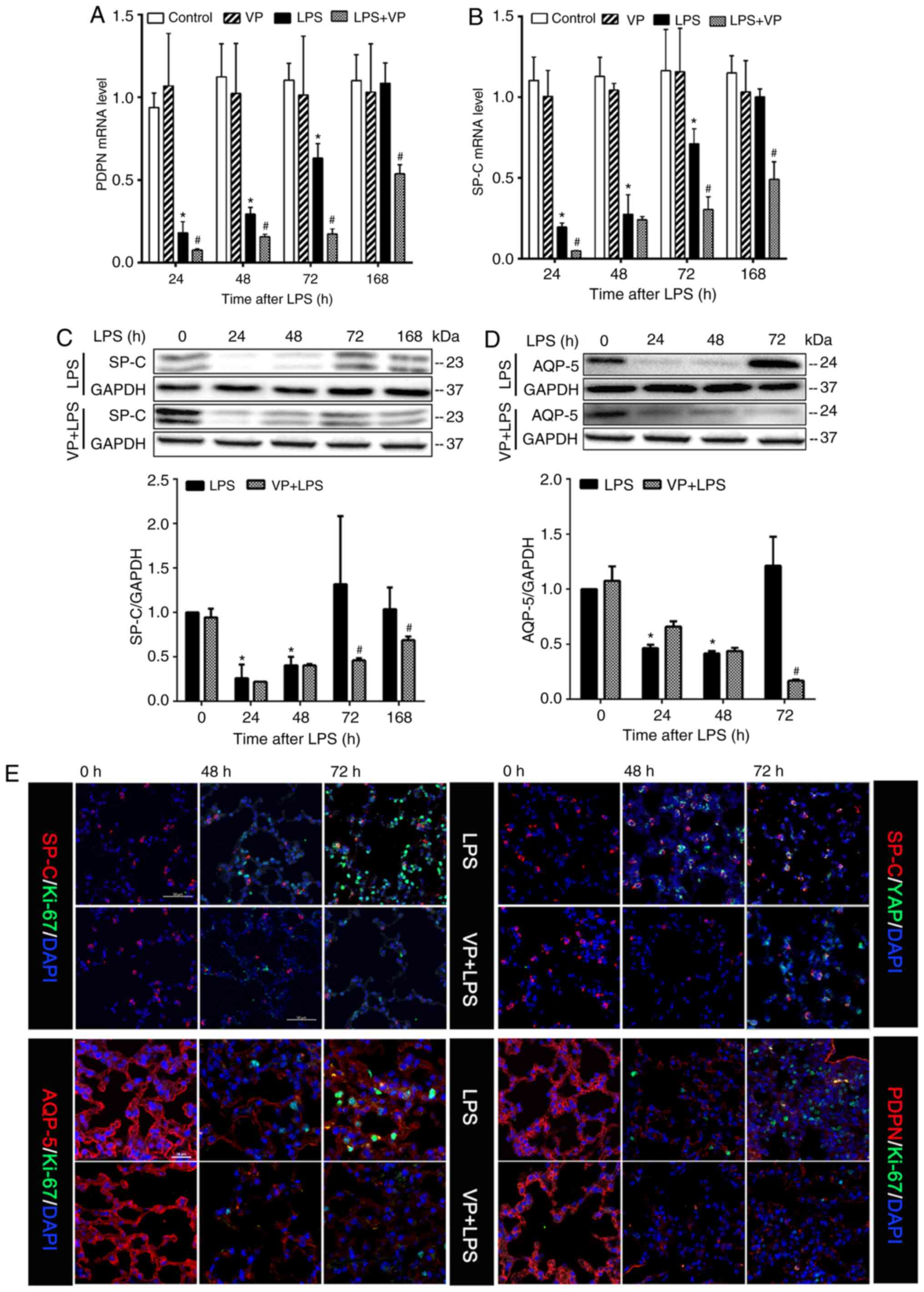 | Figure 7Inhibition of YAP activity suppresses
epithelial cell regeneration. Mice were intraperitoneally injected
with DMSO (solvent for VP), VP (100 mg/kg), LPS or LPS + VP. The
effects of inhibiting YAP activity on lung epithelial damage and
regeneration were estimated by measuring tissue levels of alveolar
type I epithelial cell-specific markers, PDPN and AQP-5, and
alveolar type II epithelial cell-specific marker, Sp-c, and by
immunofluorescence staining of lung sections. Analysis of tissue
expression levels of (A) PDPN and (B) Sp-c mRNA, and (C) AQP-5 and
(D) Sp-c protein. Data are presented as the mean ± SEM (n=3
mice/group; 2 experimental repeats/animal). *P<0.05
vs. the control group; and #P<0.05 vs. the LPS alone
group. (E) Immunofluorescence staining of lung sections. PDPN,
podoplanin; AQP-5, aquaporin-5; Sp-c, pulmonary surfactant
apoprotein C; LPS, lipopolysaccharide; VP, verteporfin; YAP,
Yes-associated protein. |
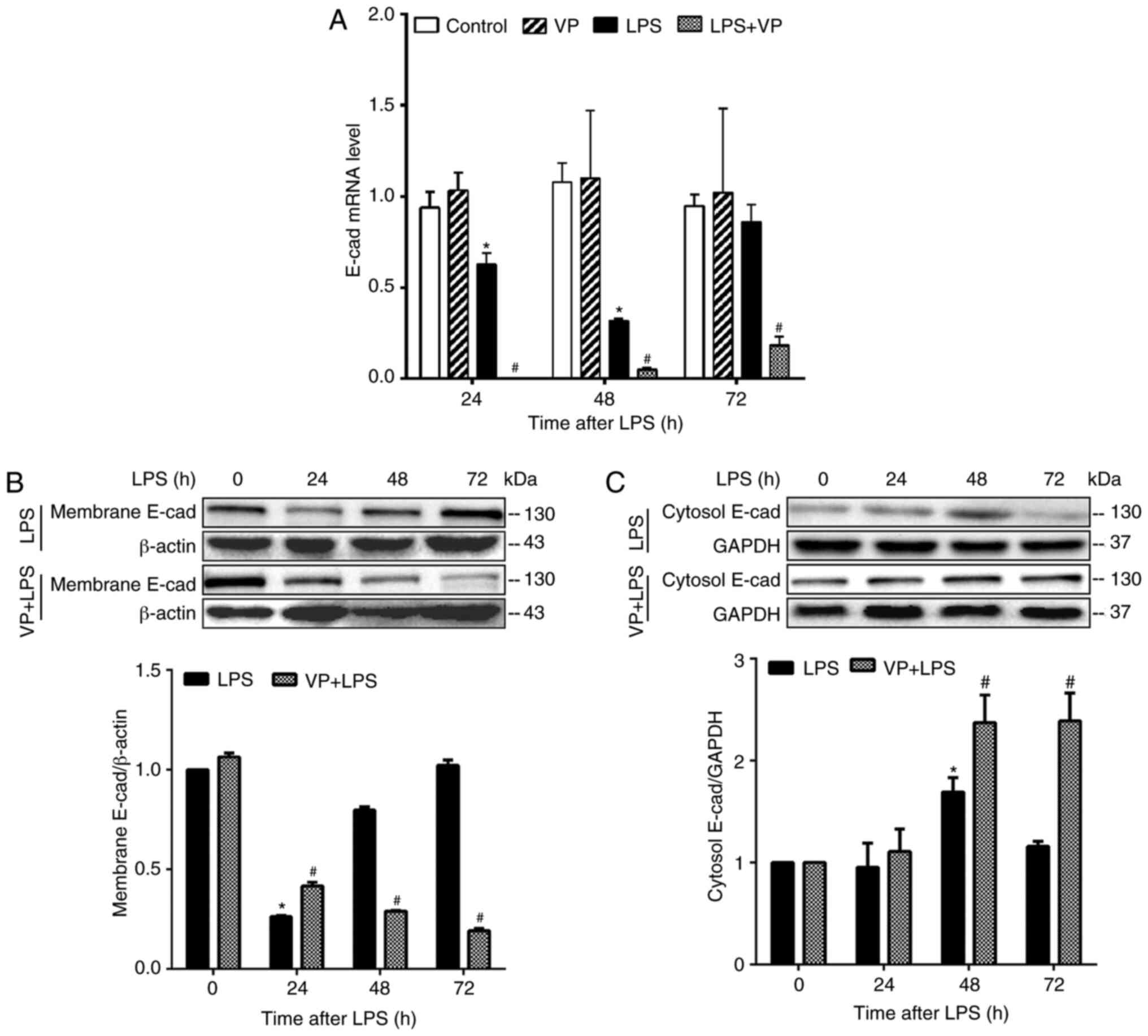
YAP regulates epithelial adherens
junction complex formation
The increase in epithelial permeability is a
hall-mark of lung injury (3,34-36). To clarify whether YAP activity
regulated epithelial adherens junction assembly and disassembly,
the present study examined the effects of inhibiting YAP activity
on the expression and intracellular translocation of membrane-bound
E-cadherin, two mechanisms that regulate the assembly and
disassembly of the epithelial adherens junction complex (34,35,37). In the LPS group, E-cadherin mRNA
expression level was significantly downregulated in the lungs at
the injury phase compared with the control group, and returned to
the control level in the lungs at the repair phase (Fig. 8A). Inhibition of YAP activity with
VP exacerbated LPS-induced downregulation of E-cadherin expression
at the injury phase and inhibited E-cadherin expression at the
repair phase (Fig. 8A). In the
LPS group, E-cadherin protein content was reduced in the membrane
fraction and increased in the cytoplasmic fraction from the same
lungs at the injury phase (Fig. 8B
and C). This shift indicates an increased subcellular
translocation of E-cadherin protein from the membrane to the
cytoplasmic compartment in the lungs at the injury phase. Compared
with control lungs, E-cadherin protein content was not altered by
LPS in both membrane and cytoplasm fractions in the lungs at the
repair phase (Fig. 8B and C),
indicating that there was little E-cadherin internalization at this
phase. Inhibition of YAP activity by VP at 48 h post LPS injection
augmented LPS-induced E-cadherin protein decrease in the membrane
fraction and resulted in an increase in the cytoplasmic fraction
compared with the LPS group (Fig. 8B
and C). Inhibition of YAP activity significantly decreased
membrane E-cadherin levels and increased cytoplasmic E-cadherin
levels in the lungs at 72 h post-LPS injection (Fig. 8B and C). These results indicated
that YAP inhibition potentiated LPS-induced E-cadherin
internalization at the injury phase and stimulated E-cadherin
internalization at the repair phase.
Discussion
This present study examined the roles of the
Hippo-YAP signaling pathway in lung injury, repair and
inflammation. The results demonstrated that lung injury was
associated with an increase in YAP phosphorylation (reduced YAP
activity) and lung repair was associated with a decrease in YAP
phosphorylation (increased YAP activity). Suppression of YAP
activity by inhibiting the formation of the YAP-TEAD complex in the
nucleus exacerbated lung inflammation, augmented the increase in
epithelial permeability and worsened lung injury. Inhibition of YAP
activity delayed lung inflammation resolution and recovery from
injury and reduced the survival rate of mice. Stimulation of YAP
activity by inhibiting Mst1/2 attenuated lung inflammation and
injury, and reduced epithelial permeability. Stimulation of YAP
activity also promoted lung inflammation resolution, accelerated
lung recovery from injury and increased the survival rate of mice.
The Hippo-YAP activity mediated cell proliferation, stimulated
epithelial cell regeneration and the formation of epithelial
adherens junctions, and improved lung repair. Collectively, the
present study demonstrated that the YAP activity was an endogenous
mechanism that protected against endotoxemic lung injury.
The Hippo-YAP activity appeared to protect against
endotoxemic lung injury by different mechanisms at different
pathological stages. Lung injury was associated with a marked
increase in lung inflammation and disassembly of epithelial
adherens junctions. However, it was not associated with cell
proliferation, epithelial cell regeneration or inflammation
resolution. Inhibition of YAP activity at the injury phase
exacerbated, whereas stimulation of YAP activity alleviated lung
injury, reduced lung inflammation and restored the disassembled
lung epithelial adherens junctions. Inhibition or stimulation of
YAP activity at the injury phase had no effect on cell
proliferation, epithelial cell regeneration or inflammation
resolution. It is likely that Hippo-YAP activity at the injury
phase protects against lung injury by inhibiting lung inflammation
and preventing epithelial adherens junction disassembly. By
contrast, lung repair was associated with a low level of lung
inflammation, as well as increased cell proliferation, epithelial
regeneration, inflammation resolution and reassembly of epithelial
adherens junctions. Stimulation of YAP activity at the repair phase
attenuated lung injury and inflammation, and accelerated
inflammation resolution and lung recovery from injury. Inhibition
of YAP activity at the repair phase exacerbated lung injury and
inflammation, inhibited cell proliferation and epithelial
regeneration, delayed inflammation resolution and lung recovery
from injury, and impeded reassembly of epithelial adherens
junctions. It is likely that at the repair phase, YAP activity
protects against lung injury by stimulating cell proliferation and
epithelial cell regeneration, promoting epithelial adherens
junction reassembly, and accelerating lung inflammation resolution
and lung recovery from injury. Thus, YAP activity may protect
against lung injury by multiple mechanisms. It inhibited
inflammation at the injury phase and promoted lung repair and
inflammation resolution at the repair phase.
The present study demonstrated that suppression of
YAP activity downregulated E-cadherin protein expression and
stimulated its translocation from the membrane to the cytoplasm,
suggesting that YAP activity upregulated E-cadherin expression and
inhibited its internalization. E-cadherin internalization and
downregulation are important mechanisms of epithelial adherens
junction disassembly during lung injury, whereas translocation of
E-cadherin from the cytoplasm to the membrane and its upregulation
are major mechanisms of epithelial adherens junction reassembly
during lung repair (35,36,47,48). The results of the present study
thus imply that YAP activity promoted epithelial adherens complex
assembly and adherens junction formation, and thereby epithelial
barrier repair. Collectively, YAP activity mediated both epithelial
proliferation (new cell generation) and epithelial cell-cell
junction formation, two key steps in epithelial barrier repair and
restoration (35,36,38,47,48). YAP/TAZ activity has been shown to
stimulate endothelial proliferation and increase VE-cadherin
turnover at inter-endothelial junctions to maintain endothelial
junction stability and barrier function (49,50). Thus, it may be hypothesized that
the Hippo-YAP signaling pathway simultaneously promotes new
epithelial/endothelial cell generation and stimulates cell-cell
junction formation and stability. The Hippo-YAP signaling pathway
might be playing a balancing role in regulating
epithelial/endothelial barrier repair following inflammatory lung
injury.
The findings of the present study may have
therapeutic implications. The steps involved in the
epithelial/endothelial barrier repair process remain to be
elucidated, but are likely to involve three phases (34,35,37,38). First, the generation of new
epithelial or endothelial cells (EpiCs or ECs) through the division
of living EpiCs or ECs adjacent to damaged or dead cells, or
through differentiation of other epithelial or endothelial
precursor cells. Second, integration of the newly generated EpiCs
or ECs into the epithelial or endothelial layer to replace the
damaged or dead EpiCs or ECs. Third, formation of stable cell-cell
junctions between the new EpiCs or ECs and the neighboring EpiCs or
ECs in the epithelium or endothelium to restore normal epithelial
or endothelial barrier function (34,35,37,38). Formation of intercellular
junctions between the new EpiCs or ECs and neighboring EpiCs or ECs
is crucial for epithelial or endothelial barrier repair, although
cell division/proliferation to generate new EpiCs or ECs is also
necessary (34,35,37,38). To facilitate epithelial or
endothelial barrier repair, cell division/proliferation and
intercellular junction formation must be regulated in a coordinated
manner. Numerous pathways have been shown to stimulate cell
division/proliferation (34,35). However, few of them also stimulate
the formation of intercellular junctions. Mechanisms and pathways
that stimulate EpiC or EC division/proliferation, but do not
stimulate the formation of intercellular junctions would impede
rather than promote epithelial or endothelial barrier repair.
Mitotic cell rounding and cytokinesis during cell division cause
extensive rear-rangement and reconstruction of epithelial or
endothelial adherens and tight junctions (51). This creates new leaky sites in the
epithelial or endothelial layer by opening new paracellular
pathways. The more EpiCs or ECs undergo cell division, the more new
leaky sites are created. This impedes rather than promotes
epithelial or endothelial barrier repair. Thus, the identification
of mechanisms and pathways that promote both cell proliferation and
cell-cell junction formation is valuable for lung regeneration. The
results of the present study indicated that the Hippo-YAP signaling
pathway stimulated EpiC proliferation and epithelial adherens
junction complex formation suggesting that stimulating YAP activity
could be a useful strategy to promote epithelial barrier
repair.
Previous studies demonstrated that selective
blockade of endothelial or type II epithelial YAP activity
exacerbates endothelial and lung inflammation (52) or impedes lung inflammation
resolution (32). The current
findings that the Hippo-YAP signaling pathway inhibits inflammation
and promotes inflammation resolution in the lungs are consistent
with these reports. The results of the present study are also in
line with a previous study which reported that epicardial YAP/TAZ
regulated an immunosuppressive response following myocardial
infarction (53). However, the
current findings are inconsistent with another study, in which it
was demonstrated that endothelial-specific YAP overexpression
exacerbates, whereas selective YAP knockdown in the endothelium
inhibits vascular wall inflammation (54). To the best of our knowledge, the
present report is the first to examine the effects of systemic
stimulation and inhibition of YAP activity on lung inflammation,
injury and repair. The present study extrapolates those prior
studies by demonstrating that YAP activity protects against lung
injury by activating different mechanisms at different pathological
stages and that it stimulates both EpiC proliferation and
epithelial adherens junction formation.
In conclusion, the results of the present study
demonstrated that YAP activity is an endogenous protective
mechanism against endotoxemic lung injury. YAP activity appears to
protect against lung injury by activating different mechanisms at
different pathological stages. At the injury phase, YAP activity
protected against lung injury by inhibiting lung inflammation and
preventing epithelial adherens junction disassembly. At the repair
phase, it promoted lung recovery by stimulating cell proliferation
and epithelial cell regeneration, promoting epithelial adherens
junction reassembly, and accelerating lung inflammation resolution.
Thus, YAP activity protects against lung injury by multiple
mechanisms. Stimulation of EpiC proliferation and epithelial
adherens junction formation suggests that stimulating YAP activity
could be a useful strategy to promote epithelial barrier
repair.
Although the present study demonstrated that YAP
activity protected against lung injury by acting at injury and
repair phases and by activating multiple mechanisms, the
therapeutic effects of stimulating YAP activity on lung injury were
not examined, and will be the focus of a future study. Mice will be
injected with VP and LPS or XMU-MP-1 and LPS simultaneously, or
administered VP or XMU-MP-1 at 6, 12 or 24 h after LPS injection.
The effects of these interventions on LPS-induced lung inflammation
and injury, on lung repair and inflammation resolution, and on
overall outcomes will be assessed. These studies will allow for
evaluation of the therapeutic value of stimulating YAP activity for
the treatment of ALI.
Acknowledgments
Not applicable.
Funding
This work was supported by the Natural Science
Foundation of Zhejiang Province of China (grant nos. LY18H010007
and LY17H010007) and the Natural Science Foundation of China (grant
no. 81370171).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
YSG, SZM and SFL contributed to the study conception
and design, supervision of all experiments, and acquisition of
funding. LYL, XQS, FKZ, XFF, YYW and JMF contributed to the study
design, and participated in data acquisition, analysis and
interpretation. All authors have read and approved the final
manuscript.
Ethics approval and consent to
participate
All animal experiments were approved by the
Institutional Animal Care and Use Committee of Wenzhou Medical
University (Wenzhou, China) and were performed in accordance with
the Guide for the Care and Use of Laboratory Animals of the
National Institute of Health.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Matthay MA and Zimmerman GA: Acute lung
injury and the acute respiratory distress syndrome: Four decades of
inquiry into pathogenesis and rational management. Am J Respir Cell
Mol Biol. 33:319–327. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matthay MA, Ware LB and Zimmerman GA: The
acute respiratory distress syndrome. J Clin Invest. 122:2731–2740.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Li G, Malinchoc M, Cartin-Ceba R, Venkata
CV, Kor DJ, Peters SG, Hubmayr RD and Gajic O: Eight-year trend of
acute respiratory distress syndrome: A population-based study in
Olmsted County, Minnesota. Am J Respir Crit Care Med. 183:59–66.
2011. View Article : Google Scholar :
|
|
5
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tsushima K, King LS, Aggarwal NR, De
Gorodo A, D'Alessio FR and Kubo K: Acute lung injury review. Intern
Med. 48:621–630. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Moya IM and Halder G: Hippo-YAP/TAZ
signalling in organ regeneration and regenerative medicine. Nat Rev
Mol Cell Biol. 20:211–226. 2019. View Article : Google Scholar
|
|
8
|
Zhao B, Tumaneng K and Guan KL: The Hippo
pathway in organ size control, tissue regeneration and stem cell
self-renewal. Nat Cell Biol. 13:877–883. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Dong J, Feldmann G, Huang J, Wu S, Zhang
N, Comerford SA, Gayyed MF, Anders RA, Maitra A and Pan D:
Elucidation of a universal size-control mechanism in Drosophila and
mammals. Cell. 130:1120–1133. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xie H, Wu L, Deng Z, Huo Y and Cheng Y:
Emerging roles of YAP/TAZ in lung physiology and diseases. Life
Sci. 214:176–183. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xin M, Kim Y, Sutherland LB, Murakami M,
Qi X, McAnally J, Porrello ER, Mahmoud AI, Tan W, Shelton JM, et
al: Hippo pathway effector Yap promotes cardiac regeneration. Proc
Natl Acad Sci USA. 110:13839–13844. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wu S, Liu Y, Zheng Y, Dong J and Pan D:
The TEAD/TEF family protein Scalloped mediates transcriptional
output of the Hippo growth-regulatory pathway. Dev Cell.
14:388–398. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang X, Freire Valls A, Schermann G, Shen
Y, Moya IM, Castro L, Urban S, Solecki GM, Winkler F, Riedemann L,
et al: YAP/TAZ Orchestrate VEGF signaling during developmental
angiogenesis. Dev Cell. 42:462–478.e7. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Elaimy AL and Mercurio AM: Convergence of
VEGF and YAP/TAZ signaling: Implications for angiogenesis and
cancer biology. Sci Signal. 11:eaau11652018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Piccolo S, Dupont S and Cordenonsi M: The
biology of YAP/TAZ: Hippo signaling and beyond. Physiol Rev.
94:1287–1312. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gumbiner BM and Kim NG: The Hippo-YAP
signaling pathway and contact inhibition of growth. J Cell Sci.
127:709–717. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Saito A and Nagase T: Hippo and TGF-β
interplay in the lung field. Am J Physiol Lung Cell Mol Physiol.
309:L756–L767. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou B, Flodby P, Luo J, Castillo DR, Liu
Y, Yu FX, McConnell A, Varghese B, Li G, Chimge NO, et al:
Claudin-18-mediated YAP activity regulates lung stem and progenitor
cell homeostasis and tumorigenesis. J Clin Invest. 128:970–984.
2018. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lange AW, Sridharan A, Xu Y, Stripp BR,
Perl AK and Whitsett JA: Hippo/Yap signaling controls epithelial
progenitor cell proliferation and differentiation in the embryonic
and adult lung. J Mol Cell Biol. 7:35–47. 2015. View Article : Google Scholar :
|
|
20
|
Volckaert T, Yuan T, Yuan J, Boateng E,
Hopkins S, Zhang JS, Thannickal VJ, Fässler R and De Langhe SP:
Hippo signaling promotes lung epithelial lineage commitment by
curbing Fgf10 and β-catenin signaling. Development.
146:dev1664542019. View Article : Google Scholar
|
|
21
|
Zhao R, Fallon TR, Saladi SV,
Pardo-Saganta A, Villoria J, Mou H, Vinarsky V, Gonzalez-Celeiro M,
Nunna N, Hariri LP, et al: Yap tunes airway epithelial size and
architecture by regulating the identity, maintenance, and
self-renewal of stem cells. Dev Cell. 30:151–165. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Azad T, Ghahremani M and Yang X: The role
of YAP and TAZ in angiogenesis and vascular mimicry. Cells.
8:4072019. View Article : Google Scholar :
|
|
23
|
Choi HJ, Zhang H, Park H, Choi KS, Lee HW,
Agrawal V, Kim YM and Kwon YG: Yes-associated protein regulates
endothelial cell contact-mediated expression of angiopoietin-2. Nat
Commun. 6:69432015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sharif GM, Schmidt MO, Yi C, Hu Z, Haddad
BR, Glasgow E, Riegel AT and Wellstein A: Cell growth density
modulates cancer cell vascular invasion via Hippo pathway activity
and CXCR2 signaling. Oncogene. 34:5879–5889. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhao B, Li L, Wang L, Wang CY, Yu J and
Guan KL: Cell detachment activates the Hippo pathway via
cytoskeleton reorganization to induce anoikis. Genes Dev. 26:54–68.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu F, Lagares D, Choi KM, Stopfer L,
Marinković A, Vrbanac V, Probst CK, Hiemer SE, Sisson TH, Horowitz
JC, et al: Mechanosignaling through YAP and TAZ drives fibroblast
activation and fibrosis. Am J Physiol Lung Cell Mol Physiol.
308:L344–L357. 2015. View Article : Google Scholar :
|
|
27
|
Chung C, Kim T, Kim M, Kim M, Song H, Kim
TS, Seo E, Lee SH, Kim H, Kim SK, et al: Hippo-Foxa2 signaling
pathway plays a role in peripheral lung maturation and surfactant
homeostasis. Proc Natl Acad Sci USA. 110:7732–7737. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Mindos T, Dun XP, North K, Doddrell RD,
Schulz A, Edwards P, Russell J, Gray B, Roberts SL, Shivane A, et
al: Merlin controls the repair capacity of Schwann cells after
injury by regulating Hippo/YAP activity. J Cell Biol. 216:495–510.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu Y, Lu T, Zhang C, Xu J, Xue Z,
Busuttil RW, Xu N, Xia Q, Kupiec-Weglinski JW and Ji H: Activation
of YAP attenuates hepatic damage and fibrosis in liver
ischemia-reperfusion injury. J Hepatol. 71:719–730. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li L, Dong L, Zhang J, Gao F, Hui J and
Yan J: Mesenchymal stem cells with downregulated Hippo signaling
attenuate lung injury in mice with lipopolysaccharide-induced acute
respiratory distress syndrome. Int J Mol Med. 43:1241–1252.
2019.PubMed/NCBI
|
|
31
|
Volckaert T, Yuan T, Chao CM, Bell H,
Sitaula A, Szimmtenings L, El Agha E, Chanda D, Majka S, Bellusci
S, et al: Fgf10-Hippo epithelial-mesenchymal crosstalk maintains
and recruits lung basal stem cells. Dev Cell. 43:48–59.e5. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
LaCanna R, Liccardo D, Zhang P, Tragesser
L, Wang Y, Cao T, Chapman HA, Morrisey EE, Shen H, Koch WJ, et al:
Yap/Taz regulate alveolar regeneration and resolution of lung
inflammation. J Clin Invest. 129:2107–2122. 2019. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hu C, Sun J, Du J, Wen D, Lu H, Zhang H,
Xue Y, Zhang A, Yang C, Zeng L and Jiang J: The Hippo-YAP pathway
regulates the proliferation of alveolar epithelial progenitors
after acute lung injury. Cell Biol Int. 43:1174–1183. 2019.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bhattacharya J and Matthay MA: Regulation
and repair of the alveolar-capillary barrier in acute lung injury.
Annu Rev Physiol. 75:593–615. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Crosby LM and Waters CM: Epithelial repair
mechanisms in the lung. Am J Physiol Lung Cell Mol Physiol.
298:L715–L731. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Frank JA: Claudins and alveolar epithelial
barrier function in the lung. Ann N Y Acad Sci. 1257:175–183. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Liu G, Ye X, Miller EJ and Liu SF:
NF-κB-to-AP-1 switch: A mechanism regulating transition from
endothelial barrier injury to repair in endotoxemic mice. Sci Rep.
4:55432014. View Article : Google Scholar
|
|
38
|
Mao SZ, Ye X, Liu G, Song D and Liu SF:
Resident endothelial cells and endothelial progenitor cells restore
endothelial barrier function after inflammatory lung injury.
Arterioscler Thromb Vasc Biol. 35:1635–1644. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
National Research Council (US) Committee
for the Update of the Guide for the Care and Use of Laboratory
Animals: Guide for the Care and Use of Laboratory Animals. 8th
edition. National Academies Press (US); Washington, DC: 2011
|
|
40
|
Fan F, He Z, Kong LL, Chen Q, Yuan Q,
Zhang S, Ye J, Liu H, Sun X, Geng J, et al: Pharmacological
targeting of kinases MST1 and MST2 augments tissue repair and
regeneration. Sci Transl Med. 8:352ra1082016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
42
|
Ye X, Ding J, Zhou X, Chen G and Liu SF:
Divergent roles of endothelial NF-kappaB in multiple organ injury
and bacterial clearance in mouse models of sepsis. J Exp Med.
205:1303–1315. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Liu SF, Ye X and Malik AB: In vivo
inhibition of nuclear factor-kappa B activation prevents inducible
nitric oxide synthase expression and systemic hypotension in a rat
model of septic shock. J Immunol. 159:3976–3983. 1997.PubMed/NCBI
|
|
44
|
Liu-Chittenden Y, Huang B, Shim JS, Chen
Q, Lee SJ, Anders RA, Liu JO and Pan D: Genetic and pharmacological
disruption of the TEAD-YAP complex suppresses the oncogenic
activity of YAP. Genes Dev. 26:1300–1305. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Williams MC: Alveolar type I cells:
Molecular phenotype and development. Annu Rev Physiol. 65:669–695.
2003. View Article : Google Scholar
|
|
46
|
Rippon HJ, Ali NN, Polak JM and Bishop AE:
Initial observations on the effect of medium composition on the
differentiation of murine embryonic stem cells to alveolar type II
cells. Cloning Stem Cells. 6:49–56. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bruser L and Bogdan S: Adherens junctions
on the move-membrane trafficking of E-cadherin. Cold Spring Harb
Perspect Biol. 9:a0291402017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Rezaee F and Georas SN: Breaking barriers.
New insights into airway epithelial barrier function in health and
disease. Am J Respir Cell Mol Biol. 50:857–869. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kim J, Kim YH, Kim J, Park DY, Bae H, Lee
DH, Kim KH, Hong SP, Jang SP, Kubota Y, et al: YAP/TAZ regulates
sprouting angiogenesis and vascular barrier maturation. J Clin
Invest. 127:3441–3461. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Neto F, Klaus-Bergmann A, Ong YT, Alt S,
Vion AC, Szymborska A, Carvalho JR, Hollfinger I, Bartels-Klein E,
Franco CA, et al: YAP and TAZ regulate adherens junction dynamics
and endothelial cell distribution during vascular development.
Elife. 7:e310372018. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Harris TJ and Tepass U: Adherens
junctions: From molecules to morphogenesis. Nat Rev Mol Cell Biol.
11:502–514. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Lv Y, Kim K, Sheng Y, Cho J, Qian Z, Zhao
YY and Hu G, Pan D, Malik AB and Hu G: YAP controls endothelial
activation and vascular inflammation through TRAF6. Circ Res.
123:43–56. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Ramjee V, Li D, Manderfield LJ, Liu F,
Engleka KA, Aghajanian H, Rodell CB, Lu W, Ho V, Wang T, et al:
Epicardial YAP/TAZ orchestrate an immunosuppressive response
following myocardial infarction. J Clin Invest. 127:899–911. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Wang L, Luo JY, Li B, Tian XY, Chen LJ,
Huang Y, Liu J, Deng D, Lau CW, Wan S, et al: Integrin-YAP/TAZ-JNK
cascade mediates atheroprotective effect of unidirectional shear
flow. Nature. 540:579–582. 2016. View Article : Google Scholar : PubMed/NCBI
|